General Experimental Details
All reagents and catalysts were purchased from Sigma-Aldrich, Acros, J&K Scientific and TCI Europe and used without further purification unless otherwise mentioned. TLC analysis was performed on Silufol chromatographic plates. For preparative chromatography, silica gel 60 (0.040–0.063 mm) was used.
1H,
13C NMR spectra were recorded on a Bruker AVANCE II 300 MHz (300.1, 75.5 MHz and 282.4 MHz, respectively) and a Bruker AMX III 400 MHz (400.1, 100.6 MHz and 376.5 MHz, respectively) spectrometers in CDCl
3, containing 0.05% Me
4Si as the internal standard. Determination and verification of structures obtained compounds and assignments of
1H and
13C signals were made using 1D and 2D DEPT, COSY, HSQC and HMBC spectra. High-resolution mass spectra were recorded on a Bruker Daltonics micrOTOF-Q II device (electrospray ionization). Measurements were carried out in positive ion mode. Samples were injected into the spray chamber of the mass spectrometer from an Agilent 1260 liquid chromatograph equipped with an Agilent Poroshell 120 EC-C18 column (3.0 × 50 mm; 2.7 μm); the flow rate was 0.4 mL min
−1; the samples of compounds were loaded using autosampler from acetonitrile solution and eluted in the following gradient of acetonitrile (A) in water: 0–6 min—0%–85% A, 6–7.5 min—85% A, 7.5–8 min—85%–0% A, 8–10 min—0% A. Preparative HPLC were performed on Thermo-Finnigan Surveyor equipped with UV-VIS detector on Supelco Ascentis C8 5 μm 250 mm × 10 mm chromatographic column. The starting material 9 was synthesized according to a literature procedure [
15].
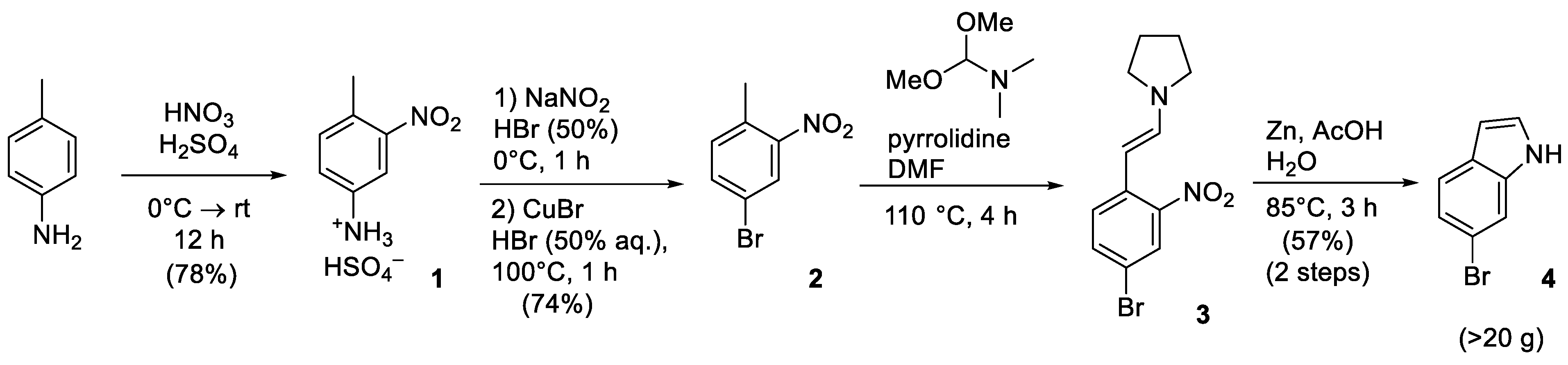
4-Bromo-1-methyl-2-nitrobenzene (2), P-toluidine (3.00 g, 28.00 mmol) was added to 6 mL of conc. H2SO4 when cooled in a water bath (10–15 °C). The water bath was replaced by an ice bath and a mixture of conc. H2SO4 (6 mL) and conc. HNO3 (1.85 g, 29.39 mmol) was added dropwise to the resulting mixture. This solution was stirred at 0 °C for 1 h and another 12 h at rt. The mixture was poured over crushed ice, and the precipitate was filtered, washed with ice-cooled water and dried in high vacuo to afford the intermediate compound 1 as a yellow solid (5.46 g, 78%).
The solution of NaNO2 (0.55 g, 7.99 mmol) in 1.3 mL of H2O was added to the ice-cooled suspension of aniline hydrosulfate (1.00 g, 3.99 mmol) in 4 mL of H2O and 1.3 mL of aq. HBr (50%). The resulting mixture was stirred at 0 °C for 1 h, followed by adding CuBr (0.63 g, 4.40 mmol) solution in 1.3 mL aq. HBr (50%) and 3 mL of H2O. The mixture was refluxed for 30 min. After cooling, the mixture was extracted with Et2O (3 × 10 mL). The combined organic phases were washed with 10% aq. ammonia, water, dried over MgSO4 and concentrated in vacuo. The target compound 2 was crystallized from MeOH as a brown solid (638 mg, 74%)
1H NMR (300 MHz, CDCl3), δ: 8.13 (d, J = 2.1 Hz, 1H), 7.63 (dd, J = 8.2, 2.1 Hz, 1H), 7.29–7.20 (m, 1H), 2.57 (s, 3H).13C NMR (75 MHz, CDCl3), δ: 149.61, 135.96, 134.12, 132.54, 127.53, 119.65, 20.03.
6-Bromoindole (4), DMF-DMA (1.65 g, 13.89 mmol) and pyrrolidine (0.53 g, 7.41 mmol) were added to the solution of 4-bromo-1-methyl-2-nitrobenzene (1.00 g, 4.63 mmol) in DMF (9 mL). The solution was stirred at 110 °C for 4 h. After cooling, the mixture was dissolved in 20 mL of Et2O and washed with 20 mL of H2O. Aq. layer was extracted with Et2O (3 × 20 mL). The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo. Crude intermediate 3 was dissolved in 30 mL of 80% aq. AcOH and heated to 80 °C. Zinc dust (2.42 g, 37.03 mmol) was added to the solution, and the suspension was stirred at this temperature for 3 h. The mixture was cooled down in an ice bath. The precipitate was filtered out and washed with EtOAc. The combined organic phases were washed with water, dried over MgSO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 15:1) and by crystallization from petroleum ether to afford the desired compound 4 as a grey solid (513 mg, 57%).
1H NMR (300 MHz, CDCl3), δ: 8.13 (br.s, 1H), 7.58–7.45 (m, 2H), 7.26–7.12 (m, 2H), 6.53 (ddd, J = 3.1, 2.1, 1.0 Hz, 1H).
13C NMR (76 MHz, CDCl3), δ: 136.60, 126.78, 124.83, 123.18, 121.98, 115.49, 113.99, 102.87.
Methyl 2-(6-bromo-1H-indol-1-yl)acetate (5), 6-Bromoindole (5.00 g, 25.50 mmol) was added to the stirred suspension of NaH (60% dispersion in oil, 1.53 g, 38.25 mmol) in dry DMF (50 mL). The mixture was stirred for 4 h followed by adding methyl bromoacetate (7.80 g, 51.00 mmol). The resulting mixture was stirred overnight, quenched with 25 mL of H2O, and extracted with AcOEt (3 × 25 mL). The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 5:1) to afford the desired compound 5 as a yellow oil (5.50 g, 80%).
1H NMR (300 MHz, CDCl3), δ: 7.51–7.45 (m, 1H, Ar), 7.39 (dt, J = 1.6, 0.6 Hz, 1H, Ar), 7.22 (dd, J = 8.4, 1.6 Hz, 1H, Ar), 7.04 (d, J = 3.2 Hz, 1H, Ar), 6.52 (dd, J = 3.3, 0.9 Hz, 1H, Ar), 4.79 (s, 2H, CH2), 3.75 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3), δ: 168.65 (COO), 137.34, 129.12, 127.52, 123.28, 122.37, 115.78, 112.08, 102.84 (Ar), 52.68 (CH2), 47.67 (CH3).
2-(6-Bromo-1H-indol-1-yl)acetic acid (6), The solution of NaOH (1.64 g, 41.03 mmol) in water (55 mL) was added to the solution of methyl 2-(6-bromo-1H-indol-1-yl) acetate 5 (5.50 g, 20.52 mmol) in MeOH (55 mL). The mixture was refluxed for 5 h and cooled to room temperature. After that MeOH was evaporated. The aqueous solution was washed with petroleum ether and treated with 10% HCl (aq.) to reach pH = 1. The solution was extracted with AcOEt (3 × 25 mL). The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo to make the target compound 6 a colorless solid (5.06 mg, 97%).
1H NMR (300 MHz, DMSO-d6), δ: 12.95 (br.s, 1H, OH)7.76–7.63 (m, 1H, Ar), 7.51 (d, J = 8.4 Hz, 1H, Ar), 7.36 (d, J = 3.2 Hz, 1H, Ar), 7.16 (dd, J = 8.4, 1.8 Hz, 1H, Ar), 6.48 (dd, J = 3.2, 0.9 Hz, 1H, Ar), 5.05 (s, 2H, CH2).
13C NMR (75 MHz, DMSO-d6), δ: 170.75 (COO), 137.81, 131.21, 127.55, 122.47, 114.57, 113.27, 101.79 (Ar), 47.58 (CH2).
Methyl 2-(2-(6-bromo-1H-indol-1-yl)acetamido)acetate (7), Glycine methyl ester hydrochloride (2.75 g, 21.92 mmol), hydroxybenzotriazole (hydrate, 80% HOBt, 3.70 g, 21.92 mmol) was added to the solution of 2-(6-bromo-1H-indol-1-yl)acetic acid 6 (5.06 g, 19.95 mmol) in CH2Cl2 (500 mL), then 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (8.77 g, 45.70 mmol) and triethylamine (10.07 mg, 99.53 mmol) were added to the reaction mixture. The mixture was stirred overnight. The solution was treated with 3% HCl (aq.) and extracted with CH2Cl2. The combined organic layers were washed with H2O and brine, dried over Na2SO4, and evaporated in vacuo. The product was purified by crystallization from methanol to afford the target compound 7 as a pink powder (4.99 mg, 77%).
1H NMR (300 MHz, CDCl3), δ: 7.58–7.43 (m, 2H, Ar), 7.37–7.21 (m, 1H, Ar), 7.09 (d, J = 3.2 Hz, 1H, Ar), 6.60 (dd, J = 3.2, 0.9 Hz, 1H, Ar), 5.88 (br.s, 1H, NH), 4.78 (s, 2H, CH2), 3.95 (d, J = 5.5 Hz, 2H, CH2), 3.69 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3), δ: 169.52 (COO), 168.10 (COO), 137.09, 128.86, 127.67, 123.90, 122.59, 116.34, 112.39, 103.88 (Ar), 52.49 (CH2), 49.76 (CH2), 40.99 (CH3).
2-(2-(6-Bromo-1H-indol-1-yl)acetamido)acetic acid (8), Lithium hydroxide hydrate was added to the suspension of methyl 2-(2-(6-bromo-1H-indol-1-yl) acetamido) acetate 7 (4.99 g, 15.38 mmol) in THF (25 mL) and water (6 mL). The mixture was stirred overnight and treated with 10% HCl (aq.). The precipitate was filtered, washed with water, and dried in high vacuo. The crude product was crystallized from MeOH to afford the target compound as a pink solid (3.50 g, 74%).
1H NMR (300 MHz, DMSO-d6), δ: 12.66 (s, 1H, OH), 8.50 (t, J = 5.9 Hz, 1H), 7.68–7.61 (m, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 3.2 Hz, 1H), 7.16 (dd, J = 8.4, 1.8 Hz, 1H), 6.48 (dd, J = 3.1, 0.9 Hz, 1H), 4.91 (s, 2H, CH2), 3.82 (d, J = 5.8 Hz, 2H, CH2).
13C NMR (75 MHz, DMSO-d6), δ: 171.42, 168.19, 137.69, 131.28, 127.63, 122.47, 114.53, 113.28, 101.65, 48.98, 41.22.
HRMS of C12H1179BrN2O3 and C12H1181BrN2O3, m/z: calcd. for [M + H]+ 311.0026 and 313.0006; found 311.0015 and 312.9999.
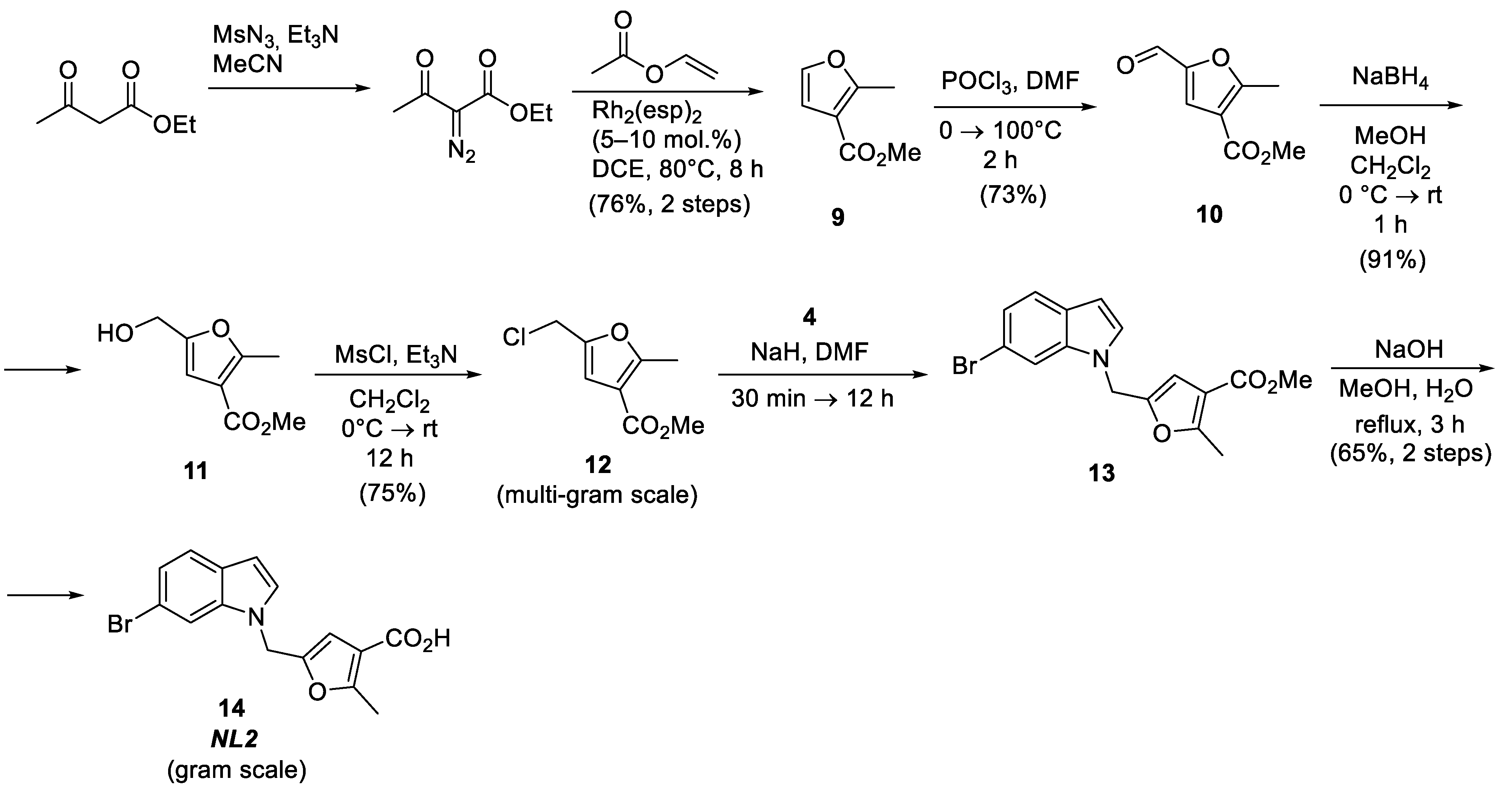
Methyl 5-formyl-2-methylfuran-3-carboxylate (10), POCl3 (5.5 g, 35.7 mmol) was carefully added to ice-cooled DMF (5 mL). The mixture was heated to room temperature and stirred for 1 h, followed by adding methyl 2-methylfuran-3-carboxylate (5.0 g, 35.7 mmol). The reaction mixture was stirred for another 2 h at 100 °C. The mixture was cooled down to 0 °C, quenched with an aqueous solution of Na2CO3 (5 g/50 mL H2O) and extracted with CH2Cl2 (3 × 50 mL). The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was crystallized from Et2O. The target compound 10 was collected as a yellow solid (4.3 g, 73%)
1H NMR (300 MHz, CDCl3), δ: 9.57 (s, 1H, CHO), 7.48 (s, 1H, CH), 3.88 (s, 3H, OCH3), 2.70 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3), δ: 177.12, 123.28, 122.30, 114.32, 112.30, 51.88, 14.34.
Methyl 5-(hydroxymethyl)-2-methylfuran-3-carboxylate (11), NaBH4 (1.16 g, 30.64 mmol) was added to the ice-cooled solution of methyl 5-formyl-2-methylfuran-3-carboxylate 10 (4.30 g, 25.59 mmol) in MeOH (70 mL) and CH2Cl2 (35 mL). The mixture was stirred at 0 °C for 30 min and at room temperature for another 2 h. The resulting solution was quenched with water and extracted with CH2Cl2. The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 2:1) to afford the target compound 11 as colorless oil (3.93 g, 91%).
1H NMR (300 MHz, CDCl3), δ: 6.52 (s, 1H, CH), 4.54 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 2.56 (s, 3H, CH3), 2.02 (br.s, 1H, OH).
13C NMR (76 MHz, CDCl3), δ: 164.57, 159.42, 152.04, 113.77, 108.49, 56.96, 51.36, 13.74.
HRMS of C8H10O4, m/z: calcd. for [M+H]+ 153.0546; found 153.0549.
Methyl 5-(chloromethyl)-2-methylfuran-3-carboxylate (12), Mesyl chloride (5.30 g, 46.31 mmol) was added dropwise to the solution of methyl 5-(hydroxymethyl)-2-methylfuran-3-carboxylate 11 (3.93 g, 23.11 mmol) and Et3N (4.68 g, 46.31 mmol) in CH2Cl2 (40 mL). The solution was stirred for 4 h, quenched with water and extracted with CH2Cl2. The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 10:1) to afford the target compound 12 as colorless oil (3.27 g, 75%).
1H NMR (300 MHz, CDCl3), δ: 6.61 (s, 1H, CH), 4.52 (s, 2H, CH2), 3.82 (s, 3H, OCH3), 2.59 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3), δ: 163.98, 160.23, 148.01, 114.33, 110.53, 51.39, 37.13, 13.81.
Methyl 5-((6-bromo-1H-indol-1-yl)methyl)-2-methylfuran-3-carboxylate (13), 6-Bromoindole (2.72 g, 13.88 mmol) was added to the stirred suspension of NaH (60% dispersion in oil, 610.7 mg, 12.23 mmol) in dry DMF (15 mL). The mixture was stirred for 4 h followed by adding methyl 5-(chloromethyl)-2-methylfuran-3-carboxylate 12 (3.27 g, 17.33 mmol). The solution was heated to 40 °C and stirred at this temperature for 3 h. The mixture was quenched with H2O and extracted with AcOEt. The combined organic phases were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 5:1) to afford the target compound 13 as pale-yellow oil (3.68 g, 76%).
1H NMR (300 MHz, CDCl3), δ: 7.51 (dt, J = 1.5, 0.6 Hz, 1H, Ar), 7.43 (dd, J = 8.4, 0.6 Hz, 1H, Ar), 7.21–7.15 (m, 1H, Ar), 7.04 (d, J = 3.2 Hz, 1H), 6.45 (dd, J = 3.2, 0.9 Hz, 1H), 6.43 (s, 1H, CH(4)), 5.02 (s, 2H, CH2), 3.75 (s, 3H, OCH3), 2.47 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3), δ: 164.16, 159.66, 147.79, 136.83, 128.36, 127.60, 123.07, 122.29, 115.55, 114.00 112.42, 109.16, 102.35, 51.41, 42.91, 13.81.
5-((6-Bromo-1H-indol-1-yl)methyl)-2-methylfuran-3-carboxylic acid (14), The solution of NaOH (846.2 mg, 21.23 mmol) in water (30 mL) was added to the solution of methyl 5-((6-bromo-1H-indol-1-yl) methyl)-2-methylfuran-3-carboxylate 13 (3.68 g, 10.47 mmol) in 30 mL of MeOH. The mixture was refluxed for 3 h and cooled to room temperature. After that MeOH was evaporated. The aqueous solution was treated with 10% HCl (aq.) to reach pH = 1. The precipitate was filtered out, washed with water, and dried in high vacuo to afford the target compound 14 as a colorless solid (2.17 g, 62%).
1H NMR (400 MHz, CDCl3), δ: 11.46 (br.s, 1H, OH), 7.52–7.48 (m, 1H), 7.43 (d, J = 8.4 Hz, 1H, Ar), 7.18 (dd, J = 8.4, 1.7 Hz, 1H, Ar), 7.05–7.00 (m, 1H, Ar), 6.47–6.40 (m, 2H, Ar), 5.02 (s, 2H, CH2), 2.43 (s, 3H, CH3).
13C NMR (101 MHz, CDCl3), δ: 169.71 (COOH), 160.95, 147.91, 136.79, 128.27, 127.57, 123.07, 122.28, 115.55, 114.22, 112.37, 109.38, 102.37 (Ar), 42.77 (CH2), 13.87 (CH3).
HRMS of C15H1279BrNO3 and C15H1281BrNO3, m/z: calcd. for [M + H]+ 334.0073 and 336.0053; found 334.0072 and 336.0058.
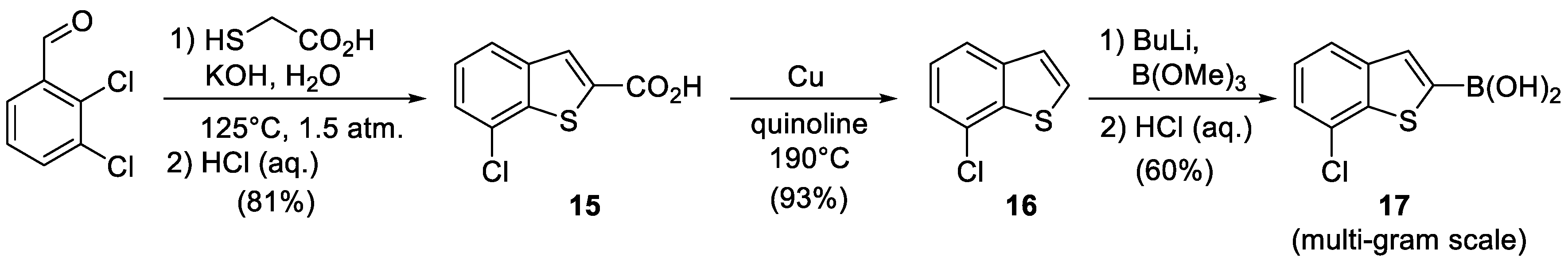
7-Chlorobenzo[b]thiophene-2-carboxylic acid (15), Potassium hydroxide solution (1.16 g, 20.74 mmol) in water (10 mL) was placed in a glass ampoule, and thioglycolic acid (868 mg, 9.43 mmol) was slowly added. 2,3-Dichlorobenzaldehyde (1.65 g, 9.43 mmol) was added to the reaction mixture. The ampoule was soldered, heated to 125 °C and kept under intense stirring for 2 h. The mixture was cooled to room temperature. The precipitate was dissolved in water. The water layer was washed with Et2O and acidified with 10% HCl (aq.). The precipitate was filtered, washed with water, and dried in high vacuo to afford the target compound 15 as a colorless powder (1.62 g, 81%).
1H NMR (300 MHz, DMSO-d6), δ: 8.20 (s, 1H), 8.01 (dd, J = 7.8, 1.0 Hz, 1H), 7.64 (dd, J = 7.8, 1.0 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H).
13C NMR (75 MHz, DMSO-d6), δ: 163.56, 140.69, 140.43, 136.20, 131.48, 127.29, 125.33.
7-Chlorobenzo[b]thiophene (16), 7-Chlorobenzothiophene-2-carboxylic acid 15 (500 mg, 2.35 mmol) was dissolved in 5 mL of quinoline. Copper powder (164 mg, 2.58 mmol) was added to the solution. The suspension was heated to 190 °C and kept under intensive stirring for 3 h. The reaction mixture was cooled to room temperature, and 20 mL of 10% HCl (aq.) solution was added. The resulting system was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The product was purified by column chromatography on silica gel in 100% petroleum ether to afford the target compound 16 as colorless liquid (385 mg, 93%).
1H NMR (300 MHz, CDCl3), δ: 7.69 (dd, J = 7.2, 1.7 Hz, 1H), 7.45 (d, J = 5.5 Hz, 1H), 7.38–7.18 (m, 3H).
13C NMR (75 MHz, CDCl3), δ: 141.07, 139.13, 127.98, 127.43, 125.43, 124.63, 123.91, 122.07.
(7-Chlorobenzo[b]thiophen-2-yl)boronic acid (17), n-Butyllithium (2.5M solution in hexane, 2.5 mL, 6.25 mmol) was added dropwise to a 7-chlorobenzothiophene 16 (530 mg, 3.14 mol) precooled to −78° in 14 mL of THF under argon atmosphere. The resulting mixture was heated to −30 °C, kept at this temperature for 10 min and cooled down again to −78 °C, and then trimethylborate was added dropwise to the solution. The solution was allowed to warm up to 0 °C and was poured into the saturated NH4Cl solution (30 mL). The resulting mixture was partitioned with Et2O (3 × 20 mL). The ether solution was collected and evaporated to dryness. The remainder was dissolved in 2 M NaOH solution (20 mL) and stirred for 20 min. The obtained solution was washed with CH2Cl2. The aqueous layer was treated with 10% HCl (aq.) to reach pH = 1. The precipitate was filtered, washed with water, and dried in high vacuo to afford the target compound 17 as a yellow powder (400 mg, 60%).
1H NMR (300 MHz, DMSO-d6), δ: 8.04 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H).
Propargyl bromide, The solution of phosphorus tribromide (53.0 g, 196.2 mmol) in CH2Cl2 (100 mL) was added dropwise to the solution of propargyl alcohol (10.0 mL, 173.40 mmol) in CH2Cl2 (100 mL) at 0 °C. The reaction mixture was stirred overnight, cooled in an ice bath, and quenched dropwise with water (125 mL). The organic layer was separated, washed with saturated NaHCO3 (aq.) and NaCl (aq.) solutions and dried over Na2SO4. The solvent was carefully evaporated, and the remainder was distilled at atmospheric pressure to afford the target compound as colorless liquid (BP = 85 °C, 17.4 g, 84%).
1H NMR (300 MHz, CDCl3), δ: 3.88 (d, J = 2.6 Hz, 2H, CH2), 2.53 (t, J = 2.6 Hz, 1H, CH).
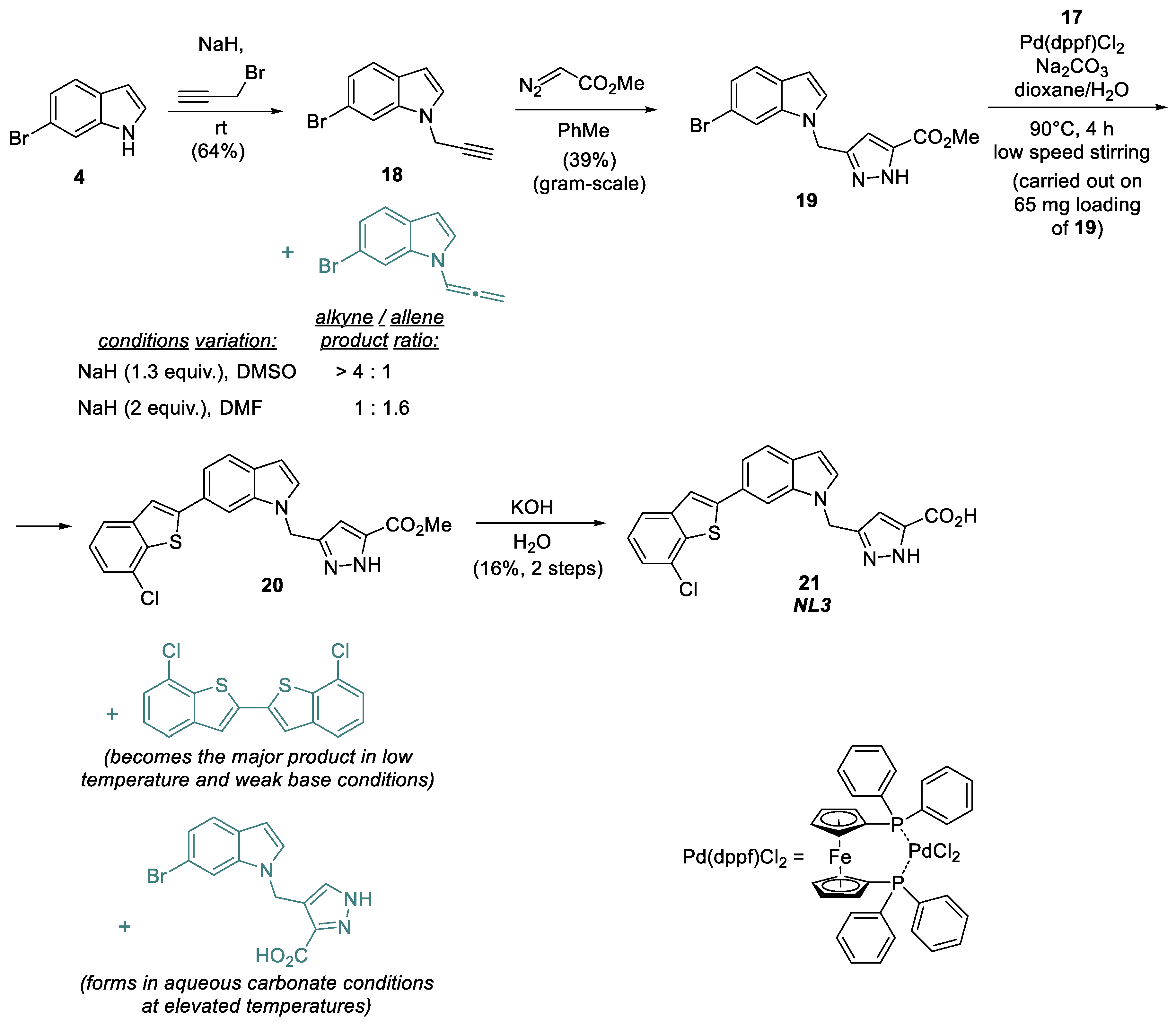
6-Bromo-1-(prop-2-yn-1-yl)-1H-indole (18), 6-Bromoindol (1.00 g, 5.10 mmol) was added to the suspension of NaH (60% in oil, 265 mg, 6.60 mmol, pre-washed with petroleum ether) in absolute DMSO (20 mL) and was kept stirring until the gas release stopped. Propargyl bromide (1.24 mL, 6.60 mmol) was added dropwise to the resulting solution under thermal control with a water bath for heat dissipation. The mixture was stirred for 3 h. Water (40 mL) was added dropwise to the resulting mixture and pre-cooled in an ice bath. The mixture was extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with H2O, with brine, dried over Na2SO4 and evaporated in vacuo. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 40:1) to afford the target compound 18 as a yellow oil (760 mg, 64%).
1H NMR (300 MHz, CDCl3), δ: 7.58–7.52 (m, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.27–7.10 (m, 2H), 6.49 (dd, J = 3.2, 0.9 Hz, 1H), 4.80 (d, J = 2.6 Hz, 2H, CH2), 2.41 (t, J = 2.6 Hz, 1H, CH).
13C NMR (75 MHz, CDCl3), δ: 136.61, 127.93, 127.78, 123.23, 122.33, 115.58, 112.47, 102.37 (Ar), 74.02 (CH), 35.95 (CH2).
Methyl 5-((6-bromo-1H-indol-1-yl)methyl)-1H-pyrazole-3-carboxylate (19), Methyl diazoacetate (632 mg, 6.32 mmol) was added dropwise to the solution of 6-bromo-1-(prop-2-yn-1-yl)-1H-indole 18 (740 mg, 3.16 mol) in 15 mL of toluene. The reaction mixture was refluxed for 6 h. The solvent was evaporated. The crude product was purified by silica gel column chromatography (eluent: petroleum ether−AcOEt, 1:1) to afford the target compound 19 as a brown solid (760 mg, 64%).
1H NMR (300 MHz, CDCl3), δ: 7.54–7.51 (m, 1H), 7.49–7.41 (m, 1H), 7.19 (dd, J = 8.4, 1.7 Hz, 1H), 7.12 (d, J = 3.2 Hz, 1H), 6.53 (s, 1H), 6.49 (dd, J = 3.2, 0.9 Hz, 1H), 5.34 (s, 2H), 3.85 (s, 3H).
13C NMR (75 MHz, CDCl3), δ: 160.35 (COO), 136.91, 128.50, 127.62, 123.05, 122.27, 115.56, 112.52, 107.39, 102.40, 52.30 (CH3), 43.43 (CH2).
HRMS of C14H1279BrN3O2 334.0186 and C14H1281BrN3O2, m/z: calcd. for [M + H]+ 336.0166; found 334.0179 and 336.0161.
Methyl 5-((6-(7-chlorobenzo[b]thiophen-2-yl)-1H-indol-1-yl)methyl)- 1H-pyrazole-3-carboxylate (20), The mixture of methyl 5-((6-bromo-1H-indol-1-yl)methyl)- 1H-pyrazole-3-carboxylate 19 (350 mg, 1.00 mmol), (7-chlorobenzo[b]thiophen-2-yl)boronic acid (256 mg, 1.20 mmol), sodium carbonate (213 mg, 2.00 mmol) and Pd(dppf)Cl2 (40 mg, 0.05 mmol) was dissolved in the mixture of dioxane (1 mL) and water (1 mL) and was stirred at 90 °C for 4 h. All volatiles were evaporated, and the residue was dissolved in water. The solution was extracted with CH2Cl2. The combined organic layers were washed with water, dried over Na2SO4 and concentrated in vacuo. The solid product 20 was used further without purification and characterization.
5-((6-(7-Chlorobenzo[b]thiophen-2-yl)-1H-indol-1-yl)methyl)-1H-pyrazole-3-carboxylic acid (21), The intermediate product from the previous step was dissolved in 10 mL of MeOH and treated with 1 mL of 30% KOH solution (aq.). The mixture was stirred overnight, treated with 10% HCl solution (aq.) and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and evaporated in vacuo. The crude product was purified by preparative HPLC (column—Supelco Ascentis C8, 5 μm, 250 mm × 10 mm, detection—220 nm, flow rate—2.5 mL/min, eluent—MeCN/H2O 25/75 containing 0.10 M of TFA, 10 mg of the crude mixture were separated per one run) to obtain the pure target compound 21 (65 mg, 16%).
1H NMR (300 MHz, DMSO-d6), δ: 8.03 (s, 1H), 7.91 (dd, J = 7.8, 1.1 Hz, 1H), 7.81 (s, 1H), 7.52–7.37 (m, 5H), 7.15 (dd, J = 8.4, 1.7 Hz, 1H), 6.54 (s, 1H), 6.48 (d, J = 3.1 Hz, 1H), 5.39 (s, 2H).
13C NMR (75 MHz, DMSO-d6), δ: 142.58, 141.74, 136.98, 133.92, 130.28, 127.71, 126.99, 126.24, 124.90, 123.57, 122.60, 122.48, 114.52, 113.35, 107.43, 101.84.
HRMS of C21H14ClN3O2S, m/z: calcd. for [M + H]+ 407.0495; found 407.0492.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
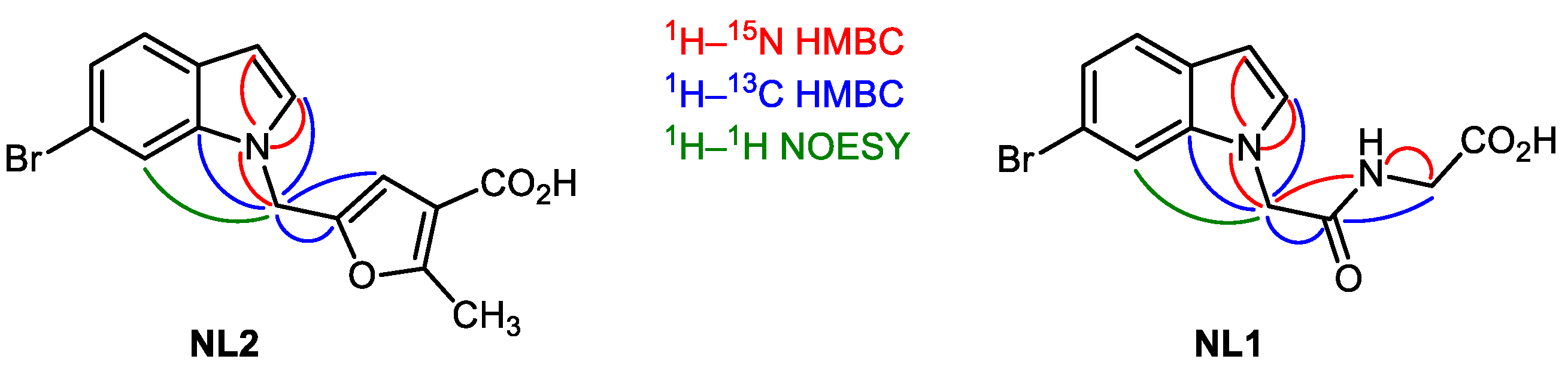{kind=link}






