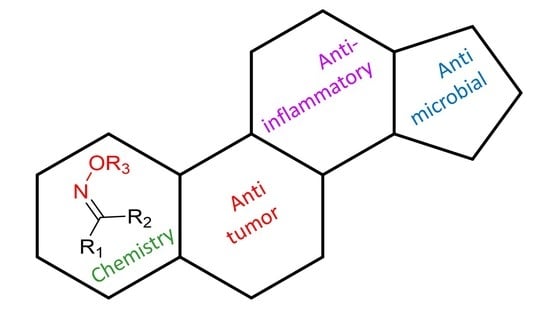
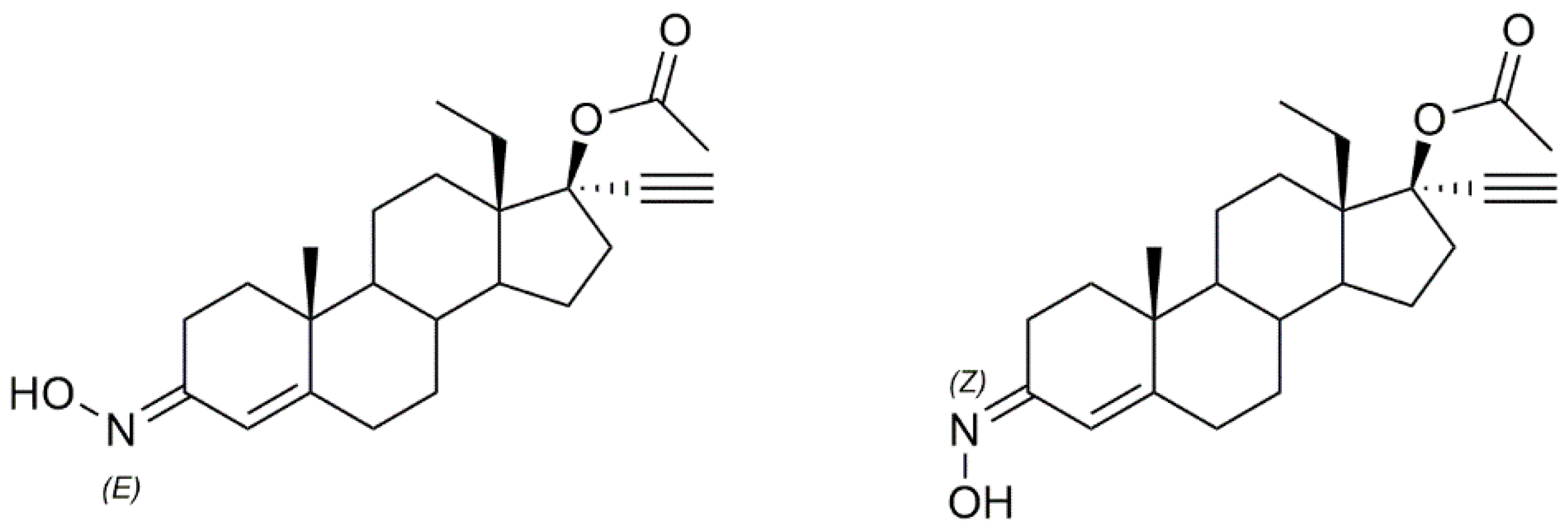
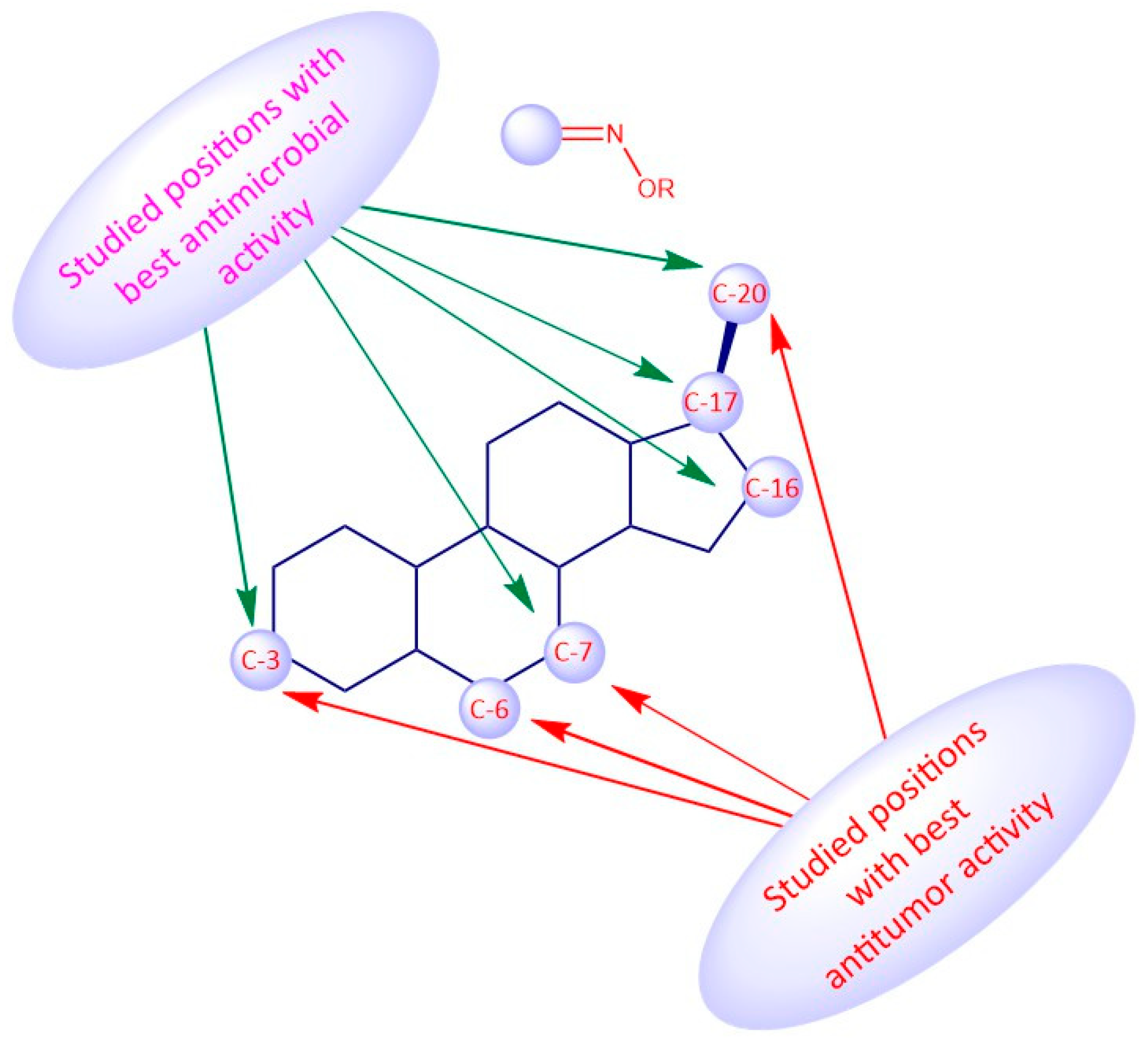
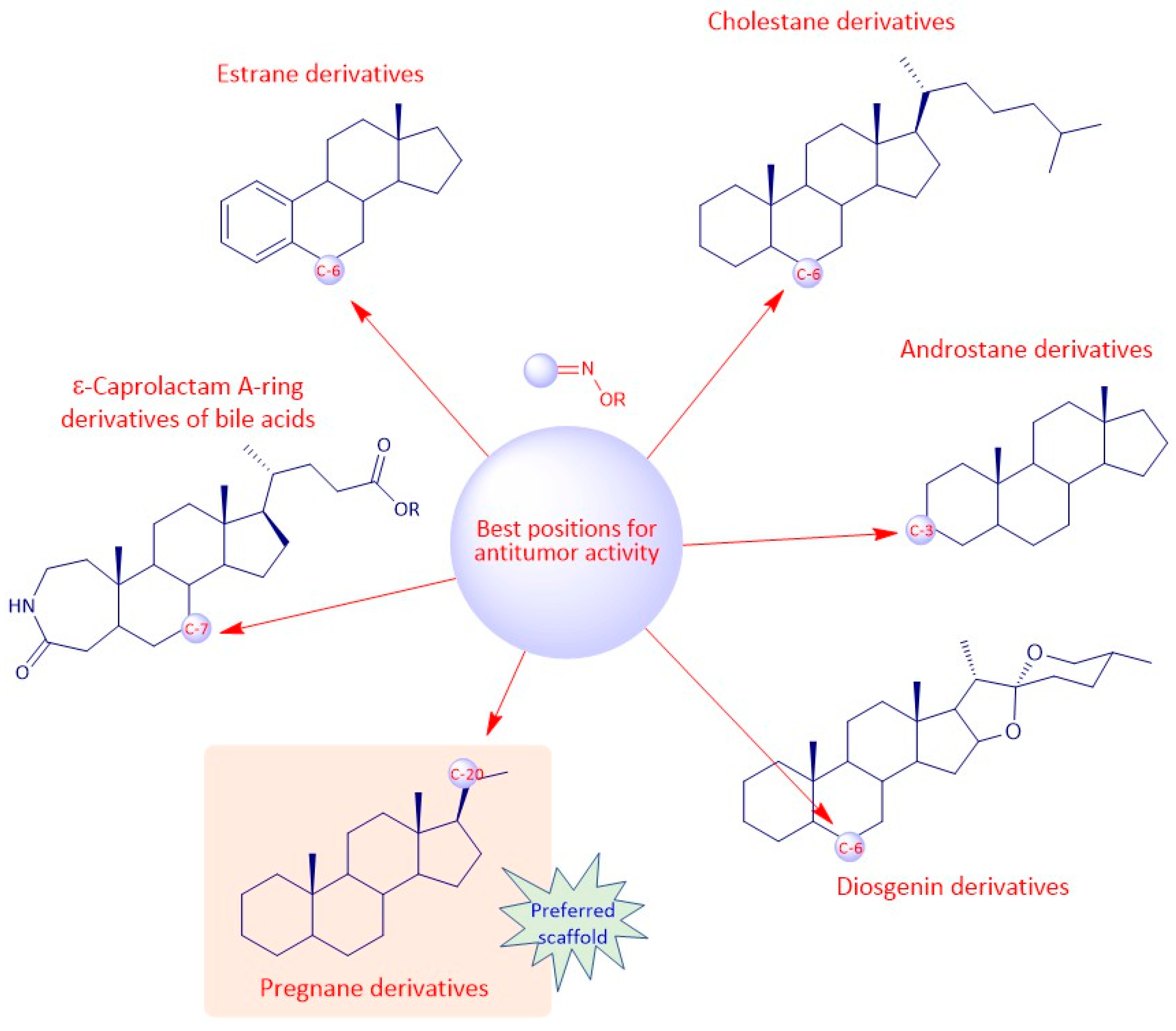
3.1. Androstane Derivatives
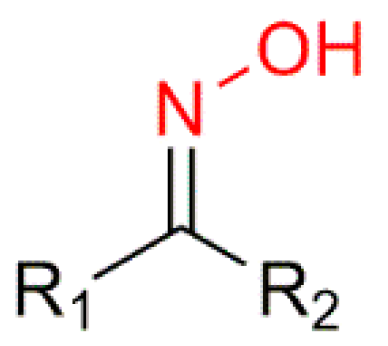
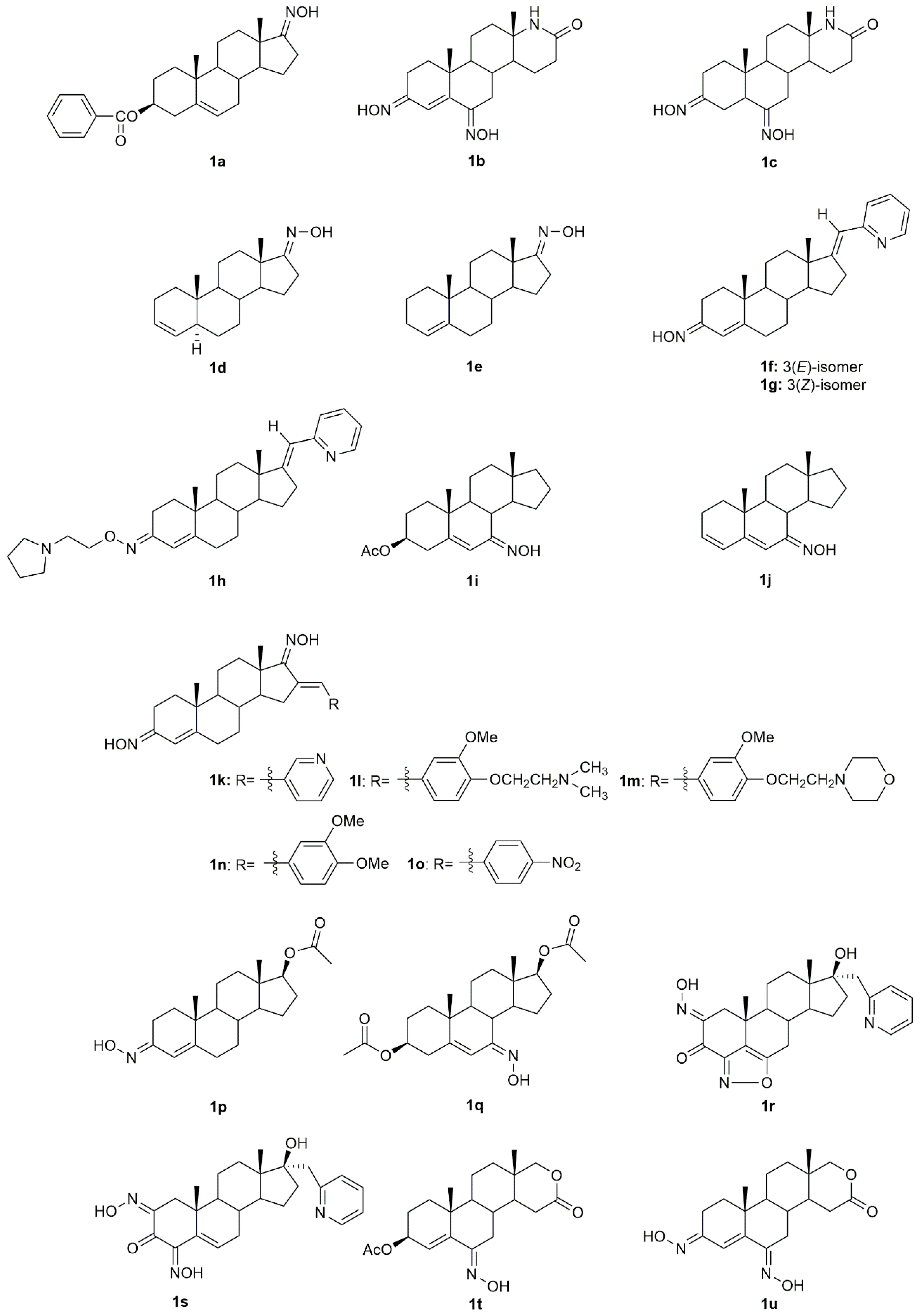
5α-Reductase inhibitors have been widely studied for the treatment of diseases that are exacerbated by 5α-dihydrotestosterone (DHT). Finasteride and dutasteride are two 5α-reductase inhibitors approved by the FDA with several therapeutic applications such as for the treatment of benign prostatic hyperplasia and prostate cancer. Bearing this in mind, Dhingra et al. developed a series of 17-oxyimino -5-androsten-3β-yl esters and evaluated their cytotoxic activity against prostate cancer cells [
35]. Compound
1a (
Figure 4) was the most active in DU145 prostate cancer cells with a percentage of growth inhibition of almost 91% at 5 µg/mL and an IC
50 value of 3.8 µM (
Table 1), being more active than finasteride (78.51% of growth inhibition and IC
50 = 3.9 µM). Moreover, the authors also evaluated the compounds’ acute toxicity using mouse macrophages, which indirectly allowed them to test for the compounds’ selectivity towards cancer cells. The toxicity index value (LC
50) presented by compound
1a was very high (LC
50 = 89.4 µM), proving that
1a was non-toxic to mouse macrophages.
The introduction of a heteroatom or the substitution of one or more carbon atoms in the steroid scaffold by a heteroatom can have a great impact on the compound’s biological activity. Aza-homosteroids are a class of compounds with unusual structures, which have been associated with a wide range of biological activities such as antiparasitic, antifungal, and anticancer [
36,
37,
38]. Following this line, Huang et al. synthesized a series of 17a-aza-D-homoandrostan-17-one derivatives, namely oximes
1b and
1c (
Figure 4) [
39]. Further cytotoxicity analysis by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method revealed that both compounds were active against HeLa and SMMC7404 cells, being that compound
1b stood out in HeLa cells with an IC
50 of 15.1 µM (
Table 1). Moreover, these oxime derivatives were in general more active than the compounds without the oxyimino group in their structure, which proves that this chemical group is important in conferring cytotoxicity.
Gomes et al. [
34] designed and synthesized two novel steroidal oxime derivatives and evaluated them and two other previously synthesized oximes [
40,
41] in several cancer cell lines to assess their antiproliferative profile. Initial screening in WiDr, PC3, HepG2, and H1299 cancer cell lines revealed that oximes
1d and
1e (
Figure 4) were able to decrease all cancer cells proliferation, being especially active against PC3 (IC
50 = 13.8 µM for
1d and 14.5 µM for
1e) and WiDr (IC
50 = 9.1 for
1d and 16.1 µM for
1e) cells (
Table 1). Moreover, both oximes were even more potent than some of the chemotherapeutic drugs currently in clinical use for these types of cancer. Both oximes were able to induce cell cycle arrest at different phases, accompanied by cell death by apoptosis/necroptosis and oxidative stress (detected by an increase in ROS production) in both cell lines. Selectivity against cancer cells was also assessed by testing the compounds in normal human colon cells. Results demonstrated that both compounds are selective toward colon cancer cells [
34].
Several novel oxime derivatives, such as (17
E)-(pyridin-2-yl)methylidene 3-oximes
1f–
1h were designed and synthesized (
Figure 4) [
42]. After the synthesis, the antiproliferative activity of both compounds was assessed in a series of several human cancer cell lines (MCF7, MDA-MB-231, PC3, HeLa, HT29, A549) and a normal human cell line, MRC5. A549, HT29, and MDA-MB-231 cancer cells were the most sensitive cell lines to both oximes being that A549 was the one with the best IC
50 values for all compounds,
1f (IC
50 = 1.5 µM)
1g (IC
50 = 1.8 µM) and
1h (IC
50 = 2.0 µM) (
Table 1). Apoptosis induction analysis showed that these oximes induced apoptosis in A549 cells, while at the same time being non-toxic to normal lung fibroblasts MRC5. These results were very encouraging since lung cancer remains one of the most difficult cancers to treat.
Some steroidal compounds with a hydroxyimino group at position C-7 conjugated with an α,β-double bond in position C-5 were designed and synthesized [
43]. After the synthesis, their antitumor activity was evaluated against several types of cancer such as cervical, gastric, epidermoid, and breast cancer. Results demonstrated that compounds
1i and
1j (
Figure 4) were both able to decrease KB, HeLa, MKN-28, and MCF7 cancer cell proliferation (
Table 1) and were more active than the corresponding parent ketones. Moreover, compound
1i was especially active against MCF7 cells presenting an IC
50 value of 10.2 µM.
Dubey et al. designed and synthesized novel dioximes of 16-benzylidene substituted derivatives [
44]. The antitumor activity of these dioximes was then evaluated in terms of the percentage of growth inhibition of NCI-H460, MCF7, and SF268 cancer cells. Results demonstrated that compounds
1k–1o (
Figure 4) were considered active against these three cell lines, which encourages the need for further and more detailed studies.
A group of investigators focused their attention on androstene oximes and their
O-alkylated derivatives [
45]. These compounds and two previously synthesized oxime derivatives, compounds
1p [
46] and
1q [
47] (
Figure 4) were evaluated in leukemia, colon, melanoma, and renal cancer cell lines. Only compounds
1p and
1q showed considerable cytotoxicity in all cell lines (percentages of growth of 10.07 to 75.01% at 10 µM), being this effect more pronounced in the leukemia cell lines, K562, HL60, and SR.
Aiming to evaluate the combined effect of the 17-heterocyclic ring and hydroxyimino function, a group of investigators designed and synthesized modified 17α-picolyl and 17(
E)-picolinylidene androstane derivatives and their antiproliferative activity against breast, prostate, cervical, colon and lung adenocarcinoma, as well as normal fetal lung fibroblasts was assessed [
48]. MTT assay results (
Table 1) demonstrated that compound
1r (
Figure 4) was the most active compound in PC3 cells (IC
50 = 6.6 µM), while compound
1s (
Figure 4) was more active, not only against PC3 cells (IC
50 = 8.7 µM) but also against MCF7 cells (IC
50 = 1.7 µM). Given these encouraging results, the authors went further ahead and studied deeply the potential mechanisms of action of compound
1s in MCF7 cells. Results showed that
1s induced apoptosis in breast cancer cells, assessed by alterations in the cells morphology, such as nuclear condensation, vacuolated cytoplasm, degradation of nuclei and cytoplasm, membrane blebbing, and apoptotic bodies formation [
48].
Savić et al., designed and synthesized some novel D-homo lactone androstane derivatives and evaluated their antiproliferative activity against several cancer cell lines [
49]. Among these compounds, the oxime derivative
1t (
Figure 4), demonstrated to have high activity (
Table 1). Moreover,
1t also revealed selectivity towards cancer cells since it presents a much higher IC
50 in the normal human cell line, MRC5. A few years later, the same group of investigators designed, synthesized and evaluated the antitumor activity of some more new D-homo lactone androstane derivatives [
50]. In vitro cytotoxicity assessment against cancer cells revealed that the steroidal oxime
1u (
Figure 4) was able to decrease the proliferation of PC3 (IC
50 = 27.94 µM) and HeLa (IC
50 = 13.86 µm) cells (
Table 1).
Table 1.
IC50 values (µM) of the synthesized androstane oxime derivatives.
Table 1.
IC50 values (µM) of the synthesized androstane oxime derivatives.
| Cell Line | Compounds |
|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | 1r | 1s | 1t | 1u |
|---|
| DU145 | 3.9 | - | - | - | - | - | - | - | - | - | - | - | - | - |
| HeLa | | 15.1 | 75.7 | - | - | >100 | >100 | 22.6 | 12.8 | 22.2 | >100 | 81.8 | 36.0 | 13.9 |
| SMMC7404 | - | >200 | 184 | - | - | - | - | - | - | - | - | - | - | - |
| WiDr | - | - | - | 9.1 | 16.1 | - | - | - | - | - | - | - | - | - |
| PC3 | - | - | - | 13.8 | 14.5 | >100 | 57.7 | 77.1 | - | - | 6.6 | 8.7 | 36.7 | 27.9 |
| HepG2 | - | - | - | 23.9 | 18.2 | - | - | - | - | - | - | - | - | - |
| H1299 | - | - | - | 18.6 | 19.2 | - | - | - | - | - | - | - | - | - |
| MCF7 | - | - | - | - | - | 41.0 | 44.9 | >100 | 10.2 | 19.8 | 50.4 | 1.7 | 81.3 | >100 |
| MDA-MB-231 | - | - | - | - | - | 47.3 | 5.2 | 4.7 | - | - | 25.3 | 40.1 | 11.9 | >100 |
| HT29 | - | - | - | - | - | 4.4 | 10.6 | 3.3 | - | - | >100 | 10.3 | 4.0 | >100 |
| A549 | - | - | - | - | - | 1.5 | 1.8 | 2.0 | - | - | >100 | 56.0 | - | - |
| MRC5 | - | - | - | - | - | >100 | >100 | >100 | - | - | >100 | >100 | >100 | >100 |
| CEM | - | - | - | - | - | >50 | >50 | 30.4 | - | - | - | - | - | - |
| G361 | - | - | - | - | - | 45.3 | 46.6 | 8.9 | - | - | - | - | - | - |
| BJ | - | - | - | - | - | >50 | >50 | 25.3 | - | - | - | - | - | - |
| KB | - | - | - | - | - | - | - | - | 26 | 28.5 | - | - | - | - |
| MKN-28 | - | - | - | - | - | - | - | - | 18.1 | 36.1 | - | - | - | - |
| Ref. | [35] | [39] | [34] | [42] | [43] | [48] | [49] | [50] |
3.2. Estrane Derivatives
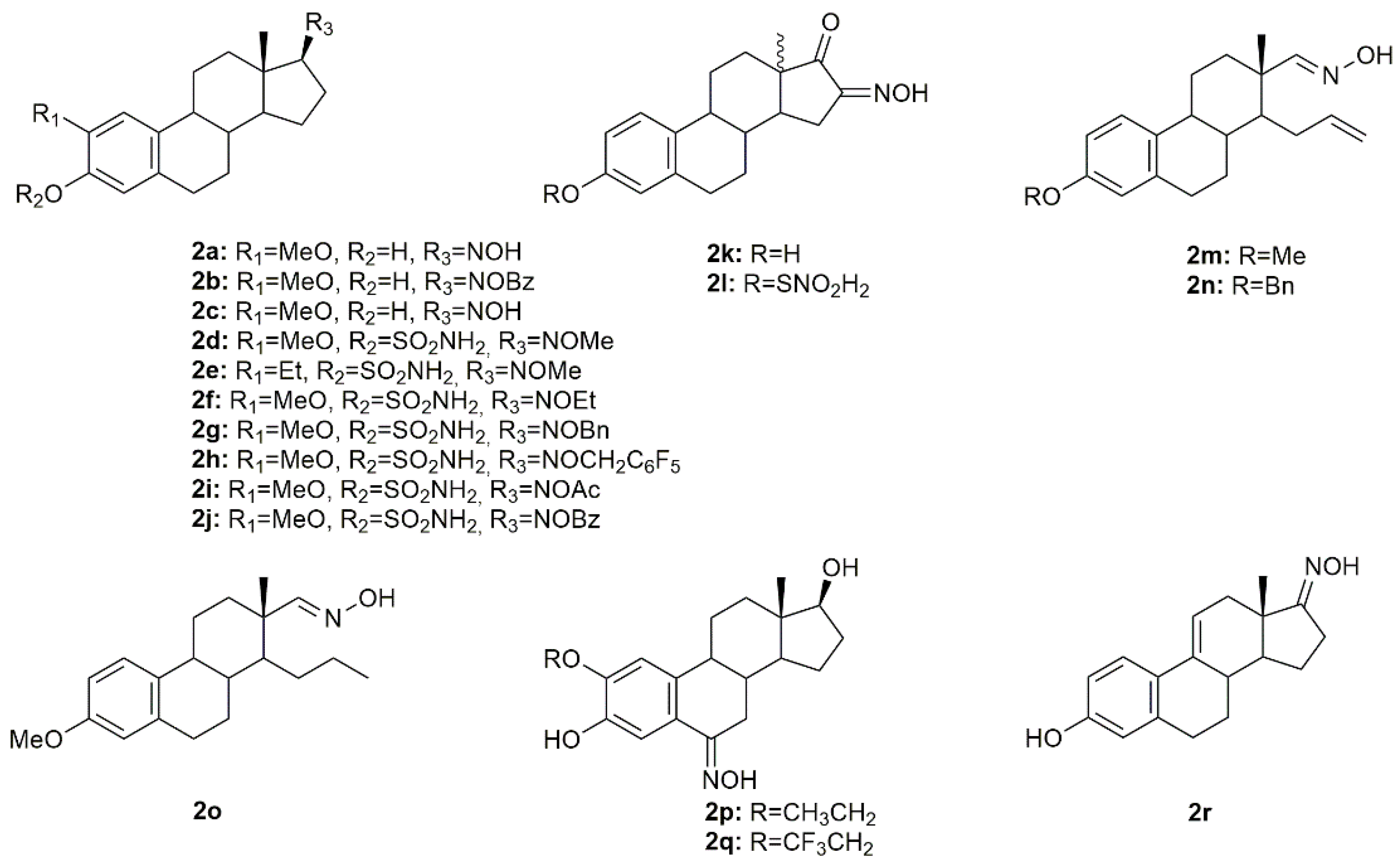
Estrogen-3-
O-sulfamates (EMATEs) are a class of steroidal compounds, which act as irreversible inhibitors of steroid sulfatase (STS), an enzyme involved in the development of estrogen-dependent breast cancer [
51]. Given this, Leese et al. decided to design and synthesize D ring-modified 2-substituted EMATEs and evaluated their in vitro anticancer activity in MCF7 cancer cells [
52]. Antiproliferative activity evaluation of the steroidal oximes
2a–
2j (
Figure 5) revealed that in general, all oximes were very effective in inhibiting MCF7 cancer cell proliferation (
Table 2). These results reinforce the importance of the modifications in the 17-position of the 2-substituted EMATEs, particularly the introduction of an oxyimino group, which increased the antiproliferative activity of this class of compounds [
52].
A series of novel estrone-16-oxime ethers were designed and synthesized, and their antitumor activity was evaluated in HeLa, MCF7, and A431 cancer cells [
53]. From all the synthesized compounds, oxime
2k and
2l (
Figure 5) decreased cancer cell proliferation in a more pronounced way, being HeLa cells the most susceptible to both compounds (IC
50 = 4.41 µM for
2k and 4.04 µM for
2l) (
Table 2). Further evaluation to characterize the mechanisms of action of these compounds revealed both of them interfered with the cell cycle at G1 phase and induced apoptosis in HeLa cells, by the activation of caspase-3. Following this study, the same authors continued their research on this topic and designed and synthesized a series of novel D-secooxime derivatives in the 13β- and 13α-methyl-estrone series [
54]. After MTT assays to assess antiproliferative activity in HeLa, MCF7, A2780, and A431 cancer cell lines, compounds
2m–
2o (
Figure 5) stand out for displaying high cytotoxicity against all cell lines and being, generally, even more active than cisplatin as it can be seen in
Table 2. Furthermore, compound
2n was selected for additional analysis in A2780 cells, namely cell cycle analysis. Results showed that
2n induced cell cycle arrest at the S phase, which in turn might be responsible for apoptosis induction [
54].
Cushman et al. investigated several estradiol analogs to improve the anticancer activity of 2-methoxyestradiol, a naturally occurring tubulin polymerization inhibitor [
55]. Starting from 2-ethoxyestradiol, an analog previously synthesized by the same group, from 2-methoxyestradiol [
56], two novel steroidal oximes
2p and
2q (
Figure 5) were designed and synthesized and their antitumor activity was assessed. Results revealed that both compounds were extremely toxic to all the cell lines studied (HOP62, HCT116, SF539, UACC62, OVCAR-3, SN12C and DU145) with GI
50 (half growth inhibition) at values ranging from 0.010 to 0.066 µM (
Table 2). Furthermore,
2p and
2q were able to inhibit tubulin polymerization. The oxime derivatives were the most active among all compounds synthesized, which points out the importance of the oxyimino functionality.
Aiming to develop new steroidal oximes with potential application in cancer treatment, a group of scientists designed and synthesized a series of estrone oxime derivatives and evaluated them in six cancer cell lines [
57]. MTT results proved that compound
2r (
Figure 5) is the most active against all cell lines, being LNCaP cells the most sensitive to this molecule (IC
50 = 3.59 µM) (
Table 2). Given this, the cytotoxicity of this steroidal oxime was mediated by a cell cycle arrest at G2/M phases accompanied by cell death by apoptosis, with evidence of condensed and fragmented nuclei. Moreover, the authors speculated that this compound might interfere with β-tubulin [
57].
Table 2.
IC50 values (µM) of the synthesized estrane oxime derivatives.
Table 2.
IC50 values (µM) of the synthesized estrane oxime derivatives.
| Cell Line | Compounds |
|---|
| 2a | 2b | 2c | 2d | 2e | 2f | 2g | 2h | 2i | 2j | 2k | 2l | 2m | 2n | 2o | 2p | 2q | 2r |
|---|
| MCF7 | 5.87 | 6.47 | 0.24 | 0.17 | 0.23 | 0.23 | 1.33 | 5.14 | 0.17 | >30 | >30 | 2.6 | 2.6 | >30 | 2.1 | - | - | 25.63 |
| HeLa | - | - | - | - | - | - | - | - | - | - | - | - | 1.2 | 7.1 | 1.7 | - | - | - |
| A431 | - | - | - | - | - | - | - | - | - | - | - | - | 0.8 | 0.9 | 0.9 | - | - | - |
| A2780 | - | - | - | - | - | - | - | - | - | - | - | - | 0.9 | 1.4 | 0.7 | - | - | - |
| HOP62 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.017 | 1.2 | - |
| HCT116 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.031 | 2.1 | - |
| SF539 | | | | | | | | | | | | | | | | 0.021 | 2.8 | |
| UACC62 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.015 | 0.88 | - |
| OVCAR3 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.016 | 5.4 | - |
| SN12C | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.045 | 17 | - |
| DU145 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.049 | 17 | - |
| MDA-MB-231 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.010 | 6.5 | - |
| MGM | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.066 | 4.2 | - |
| T47D | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 43.45 |
| LNCaP | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 3.59 |
| HepaRG | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 18.35 |
| Caco | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 24.33 |
| NHDF | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 30.84 |
| Ref. | [52] | [53] | [54] | [55] | [57] |
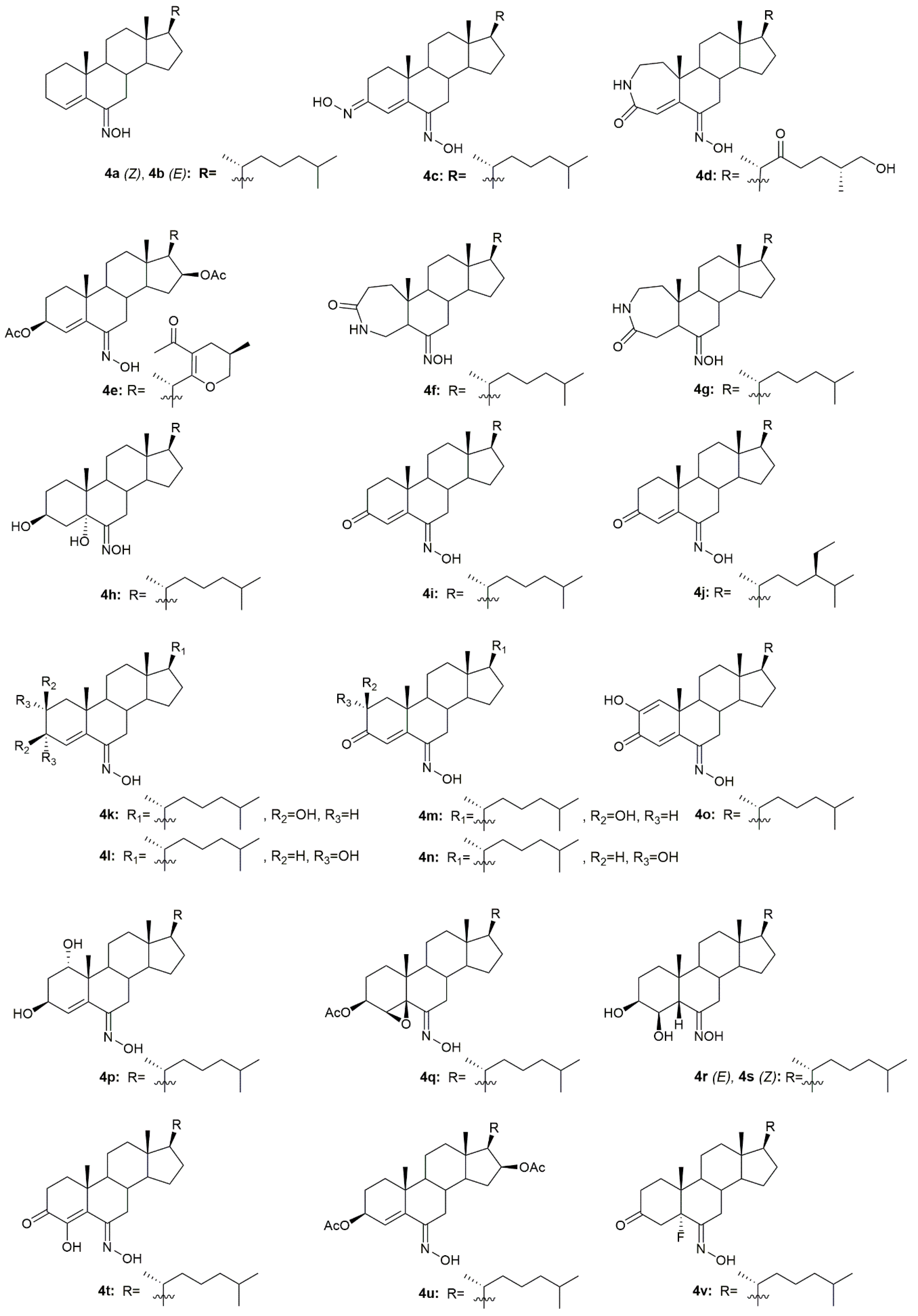
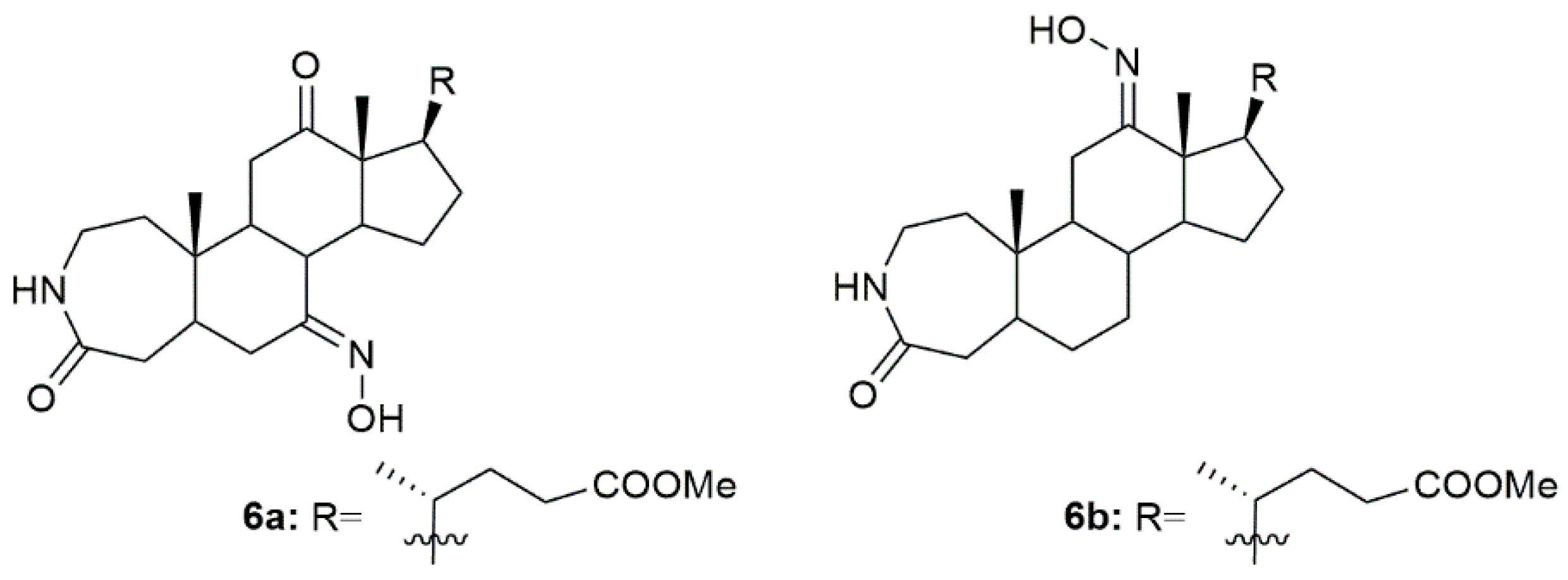
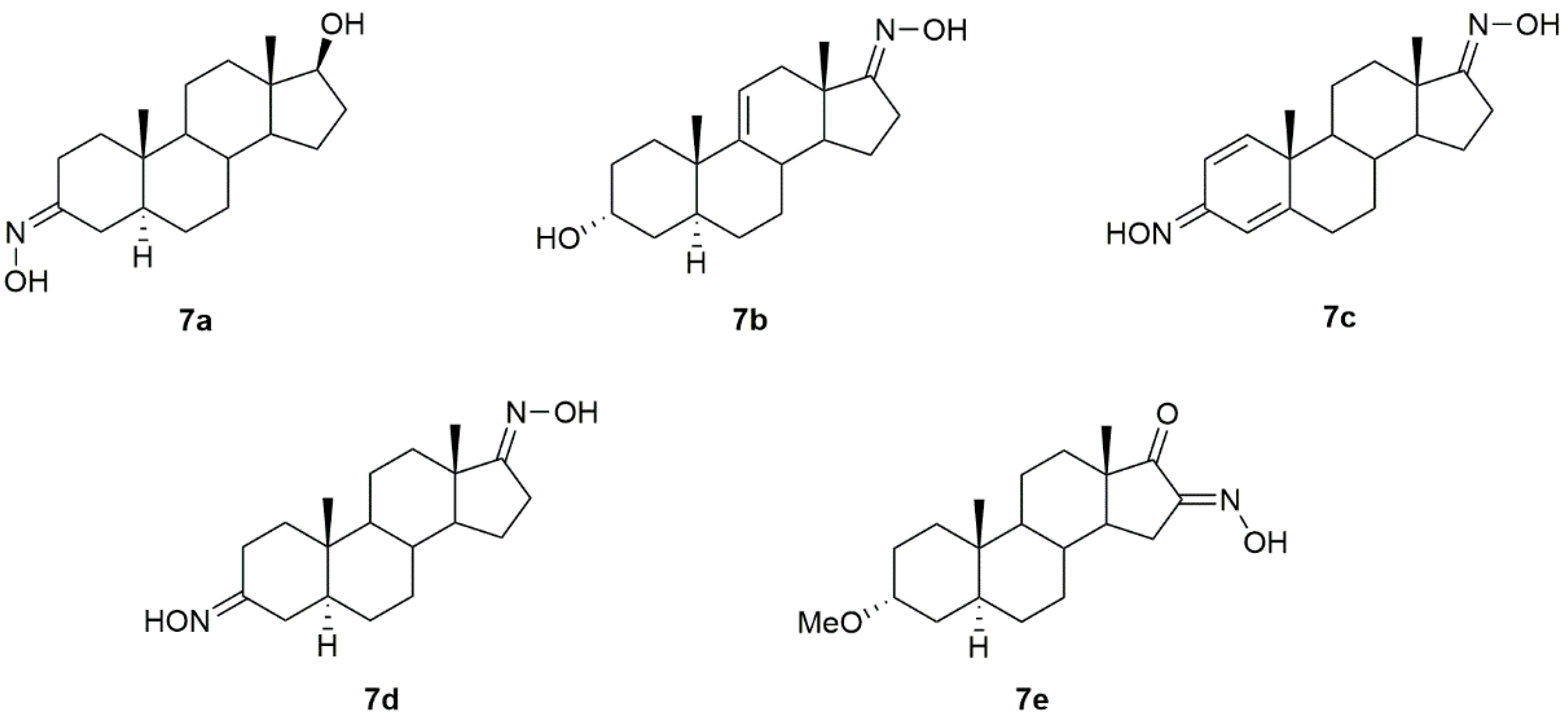
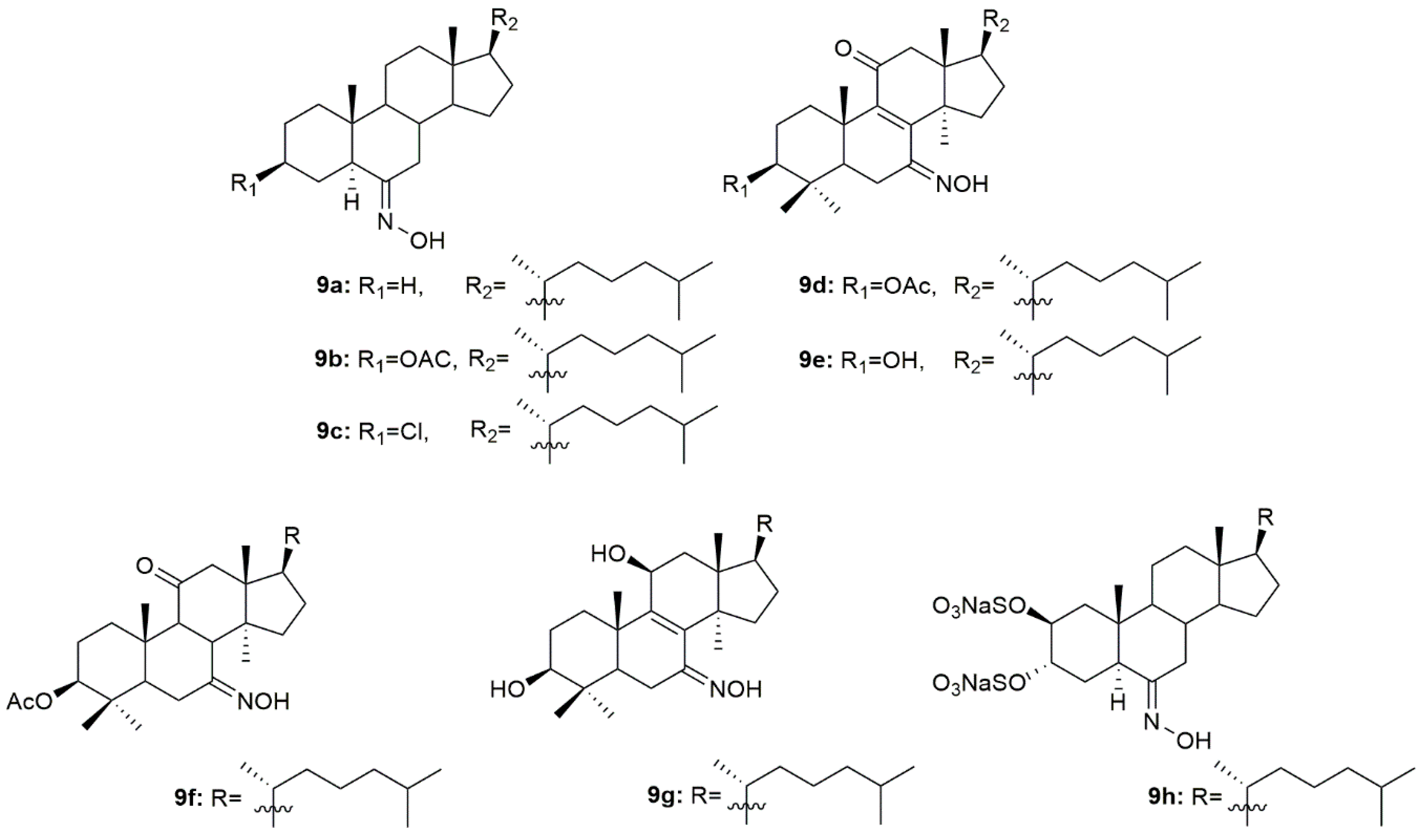
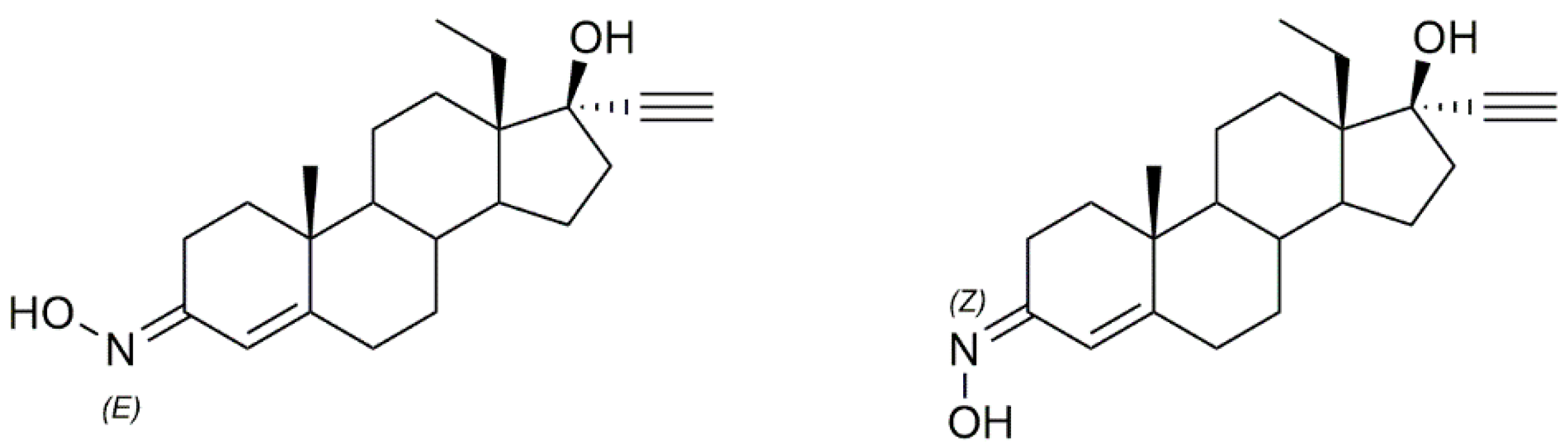
3.4. Cholestane Derivatives
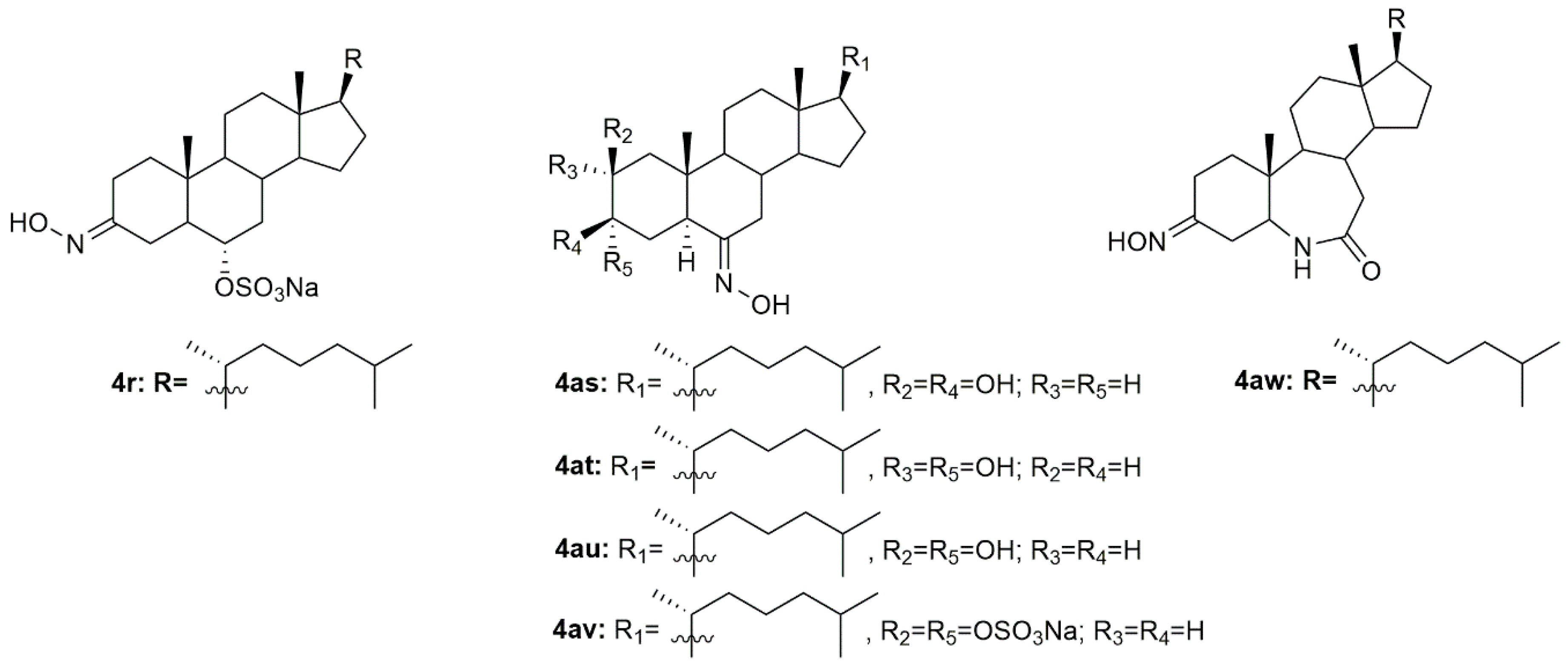
Krstić et al. reported the design and synthesis of two novel steroidal oximes from cholesterol, compounds
4a and
4b (
Figure 7) [
60]. A few years later, the same group of scientists decided to evaluate the potential antitumor activity of these two oximes [
61] against two human cancer cell lines (HeLa and K-562) and against non-stimulated and PHA-stimulated peripheral blood mononuclear cells (PBMC’s) from healthy donors to assess selectivity. Both compounds showed a dose-dependent decrease in the proliferation of HeLa cancer cells (IC
50 = 35.24 ± 4.09 for
4a and IC
50 = 20.68 ± 3.10 for
4b) and K-562 cancer cells (IC
50 = 28.05 ± 10.18 for
4a and IC
50 = 11.16 ± 1.24 for
4b), while showing almost no effects in normal immunocompetent cells (
Table 4). Further evaluation revealed that compound
4b exerted its cytotoxicity by inducing apoptosis in HeLa cells. On the contrary, compound
4a showed no evidence of apoptosis or necrosis, which can be explained by the difference in the
Z/E stereochemistry in the hydroxyimino group of both compounds [
61].
A group of researchers investigated in aza-homosteroids derived from diosgenin and cholesterol-containing hydroxyimino and lactam groups in the A/B ring with four types of side chains: cholestane, spirostane, 22-oxocholestane and 22,26-epoxycholestene [
62]. These compounds were further evaluated as potential antitumor agents in MCF7 cancer cells. Antitumor activity assessment revealed that from all the synthesized compounds, oximes
4c–
4e (
Figure 7) were the most promising ones with an IC
50 of 8.2, 9.5, and 7.9 µM, respectively (
Table 4). MCF7 cancer cells were more sensitive to the compounds containing a hydroxyimino group in comparison with the compounds without this chemical function. Moreover, Mora-Medina et al., evaluated these three oximes against PBMCs to test for the selectivity index of these compounds. Results showed that
4c-4e were all remarkably selective for MCF-7 cells, which makes these compounds very promising [
62].
Huang et al. synthesized a series of 6-hydroxyimino-substituted-3-aza- and 4-aza-A-homo-3-oxycholestanes using cholesterol as starting material [
63]. Oximes
4f and
4g (
Figure 7) were further evaluated in three different cancer cell lines, namely HeLa, SMMC7404, and MGC7901. Results showed that both compounds displayed cytotoxicity against these cell lines (
Table 4), being more active than the referenced drug, cisplatin, in HeLa and SMMC7404 cells. Altogether, these results demonstrated that the introduction of the hydroxyimino group at position 6 was crucial for the compounds’ cytotoxicity against cancer cells. Given the good outcomes obtained, these investigators decided to further analyze compound
4f (
Figure 7) and evaluated its antitumor activity in five more cancer cell lines (GNE2, SPC-A, Tu686, PC3, and HT29) [
64]. Results indicate that oxime
4f was also quite active in all cell lines with IC
50 values ranging from 10.6 to 74.5 µM (
Table 4). Furthermore, the molecular mechanisms by which
4f decreased cancer cell proliferation were studied. Results unveiled that compound
4f induced cancer cell apoptosis by activation of the intrinsic pathway, which was demonstrated by the annexin V labeling, activation of caspase-3, and release of cytochrome C. Moreover, this oxime was also evaluated in an in vivo model and proved to be able to inhibit tumor growth [
64].
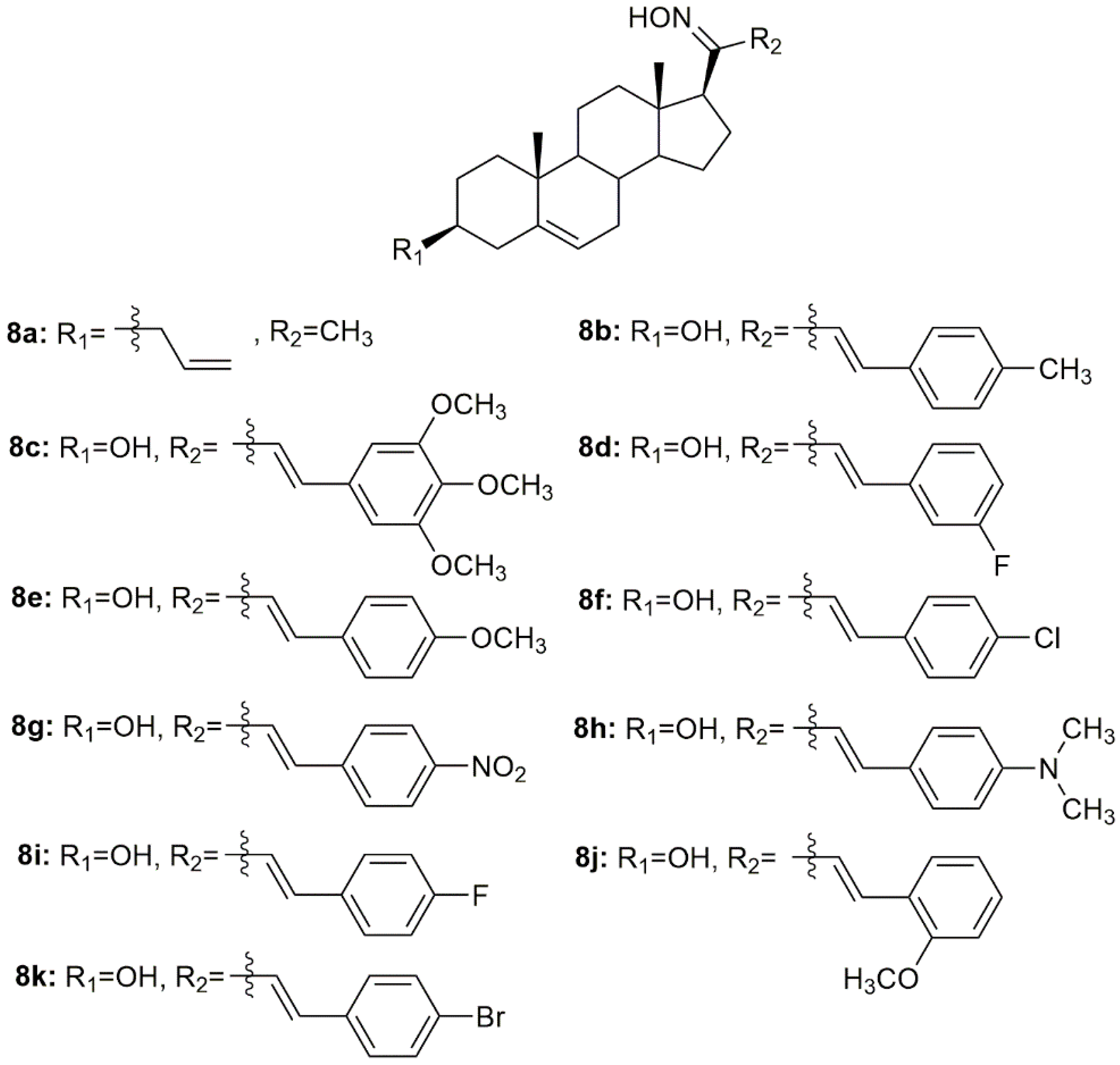
Oxysterols are a group of lipids derived from cholesterol, particularly interesting in the medicinal chemistry field due to their diverse biological effects [
65]. Given this, Carvalho et al. designed and synthesized a series of oxysterols in which oxime
4h (
Figure 7), a 3β,5α,6β-trihydroxycholestanol derivative, was included [
66]. After the synthesis of
4h, the compound’s cytotoxicity was studied in five human cancer cell lines (HT29, SH-SY5Y, HepG2, A549, PC3) and two human normal cell lines (ARPE-19 and BJ). Oxime
4h was quite active in all cancer cell lines being that HT29 was the most sensitive (IC
50 = 11.9 µM). Moreover, the compound revealed some selectivity towards HT29 cells (selectivity index of 1.99) since the IC
50 displayed was higher in the normal cell lines (
Table 4). This work contributed to deepening the understanding of oxysterols’ cytotoxicity and shed some light on the SAR of these classes of compounds.
In 1997, two steroidal molecules with very interesting and unusual structures, (6
E)- hydroxyiminocholest-4-en-3-one (
4i,
Figure 7) and its 24-ethyl analog (
4j,
Figure 7), were isolated from
Cinachyrella marine sponges [
67]. Analysis of their antitumor activity revealed that only compound
4i was able to decrease the proliferation of P388 (IC
50 = 1.25 µg/mL), A549 (IC
50 = 1.25 µg/mL), HT29 (IC
50 = 1.25 µg/mL), and MEL28 (IC
50 = 2.5 µg/mL) cells (
Table 5). Following this discovery, Deive et al. [
68] further explored the SAR of this type of compound and prepared several derivatives of
4i and
4j with different structural features, namely with different side chains and degrees of unsaturation on ring A. From all the synthesized compounds,
4k–
4o (
Figure 7) stood out with an IC
50 ranging from 0.125 to 1.25 µg/mL (
Table 5) in the above-mentioned cancer cell lines. These results allowed to shed some light regarding SAR analysis and demonstrated that the presence of a cholesterol-type side chain, a ketone group at C-3 and a high degree of oxidation in ring A might play a major role in the compounds’ cytotoxicity [
68]. A few years later, the same group continued their investigation in 6-hydroxyiminosteroids [
69] and, bearing in mind the information about the SAR of these compounds, a series of new steroidal oximes were synthesized and evaluated as potential antitumor agents. After biological evaluation in four different cancer cell lines (A549, HCT116, PSN1, T98G), compounds
4p–
4w (
Figure 7) were the most effective in decreasing cancer cell proliferation, demonstrating good IC
50 values (
Table 4). Once again, the oxygenation of ring A turned out to be very important in the increased cytotoxicity of the compounds against cancer cells [
69].
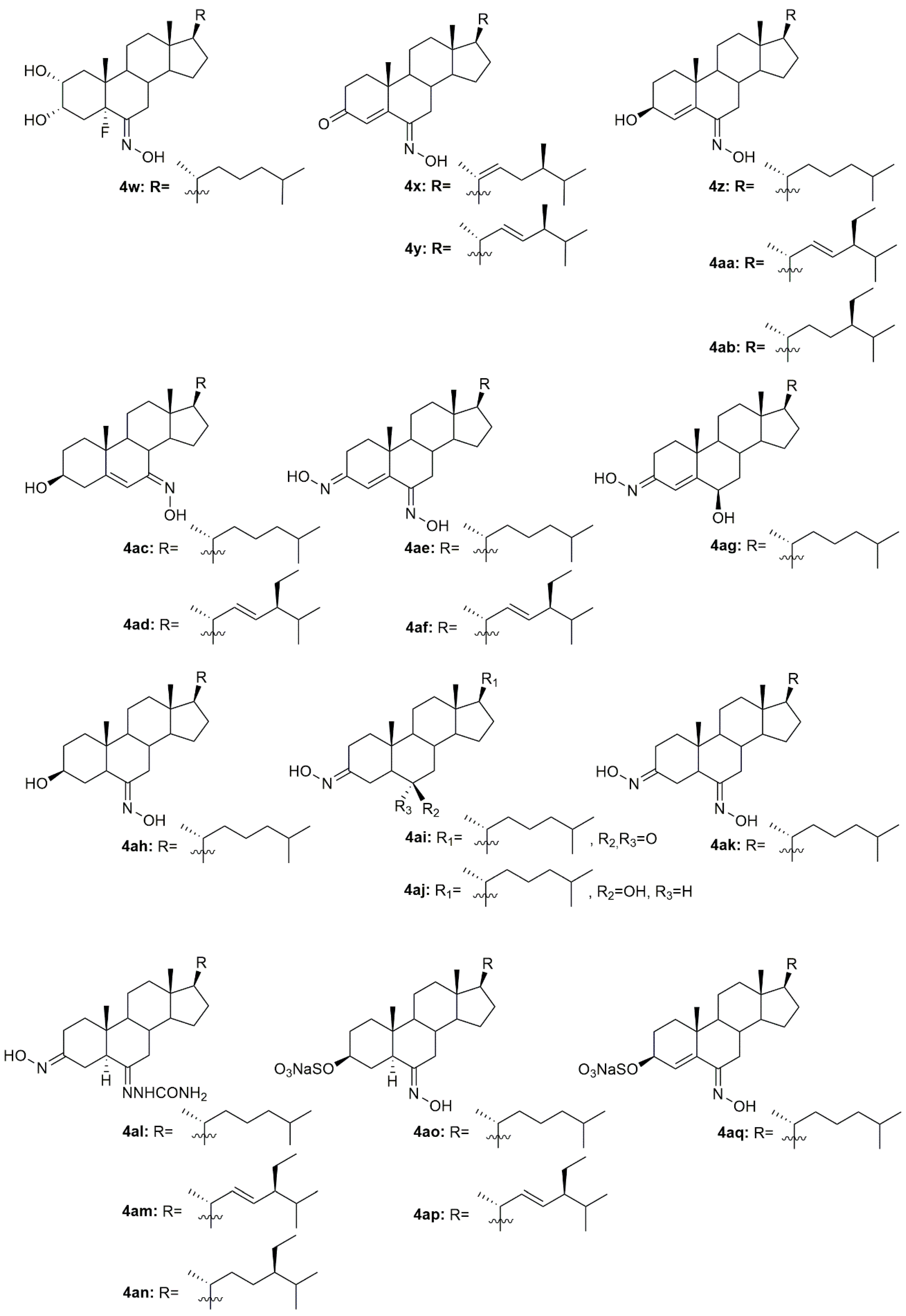
Another study by Cui et al. [
70] also reported the synthesis and further biological evaluation of 6-hydroxyiminosteroidal cholestane derivatives. They not only developed a facile and efficient synthetic method for the synthesis of the natural compounds
4i and
4j (
Figure 7) but also designed and synthesized a new steroidal oxime (
4x,
Figure 7). Further antitumor activity evaluation in four human cancer cell lines, namely Sk-Hep-1, H292, PC3, and Hey1B revealed that compound
4x presented modest cytotoxicity against these cell lines with IC
50 values ranging from 37 to 59.5 µg/mL (
Table 5). Once again and compared to the cytotoxicity displayed by compounds
4i and
4j, the structure of the side chains impacts the cytotoxicity of the compounds. The cholesterol-type side chain seems to be important for biological activity, which goes accordingly to the results obtained by other research groups [
68]. The same group of investigators continued their research on this topic and synthesized more steroidal oximes with the hydroxyimino groups in different locations (A ring or B ring) and with different types of side chains at position 17 [
71]. From all the derivatives, compounds
4y–
4ag (
Figure 7) were the ones with the best antitumor activity against Sk-Hep-1, H292, PC3, and Hey1B (
Table 5). This study reinforced the conclusions obtained in the previous ones [
68,
69,
70], where it is stated that for enhanced cytotoxicity the compounds must have a cholesterol-type side chain, a hydroxyimino group on the B ring, and a hydroxy group on the A or B ring. The same authors went further ahead in the SAR and synthesized a series of derivatives similar to the ones synthesized in the previous study but without the 4,5-double bond [
72]. After biological activity evaluation, compounds
4ah–
4ak (
Figure 7) proved to be the most effective in decreasing Sk-Hep-1, H292, PC3, and Hey1B cancer cells proliferation with IC
50 values ranging from 35.4 to 103 µg/mL (
Table 5). These compounds (without the 4,5-double bond) were more active than the equivalent ones with the 4,5-double bond [
71], which indicates that the double bond in this particular position confers a negative effect in the biological activity displayed by these derivatives. All these studies were very important in unraveling the SAR of 6-hydroxyiminosteroids and might help shed some light on the design of novel chemotherapeutic drugs for the treatment of different types of cancer.
Gan et al. designed and synthesized some steroidal hydrazone derivatives with 3,6-disubstituted structure and different side chains at 17-position, being the hydroxyimino group one of these substitutions, namely in the 3-position [
73]. Antiproliferative activity evaluation was carried out in vitro against gastric and liver cancer cells after 72 h of incubation. The synthetic routes gave rise to a series of novel molecules among them, compounds
4al–
4an (
Figure 7), which were remarkably active against the two cancer cell lines used, Bel7404 and SGC7901 (
Table 6). Compound
4am was particularly active to Bel7404 cells (IC
50 = 7.4 µM) being three times more active than cisplatin (IC
50 = 22.3 µM), which makes
4am a potential antitumor drug.
Huang et al. published a study describing the synthesis of new sulfated hydroxyiminosterols as potential antitumor agents [
74]. In vitro antiproliferative activity, assessed in HeLa, SMMC7404 and MGC7901 cancer cell lines revealed that compounds
4ao–
4ar (
Figure 7) were all able to decrease all cancer cell lines proliferation (
Table 6). Please note that compound
4ao was even more cytotoxic than cisplatin against all the cancer cell lines studied.
A group of investigators from Argentina synthesized three new 6
E-hydroxyiminosteroids and then assessed their antitumor activity against prostate cancer cells (PC3 and LNCaP) [
75]. After antiproliferative activity evaluation, compounds
4as–
4av (
Figure 7) proved to be quite active in both cancer cell lines with IC
50 values ranging from 10.8 to 44.8 µM (
Table 6).
Using analogs of compounds
4i and
4j as precursors, Huang et al. designed and synthesized novel steroidal oximes and then evaluated their anticancer efficacy against a panel of six cancer cell lines [
76]. Of all the synthesized compounds, oxime
4aw (
Figure 7) was the most powerful oxime, especially in HeLa and GNE2 cancer cell lines with IC
50 values of 9.1 and 11.3 µM, respectively (
Table 6). Remarkably, this compound was even more active than cisplatin (IC
50 = 10.1 and 16.8 µM in HeLa and GNE2 cells, respectively).
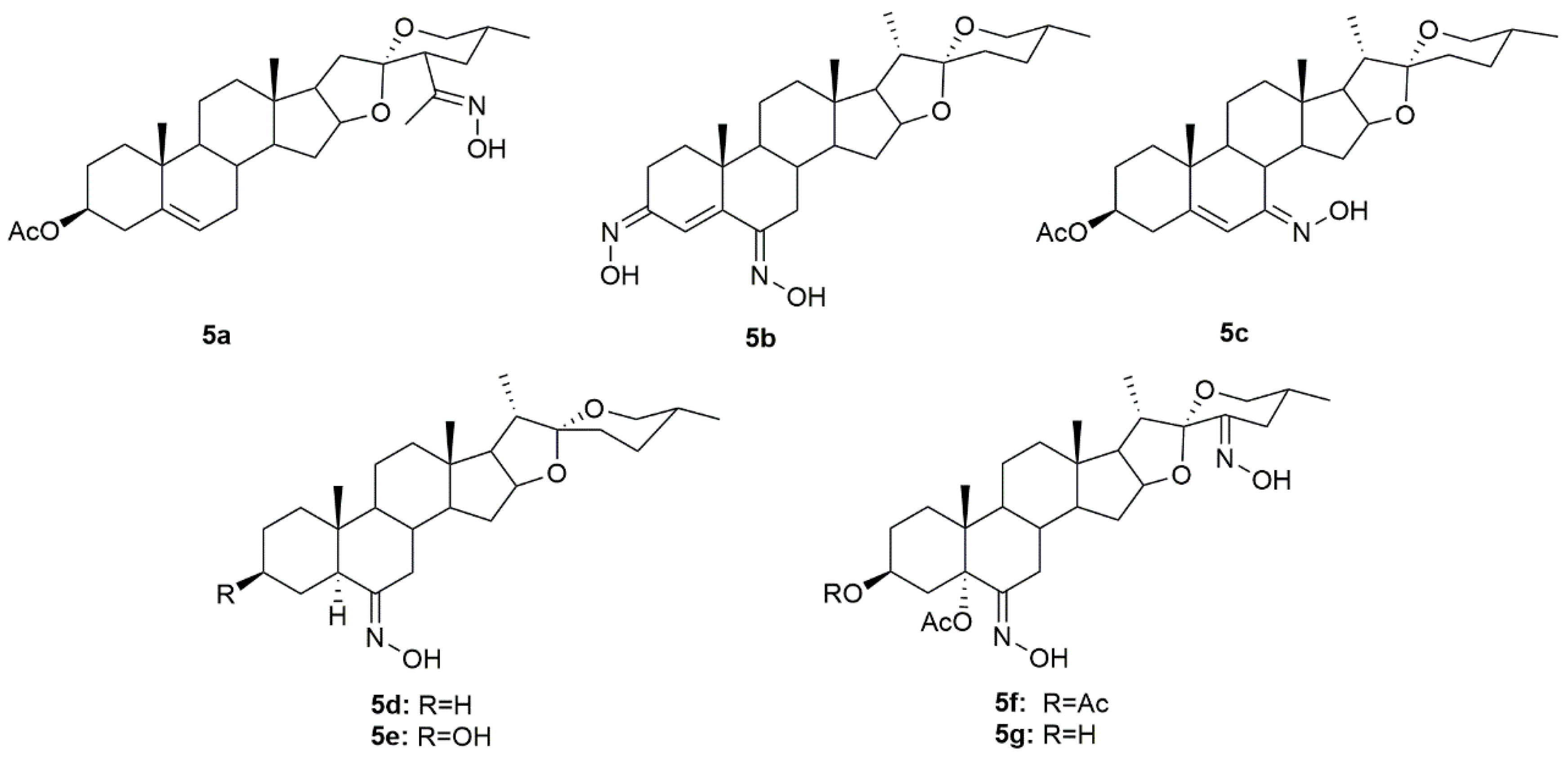
3.5. Diosgenin Derivatives
Diosgenin is a natural steroidal sapogenin, which was first isolated from
Discorea tokoro by Takeo Tsukamato in 1936 [
77]. This molecule is widely used in the pharmaceutical industry as the main precursor in the synthesis of steroids. Given this, several investigators have been focusing their attention on diosgenin derivatives. Sánchez et al. designed and synthesized two novel oxime derivatives (
5a and
5b,
Figure 8) using diosgenin as a precursor and then tested their antitumor activity against HeLa and CaSki cancer cell lines [
78]. Results revealed that both compounds caused a dose-dependent decrease in HeLa and CaSki cell proliferation with IC
50 ranging from 10.9 to 48.18 µM (
Table 7) and compound
5a was even more active than diosgenin, which reinforces the importance of the hydroxyimino group, positioned in the side chain, in conferring cytotoxicity. Further analysis of both compounds demonstrated that they exerted their antitumor activity by interfering with the cell cycle, and causing cell death by apoptosis mediated by the activation of caspase-3, which, in turn, is responsible for DNA fragmentation [
78].
Another group of investigators synthesized a series of diosgenin derivatives by introducing new modifications in rings A and B [
79]. Among all these compounds, oxime
5c (
Figure 8) was evaluated in HeLa, MDA-MB-231, and HCT-15 cancer cells to assess its antitumor activity. Results showed that
5c was quite active in all cell lines with IC
50 of 18.23, 10.83, and 17.56 µg/mL in HeLa, MDA-MB-231, and HCT-15 (
Table 7), respectively. Furthermore, this compound was not toxic to PBMC which can point to a selectivity towards cancer cells.
Carballo et al. designed and synthesized a series of novel hydroxyimino steroidal derivatives and evaluated their antiproliferative activity in MCF7 and MDA-MB-231 breast cancer cells [
80]. Compounds
5d–
5e (
Figure 8) were able to decrease cell proliferation with IC
50 values ranging from 9.3 to 11.8 µM (
Table 7). Interestingly, compounds
5d and
5e (spirostan derivatives) were more active against the triple-negative cells, a subtype of breast cancer associated with a worse prognosis and fewer therapeutic options.
More recently, a group of scientists designed and synthesized a series of steroidal oximes from diosgenin and evaluated their antitumor activity against six cancer cell lines (A549, HBL100, HeLa, SW1573, T47D, and WiDr) [
81]. Of all the compounds synthesized,
5f and
5g (
Figure 8) were the most active against all cancer cell lines tested (
Table 7). Moreover, compound
5f was more active than cisplatin in T47D and WiDr cells and
5g was more active only in WiDr cells. These results encourage more research to unravel the mechanisms of action behind the cytotoxicity displayed by these compounds against cancer cells.







 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















