
Synthesis and Characterization of Graft Copolymers with Poly(ε-caprolactone) Side Chain Using Hydroxylated Poly(β-myrcene-co-α-methyl styrene)


Abstract

1. Introduction
2. Results
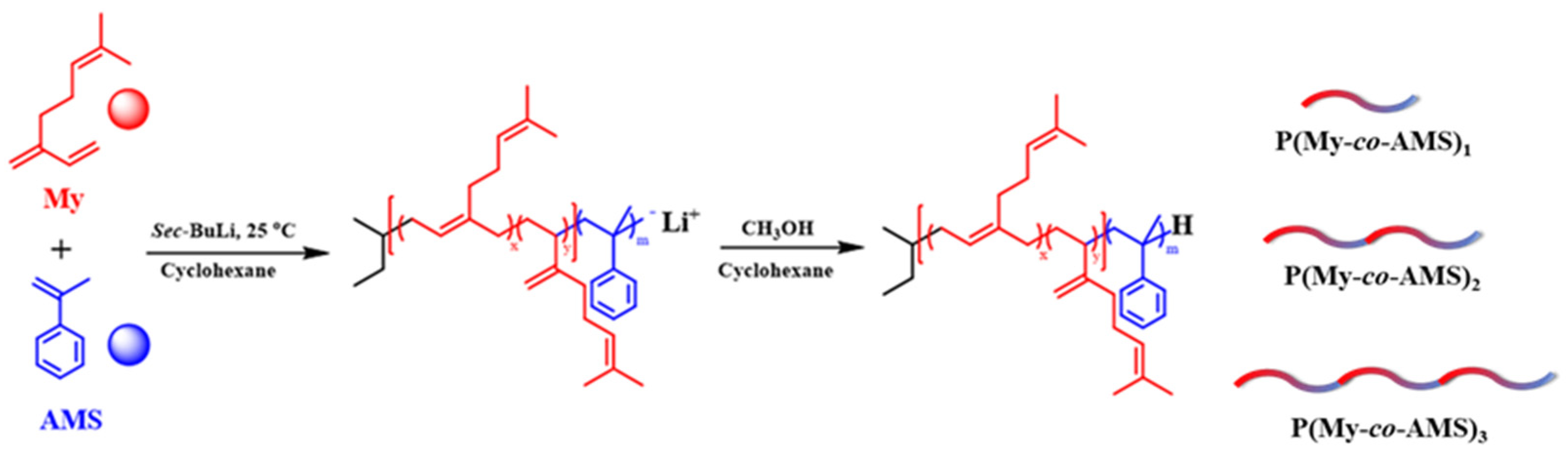
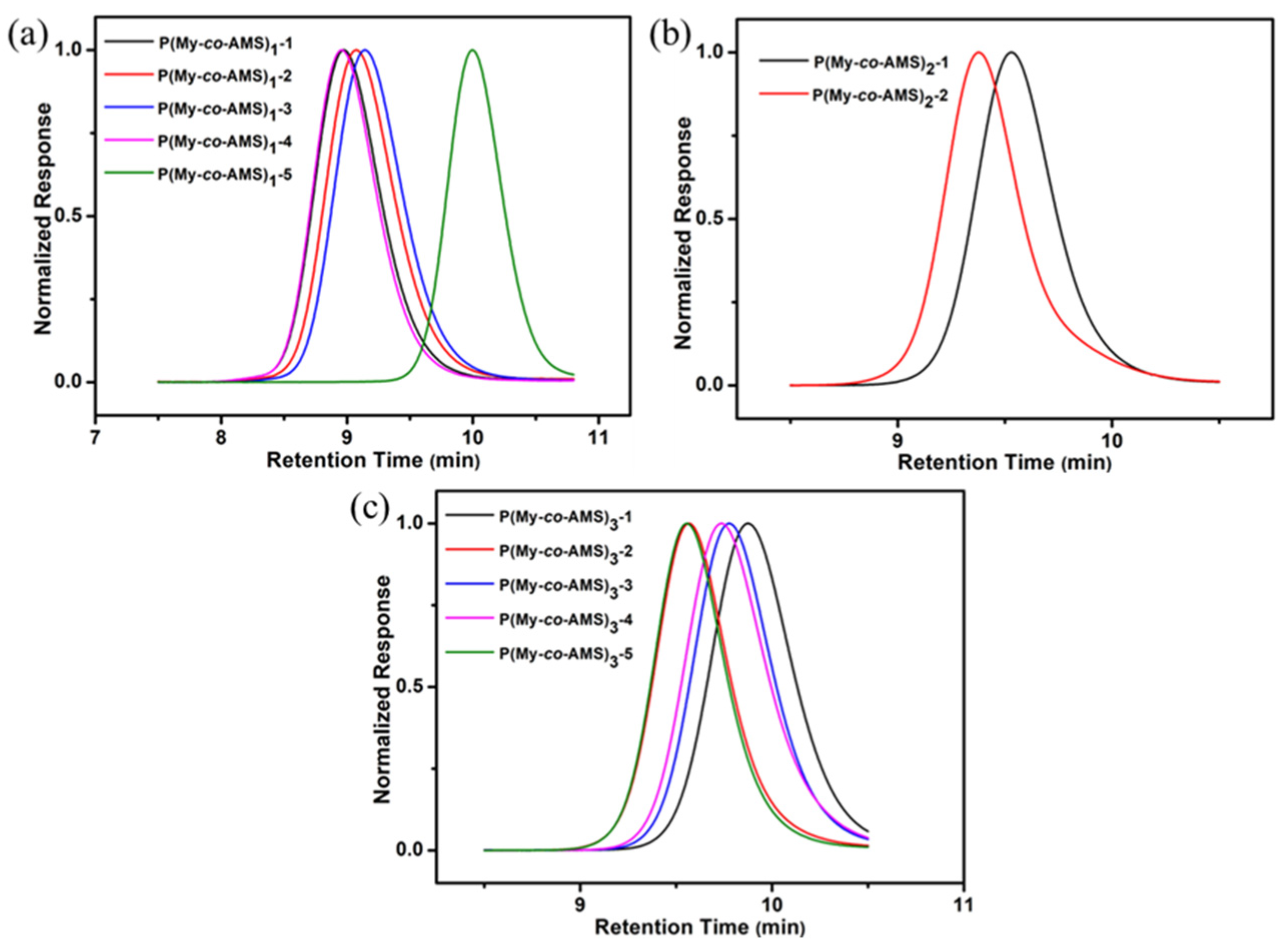
2.1. Synthesis and Characterization of P(My-co-AMS)k Copolymers
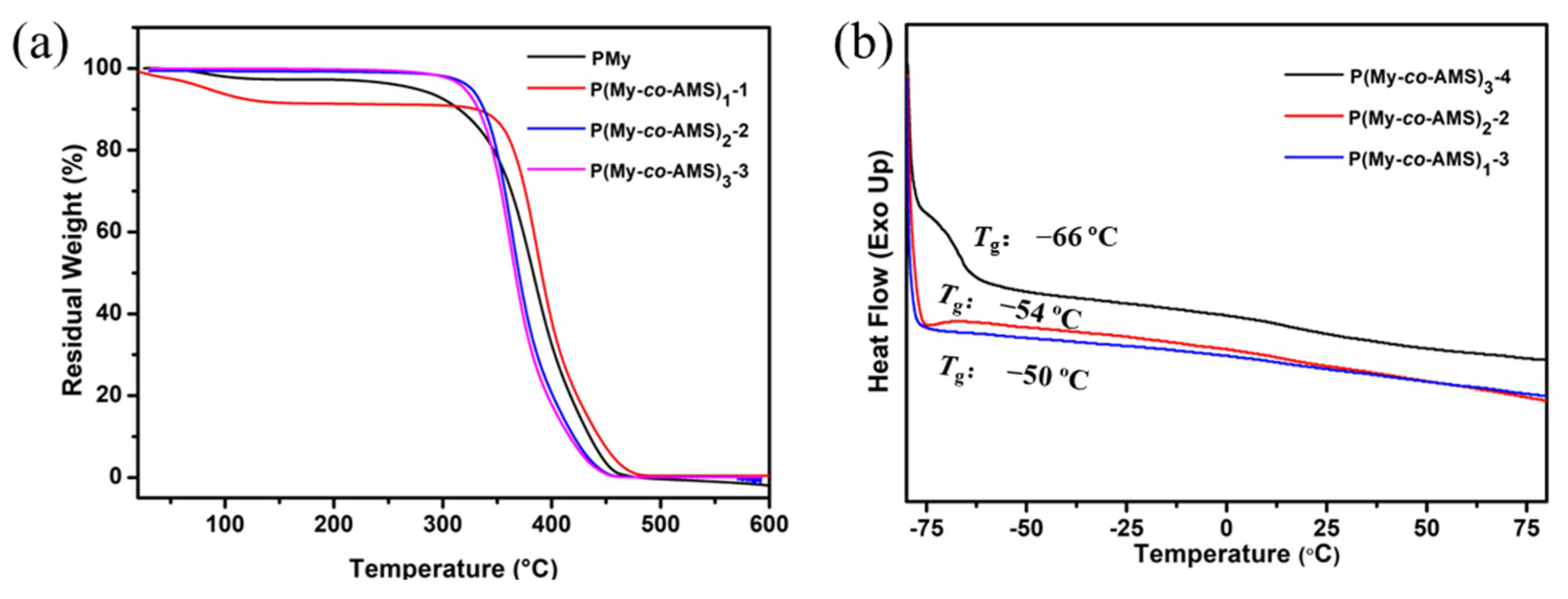
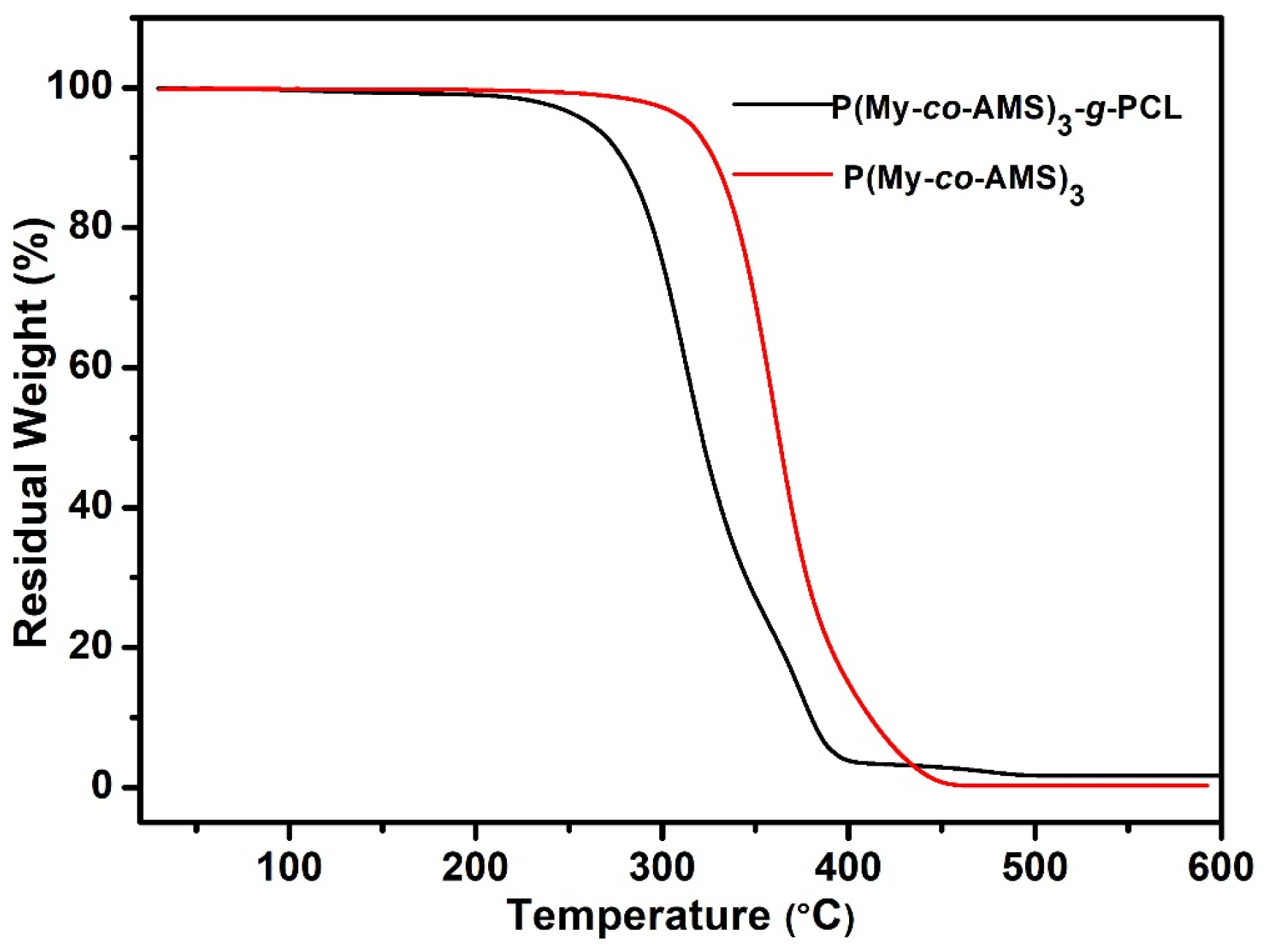
2.2. Thermal Degradation of P(My-co-AMS)k and PMy
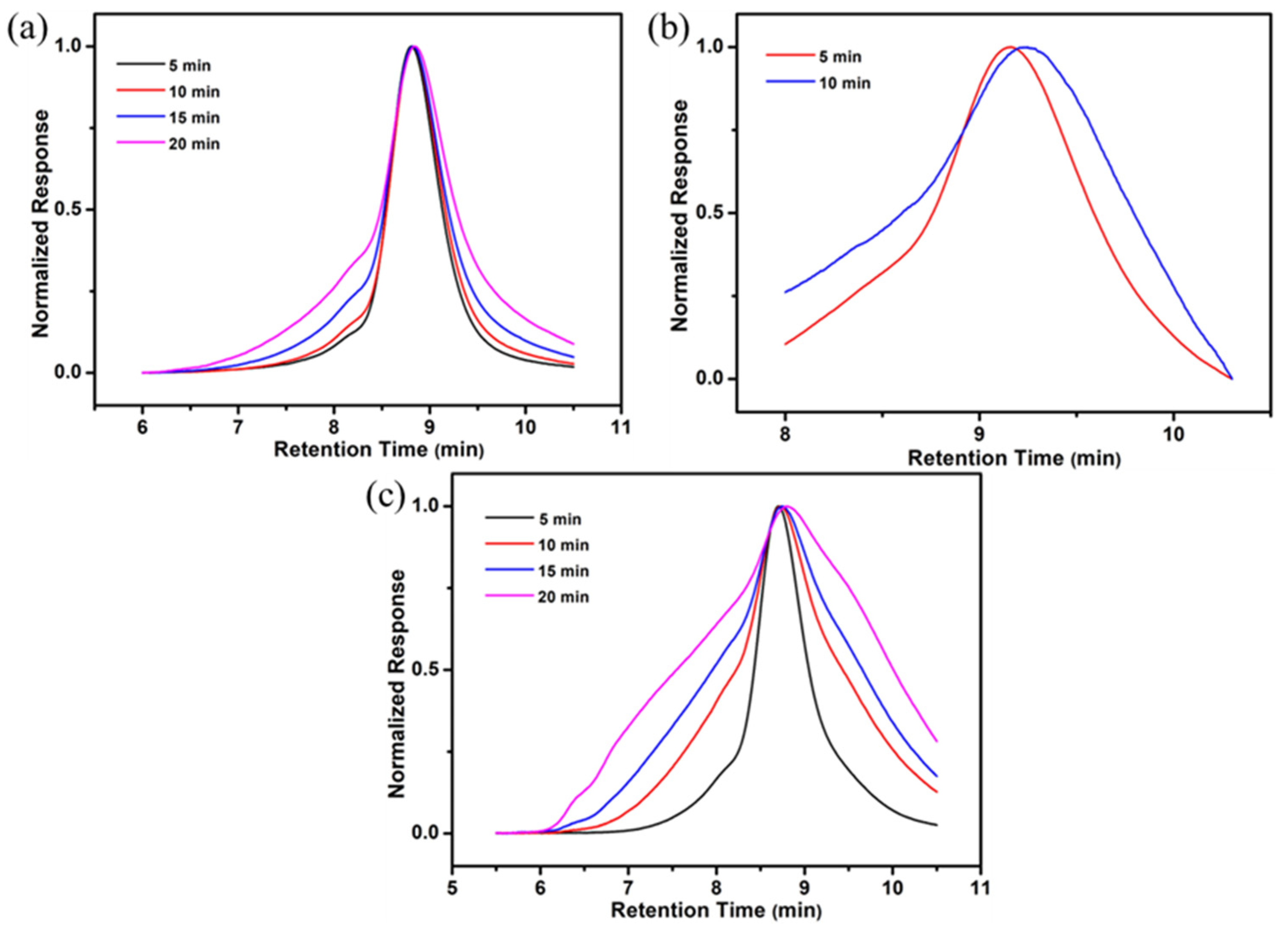
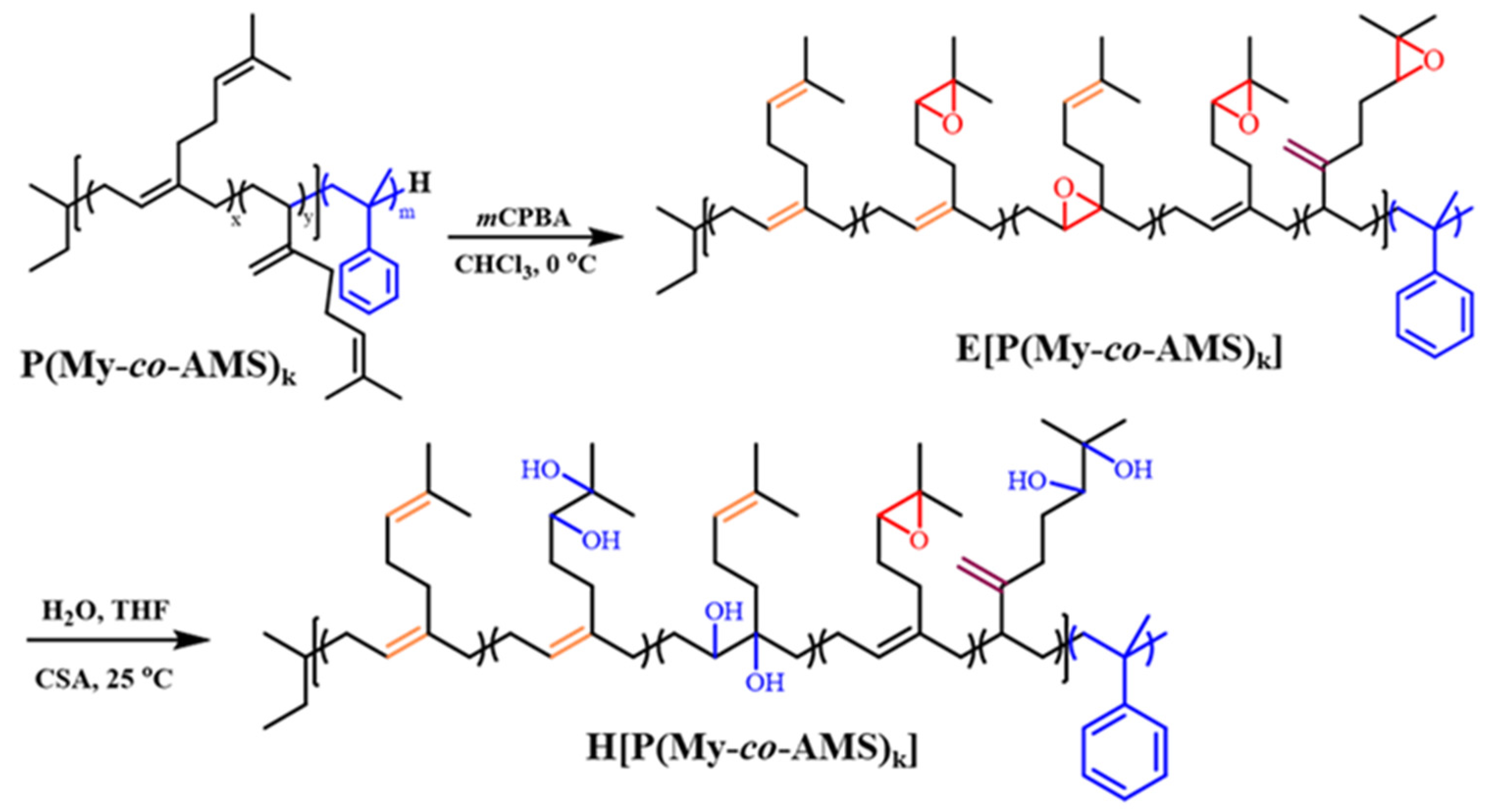
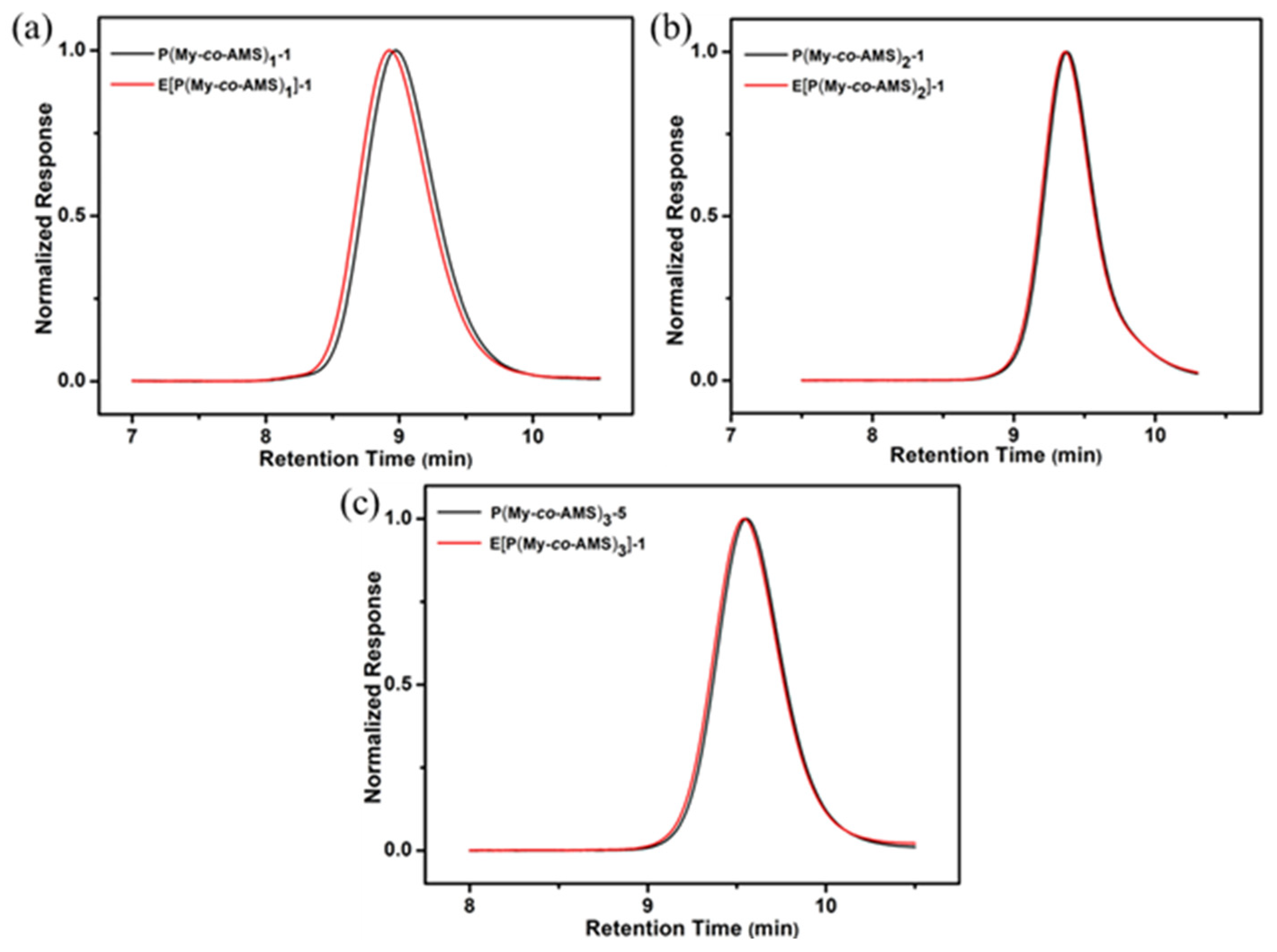
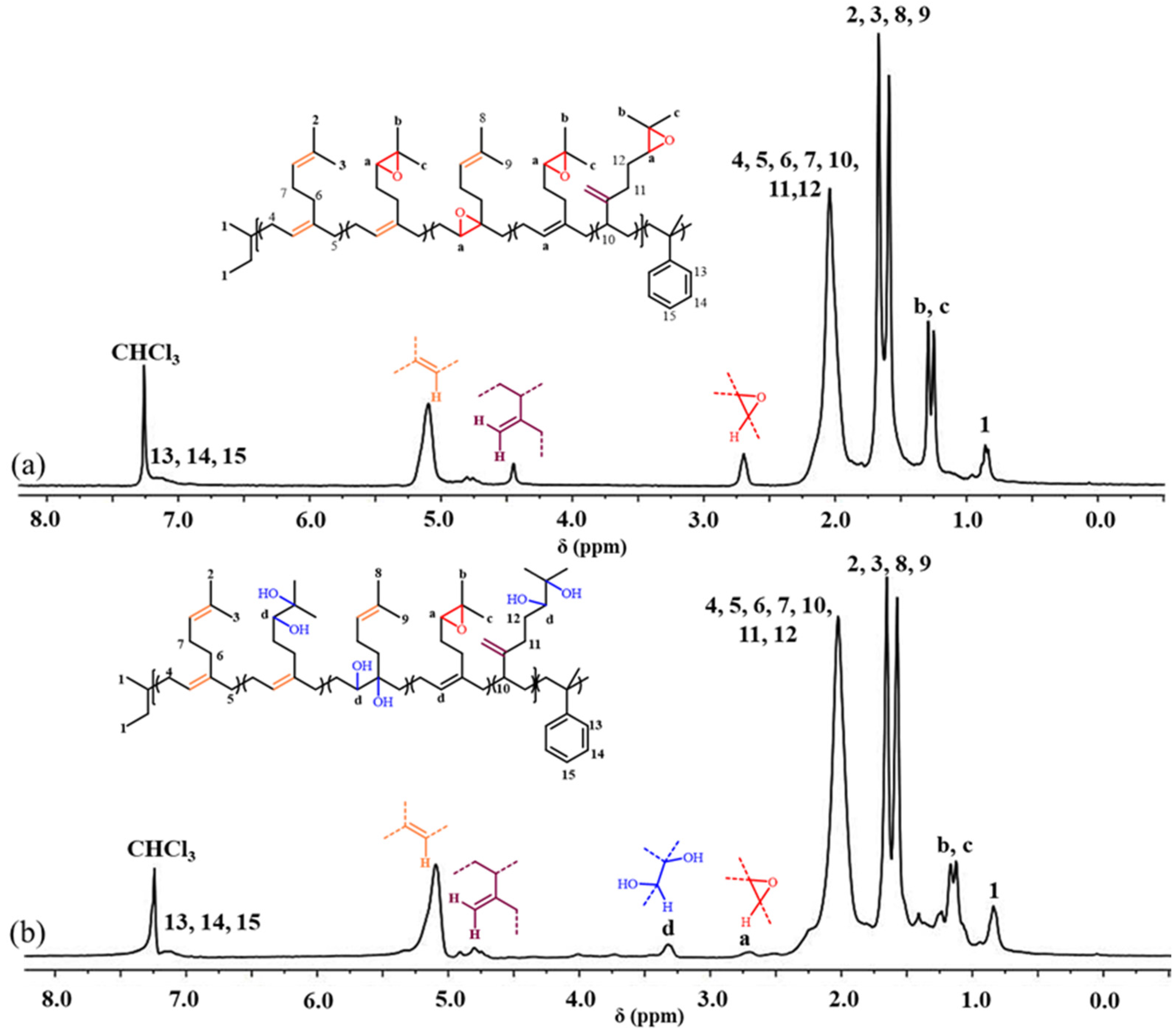
2.3. Synthesis and Characterization of E[P(My-co-AMS)k]
2.4. Synthesis and Characterization of HP(My-co-AMS)3
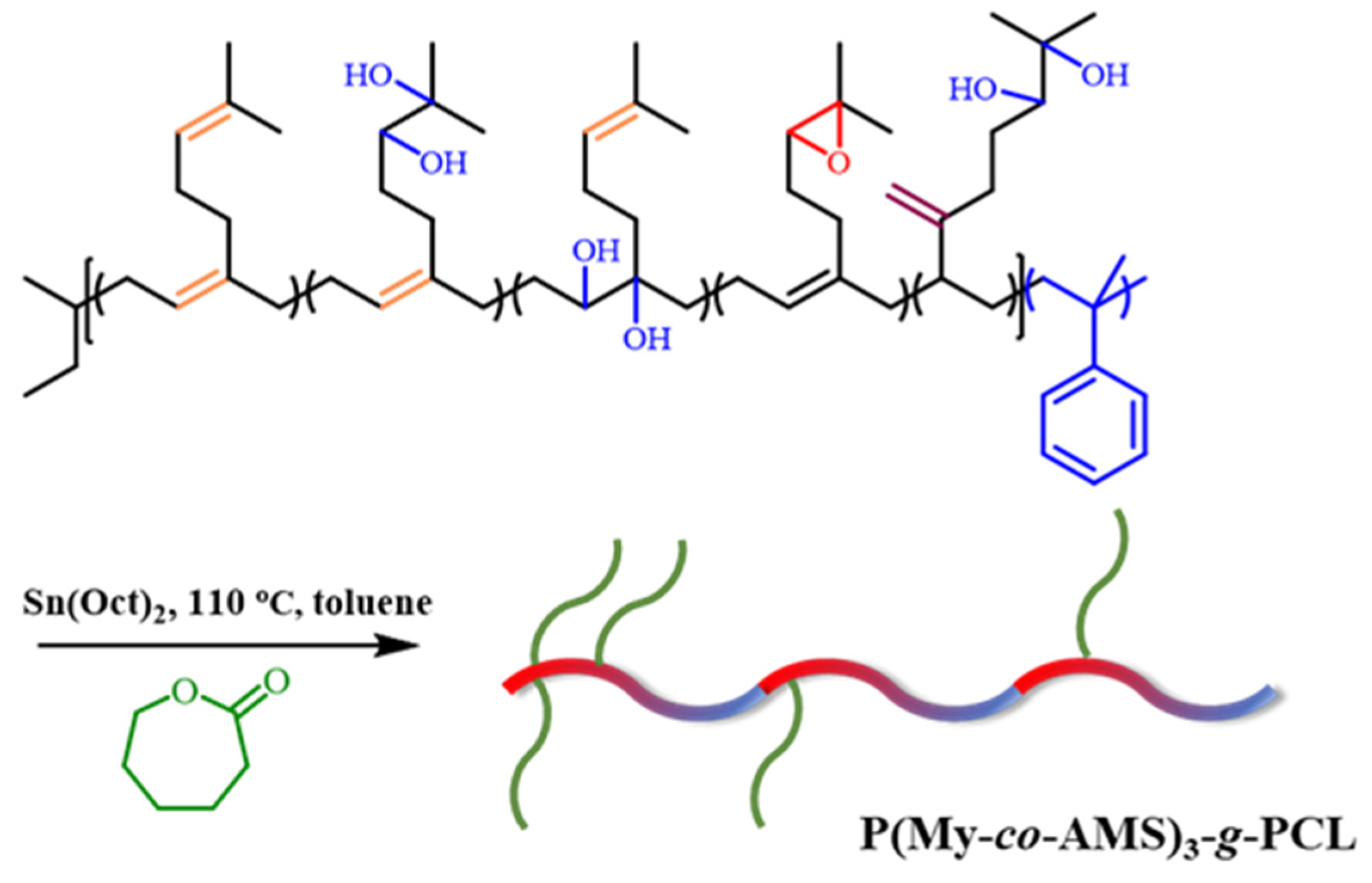
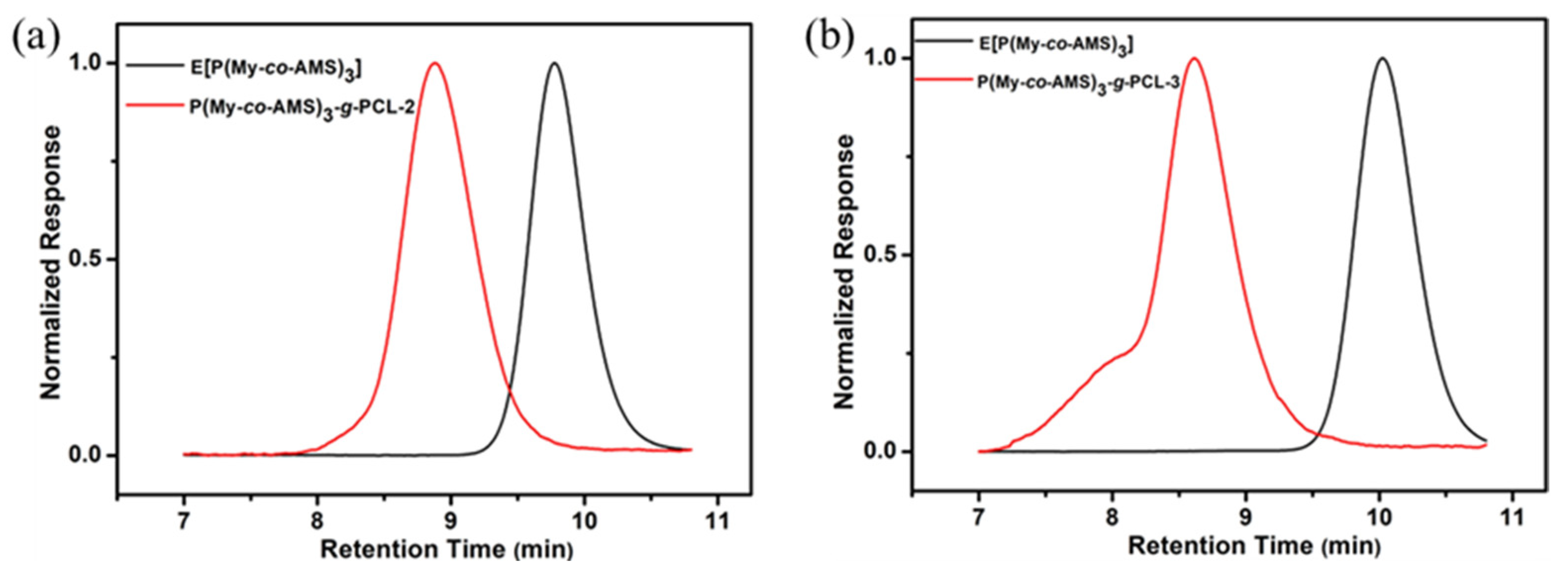
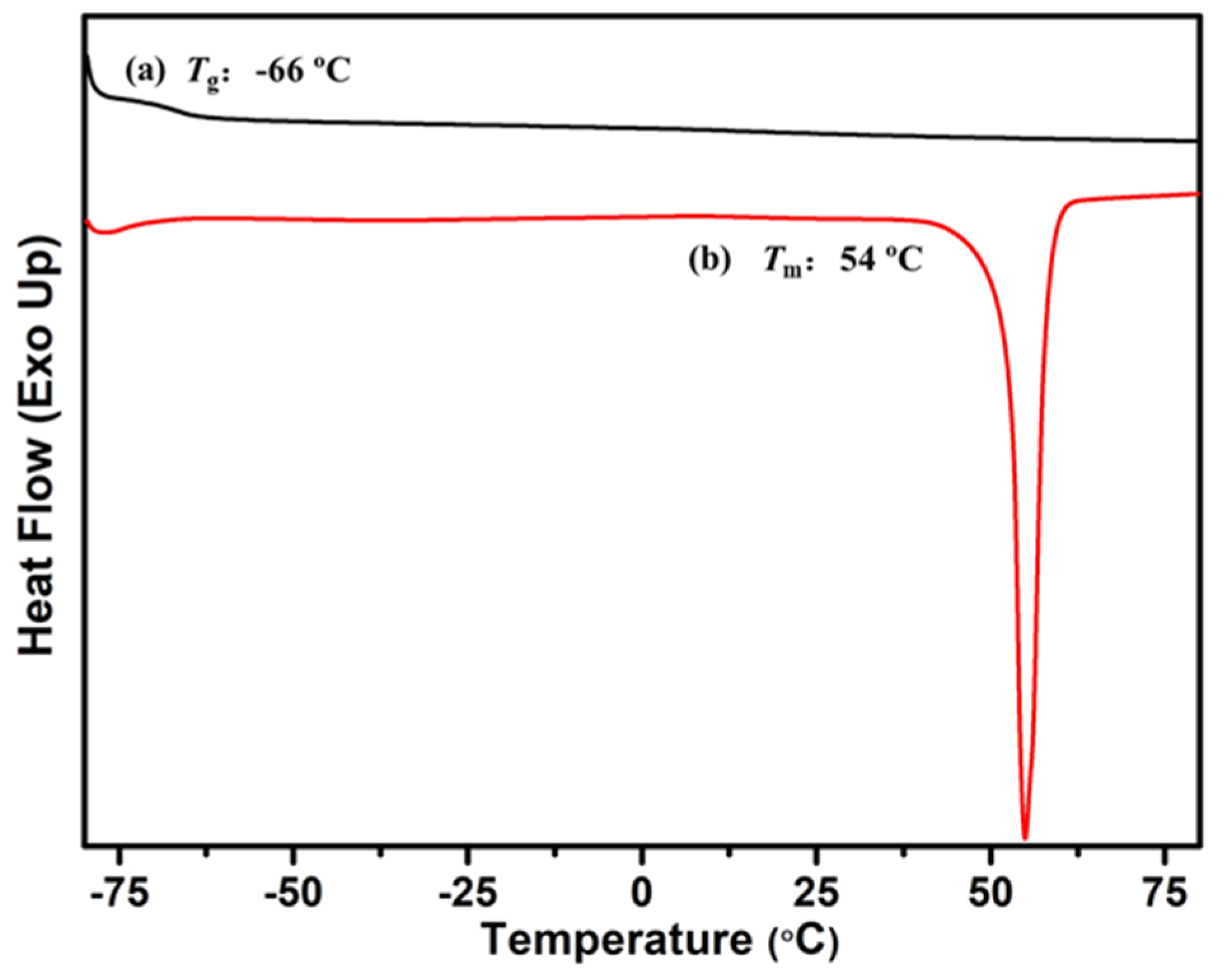
2.5. Synthesis and Characterization of P(My-co-AMS)3-g-PCL
3. Materials and Methods
3.1. Starting Materials
3.2. Synthesis of P(My-co-AMS)k Copolymers
3.3. Thermal Degradation of P(My-co-AMS)k Copolymers
3.4. Epoxidation of P(My-co-AMS)k
3.5. Hydrolysis of EP(My-co-AMS)3
3.6. Ring-Opening Polymerization of ε-CL with H[P(My-co-AMS)3]
3.7. Characterization
3.7.1. Nuclear Magnetic Resonance (NMR) Analysis
3.7.2. Gel Permeation Chromatography (GPC)
3.7.3. Thermogravimetry Analysis (TGA)
3.7.4. Differential Scanning Calorimeter (DSC)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Macromolecular Architectures by Living and Controlled/living Polymerizations. Prog. Polym. Sci. 2006, 31, 1068–1132. [Google Scholar] [CrossRef]
- Kilbinger, A.F.M. Controlled and Living Polymerizations. Angew. Chem. Int. Ed. 2010, 49, 1191–1192. [Google Scholar] [CrossRef]
- Magana, I.; Lopez, R.; Enríquez-Medrano, F.J.; Kumar, S.; Aguilar-Sanchez, A.; Handa, R.; Leon, R.D.D.; Valencia, L. Bioelastomers: Current State of Development. J. Mater. Chem. A 2022, 10, 5019–5043. [Google Scholar] [CrossRef]
- Hirao, A.; Goseki, R.; Ishizone, T. Advances in Living Anionic Polymerization: From Functional Monomers, Polymerization Systems, to Macromolecular Architectures. Macromolecules 2014, 47, 1883–1905. [Google Scholar] [CrossRef]
- Ito, S.; Goseki, R.; Ishizone, T.; Hirao, A. Successive Synthesis of Well-Defined Multiarmed Miktoarm Star Polymers by Iterative Methodology Using Living Anionic Polymerization. Eur. Polym. J. 2013, 49, 2545–2566. [Google Scholar] [CrossRef]
- Hahn, C.; Rauschenbach, M.; Frey, H. Merging Styrene and Diene Structures to a Cyclic Diene: Anionic Polymerization of 1-Vinylcyclohexene (VCH). Angew. Chem. Int. Ed. 2023, 62, e202302907. [Google Scholar] [CrossRef] [PubMed]
- Ntetsikas, K.; Ladelta, V.; Bhaumik, S.; Hadjichristidis, N. Quo Vadis Carbanionic Polymerization? ACS Polym. Au. 2023, 3, 158–181. [Google Scholar] [CrossRef]
- Patel, R.M.; Hahn, S.F.; Esneault, C.; Bensason, S. Processing and Properties of Polyolefin Elastomers and Fully Hydrogenated Styrenic Block Copolymer Elastomers. Adv. Mater. 2000, 23, 1813–1817. [Google Scholar] [CrossRef]
- Maji, P.; Naskar, K. Styrenic Block Copolymer-based Thermoplastic Elastomers in Smart Applications: Advances in Synthesis, Microstructure, and Structure-property Relationships—A Review. J. Appl. Polym. Sci. 2022, 139, e52942. [Google Scholar] [CrossRef]
- Wang, C.M.; Wu, Y.B.; Zhu, Y.H.; Ma, H.B.; Zhang, M.Z.; Liu, G.X.; He, J.L.; Ni, P.H. Investigation of Eight-arm Tapered Star Copolymers Prepared by Anionic Copolymerization and Coupling Reaction. Polym. Chem. 2022, 13, 3938–3948. [Google Scholar] [CrossRef]
- Mai, Y.Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Wang, L.Y.; Yan, Q.; Liu, F.S.; Shen, Y.; Li, Z.B. Rapid and Controlled Polymerization of Bio-sourced δ-Caprolactone toward Fully Recyclable Polyesters and Thermoplastic Elastomers. Angew. Chem. Int. Ed. 2022, 61, e202201407. [Google Scholar] [CrossRef] [PubMed]
- Murataj, I.; Channab, M.; Cara, E.; Pirri, C.F.; Boarino, L.; Angelini, A.; Lupi, F. Hyperbolic Metamaterials via Hierarchical Block Copolymer Nanostructures. Adv. Optical. Mater. 2021, 9, 2001933. [Google Scholar] [CrossRef]
- Chountoulesi, M.; Selianitis, D.; Pispas, S.; Pippa, N. Recent Advances on PEO-PCL Block and Graft Copolymers as Nanocarriers for Drug Delivery Applications. Materials 2023, 16, 2298. [Google Scholar] [CrossRef] [PubMed]
- Ferrentino, N.; Romano, M.P.; Zappavigna, S.; Abate, M.; Vecchio, V.D.; Romano, D.; Germinario, C.; Grifa, C.; Filosa, R.; Pappalardo, D. Poly(ε-caprolactone)-poly(ethylene glycol) Tri-block Copolymer as Quercetin Delivery System for Human Colorectal Carcinoma Cells: Synthesis, Characterization and In Vitro Study. Polymers 2023, 15, 1179. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, J.; Bang, J.; Jang, Y.J.; Park, J.; Kim, D.H. Simultaneously Achieving Room-temperature Circularly Polarized Luminescence and High Stability in Chiral Perovskite Nanocrystals Via Block Copolymer Micellar Nanoreactors. J. Mater. Chem. A 2023, 11, 12876–12884. [Google Scholar] [CrossRef]
- Wang, W.Y.; Lu, W.; Goodwin, A.; Wang, H.Q.; Yin, P.; Kang, N.G.; Hong, K.L.; Mays, J.W. Recent Advances in Thermoplastic Elastomers from Living Polymerizations: Macromolecular Architectures and Supramolecular Chemistry. Prog. Polym. Sci. 2019, 95, 1–31. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.H.; Yang, L.; Chu, C.Z.; Cao, S.C.; Wang, Z.; Xue, J.J.; You, Z.W. Biodegradable elastomers for biomedical applications. Prog. Polym. Sci. 2023, 147, 101763. [Google Scholar] [CrossRef]
- Dau, H.; Jones, G.R.; Tsogtgerel, E.; Nguyen, D.; Keyes, A.; Liu, Y.S.; Rauf, H.; Ordonez, O.; Ordonez, V.; Alhan, H.B.; et al. Linear Block Copolymer Synthesis. Chem. Rev. 2022, 122, 14471–14553. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, S.; Lu, G.L.; Huang, X.Y. Constructing well-defined star graft copolymers. Polym. Chem. 2013, 4, 1289–1299. [Google Scholar] [CrossRef]
- Haque, M.F.; Grayson, M.S. The synthesis, properties and potential applications of cyclic polymers. Nat. Chem. 2020, 12, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, F.; Niebuur, B.J.; Koch, M.; Kraus, T.; Gallei, M. Synthesis and Microphase Separation of Dendrimer-like Block Copolymers by Anionic Polymerization Strategies. Eur. Polym. J. 2023, 187, 111894. [Google Scholar] [CrossRef]
- Steube, M.; Johann, T.; Barent, R.D.; Müller, A.H.E.; Frey, H. Rational design of tapered multiblock copolymers for thermoplastic elastomers. Prog. Polym. Sci. 2022, 124, 101488. [Google Scholar] [CrossRef]
- Feng, C.; Li, Y.J.; Yang, D.; Hu, J.H.; Zhang, X.H.; Huang, X.Y. Well-defined Graft Copolymers: From Controlled Synthesis to Multipurpose Applications. Chem. Soc. Rev. 2011, 40, 1282–1295. [Google Scholar] [CrossRef]
- Zang, M.F.; Müller, A.H.E. Cylindrical Polymer Brushes. J. Polym. Sci. A Polym. Chem. 2005, 43, 3461–3481. [Google Scholar] [CrossRef]
- Maher, M.J.; Jones, S.D.; Zografos, A.; Xu, J.; Schibur, H.J.; Bates, F.S. The Order-Disorder Transition in Graft Block Copolymers. Macromolecules 2018, 51, 232–241. [Google Scholar] [CrossRef]
- Liu, J.R.; Lu, B.K.; Xiao, R. Comparative Study on Annealing-induced High-Impact Toughness of Linear and Grafted Polypropylene Random Copolymer. Polym. Adv. Technol. 2023, 34, 1467–1478. [Google Scholar] [CrossRef]
- Monica, F.D.; Kleij, A.W. From Terpenes to Sustainable and Functional Polymers. Polym. Chem. 2020, 11, 5109–5127. [Google Scholar] [CrossRef]
- Cywar, R.M.; Rorrer, N.A.; Hoyt, C.B.; Beckham, G.T.; Chen, E.Y.X. Bio-based Polymers with Performance-advantaged Properties. Nat. Rev. Mater. 2022, 7, 83–103. [Google Scholar] [CrossRef]
- Burelo, M.; Martínez, A.; Hernández-Varela, J.D.; Stringer, T.; Ramírez-Melgarejo, M.; Yau, A.Y.; Luna-Bárcenas, G.; Treviño-Quintanilla, C.D. Recent Developments in Synthesis, Properties, Applications and Recycling of Bio-Based. Molecules 2024, 29, 387. [Google Scholar] [CrossRef]
- Jia, B.; Huang, H.Y.; Dong, Z.C.; Ren, X.Y.; Lu, Y.Y.; Wang, W.Z.; Zhou, S.W.; Zhao, X.; Guo, B.L. Degradable Biomedical Elastomers: Paving the Future of Tissue Repair and Regenerative Medicine. Chem. Soc. Rev. 2024, 53, 4086–4153. [Google Scholar] [CrossRef] [PubMed]
- John, G.; Nagarajan, S.; Vemula, P.K.; Silverman, J.R.; Pillai, C.K.S. Natural Monomers: A Mine for Functional and Sustainable Materials-Occurrence, Chemical Modification and Polymerization. Prog. Polym. Sci. 2019, 92, 158–209. [Google Scholar] [CrossRef]
- Coudane, J.; Berghe, H.V.D.; Mouton, J.; Garric, X.; Nottelet, B. Poly(Lactic Acid)-Based Graft Copolymers: Syntheses Strategies and Improvement of Properties for Biomedical and Environmentally Friendly Applications: A Review. Molecules 2022, 27, 4135. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.K.; Yu, L.; Cao, Y.T.; Zhang, X.; Zhang, Y.Y. Progresses in Synthetic Technology Development for the Production of L-lactide. Org. Biomol. Chem. 2021, 19, 10288–10295. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rana, A.; Karak, N.; Kumar, I.; Rana, S.; Kumar, P. Sustainable Smart Anti-corrosion Coating Materials Derived from Vegetable Oil Derivatives: A Review. RSC Adv. 2023, 13, 3910–3941. [Google Scholar] [CrossRef] [PubMed]
- Chek, Y.W.; Ang, T.C. Progress of Bio-based Coatings in Waterborne System: Synthesis Routes and Monomers from Renewable Resources. Prog. Org. Coat. 2024, 188, 108190. [Google Scholar] [CrossRef]
- Wahlen, C.; Frey, H. Anionic Polymerization of Terpene Monomers: New Options for Bio-Based Thermoplastic Elastomers. Macromolecules 2021, 54, 7323–7336. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, L.Y.; Ma, Y.H.; Zhao, C.W.; Yang, W.T. Copolymerization of α-Methyl styrene and Styrene. Chin. J. Polym. Sci. 2014, 32, 986–995. [Google Scholar] [CrossRef]
- Chin, M.T.; Yang, T.G.; Quirion, K.P.; Lian, C.; He, J.; Diao, T.N. Implementing a Doping Approach for Poly(methyl methacrylate) Recycling in a Circular Economy. J. Am. Chem. Soc. 2024, 146, 5786–5792. [Google Scholar] [CrossRef]
- Coudane, J.; Nottelet, B.; Mouton, J.; Garric, X.; Berghe, H.V.D. Poly(ε-caprolactone)-based Graft Copolymers: Synthesis Methods and Applications in the Biomedical Field: A Review. Molecules 2022, 27, 7339. [Google Scholar] [CrossRef]
- Matic, A.; Hess, A.; Schanzenbach, D.; Schlaad, H. Epoxidized 1,4-polymyrcene. Polym. Chem. 2020, 11, 1364–1368. [Google Scholar] [CrossRef]
- Ren, Y.Y.; Gao, Q.; Zhou, C.; Wei, Z.Y.; Zhang, Y.; Li, Y. Facile Synthesis of Well-defined Linear-comb Highly Branched Poly(ε-caprolactone) Using Hydroxylated Polybutadiene and Organocatalyst. RSC Adv. 2015, 5, 27421–27430. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Entry | n, GPC (kg mol−1) a | w, GPC (kg mol−1) a | Ð a |
|---|---|---|---|---|
| P(My-co-AMS)1-1 | 20231127-1 | 3.70 | 4.10 | 1.09 |
| P(My-co-AMS)1-2 | 20231127-2 | 4.10 | 4.50 | 1.08 |
| P(My-co-AMS)1-3 | 20231123-1 | 4.20 | 4.60 | 1.08 |
| P(My-co-AMS)1-4 | 20231123-2 | 3.40 | 3.80 | 1.09 |
| P(My-co-AMS)1-5 | 20230425 | 1.80 | 2.00 | 1.12 |
| P(My-co-AMS)2-1 | 20230516-2 | 3.90 | 4.40 | 1.12 |
| P(My-co-AMS)2-2 | 20230504-2 | 4.80 | 5.60 | 1.15 |
| P(My-co-AMS)3-1 | 20230523-1 | 2.20 | 2.50 | 1.13 |
| P(My-co-AMS)3-2 | 20230523-2 | 3.70 | 4.10 | 1.13 |
| P(My-co-AMS)3-3 | 20230516-1 | 2.50 | 2.90 | 1.13 |
| P(My-co-AMS)3-4 | 20230530-2 | 2.70 | 3.10 | 1.14 |
| P(My-co-AMS)3-5 | 20230509-2 | 3.80 | 4.20 | 1.12 |
| Sample | Entry | Td (°C) |
|---|---|---|
| PMy | 20230504-1 | 375 |
| P(My-co-AMS)1-1 | 20231127-1 | 381 |
| P(My-co-AMS)2-2 | 20230504-2 | 370 |
| P(My-co-AMS)3-3 | 20230516-1 | 362 |
| Sample | Entry | n, GPC (kg mol−1) a | w, GPC (kg mol−1) a | Ð a |
|---|---|---|---|---|
| E[P(My-co-AMS)1]-1 | 20231201 | 4.30 | 4.80 | 1.08 |
| E[P(My-co-AMS)2]-1 | 20230515 | 4.80 | 5.70 | 1.12 |
| E[P(My-co-AMS)3]-1 | 20230517 | 3.9 | 4.4 | 1.11 |
| E[P(My-co-AMS)3]-2 | 20230530 | 3.7 | 4.2 | 1.13 |
| E[P(My-co-AMS)3]-3 | 20230531 | 2.2 | 2.5 | 1.12 |
| E[P(My-co-AMS)3]-4 | 20230609 | 2.7 | 3.1 | 1.12 |
| Sample | Entry | n, GPC (kg mol−1) a | w, GPC (kg mol−1) a | Ð a |
|---|---|---|---|---|
| E[P(My-co-AMS)3] | 20230822 | 2.50 | 2.90 | 1.13 |
| P(My-co-AMS)3-g-PCL-2 | 20231230-1 | 4.70 | 5.20 | 1.10 |
| E[P(My-co-AMS)3] | 20230609 | 2.70 | 3.10 | 1.15 |
| P(My-co-AMS)3-g-PCL-3 | 20231227 | 6.70 | 8.10 | 1.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zhang, M.; He, J.; Ni, P. Synthesis and Characterization of Graft Copolymers with Poly(ε-caprolactone) Side Chain Using Hydroxylated Poly(β-myrcene-co-α-methyl styrene). Molecules 2024, 29, 2363. https://doi.org/10.3390/molecules29102363
Li T, Zhang M, He J, Ni P. Synthesis and Characterization of Graft Copolymers with Poly(ε-caprolactone) Side Chain Using Hydroxylated Poly(β-myrcene-co-α-methyl styrene). Molecules. 2024; 29(10):2363. https://doi.org/10.3390/molecules29102363
Chicago/Turabian StyleLi, Tao, Mingzu Zhang, Jinlin He, and Peihong Ni. 2024. "Synthesis and Characterization of Graft Copolymers with Poly(ε-caprolactone) Side Chain Using Hydroxylated Poly(β-myrcene-co-α-methyl styrene)" Molecules 29, no. 10: 2363. https://doi.org/10.3390/molecules29102363
APA StyleLi, T., Zhang, M., He, J., & Ni, P. (2024). Synthesis and Characterization of Graft Copolymers with Poly(ε-caprolactone) Side Chain Using Hydroxylated Poly(β-myrcene-co-α-methyl styrene). Molecules, 29(10), 2363. https://doi.org/10.3390/molecules29102363