
Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
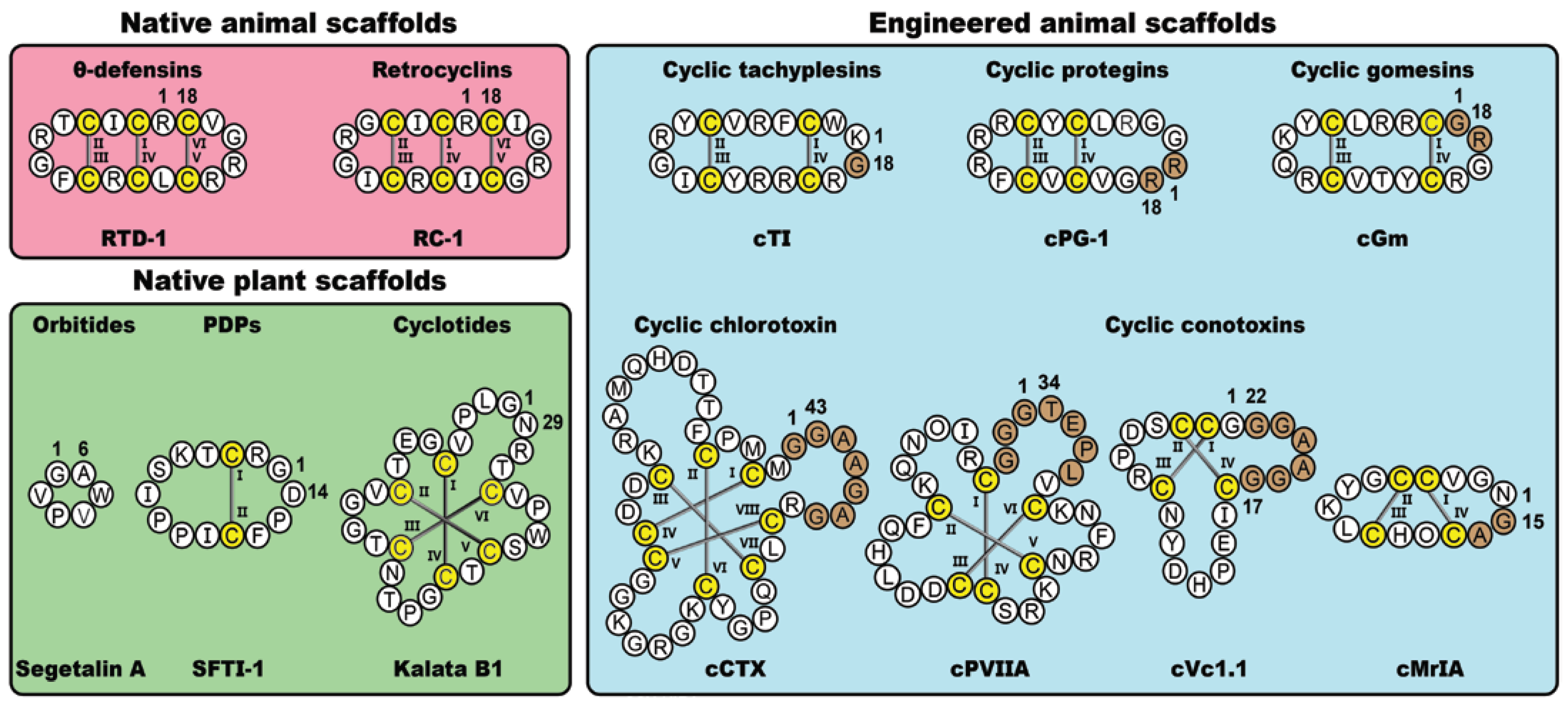
1. Introduction
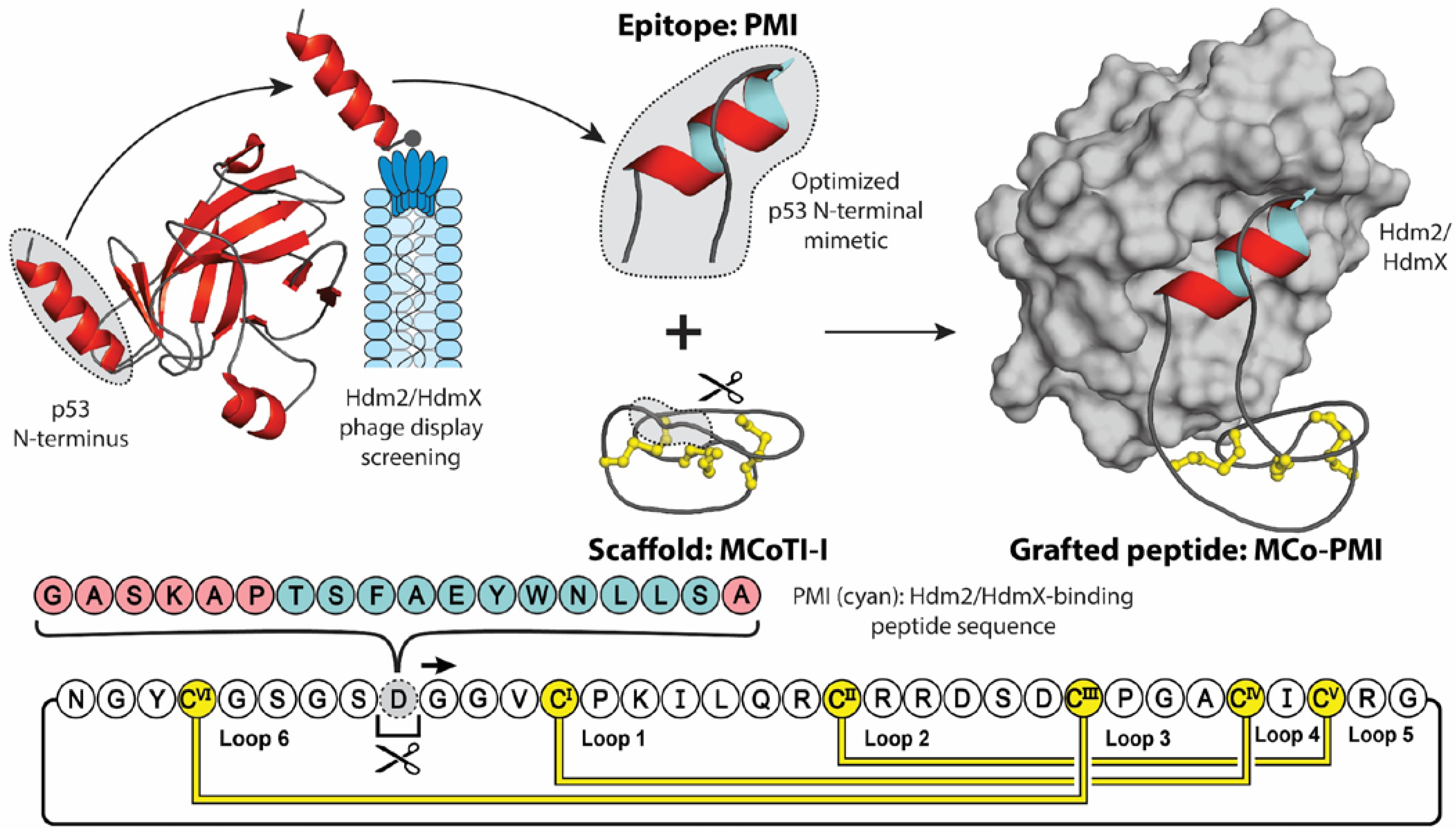
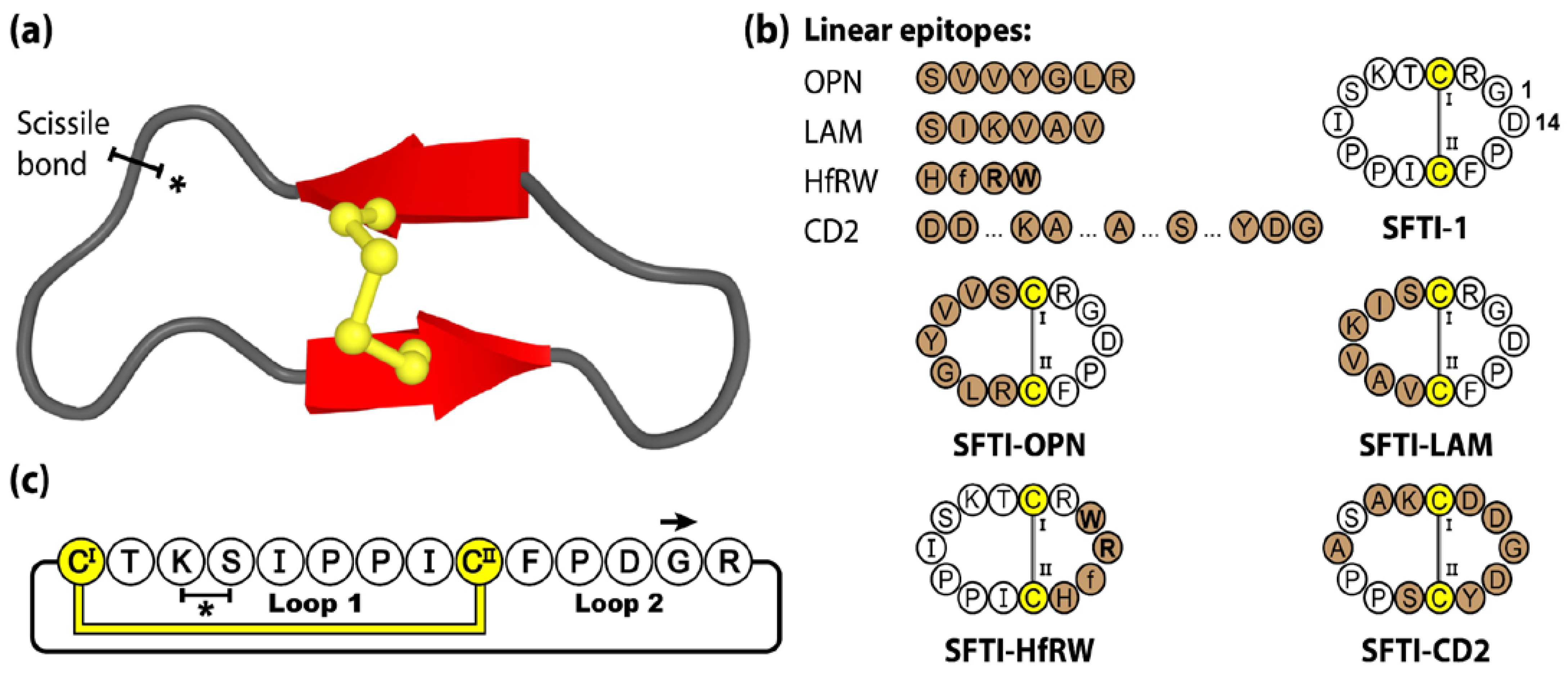
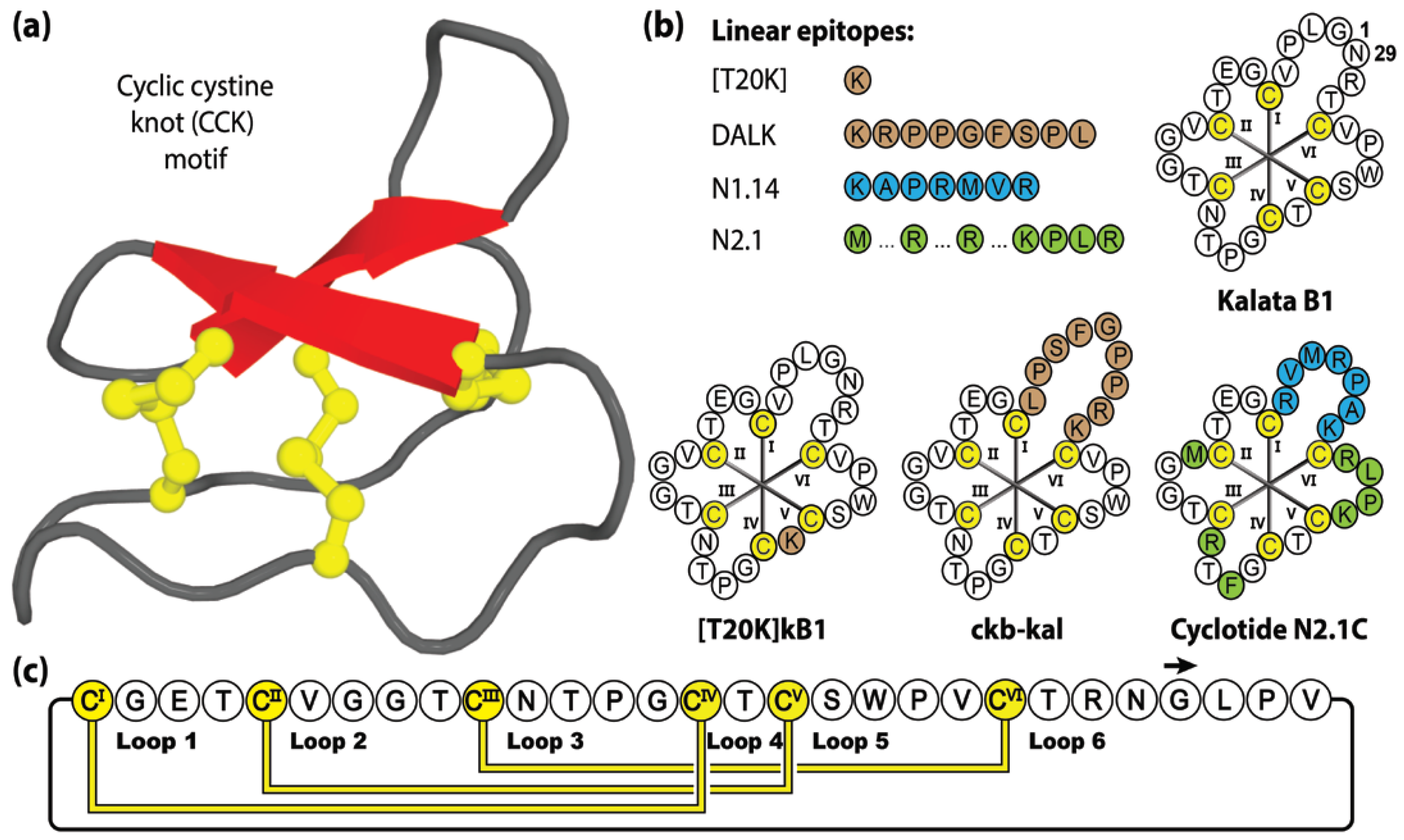
2. Molecular Grafting
3. Plant-Derived Cyclic Peptides
3.1. Orbitides
3.2. PawS-Derived Peptides (PDPs)
3.3. Cyclotides
4. Animal-Derived Cyclic Peptides
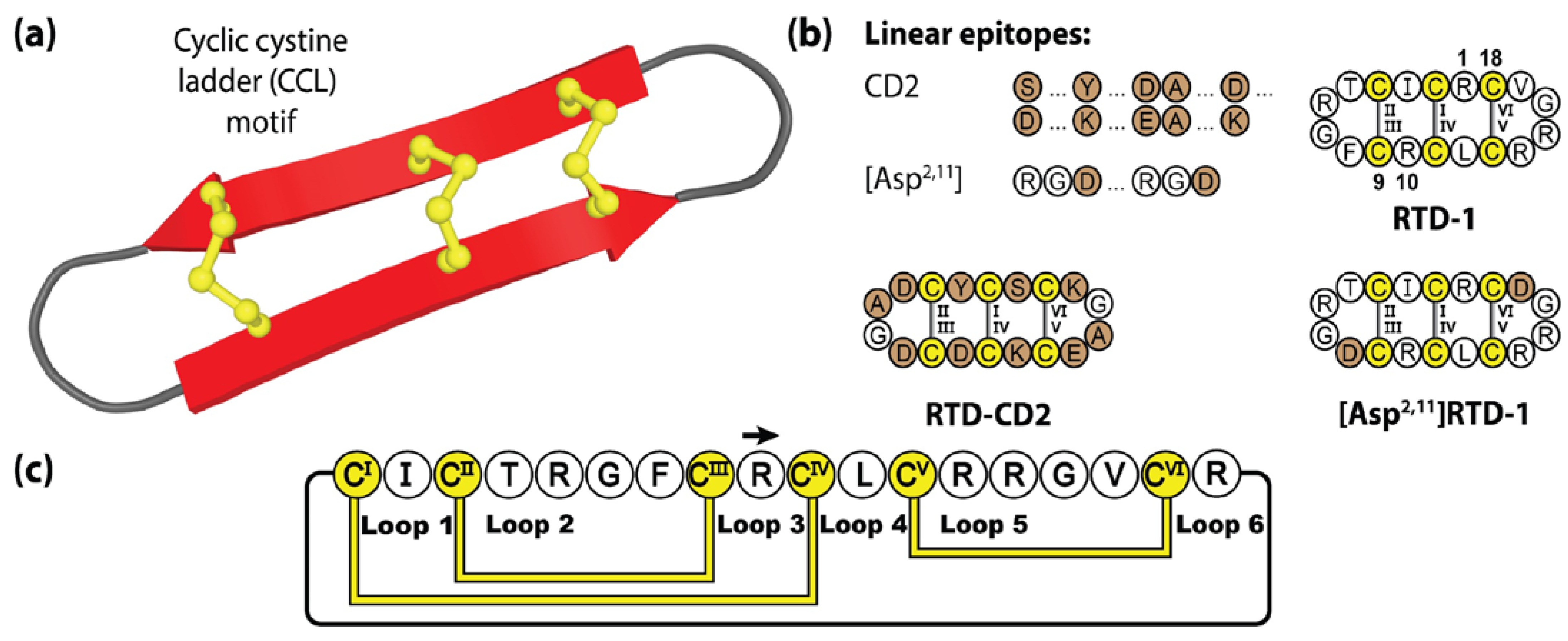
4.1. θ-Defensins
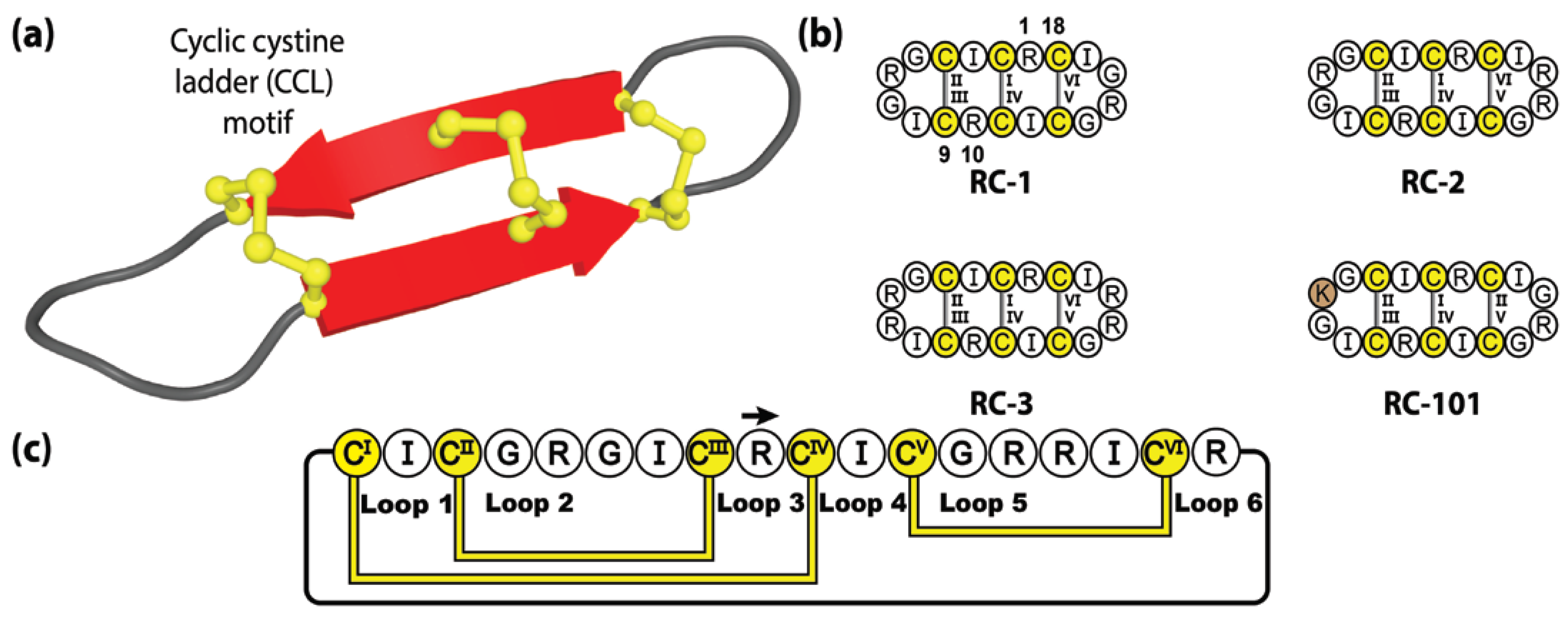
4.2. Retrocyclins
5. Engineered Cyclic Peptides
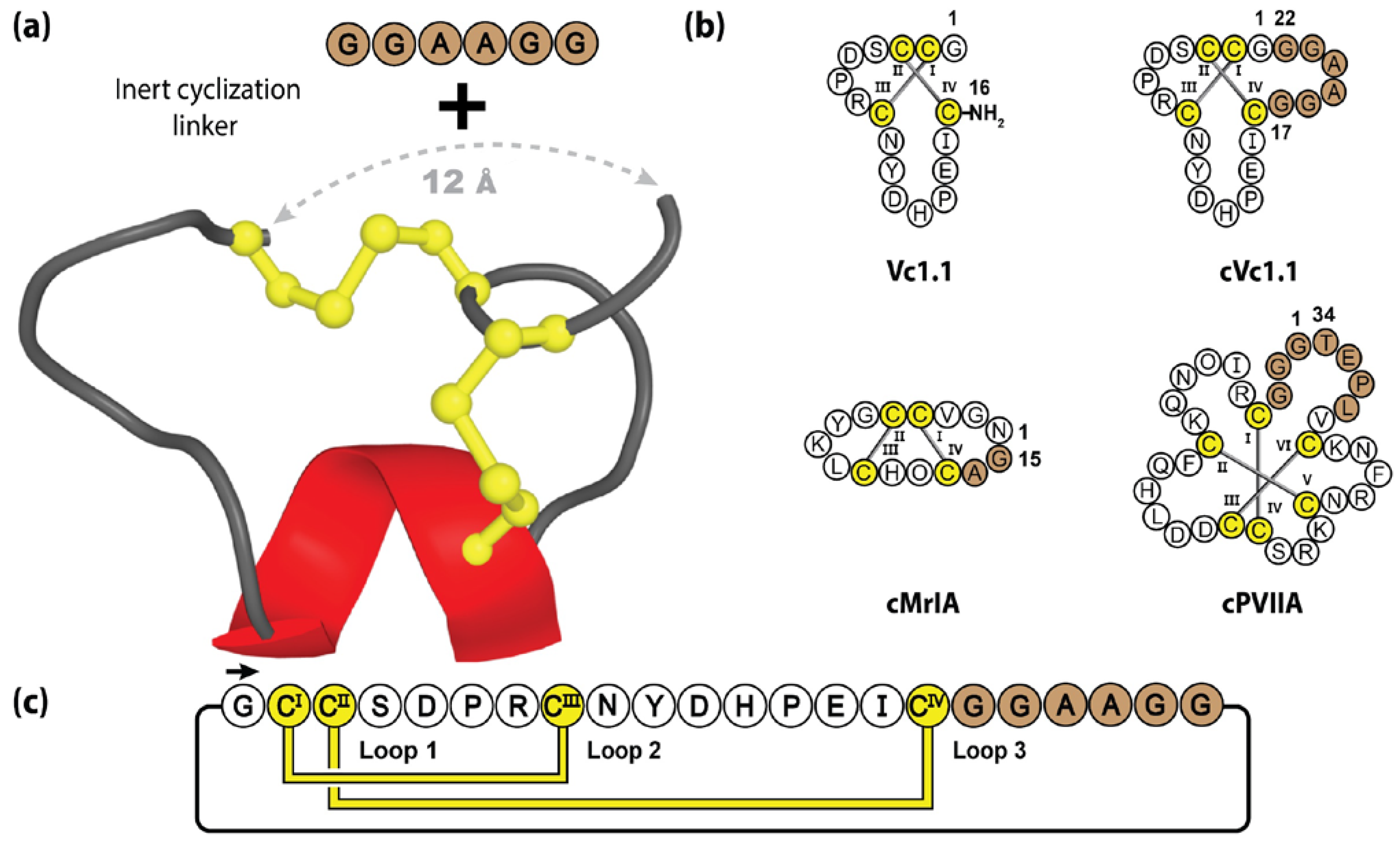
5.1. Cyclic Conotoxins
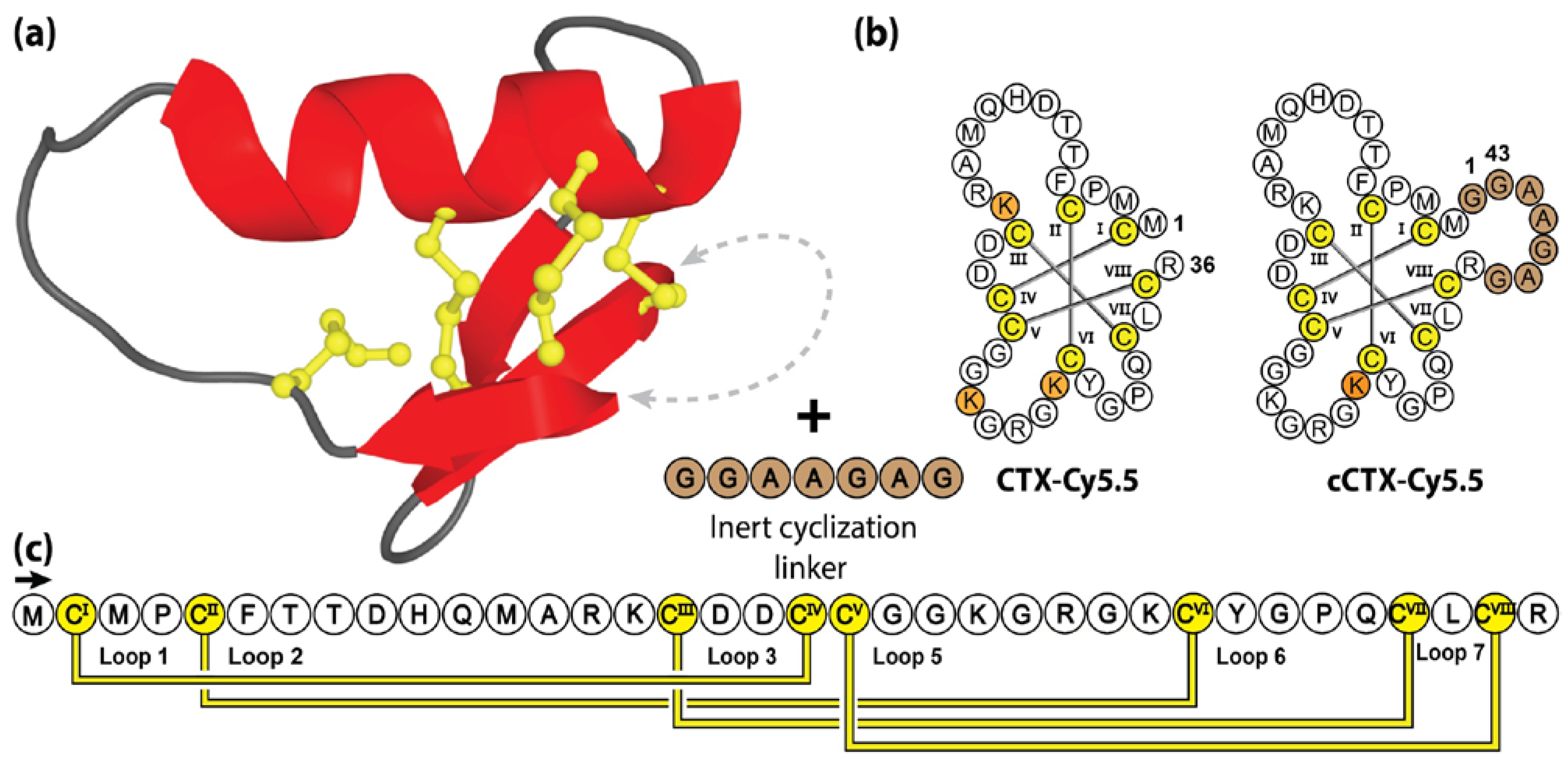
5.2. Cyclic Chlorotoxin
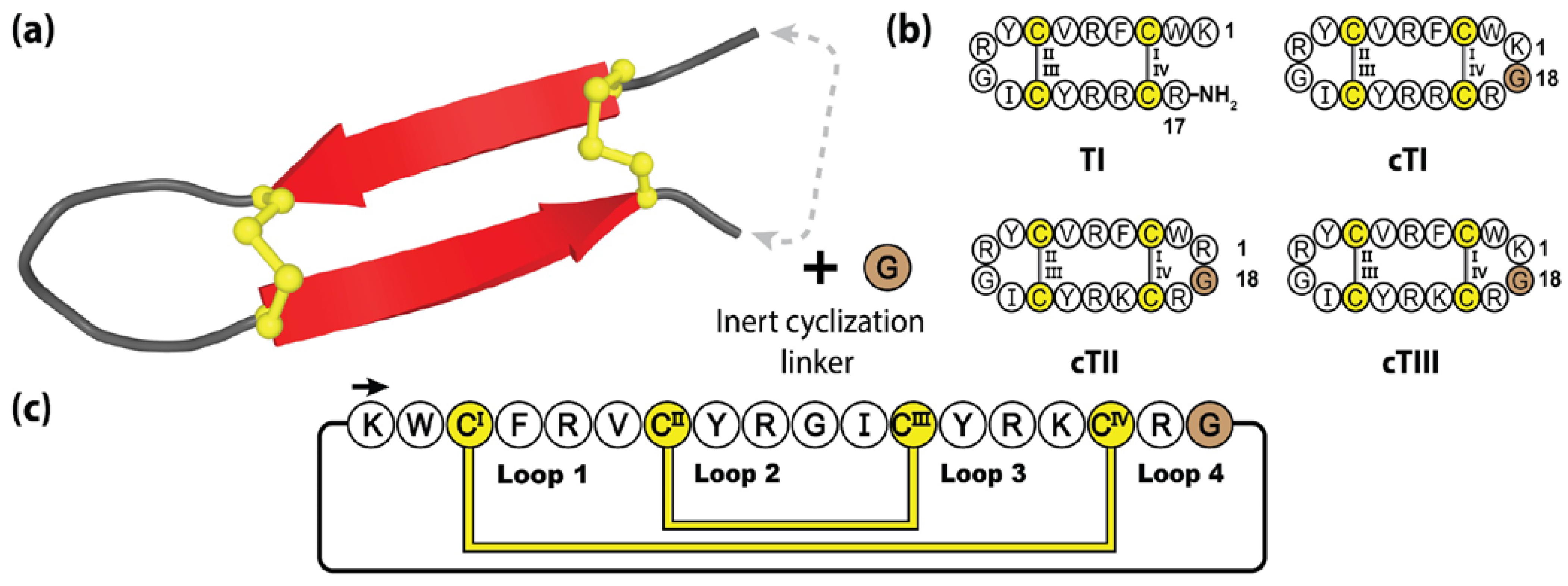
5.3. Cyclic Tachyplesins
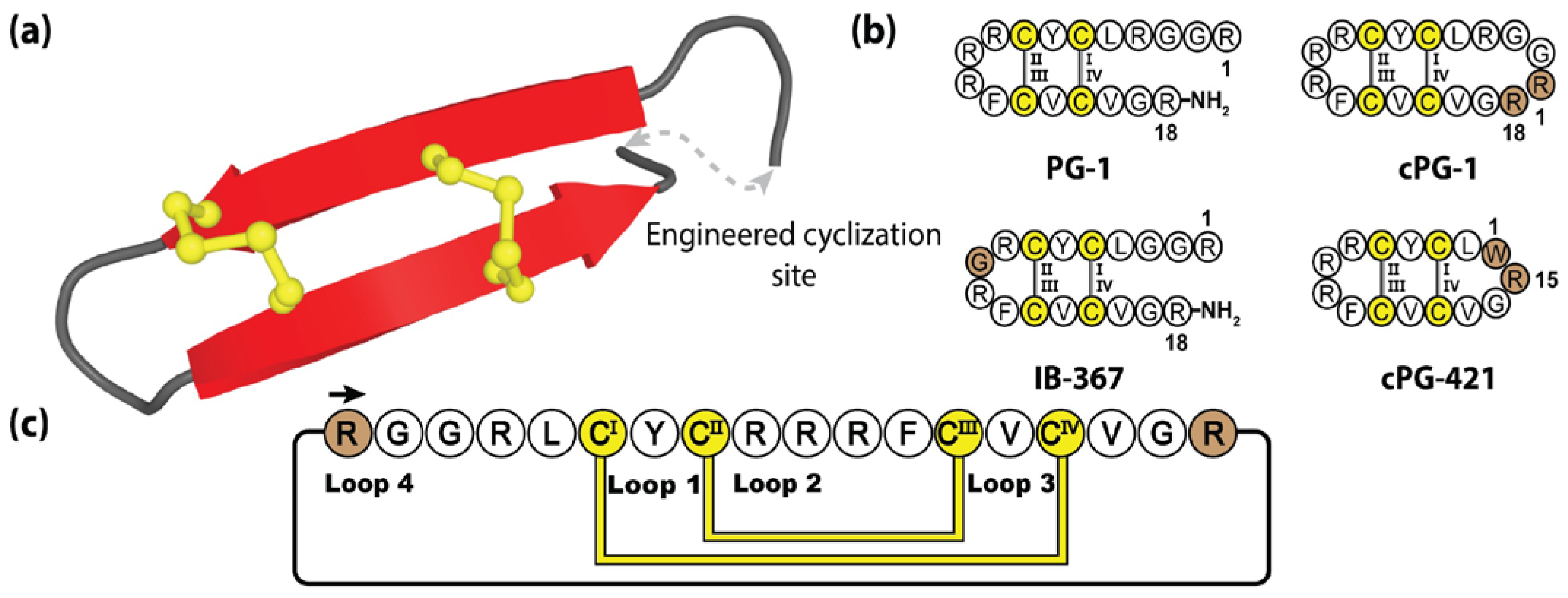
5.4. Cyclic Protegrins
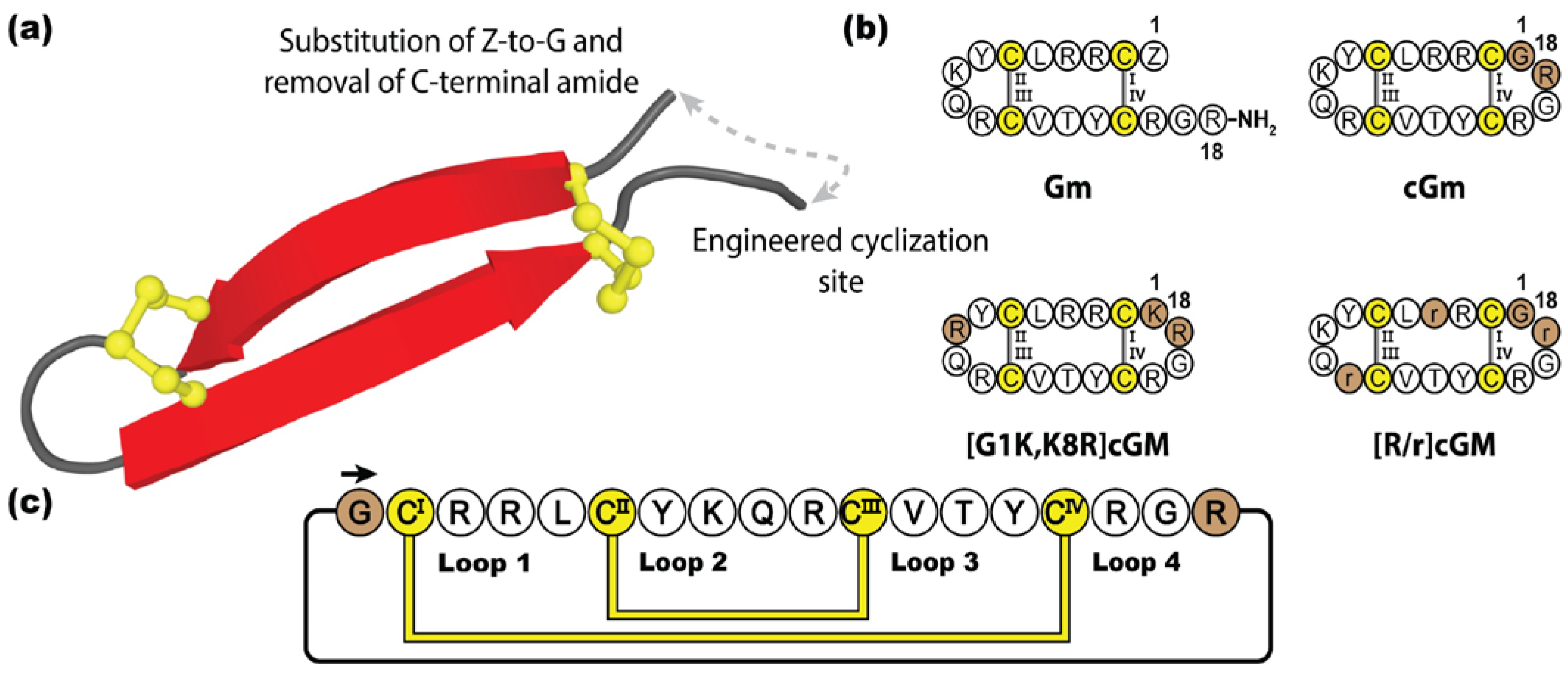
5.5. Cyclic Gomesins
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Craik, D.J.; Kan, M.W. How can we improve peptide drug discovery? Learning from the past. Expert Opin. Drug Discov. 2021, 16, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Philippe, G.J.B.; Craik, D.J.; Henriques, S.T. Converting peptides into drugs targeting intracellular protein-protein interactions. Drug Discov. Today 2021, 26, 1521–1531. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Tapeinou, A.; Matsoukas, M.T.; Simal, C.; Tselios, T. Review cyclic peptides on a merry-go-round; towards drug design. Biopolymers 2015, 104, 453–461. [Google Scholar] [CrossRef]
- Wang, W.; Khojasteh, S.C.; Su, D. Biosynthetic Strategies for Macrocyclic Peptides. Molecules 2021, 26, 3338. [Google Scholar] [CrossRef]
- McHugh, S.M.; Rogers, J.R.; Yu, H.; Lin, Y.-S. Insights into how cyclic peptides switch conformations. J. Chem. Theory Comput. 2016, 12, 2480–2488. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef]
- Räder, A.F.B.; Reichart, F.; Weinmüller, M.; Kessler, H. Improving oral bioavailability of cyclic peptides by N-methylation. Bioorgan. Med. Chem. 2018, 26, 2766–2773. [Google Scholar] [CrossRef]
- Chow, H.Y.; Zhang, Y.; Matheson, E.; Li, X. Ligation technologies for the synthesis of cyclic peptides. Chem. Rev. 2019, 119, 9971–10001. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Chia, L.Y.; Kumar, P.V.; Maki, M.A.A.; Ravichandran, G.; Thilagar, S. A Review: The Antiviral Activity of Cyclic Peptides. Int. J. Pept. Res. Ther. 2023, 29, 7. [Google Scholar] [CrossRef]
- Ramadhani, D.; Maharani, R.; Gazzali, A.M.; Muchtaridi, M. Cyclic Peptides for the Treatment of Cancers: A Review. Molecules 2022, 27, 4428. [Google Scholar] [CrossRef]
- Lai, S.; Zhang, Q.; Jin, L. Natural and Man-Made Cyclic Peptide-Based Antibiotics. Antibiotics 2022, 12, 42. [Google Scholar] [CrossRef]
- Barkan, D.T.; Cheng, X.L.; Celino, H.; Tran, T.T.; Bhandari, A.; Craik, C.S.; Sali, A.; Smythe, M.L. Clustering of disulfide-rich peptides provides scaffolds for hit discovery by phage display: Application to interleukin-23. BMC Bioinform. 2016, 17, 481–497. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Fass, D. Disulfide bonding in protein biophysics. Annu. Rev. Biophys. 2012, 41, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Manteca, A.; Alonso-Caballero, A.; Fertin, M.; Poly, S.; De Sancho, D.; Perez-Jimenez, R. The influence of disulfide bonds on the mechanical stability of proteins is context dependent. J. Biol. Chem. 2017, 292, 13374–13380. [Google Scholar] [CrossRef]
- Li, J.; Hu, S.; Jian, W.; Xie, C.; Yang, X. Plant antimicrobial peptides: Structures, functions, and applications. Bot. Stud. 2021, 62, 5. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ding, J.; Liao, C.; Xu, J.; Liu, X.; Lu, W. Defensins: The natural peptide antibiotic. Adv. Drug Deliv. Rev. 2021, 179, 114008. [Google Scholar] [CrossRef] [PubMed]
- Heilbronner, S.; Krismer, B.; Brotz-Oesterhelt, H.; Peschel, A. The microbiome-shaping roles of bacteriocins. Nat. Rev. Microbiol. 2021, 19, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Cristofori-Armstrong, B.; Israel, M.R.; Nixon, S.A.; Vetter, I.; King, G.F. Animal toxins—Nature’s evolutionary-refined toolkit for basic research and drug discovery. Biochem. Pharmacol. 2020, 181, 114096. [Google Scholar] [CrossRef]
- Morrison, C. Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018, 17, 531–533. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Zhang, W.D.; Hu, H.G. Stapled Helical Peptides Bearing Different Anchoring Residues. Chem. Rev. 2020, 120, 10079–10144. [Google Scholar] [CrossRef]
- Bluntzer, M.T.J.; O’Connell, J.; Baker, T.S.; Michel, J.; Hulme, A.N. Designing stapled peptides to inhibitprotein-protein interactions: An analysis of successes in a rapidly changing field. Pept. Sci. 2021, 113, e24191. [Google Scholar] [CrossRef]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Domling, A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Moiola, M.; Memeo, M.G.; Quadrelli, P. Stapled Peptides—A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. [Google Scholar] [CrossRef]
- Wang, C.K.; Craik, D.J. Linking molecular evolution to molecular grafting. J. Biol. Chem. 2021, 296, 100425. [Google Scholar] [CrossRef]
- Kashmiri, S.V.; De Pascalis, R.; Gonzales, N.R.; Schlom, J. SDR grafting—A new approach to antibody humanization. Methods 2005, 36, 25–34. [Google Scholar] [CrossRef]
- Jones, N.C. Gene expression: Negative regulation of enhancers. Nature 1986, 321, 202–223. [Google Scholar] [CrossRef]
- Wang, C.K.; Craik, D.J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Craik, D.J.; Lee, M.-H.; Rehm, F.B.H.; Tombling, B.; Doffek, B.; Peacock, H. Ribosomally-synthesised cyclic peptides from plants as drug leads and pharmaceutical scaffolds. Bioorganic Med. Chem. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Ji, Y.; Majumder, S.; Millard, M.; Borra, R.; Bi, T.; Elnagar, A.Y.; Neamati, N.; Shekhtman, A.; Camarero, J.A. In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. [Google Scholar] [CrossRef]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Rosal, R.; Brandt-Rauf, P.; Pincus, M.R.; Wang, H.; Mao, Y.; Li, Y.; Fine, R.L. The role of alpha-helical structure in p53 peptides as a determinant for their mechanism of cell death: Necrosis versus apoptosis. Adv. Drug Deliv. Rev. 2005, 57, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.; Elnagar, A.Y.O.; Hamm-Alvarez, S.F.; Camarero, J.A. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J. Control. Release 2011, 155, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Pazgier, M.; Liu, M.; Lu, W.Y.; Lu, W. Apamin as a template for structure-based rational design of potent peptide activators of p53. Angew. Chem. Int. Ed. Engl. 2009, 48, 8712–8715. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Ramalho, S.D.; Wang, C.K.; King, G.J.; Byriel, K.A.; Huang, Y.; Bolzani, V.S.; Craik, D.J. Synthesis, racemic X-ray crystallographic, and permeability studies of bioactive orbitides from jatropha species. J. Nat. Prod. 2018, 81, 2436–2445. [Google Scholar] [CrossRef]
- Shim, Y.Y.; Young, L.W.; Arnison, P.G.; Gilding, E.; Reaney, M.J. Proposed systematic nomenclature for orbitides. J. Nat. Prod. 2015, 78, 645–652. [Google Scholar] [CrossRef]
- Ramalho, S.D.; Pinto, M.E.F.; Ferreira, D.; Bolzani, V.S. Biologically active orbitides from the euphorbiaceae family. Planta Med. 2018, 84, 558–567. [Google Scholar] [CrossRef]
- Chekan, J.R.; Estrada, P.; Covello, P.S.; Nair, S.K. Characterization of the macrocyclase involved in the biosynthesis of RiPP cyclic peptides in plants. Proc. Natl. Acad. Sci. USA 2017, 114, 6551–6556. [Google Scholar] [CrossRef]
- Barbosa, S.C.; Nobre, T.M.; Volpati, D.; Ciancaglini, P.; Cilli, E.M.; Lorenzon, E.N.; Oliveira, O.N., Jr. The importance of cyclic structure for labaditin on its antimicrobial activity against Staphylococcus aureus. Colloids Surf. B Biointerfaces 2016, 148, 453–459. [Google Scholar] [CrossRef]
- Okinyo-Owiti, D.P.; Dong, Q.; Ling, B.; Jadhav, P.D.; Bauer, R.; Maley, J.M.; Reaney, M.J.T.; Yang, J.; Sammynaiken, R. Evaluating the cytotoxicity of flaxseed orbitides for potential cancer treatment. Toxicol. Rep. 2015, 2, 1014–1018. [Google Scholar] [CrossRef]
- Pinto, M.E.; Batista, J.M., Jr.; Koehbach, J.; Gaur, P.; Sharma, A.; Nakabashi, M.; Cilli, E.M.; Giesel, G.M.; Verli, H.; Gruber, C.W.; et al. Ribifolin, an orbitide from Jatropha ribifolia, and its potential antimalarial activity. J. Nat. Prod. 2015, 78, 374–380. [Google Scholar] [CrossRef]
- Altei, W.F.; Picchi, D.G.; Abissi, B.M.; Giesel, G.M.; Flausino, O., Jr.; Reboud-Ravaux, M.; Verli, H.; Crusca, E., Jr.; Silveira, E.R.; Cilli, E.M.; et al. Jatrophidin I, a cyclic peptide from Brazilian Jatropha curcas L.: Isolation, characterization, conformational studies and biological activity. Phytochemistry 2014, 107, 91–96. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Cebrat, M.; Siemion, I.Z.; Kay, J.E. Cyclolinopeptide A (CLA) mediates its immunosuppressive activity through cyclophilin-dependent calcineurin inactivation. FEBS Lett. 1997, 418, 224–227. [Google Scholar] [CrossRef]
- Thell, K.; Hellinger, R.; Schabbauer, G.; Gruber, C.W. Immunosuppressive peptides and their therapeutic applications. Drug Discov. Today 2014, 19, 645–653. [Google Scholar] [CrossRef]
- Katarzyńska, J.; Mazur, A.; Rudzinska, E.; Artym, J.; Zimecki, M.; Jankowski, S.; Zabrocki, J. Cyclolinopeptide derivatives modify methotrexate-induced suppression of the humoral immune response in mice. Eur. J. Med. Chem. 2011, 46, 4608–4617. [Google Scholar] [CrossRef]
- Morita, H.; Eda, M.; Iizuka, T.; Hirasawa, Y.; Sekiguchi, M.; Yun, Y.S.; Itokawa, H.; Takeya, K. Structure of a new cyclic nonapeptide, segetalin F, and vasorelaxant activity of segetalins from Vaccaria segetalis. Bioorganic Med. Chem. Lett. 2006, 16, 4458–4461. [Google Scholar] [CrossRef]
- Shim, Y.Y.; Song, Z.L.; Jadhav, P.D.; Reaney, M.J.T. Orbitides from flaxseed (Linum usitatissimum L.): A comprehensive review. Trends Food Sci. Technol. 2019, 93, 197–211. [Google Scholar] [CrossRef]
- Franke, B.; Mylne, J.S.; Rosengren, K.J. Buried treasure: Biosynthesis, structures and applications of cyclic peptides hidden in seed storage albumins. Nat. Prod. Rep. 2018, 35, 137–146. [Google Scholar] [CrossRef]
- Mylne, J.S.; Colgrave, M.L.; Daly, N.L.; Chanson, A.H.; Elliott, A.G.; McCallum, E.J.; Jones, A.; Craik, D.J. Albumins and their processing machinery are hijacked for cyclic peptides in sunflower. Nat. Chem. Biol. 2011, 7, 257–259. [Google Scholar] [CrossRef]
- Payne, C.D.; Franke, B.; Fisher, M.F.; Hajiaghaalipour, F.; McAleese, C.E.; Song, A.; Eliasson, C.; Zhang, J.; Jayasena, A.S.; Vadlamani, G.; et al. A chameleonic macrocyclic peptide with drug delivery applications. Chem. Sci. 2021, 12, 6670–6683. [Google Scholar] [CrossRef] [PubMed]
- de Veer, S.J.; White, A.M.; Craik, D.J. Sunflower Trypsin Inhibitor-1 (SFTI-1): Sowing Seeds in the Fields of Chemistry and Biology. Angew. Chem. Int. Ed. Engl. 2021, 60, 8050–8071. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.G.; Franke, B.; Armstrong, D.A.; Craik, D.J.; Mylne, J.S.; Rosengren, K.J. Natural structural diversity within a conserved cyclic peptide scaffold. Amino Acids 2017, 49, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Luckett, S.; Garcia, R.S.; Barker, J.J.; Konarev, A.V.; Shewry, P.R.; Clarke, A.R.; Brady, R.L. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 1999, 290, 525–533. [Google Scholar] [CrossRef]
- Qi, R.F.; Song, Z.W.; Chi, C.W. Structural features and molecular evolution of Bowman-Birk protease inhibitors and their potential application. Acta Biochim. Biophys. Sin. 2005, 37, 283–292. [Google Scholar] [CrossRef]
- Birk, Y. The Bowman-Birk inhibitor trypsin- and chymotrypsin-inhibitor from soybeans. Int. J. Pept. Protein Res. 1985, 25, 113–131. [Google Scholar] [CrossRef]
- Jaulent, A.M.; Leatherbarrow, R.J. Design, synthesis and analysis of novel bicyclic and bifunctional protease inhibitors. Protein Eng. Des. Sel. 2004, 17, 681–687. [Google Scholar] [CrossRef]
- Laskowski, M.; Qasim, M.A. What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes? Biochim. Biophys. Acta 2000, 1477, 324–337. [Google Scholar] [CrossRef]
- de Veer, S.J.; Swedberg, J.E.; Akcan, M.; Rosengren, K.J.; Brattsand, M.; Craik, D.J.; Harris, J.M. Engineered protease inhibitors based on sunflower trypsin inhibitor-1 (SFTI-1) provide insights into the role of sequence and conformation in Laskowski mechanism inhibition. Biochem. J. 2015, 469, 243–253. [Google Scholar] [CrossRef]
- de Veer, S.J.; Ukolova, S.S.; Munro, C.A.; Swedberg, J.E.; Buckle, A.M.; Harris, J.M. Mechanism-based selection of a potent kallikrein-related peptidase 7 inhibitor from a versatile library based on the sunflower trypsin inhibitor SFTI-1. Pept. Sci. 2013, 100, 510–518. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Chen, C.C.; He, Y.; Yu, M.; Xu, L.; Tian, C.L.; Guo, Q.X.; Shi, J.; Zhang, M.; Li, Y.M. Efficient synthesis of trypsin inhibitor SFTI-1 via intramolecular ligation of peptide hydrazide. Tetrahedron Lett. 2014, 55, 2883–2886. [Google Scholar] [CrossRef]
- Li, Y.; Aboye, T.; Breindel, L.; Shekhtman, A.; Camarero, J.A. Efficient recombinant expression of SFTI-1 in bacterial cells using intein-mediated protein trans-splicing. Biopolymers 2016, 106, 818–824. [Google Scholar] [CrossRef]
- Jackson, M.A.; Yap, K.; Poth, A.G.; Gilding, E.K.; Swedberg, J.E.; Poon, S.; Qu, H.; Durek, T.; Harris, K.; Anderson, M.A.; et al. Rapid and Scalable Plant-Based Production of a Potent Plasmin Inhibitor Peptide. Front. Plant Sci. 2019, 10, 602. [Google Scholar] [CrossRef]
- Northfield, S.E.; Wang, C.K.; Schroeder, C.I.; Durek, T.; Kan, M.W.; Swedberg, J.E.; Craik, D.J. Disulfide-rich macrocyclic peptides as templates in drug design. Eur. J. Med. Chem. 2014, 77, 248–257. [Google Scholar] [CrossRef]
- Sable, R.; Durek, T.; Taneja, V.; Craik, D.J.; Pallerla, S.; Gauthier, T.; Jois, S. Constrained cyclic peptides as immunomodulatory inhibitors of the CD2:CD58 protein-protein interaction. ACS Chem. Biol. 2016, 11, 2366–2374. [Google Scholar] [CrossRef]
- Swedberg, J.E.; Li, C.Y.; de Veer, S.J.; Wang, C.K.; Craik, D.J. Design of potent and selective cathepsin g inhibitors based on the sunflower trypsin inhibitor-1 scaffold. J. Med. Chem. 2017, 60, 658–667. [Google Scholar] [CrossRef]
- Quimbar, P.; Malik, U.; Sommerhoff, C.P.; Kaas, Q.; Chan, L.Y.; Huang, Y.H.; Grundhuber, M.; Dunse, K.; Craik, D.J.; Anderson, M.A.; et al. High-affinity cyclic peptide matriptase inhibitors. J. Biol. Chem. 2013, 288, 13885–13896. [Google Scholar] [CrossRef]
- Fittler, H.; Avrutina, O.; Empting, M.; Kolmar, H. Potent inhibitors of human matriptase-1 based on the scaffold of sunflower trypsin inhibitor. J. Pept. Sci. 2014, 20, 415–420. [Google Scholar] [CrossRef]
- Chan, L.Y.; Craik, D.J.; Daly, N.L. Cyclic thrombospondin-1 mimetics: Grafting of a thrombospondin sequence into circular disulfide-rich frameworks to inhibit endothelial cell migration. Biosci. Rep. 2015, 35, e00270. [Google Scholar] [CrossRef]
- Jendrny, C.; Beck-Sickinger, A.G. Inhibition of kallikrein-related peptidases 7 and 5 by grafting serpin reactive-center loop sequences onto sunflower trypsin inhibitor-1 (SFTI-1). ChemBioChem 2016, 17, 719–726. [Google Scholar] [CrossRef]
- Chan, L.Y.; Gunasekera, S.; Henriques, S.T.; Worth, N.F.; Le, S.J.; Clark, R.J.; Campbell, J.H.; Craik, D.J.; Daly, N.L. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood 2011, 118, 6709–6717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yamaguchi, S.; Nagamune, T. Sortase A-mediated synthesis of ligand-grafted cyclized peptides for modulating a model protein-protein interaction. Biotechnol. J. 2015, 10, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Northfield, S.E.; Huang, Y.H.; Ramos, M.C.; Craik, D.J. Inhibition of tau aggregation using a naturally-occurring cyclic peptide scaffold. Eur. J. Med. Chem. 2016, 109, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Cobos Caceres, C.; Bansal, P.S.; Navarro, S.; Wilson, D.; Don, L.; Giacomin, P.; Loukas, A.; Daly, N.L. An engineered cyclic peptide alleviates symptoms of inflammation in a murine model of inflammatory bowel disease. J. Biol. Chem. 2017, 292, 10288–10294. [Google Scholar] [CrossRef]
- Qiu, Y.; Taichi, M.; Wei, N.; Yang, H.; Luo, K.Q.; Tam, J.P. An orally active bradykinin B1 receptor antagonist engineered as a bifunctional chimera of sunflower trypsin inhibitor. J. Med. Chem. 2017, 60, 504–510. [Google Scholar] [CrossRef]
- Durek, T.; Cromm, P.M.; White, A.M.; Schroeder, C.I.; Kaas, Q.; Weidmann, J.; Ahmad Fuaad, A.; Cheneval, O.; Harvey, P.J.; Daly, N.L.; et al. Development of Novel Melanocortin Receptor Agonists Based on the Cyclic Peptide Framework of Sunflower Trypsin Inhibitor-1. J. Med. Chem. 2018, 61, 3674–3684. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef]
- de Veer, S.J.; Kan, M.W.; Craik, D.J. Cyclotides: From structure to function. Chem. Rev. 2019, 119, 12375–12421. [Google Scholar] [CrossRef]
- Huang, Y.H.; Du, Q.; Craik, D.J. Cyclotides: Disulfide-rich peptide toxins in plants. Toxicon 2019, 172, 33–44. [Google Scholar] [CrossRef]
- Poth, A.G.; Colgrave, M.L.; Philip, R.; Kerenga, B.; Daly, N.L.; Anderson, M.A.; Craik, D.J. Discovery of cyclotides in the Fabaceae plant family provides new insights into the cyclization, evolution, and distribution of circular proteins. ACS Chem. Biol. 2011, 6, 345–355. [Google Scholar] [CrossRef]
- Gruber, C.W.; Elliott, A.G.; Ireland, D.C.; Delprete, P.G.; Dessein, S.; Göransson, U.; Trabi, M.; Wang, C.K.; Kinghorn, A.B.; Robbrecht, E.; et al. Distribution and evolution of circular miniproteins in flowering plants. Plant Cell 2008, 20, 2471–2483. [Google Scholar] [CrossRef]
- Poth, A.G.; Mylne, J.S.; Grassl, J.; Lyons, R.E.; Millar, A.H.; Colgrave, M.L.; Craik, D.J. Cyclotides associate with leaf vasculature and are the products of a novel precursor in Petunia (Solanaceae). J. Biol. Chem. 2012, 287, 27033–27046. [Google Scholar] [CrossRef]
- Kaas, Q.; Craik, D.J. Analysis and classification of circular proteins in CyBase. Biopolym. Pept. Sci. 2010, 94, 584–591. [Google Scholar] [CrossRef]
- Burman, R.; Gruber, C.W.; Rizzardi, K.; Herrmann, A.; Craik, D.J.; Gupta, M.P.; Göransson, U. Cyclotide proteins and precursors from the genus Gloeospermum: Filling a blank spot in the cyclotide map of Violaceae. Phytochemistry 2010, 71, 13–20. [Google Scholar] [CrossRef]
- Craik, D.J.; Cemazar, M.; Wang, C.K.; Daly, N.L. The cyclotide family of circular miniproteins: Nature’s combinatorial peptide template. Biopolymers 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Wang, C.K.; Colgrave, M.L.; Gustafson, K.R.; Ireland, D.C.; Göransson, U.; Craik, D.J. Anti-HIV cyclotides from the Chinese medicinal herb Viola yedoensis. J. Nat. Prod. 2008, 71, 47–52. [Google Scholar] [CrossRef]
- Daly, N.L.; Gustafson, K.R.; Craik, D.J. The role of the cyclic peptide backbone in the anti-HIV activity of the cyclotide kalata B1. FEBS Lett. 2004, 574, 69–72. [Google Scholar] [CrossRef]
- Daly, N.L.; Clark, R.J.; Plan, M.R.; Craik, D.J. Kalata B8, a novel antiviral circular protein, exhibits conformational flexibility in the cystine knot motif. Biochem. J. 2006, 393, 619–626. [Google Scholar] [CrossRef]
- Gerlach, S.L.; Yeshak, M.; Göransson, U.; Roy, U.; Izadpanah, R.; Mondal, D. Cycloviolacin O2 (CyO2) suppresses productive infection and augments the antiviral efficacy of nelfinavir in HIV-1 infected monocytic cells. Biopolym. Pept. Sci. 2013, 100, 471–479. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Sowder, R.C.; Pannell, L.K.; Hughes, C.B.; Johnson, D.G.; Gulakowski, R.; Cardellina, J.H.; Boyd, M.R. Cycloviolins A-D, anti-HIV macrocyclic peptides from Leonia cymosa. J. Org. Chem. 2000, 65, 124–128. [Google Scholar] [CrossRef]
- Bokesch, H.R.; Pannell, L.K.; Cochran, P.K.; Sowder, R.C.; McKee, T.C.; Boyd, M.R. A novel anti-HIV macrocyclic peptide from Palicourea condensata. J. Nat. Prod. 2001, 64, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Colgrave, M.L.; Daly, N.L.; Rosengren, K.J.; Gustafson, K.R.; Craik, D.J. Isolation and characterization of novel cyclotides from Viola hederaceae: Solution structure and anti-HIV activity of vhl-1, a leaf-specific expressed cyclotide. J. Biol. Chem. 2005, 280, 22395–22405. [Google Scholar] [CrossRef] [PubMed]
- Ireland, D.C.; Wang, C.K.; Wilson, J.A.; Gustafson, K.R.; Craik, D.J. Cyclotides as natural anti-HIV agents. Biopolym. Pept. Sci. 2008, 90, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, K.R.; Sowder, R.C.; Henderson, L.E.; Parsons, I.C.; Kashman, Y.; Cardellina, J.H.; McMahon, J.B.; Buckheit, R.W.; Pannell, L.K.; Boyd, M.R. Circulins A and B: Novel HIV-inhibitory macrocyclic peptides from the tropical tree Chassalia parvifolia. J. Am. Chem. Soc. 1994, 116, 9337–9338. [Google Scholar] [CrossRef]
- Liu, M.Z.; Yang, Y.; Zhang, S.X.; Tang, L.; Wang, H.M.; Chen, C.J.; Shen, Z.F.; Cheng, K.D.; Kong, J.Q.; Wang, W. A cyclotide against influenza A H1N1 virus from Viola yedoensis. Acta Pharm. Sin. 2014, 49, 905–912. [Google Scholar]
- Fensterseifer, I.C.; Silva, O.N.; Malik, U.; Ravipati, A.S.; Novaes, N.R.F.; Miranda, P.R.R.; Rodrigues, E.A.; Moreno, S.E.; Craik, D.J.; Franco, O.L. Effects of cyclotides against cutaneous infections caused by Staphylococcus aureus. Peptides 2015, 63, 38–42. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.A.; Yang, J.L.; Chiu, K.W. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. USA 1999, 96, 8913–8918. [Google Scholar] [CrossRef]
- Gran, L.; Sletten, K.; Skjeldal, L. Cyclic peptides from Oldenlandia affinis DC. Molecular and biological properties. Chem. Biodivers. 2008, 5, 2014–2022. [Google Scholar] [CrossRef]
- Pränting, M.; Lööv, C.; Burman, R.; Göransson, U.; Andersson, D.I. The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 1964–1971. [Google Scholar] [CrossRef]
- Gran, L. On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. 1973, 33, 400–408. [Google Scholar] [CrossRef]
- Koehbach, J.; O’Brien, M.; Muttenthaler, M.; Miazzo, M.; Akcan, M.; Elliott, A.G.; Daly, N.L.; Harvey, P.J.; Arrowsmith, S.; Gunasekera, S.; et al. Oxytocic plant cyclotides as templates for peptide G protein-coupled receptor ligand design. Proc. Natl. Acad. Sci. USA 2013, 110, 21183–21188. [Google Scholar] [CrossRef]
- Gerlach, S.L.; Rathinakumar, R.; Chakravarty, G.; Göransson, U.; Wimley, W.C.; Darwin, S.P.; Mondal, D. Anticancer and chemosensitizing abilities of cycloviolacin O2 from Viola odorata and psyle cyclotides from Psychotria leptothyrsa. Biopolym. Pept. Sci. 2010, 94, 617–625. [Google Scholar] [CrossRef]
- Tang, J.; Wang, C.K.; Pan, X.; Yan, H.; Zeng, G.; Xu, W.; He, W.; Daly, N.L.; Craik, D.J.; Tan, N. Isolation and characterization of cytotoxic cyclotides from Viola tricolor. Peptides 2010, 31, 1434–1440. [Google Scholar] [CrossRef]
- Svangård, E.; Göransson, U.; Hocaoglu, Z.; Gullbo, J.; Larsson, R.; Claeson, P.; Bohlin, L. Cytotoxic cyclotides from Viola tricolor. J. Nat. Prod. 2004, 67, 144–147. [Google Scholar] [CrossRef]
- Herrmann, A.; Burman, R.; Mylne, J.S.; Karlsson, G.; Gullbo, J.; Craik, D.J.; Clark, R.J.; Göransson, U. The alpine violet, Viola biflora, is a rich source of cyclotides with potent cytotoxicity. Phytochemistry 2008, 69, 939–952. [Google Scholar] [CrossRef]
- Gerlach, S.L.; Burman, R.; Bohlin, L.; Mondal, D.; Göransson, U. Isolation, characterization, and bioactivity of cyclotides from the Micronesian plant Psychotria leptothyrsa. J. Nat. Prod. 2010, 73, 1207–1213. [Google Scholar] [CrossRef]
- Gründemann, C.; Koehbach, J.; Huber, R.; Gruber, C.W. Do plant cyclotides have potential as immunosuppressant peptides? J. Nat. Prod. 2012, 75, 167–174. [Google Scholar] [CrossRef]
- Grundemann, C.; Thell, K.; Lengen, K.; Garcia-Kaufer, M.; Huang, Y.H.; Huber, R.; Craik, D.J.; Schabbauer, G.; Gruber, C.W. Cyclotides suppress human T-lymphocyte proliferation by an interleukin 2-dependent mechanism. PLoS ONE 2013, 8, e68016. [Google Scholar] [CrossRef]
- Wieczorek, Z.; Siemion, I.Z.; Zimecki, M.; Bolewska-Pedyczak, E.; Wieland, T. Immunosuppressive activity in the series of cycloamanide peptides from mushrooms. Peptides 1993, 14, 1–5. [Google Scholar] [CrossRef]
- Hellinger, R.; Koehbach, J.; Fedchuk, H.; Sauer, B.; Huber, R.; Gruber, C.W.; Gründemann, C. Immunosuppressive activity of an aqueous Viola tricolor herbal extract. J. Ethnopharmacol. 2014, 151, 299–306. [Google Scholar] [CrossRef]
- Li, Y.; Bi, T.; Camarero, J.A. Chemical and biological production of cyclotides. Adv. Bot. Res. 2015, 76, 271–303. [Google Scholar] [PubMed]
- Yap, K.; Du, J.; Rehm, F.B.H.; Tang, S.R.; Zhou, Y.; Xie, J.; Wang, C.K.; de Veer, S.J.; Lua, L.H.L.; Durek, T.; et al. Yeast-based bioproduction of disulfide-rich peptides and their cyclization via asparaginyl endopeptidases. Nat. Protoc. 2021, 16, 1740–1760. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.A.; Xie, J.; Nguyen, L.T.T.; Wang, X.; Yap, K.; Harvey, P.J.; Gilding, E.K.; Craik, D.J. Plant-based production of an orally active cyclotide for the treatment of multiple sclerosis. Transgenic Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Camarero, J.A.; Campbell, M.J. The potential of the cyclotide scaffold for drug development. Biomedicines 2019, 7, 31. [Google Scholar] [CrossRef]
- Huang, Y.H.; Colgrave, M.L.; Clark, R.J.; Kotze, A.C.; Craik, D.J. Lysine-scanning mutagenesis reveals a previously unidentified amendable face of the cyclotide kalata B1 for the optimisation of nematocidal activity. J. Biol. Chem. 2010, 285, 10797–10805. [Google Scholar] [CrossRef]
- Wong, C.T.T.; Rowlands, D.K.; Wong, C.H.; Lo, T.W.C.; Nguyen, G.K.T.; Li, H.Y.; Tam, J.P. Orally active peptidic bradykinin B-1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. 2012, 51, 5620–5624. [Google Scholar] [CrossRef]
- Getz, J.A.; Cheneval, O.; Craik, D.J.; Daugherty, P.S. Design of a cyclotide antagonist of neuropilin-1 and -2 that potently inhibits endothelial cell migration. ACS Chem. Biol. 2013, 8, 1147–1154. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef]
- Conibear, A.C.; Craik, D.J. The chemistry and biology of theta defensins. Angew. Chem. Int. Ed. 2014, 53, 10612–10623. [Google Scholar] [CrossRef]
- Conibear, A.C.; Rosengren, K.J.; Harvey, P.J.; Craik, D.J. Structural characterization of the cyclic cystine ladder motif of theta-defensins. Biochemistry 2012, 51, 9718–9726. [Google Scholar] [CrossRef]
- Conibear, A.C.; Rosengren, K.J.; Daly, N.L.; Henriques, S.T.; Craik, D.J. The cyclic cystine ladder in theta-defensins is important for structure and stability, but not antibacterial activity. J. Biol. Chem. 2013, 288, 10830–10840. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Cole, A.M.; Selsted, M.E. θ-Defensins: Cyclic peptides with endless potential. J. Biol. Chem. 2012, 287, 27014–27019. [Google Scholar] [CrossRef]
- Garcia, A.E.; Osapay, G.; Tran, P.A.; Yuan, J.; Selsted, M.E. Isolation, synthesis, and antimicrobial activities of naturally occurring theta-defensin isoforms from baboon leukocytes. Infect. Immun. 2008, 76, 5883–5891. [Google Scholar] [CrossRef]
- Bensman, T.J.; Jayne, J.G.; Sun, M.; Kimura, E.; Meinert, J.; Wang, J.C.; Schaal, J.B.; Tran, D.; Rao, A.P.; Akbari, O.; et al. Efficacy of rhesus theta-defensin-1 in experimental models of Pseudomonas aeruginosa lung infection and inflammation. Antimicrob. Agents Chemother. 2017, 61, e00154-17. [Google Scholar] [CrossRef]
- Schaal, J.B.; Tran, D.; Tran, P.; Osapay, G.; Trinh, K.; Roberts, K.D.; Brasky, K.M.; Tongaonkar, P.; Ouellette, A.J.; Selsted, M.E. Rhesus macaque theta defensins suppress inflammatory cytokines and enhance survival in mouse models of bacteremic sepsis. PLoS ONE 2012, 7, e51337. [Google Scholar] [CrossRef]
- Wilmes, M.; Stockem, M.; Bierbaum, G.; Schlag, M.; Gotz, F.; Tran, D.Q.; Schaal, J.B.; Ouellette, A.J.; Selsted, M.E.; Sahl, H.G. Killing of staphylococci by theta-defensins involves membrane impairment and activation of autolytic enzymes. Antibiotics 2014, 3, 617–631. [Google Scholar] [CrossRef]
- Tongaonkar, P.; Trinh, K.K.; Schaal, J.B.; Tran, D.; Gulko, P.S.; Ouellette, A.J.; Selsted, M.E. Rhesus macaque theta-defensin RTD-1 inhibits proinflammatory cytokine secretion and gene expression by inhibiting the activation of NF-kappaB and MAPK pathways. J. Leukoc. Biol. 2015, 98, 1061–1070. [Google Scholar] [CrossRef]
- Tran, D.; Tran, P.; Roberts, K.; Osapay, G.; Schaal, J.; Ouellette, A.; Selsted, M.E. Microbicidal properties and cytocidal selectivity of rhesus macaque theta defensins. Antimicrob. Agents Chemother. 2008, 52, 944–953. [Google Scholar] [CrossRef]
- Wohlford-Lenane, C.L.; Meyerholz, D.K.; Perlman, S.; Zhou, H.; Tran, D.; Selsted, M.E.; McCray, P.B., Jr. Rhesus theta-defensin prevents death in a mouse model of severe acute respiratory syndrome coronavirus pulmonary disease. J. Virol. 2009, 83, 11385–11390. [Google Scholar] [CrossRef]
- Negahdaripour, M.; Rahbar, M.R.; Mosalanejad, Z.; Gholami, A. Theta-Defensins to Counter COVID-19 as Furin Inhibitors: In Silico Efficiency Prediction and Novel Compound Design. Comput. Math. Methods Med. 2022, 2022, 9735626. [Google Scholar] [CrossRef]
- Oh, Y.T.; Tran, D.; Buchanan, T.A.; Selsted, M.E.; Youn, J.H. theta-Defensin RTD-1 improves insulin action and normalizes plasma glucose and FFA levels in diet-induced obese rats. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E154–E160. [Google Scholar] [CrossRef] [PubMed]
- Penberthy, W.T.; Chari, S.; Cole, A.L.; Cole, A.M. Retrocyclins and their activity against HIV-1. Cell. Mol. Life Sci. 2011, 68, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Bochen, A.; Rosengren, K.J.; Stupar, P.; Wang, C.; Kessler, H.; Craik, D.J. The cyclic cystine ladder of theta-defensins as a stable, bifunctional scaffold: A proof-of-concept study using the integrin-binding RGD motif. ChemBioChem 2014, 15, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, N.; Cole, A.L.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Stuchlik, O.; Pohl, J.; Cole, A.M. Reawakening retrocyclins: Ancestral human defensins active against HIV-1. PLoS Biol. 2009, 7, e95. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Hong, T.; Boo, L.M.; Nguyen, T.; Zhao, C.; Bristol, G.; Zack, J.A.; Waring, A.J.; Yang, O.O.; Lehrer, R.I. Retrocyclin: A primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc. Natl. Acad. Sci. USA 2002, 99, 1813–1818. [Google Scholar] [CrossRef]
- Owen, S.M.; Rudolph, D.L.; Wang, W.; Cole, A.M.; Waring, A.J.; Lal, R.B.; Lehrer, R.I. RC-101, a retrocyclin-1 analogue with enhanced activity against primary HIV type 1 isolates. AIDS Res. Hum. Retrovir. 2004, 20, 1157–1165. [Google Scholar] [CrossRef]
- Kudryashova, E.; Zani, A.; Vilmen, G.; Sharma, A.; Lu, W.; Yount, J.S.; Kudryashov, D.S. Inhibition of SARS-CoV-2 Infection by Human Defensin HNP1 and Retrocyclin RC-101. J. Mol. Biol. 2022, 434, 167225. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Bjorn-Yoshimoto, W.E.; Ramiro, I.B.L.; Yandell, M.; McIntosh, J.M.; Olivera, B.M.; Ellgaard, L.; Safavi-Hemami, H. Curses or Cures: A Review of the Numerous Benefits Versus the Biosecurity Concerns of Conotoxin Research. Biomedicines 2020, 8, 235. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Expert Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone Snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef]
- Clark, R.J.; Fischer, H.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The synthesis, structural characterization, and receptor specificity of the alpha-conotoxin Vc1.1. J. Biol. Chem. 2006, 281, 23254–23263. [Google Scholar] [CrossRef]
- Sandall, D.W.; Satkunanathan, N.; Keays, D.A.; Polidano, M.A.; Liping, X.; Pham, V.; Down, J.G.; Khalil, Z.; Livett, B.G.; Gayler, K.R. A novel alpha-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 2003, 42, 6904–6911. [Google Scholar] [CrossRef]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef]
- Clark, R.J.; Akcan, M.; Kaas, Q.; Daly, N.L.; Craik, D.J. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 2012, 59, 446–455. [Google Scholar] [CrossRef]
- Castro, J.; Grundy, L.; Deiteren, A.; Harrington, A.M.; O’Donnell, T.; Maddern, J.; Moore, J.; Garcia-Caraballo, S.; Rychkov, G.Y.; Yu, R.; et al. Cyclic analogues of alpha-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. Br. J. Pharmacol. 2018, 175, 2384–2398. [Google Scholar] [CrossRef]
- Poth, A.G.; Chiu, F.C.K.; Stalmans, S.; Hamilton, B.R.; Huang, Y.-H.; Shackleford, D.M.; Patil, R.; Le, T.T.; Kan, M.-W.; Durek, T.; et al. Effects of backbone cyclization on the pharmacokinetics and drug efficiency of the orally active analgesic conotoxin cVc1.1. Med. Drug Discov. 2021, 10, 100087. [Google Scholar] [CrossRef]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar] [CrossRef]
- DeBin, J.A.; Maggio, J.E.; Strichartz, G.R. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am. J. Physiol. 1993, 264, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Dardevet, L.; Rani, D.; Aziz, T.A.; Bazin, I.; Sabatier, J.M.; Fadl, M.; Brambilla, E.; De Waard, M. Chlorotoxin: A helpful natural scorpion peptide to diagnose glioma and fight tumor invasion. Toxins 2015, 7, 1079–1101. [Google Scholar] [CrossRef] [PubMed]
- Čemažar, M.; Kwon, S.; Mahatmanto, T.; Ravipati, A.S.; Craik, D.J. Discovery and applications of disulfide-rich cyclic peptides. Curr. Top. Med. Chem. 2012, 12, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Fu, Y.; An, N.; Li, K.; Zheng, Y.; Liang, A. Chlorotoxin-conjugated nanoparticles as potential glioma-targeted drugs. J. Neurooncol. 2012, 107, 457–462. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, J.; Qiao, W.; Chen, K. Recent advances in diagnosis and treatment of gliomas using chlorotoxin-based bioconjugates. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 385–405. [Google Scholar]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef]
- Veiseh, M.; Gabikian, P.; Bahrami, S.B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar] [CrossRef]
- Butte, P.V.; Mamelak, A.; Parrish-Novak, J.; Drazin, D.; Shweikeh, F.; Gangalum, P.R.; Chesnokova, A.; Ljubimova, J.Y.; Black, K. Near-infrared imaging of brain tumors using the Tumor Paint BLZ-100 to achieve near-complete resection of brain tumors. Neurosurg. Focus 2014, 36, E1. [Google Scholar] [CrossRef]
- Fidel, J.; Kennedy, K.C.; Dernell, W.S.; Hansen, S.; Wiss, V.; Stroud, M.R.; Molho, J.I.; Knoblaugh, S.E.; Meganck, J.; Olson, J.M.; et al. Preclinical Validation of the Utility of BLZ-100 in Providing Fluorescence Contrast for Imaging Spontaneous Solid Tumors. Cancer Res. 2015, 75, 4283–4291. [Google Scholar] [CrossRef]
- Akcan, M.; Stroud, M.R.; Hansen, S.J.; Clark, R.J.; Daly, N.L.; Craik, D.J.; Olson, J.M. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J. Med. Chem. 2011, 54, 782–787. [Google Scholar] [CrossRef]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus), isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar] [CrossRef]
- Vernen, F.; Harvey, P.J.; Dias, S.A.; Veiga, A.S.; Huang, Y.H.; Craik, D.J.; Lawrence, N.; Troeira Henriques, S. Characterization of tachyplesin peptides and their cyclized analogues to improve antimicrobial and anticancer properties. Int. J. Mol. Sci. 2019, 20, 4184. [Google Scholar] [CrossRef]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Liu, C.; Qi, J.; Shan, B.; Ma, Y. Tachyplesin causes membrane instability that kills multidrug-resistant bacteria by inhibiting the 3-ketoacyl carrier protein reductase FabG. Front. Microbiol. 2018, 9, 825–838. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.A.; Yang, J.L. Marked increase in membranolytic selectivity of novel cyclic tachyplesins constrained with an antiparallel two-beta strand cystine knot framework. Biochem. Biophys. Res. Commun. 2000, 267, 783–790. [Google Scholar] [CrossRef]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Steinberg, D.A.; Hurst, M.A.; Fujii, C.A.; Kung, A.H.; Ho, J.F.; Cheng, F.C.; Loury, D.J.; Fiddes, J.C. Protegrin-1: A broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997, 41, 1738–1742. [Google Scholar] [CrossRef]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef]
- Sokolov, Y.; Mirzabekov, T.; Martin, D.W.; Lehrer, R.I.; Kagan, B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta 1999, 1420, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Bellm, L.; Lehrer, R.I.; Ganz, T. Protegrins: New antibiotics of mammalian origin. Expert Opin. Investig. Drugs 2000, 9, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Miller, C.B.; Hurd, D.D.; Wingard, J.R.; Fleming, T.R.; Sonis, S.T.; Bradford, W.Z.; Pulliam, J.G.; Anaissie, E.J.; Beveridge, R.A.; et al. A phase III, randomized, double-blind, placebo-controlled, multinational trial of iseganan for the prevention of oral mucositis in patients receiving stomatotoxic chemotherapy (PROMPT-CT trial). Leuk. Lymphoma 2003, 44, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Elad, S.; Epstein, J.B.; Raber-Durlacher, J.; Donnelly, P.; Strahilevitz, J. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: Analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J. Oral Pathol. Med. 2012, 41, 229–234. [Google Scholar] [CrossRef]
- Tam, J.P.; Wu, C.; Yang, J.L. Membranolytic selectivity of cystine-stabilized cyclic protegrins. Eur. J. Biochem. 2000, 267, 3289–3300. [Google Scholar] [CrossRef]
- Chen, J.; Falla, T.J.; Liu, H.; Hurst, M.A.; Fujii, C.A.; Mosca, D.A.; Embree, J.R.; Loury, D.J.; Radel, P.A.; Chang, C.C.; et al. Development of protegrins for the treatment and prevention of oral mucositis: Structure-activity relationships of synthetic protegrin analogues. Biopolymers 2004, 55, 88–98. [Google Scholar] [CrossRef]
- Silva, P.I., Jr.; Daffre, S.; Bulet, P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000, 275, 33464–33470. [Google Scholar] [CrossRef]
- Fukuzawa, A.H.; Vellutini, B.C.; Lorenzini, D.M.; Silva, P.I., Jr.; Mortara, R.A.; da Silva, J.M.; Daffre, S. The role of hemocytes in the immunity of the spider Acanthoscurria gomesiana. Dev. Comp. Immunol. 2008, 32, 716–725. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.; Cooper, M.A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of beta-hairpin peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Moreira, C.K.; Rodrigues, F.G.; Ghosh, A.; Varotti Fde, P.; Miranda, A.; Daffre, S.; Jacobs-Lorena, M.; Moreira, L.A. Effect of the antimicrobial peptide gomesin against different life stages of Plasmodium spp. Exp. Parasitol. 2007, 116, 346–353. [Google Scholar] [CrossRef]
- Barbosa, F.M.; Daffre, S.; Maldonado, R.A.; Miranda, A.; Nimrichter, L.; Rodrigues, M.L. Gomesin, a peptide produced by the spider Acanthoscurria gomesiana, is a potent anticryptococcal agent that acts in synergism with fluconazole. FEMS Microbiol. Lett. 2007, 274, 279–286. [Google Scholar] [CrossRef]
- Fazio, M.A.; Jouvensal, L.; Vovelle, F.; Bulet, P.; Miranda, M.T.; Daffre, S.; Miranda, A. Biological and structural characterization of new linear gomesin analogues with improved therapeutic indices. Biopolymers 2007, 88, 386–400. [Google Scholar] [CrossRef]
- Ikonomopoulou, M.P.; Fernandez-Rojo, M.A.; Pineda, S.S.; Cabezas-Sainz, P.; Winnen, B.; Morales, R.A.V.; Brust, A.; Sanchez, L.; Alewood, P.F.; Ramm, G.A.; et al. Gomesin inhibits melanoma growth by manipulating key signaling cascades that control cell death and proliferation. Sci. Rep. 2018, 8, 11519–11633. [Google Scholar] [CrossRef]
- Mandard, N.; Bulet, P.; Caille, A.; Daffre, S.; Vovelle, F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 2002, 269, 1190–1198. [Google Scholar] [CrossRef]
- Tanner, J.D.; Deplazes, E.; Mancera, R.L. The biological and biophysical properties of the spider peptide gomesin. Molecules 2018, 23, 1733. [Google Scholar] [CrossRef]
- Chan, L.Y.; Zhang, V.M.; Huang, Y.H.; Waters, N.C.; Bansal, P.S.; Craik, D.J.; Daly, N.L. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: Influences on stability and bioactivity. ChemBioChem 2013, 14, 617–624. [Google Scholar] [CrossRef]
- Henriques, S.T.; Lawrence, N.; Chaousis, S.; Ravipati, A.S.; Cheneval, O.; Benfield, A.H.; Elliott, A.G.; Kavanagh, A.M.; Cooper, M.A.; Chan, L.Y.; et al. Redesigned spider peptide with improved antimicrobial and anticancer properties. ACS Chem. Biol. 2017, 12, 2324–2334. [Google Scholar] [CrossRef]
- Dias, S.A.; Pinto, S.N.; Silva-Herdade, A.S.; Cheneval, O.; Craik, D.J.; Coutinho, A.; Castanho, M.; Henriques, S.T.; Veiga, A.S. A designed cyclic analogue of gomesin has potent activity against Staphylococcus aureus biofilms. J. Antimicrob. Chemother. 2022, 77, 3256–3264. [Google Scholar] [CrossRef]
- Benfield, A.H.; Defaus, S.; Lawrence, N.; Chaousis, S.; Condon, N.; Cheneval, O.; Huang, Y.H.; Chan, L.Y.; Andreu, D.; Craik, D.J.; et al. Cyclic gomesin, a stable redesigned spider peptide able to enter cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183480. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyler, T.J.; Durek, T.; Craik, D.J. Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules 2023, 28, 3189. https://doi.org/10.3390/molecules28073189
Tyler TJ, Durek T, Craik DJ. Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules. 2023; 28(7):3189. https://doi.org/10.3390/molecules28073189
Chicago/Turabian StyleTyler, Tristan J., Thomas Durek, and David J. Craik. 2023. "Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads" Molecules 28, no. 7: 3189. https://doi.org/10.3390/molecules28073189
APA StyleTyler, T. J., Durek, T., & Craik, D. J. (2023). Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules, 28(7), 3189. https://doi.org/10.3390/molecules28073189