
Roles of G4-DNA and G4-RNA in Class Switch Recombination and Additional Regulations in B-Lymphocytes


{kind=link}
{kind=link}
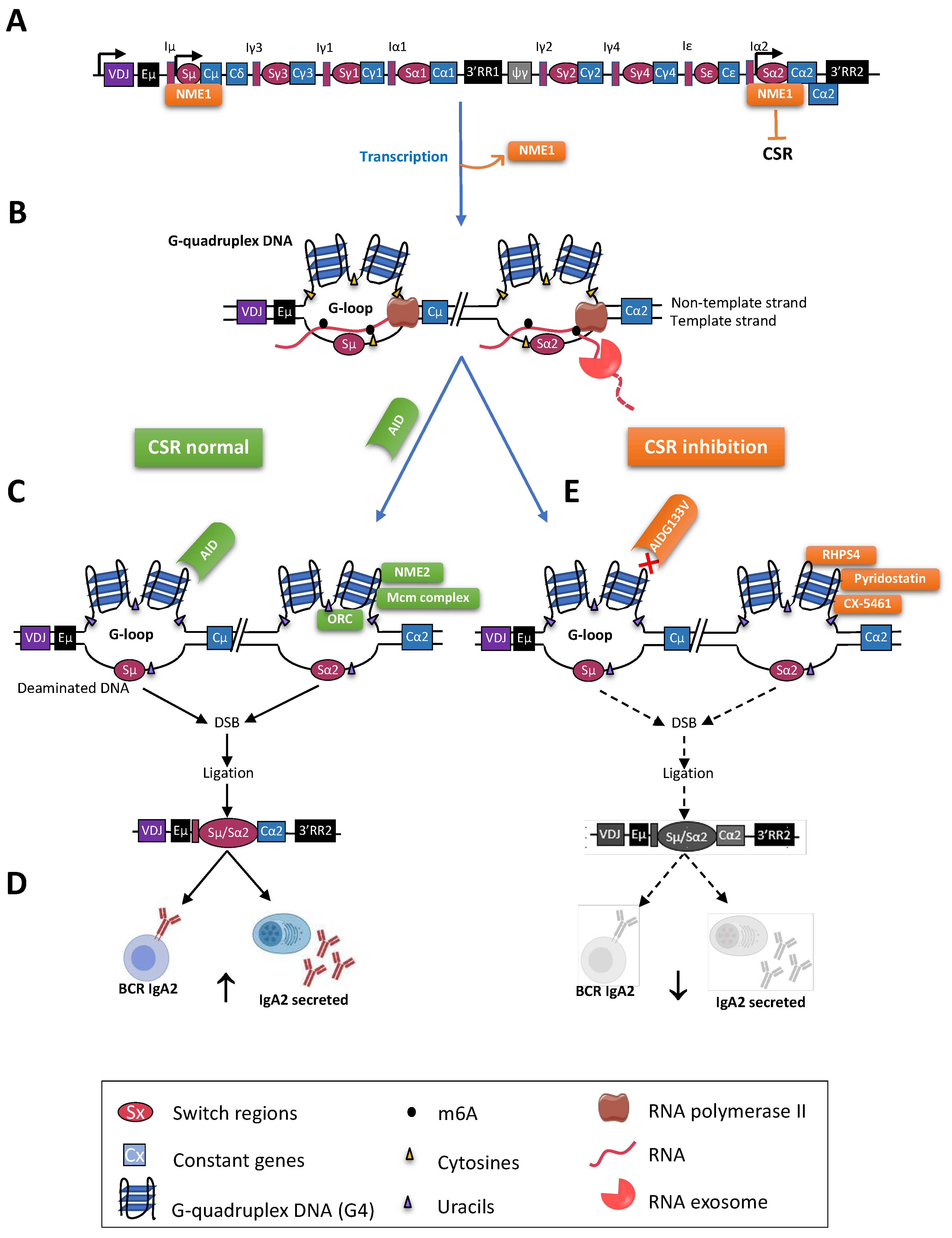{kind=link}
Abstract
1. Introduction
2. General Role of G4s in DNA Accessibility and Gene Regulations, with Pertinence to B-Cells
2.1. Unless Unwinded by Helicases, G4s Restrict DNA Accessibility to Various Factors, Notably for Replication and Maintenance of Telomeres
2.2. G4-DNA and Regulation of Transcription
2.3. Links between Transcribed G4s and DNA Breaks in R-Loops
3. The Context of CSR and Its Regulation
3.1. The CSR Machinery
3.2. S Region Structures
3.3. Transcriptional Regulation of CSR and the Role of the IgH 3′RR
3.4. Structure and Role of Germline Transcripts
3.5. Post-Transcriptional Regulation of GLTs and Their Processing by the RNA Exosome
4. Connection between G4s and CSR
4.1. Role of G4-DNA at Transcribed S Regions and in the Resolution of G4s/R-Loops Conflicts
4.2. Role of G4-RNA Structures within S Region Transcripts
4.3. Connections between CSR and DNA Replication
4.4. A Role of G4-DNA in IgH Locus High-Dimensional Organization
5. A Role of G4s in the Regulation of Locus Suicide Recombination (LSR)
6. G4s and Illegitimate Recombination
7. Mutations of Various Nuclear Factors with G4-Dependence
8. Pharmacological G4 Targeting and Potential Implications for B Cells and CSR
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williamson, J.R.; Raghuraman, M.K.; Cech, T.R. Monovalent cation-induced structure of telomeric DNA: The G-quartet model. Cell 1989, 59, 871–880. [Google Scholar] [CrossRef]
- Xu, Y.; Noguchi, Y.; Sugiyama, H. The new models of the human telomere d[AGGG(TTAGGG)3] in K+ solution. Bioorg. Med. Chem. 2006, 14, 5584–5591. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; MacCarthy, T. Characterization of DNA G-Quadruplex Structures in Human Immunoglobulin Heavy Variable (IGHV) Genes. Front. Immunol. 2021, 12, 671944. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Bartel, D.P. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 2016, 353, aaf5371. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Marsico, G.; Sahakyan, A.B.; Chambers, V.S.; Balasubramanian, S. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 2016, 13, 841–844. [Google Scholar] [CrossRef]
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef]
- Haensel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef]
- Ceschi, S.; Berselli, M.; Cozzaglio, M.; Giantin, M.; Toppo, S.; Spolaore, B.; Sissi, C. Vimentin binds to G-quadruplex repeats found at telomeres and gene promoters. Nucleic Acids Res. 2022, 50, 1370–1381. [Google Scholar] [CrossRef]
- Evans, T.; Schon, E.; Gora-Maslak, G.; Patterson, J.; Efstratiadis, A. S1-hypersensitive sites in eukaryotic promoter regions. Nucleic Acids Res. 1984, 12, 8043–8058. [Google Scholar] [CrossRef]
- Kilpatrick, M.W.; Torri, A.; Kang, D.S.; Engler, J.A.; Wells, R.D. Unusual DNA structures in the adenovirus genome. J. Biol. Chem. 1986, 261, 11350–11354. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, A.; Honjo, T. Immunoglobulin class switching. Cell 1984, 36, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.W. NMR methods for studying quadruplex nucleic acids. Methods 2007, 43, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.H.; Parkinson, G.N. Crystallographic studies of quadruplex nucleic acids. Methods 2007, 43, 252–263. [Google Scholar] [CrossRef]
- Paramasivan, S.; Rujan, I.; Bolton, P.H. Circular dichroism of quadruplex DNAs: Applications to structure, cation effects and ligand binding. Methods 2007, 43, 324–331. [Google Scholar] [CrossRef]
- Fojtik, P.; Kejnovská, I.; Vorlícková, M. The guanine-rich fragile X chromosome repeats are reluctant to form tetraplexes. Nucleic Acids Res. 2004, 32, 298–306. [Google Scholar] [CrossRef]
- Giraldo, R.; Suzuki, M.; Chapman, L.; Rhodes, D. Promotion of parallel DNA quadruplexes by a yeast telomere binding protein: A circular dichroism study. Proc. Natl. Acad. Sci. USA 1994, 91, 7658–7662. [Google Scholar] [CrossRef]
- Rachwal, P.A.; Fox, K.R. Quadruplex melting. Methods 2007, 43, 291–301. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Phan, A.T.; Lacroix, L. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998, 435, 74–78. [Google Scholar] [CrossRef]
- Puig Lombardi, E.; Londoño-Vallejo, A. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2020, 48, 1603. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Tanaka, T. G-quadruplex DNA and RNA: Their roles in regulation of DNA replication and other biological functions. Biochem. Biophys. Res. Commun. 2020, 531, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Valton, A.; Hassan-Zadeh, V.; Lema, I.; Boggetto, N.; Alberti, P.; Saintomé, C.; Riou, J.-F.; Prioleau, M. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 2014, 33, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Callegaro, S.; Perrone, R.; Scalabrin, M.; Doria, F.; Palù, G.; Richter, S.N. A core extended naphtalene diimide G-quadruplex ligand potently inhibits herpes simplex virus 1 replication. Sci. Rep. 2017, 7, 2341. [Google Scholar] [CrossRef]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef]
- Bryan, T.M. Mechanisms of DNA Replication and Repair: Insights from the Study of G-Quadruplexes. Molecules 2019, 24, 3439. [Google Scholar] [CrossRef]
- Bugaut, A.; Balasubramanian, S. 5’-UTR RNA G-quadruplexes: Translation regulation and targeting. Nucleic Acids Res. 2012, 40, 4727–4741. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Klug, A. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef]
- Lipps, H.J.; Gruissem, W.; Prescott, D.M. Higher order DNA structure in macronuclear chromatin of the hypotrichous ciliate Oxytricha nova. Proc. Natl. Acad. Sci. USA 1982, 79, 2495–2499. [Google Scholar] [CrossRef]
- Bao, H.-L.; Liu, H.-S.; Xu, Y. Hybrid-type and two-tetrad antiparallel telomere DNA G-quadruplex structures in living human cells. Nucleic Acids Res. 2019, 47, 4940–4947. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.L.; Vaughan, A.T. A systematic description of MLL fusion gene formation. Crit. Rev. Oncol. Hematol. 2014, 91, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.J.; Chai, W.; Shay, J.W.; Wright, W.E. Telomere-End Processing: The Terminal Nucleotidesof Human Chromosomes. Mol. Cell 2005, 18, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Roach, R.J.; Garavís, M.; González, C.; Jameson, G.B.; Filichev, V.V.; Hale, T.K. Heterochromatin protein 1α interacts with parallel RNA and DNA G-quadruplexes. Nucleic Acids Res. 2020, 48, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-Quadruplex-Interactive Molecule BRACO-19 Inhibits Tumor Growth, Consistent with Telomere Targeting and Interference with Telomerase Function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of Human Telomerase by a G-Quadruplex-Interactive Compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844–4849. [Google Scholar] [CrossRef]
- Sette, M.; D’Addabbo, P.; Kelly, G.; Cicconi, A.; Micheli, E.; Cacchione, S.; Poma, A.; Gargioli, C.; Giambra, V.; Frezza, D. Evidence for a quadruplex structure in the polymorphic hs1.2 enhancer of the immunoglobulin heavy chain 3′ regulatory regions and its conservation in mammals. Biopolymers 2016, 105, 768–778. [Google Scholar] [CrossRef]
- Teng, F.-Y.; Jiang, Z.-Z.; Guo, M.; Tan, X.-Z.; Chen, F.; Xi, X.-G.; Xu, Y. G-quadruplex DNA: A novel target for drug design. Cell. Mol. Life Sci. 2021, 78, 6557–6583. [Google Scholar] [CrossRef]
- Yang, S.Y.; Chang, E.Y.C.; Lim, J.; Kwan, H.H.; Monchaud, D.; Yip, S.; Stirling, P.C.; Wong, J.M.Y. G-quadruplexes mark alternative lengthening of telomeres. NAR Cancer 2021, 3, zcab031. [Google Scholar] [CrossRef]
- Koirala, D.; Dhakal, S.; Ashbridge, B.; Sannohe, Y.; Rodriguez, R.; Sugiyama, H.; Balasubramanian, S.; Mao, H. A single-molecule platform for investigation of interactions between G-quadruplexes and small-molecule ligands. Nat. Chem. 2011, 3, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Schertzer, M.; Wu, X.; Gertsenstein, M.; Selig, S.; Kammori, M.; Pourvali, R.; Poon, S.; Vulto, I.; Chavez, E.; et al. Regulation of Murine Telomere Length by Rtel: An Essential Gene Encoding a Helicase-like Protein. Cell 2004, 117, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In Vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; McDonald, K.R.; Zakian, V.A. Telomeres: Structures in need of unwinding. FEBS Lett. 2010, 584, 3760–3772. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective Telomere Lagging Strand Synthesis in Cells Lacking WRN Helicase Activity. Science 2004, 306, 1951–1953. [Google Scholar] [CrossRef]
- Babbe, H.; McMenamin, J.; Hobeika, E.; Wang, J.; Rodig, S.J.; Reth, M.; Leder, P. Genomic Instability Resulting from Blm Deficiency Compromises Development, Maintenance, and Function of the B Cell Lineage. J. Immunol. 2009, 182, 347–360. [Google Scholar] [CrossRef]
- Odermatt, D.C.; Lee, W.T.C.; Wild, S.; Jozwiakowski, S.K.; Rothenberg, E.; Gari, K. Cancer-associated mutations in the iron-sulfur domain of FANCJ affect G-quadruplex metabolism. PLoS Genet. 2020, 16, e1008740. [Google Scholar] [CrossRef] [PubMed]
- Van Schie, J.J.M.; Faramarz, A.; Balk, J.A.; Stewart, G.S.; Cantelli, E.; Oostra, A.B.; Rooimans, M.A.; Parish, J.L.; de Almeida Estéves, C.; Dumic, K.; et al. Warsaw breakage syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nat. Commun. 2020, 11, 4287. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.-C.; Sundaresan, A.; O’Hara, R.; Gant, V.U.; Li, M.; Martire, S.; Warshaw, J.N.; Basu, A.; Banaszynski, L.A. ATRX promotes heterochromatin formation to protect cells from G-quadruplex DNA-mediated stress. Nat. Commun. 2021, 12, 3887. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.-M.; Lemaitre, J.-M. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Broxson, C.; Beckett, J.; Tornaletti, S. Transcription Arrest by a G Quadruplex Forming-Trinucleotide Repeat Sequence from the Human c-myb Gene. Biochemistry 2011, 50, 4162–4172. [Google Scholar] [CrossRef]
- Smestad, J.A.; Maher, L.J. Relationships between putative G-quadruplex-forming sequences, RecQ helicases, and transcription. BMC Med Genet. 2015, 16, 91. [Google Scholar] [CrossRef]
- Mirkin, S.M. DNA structures, repeat expansions and human hereditary disorders. Curr. Opin. Struct. Biol. 2006, 16, 351–358. [Google Scholar] [CrossRef]
- Belotserkovskii, B.P.; Liu, R.; Tornaletti, S.; Krasilnikova, M.M.; Mirkin, S.M.; Hanawalt, P.C. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 12816–12821. [Google Scholar] [CrossRef]
- Belotserkovskii, B.P.; Neil, A.J.; Saleh, S.S.; Shin, J.H.S.; Mirkin, S.M.; Hanawalt, P.C. Transcription blockage by homopurine DNA sequences: Role of sequence composition and single-strand breaks. Nucleic Acids Res. 2013, 41, 1817–1828. [Google Scholar] [CrossRef]
- Belotserkovskii, B.P.; Shin, J.H.S.; Hanawalt, P.C. Strong transcription blockage mediated by R-loop formation within a G-rich homopurine–homopyrimidine sequence localized in the vicinity of the promoter. Nucleic Acids Res. 2017, 45, 6589–6599. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Xia, Y.; Hao, Y.-H.; Tan, Z. DNA:RNA hybrid G-quadruplex formation upstream of transcription start site. Sci. Rep. 2020, 10, 7429. [Google Scholar] [CrossRef] [PubMed]
- Szlachta, K.; Thys, R.; Atkin, N.D.; Pierce, L.C.T.; Bekiranov, S.; Wang, Y.-H. Alternative DNA secondary structure formation affects RNA polymerase II promoter-proximal pausing in human. Genome Biol. 2018, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, Z.; Zhao, X. Targeting Bcl-2 for cancer therapy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1876, 188569. [Google Scholar] [CrossRef]
- Shklover, J.; Weisman-Shomer, P.; Yafe, A.; Fry, M. Quadruplex structures of muscle gene promoter sequences enhance in vivo MyoD-dependent gene expression. Nucleic Acids Res. 2010, 38, 2369–2377. [Google Scholar] [CrossRef]
- Valton, A.-L.; Venev, S.V.; Mair, B.; Khokhar, E.S.; Tong, A.H.Y.; Usaj, M.; Chan, K.; Pai, A.A.; Moffat, J.; Dekker, J. A cohesin traffic pattern genetically linked to gene regulation. Nat. Struct. Mol. Biol. 2022, 29, 1239–1251. [Google Scholar] [CrossRef]
- Duquette, M.L.; Huber, M.D.; Maizels, N. G-Rich Proto-Oncogenes Are Targeted for Genomic Instability in B-Cell Lymphomas. Cancer Res. 2007, 67, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Phang, T.; Edwards, M.G.; Geraci, M.W.; Gillespie, M.N. Promoter G-quadruplex sequences are targets for base oxidation and strand cleavage during hypoxia-induced transcription. Free Radic. Biol. Med. 2012, 53, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Thomas, C.J.F.; Petersen-Mahrt, S.K.; Neuberger, M.S. Evolution of the AID/APOBEC Family of Polynucleotide (Deoxy)cytidine Deaminases. Mol. Biol. Evol. 2005, 22, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, I.; Laffleur, B.; Dalloul, Z.; Wehbi, B.; Jouan, F.; Brauge, B.; Derouault, P.; Moreau, J.; Kracker, S.; Fischer, A.; et al. UnAIDed Class Switching in Activated B-Cells Reveals Intrinsic Features of a Self-Cleaving IgH Locus. Front. Immunol. 2021, 12, 737427. [Google Scholar] [CrossRef]
- Wang, L.; Wuerffel, R.; Feldman, S.; Khamlichi, A.A.; Kenter, A.L. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J. Exp. Med. 2009, 206, 1817–1830. [Google Scholar] [CrossRef]
- Matthews, A.J.; Zheng, S.; DiMenna, L.J.; Chaudhuri, J. Chapter One-Regulation of Immunoglobulin Class-Switch Recombination: Choreography of Noncoding Transcription, Targeted DNA Deamination, and Long-Range DNA Repair. Adv. Immunol. 2014, 122, 1–57. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Jung, S.; Radbruch, A. Switch Transcripts in Immunoglobulin Class Switching. Science 1995, 267, 1825–1828. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.; Lorenz, M.G.; Siebenkotten, G.; Petry, K.; Christine, R.; Radbruch, A. Processing of Switch Transcripts Is Required for Targeting of Antibody Class Switch Recombination. J. Exp. Med. 1998, 188, 2369–2374. [Google Scholar] [CrossRef]
- Saintamand, A.; Rouaud, P.; Saad, F.; Rios, G.; Cogné, M.; Denizot, Y. Elucidation of IgH 3′ region regulatory role during class switch recombination via germline deletion. Nat. Commun. 2015, 6, 7084. [Google Scholar] [CrossRef] [PubMed]
- Fitz, J.; Neumann, T.; Steininger, M.; Wiedemann, E.-M.; Garcia, A.C.; Athanasiadis, A.; Schoeberl, U.E.; Pavri, R. Spt5-mediated enhancer transcription directly couples enhancer activation with physical promoter interaction. Nat. Genet. 2020, 52, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bottaro, A.; Li, S.; Stewart, V.; Alt, F.W. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. EMBO J. 1993, 12, 3529–3537. [Google Scholar] [CrossRef] [PubMed]
- Dunnick, W.; Hertz, G.Z.; Scappino, L.; Gritzmacher, C. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 1993, 21, 365–372. [Google Scholar] [CrossRef]
- Stavnezer, J. Immunoglobulin class switching. Curr. Opin. Immunol. 1996, 8, 199–205. [Google Scholar] [CrossRef]
- Reaban, M.E.; Griffin, J.A. Induction of RNA-stabilized DMA conformers by transcription of an immunoglobulin switch region. Nature 1990, 348, 342–344. [Google Scholar] [CrossRef]
- Shinkura, R.; Tian, M.; Smith, M.; Chua, K.; Fujiwara, Y.; Alt, F.W. The influence of transcriptional orientation on endogenous switch region function. Nat. Immunol. 2003, 4, 435–441. [Google Scholar] [CrossRef]
- Chauveau, C.; Cogné, M. Palindromic structure of the IgH 3′locus control region. Nat. Genet. 1996, 14, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Pinaud, E.; Cogne, M. Synergies between Regulatory Elements of the Immunoglobulin Heavy Chain Locus and Its Palindromic 3′ Locus Control Region. Eur. J. Immunol. 1998, 28, 3048–3056. [Google Scholar] [CrossRef]
- Le Noir, S.; Boyer, F.; Lecardeur, S.; Brousse, M.; Oruc, Z.; Cook-Moreau, J.; Denizot, Y.; Cogné, M. Functional anatomy of the immunoglobulin heavy chain 3′ super-enhancer needs not only core enhancer elements but also their unique DNA context. Nucleic Acids Res. 2017, 45, 5829–5837. [Google Scholar] [CrossRef]
- Saintamand, A.; Vincent-Fabert, C.; Garot, A.; Rouaud, P.; Oruc, Z.; Magnone, V.; Cogné, M.; Denizot, Y. Deciphering the importance of the palindromic architecture of the immunoglobulin heavy-chain 3’ regulatory region. Nat. Commun. 2016, 7, 10730. [Google Scholar] [CrossRef]
- Pinaud, E.; Marquet, M.; Fiancette, R.; Péron, S.; Vincent-Fabert, C.; Denizot, Y.; Cogné, M. The IgH Locus 3′ Regulatory Region: Pulling the Strings from Behind. Adv. Immunol. 2011, 110, 27–70. [Google Scholar] [CrossRef] [PubMed]
- Péron, S.; Laffleur, B.; Denis-Lagache, N.; Cook-Moreau, J.; Tinguely, A.; Delpy, L.; Denizot, Y.; Pinaud, E.; Cogné, M. AID-Driven Deletion Causes Immunoglobulin Heavy Chain Locus Suicide Recombination in B Cells. Science 2012, 336, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Cogné, M.; Lansford, R.; Bottaro, A.; Zhang, J.; Gorman, J.; Young, F.; Cheng, H.-L.; Alt, F.W. A class switch control region at the 3′ end of the immunoglobulin heavy chain locus. Cell 1994, 77, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Pinaud, E.; Khamlichi, A.A.; Le Morvan, C.; Drouet, M.; Nalesso, V.; Le Bert, M.; Cogné, M. Localization of the 3′ IgH Locus Elements that Effect Long-Distance Regulation of Class Switch Recombination. Immunity 2001, 15, 187–199. [Google Scholar] [CrossRef]
- Wuerffel, R.; Wang, L.; Grigera, F.; Manis, J.; Selsing, E.; Perlot, T.; Alt, F.W.; Cogne, M.; Pinaud, E.; Kenter, A.L. S-S Synapsis during Class Switch Recombination Is Promoted by Distantly Located Transcriptional Elements and Activation-Induced Deaminase. Immunity 2007, 27, 711–722. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Ba, Z.; Kyritsis, N.; Casellas, R.; Alt, F.W. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 2019, 575, 385–389. [Google Scholar] [CrossRef]
- Vian, L.; Pękowska, A.; Rao, S.S.; Kieffer-Kwon, K.-R.; Jung, S.; Baranello, L.; Huang, S.-C.; El Khattabi, L.; Dose, M.; Pruett, N.; et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 2018, 173, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, A.C.; Shen, A.; Richards, J.E.; Blattner, F.R.; Mushinski, J.F.; Tucker, P.W. Illegitimate recombination generates a class switch from C mu to C delta in an IgD-secreting plasmacytoma. Proc. Natl. Acad. Sci. USA 1984, 81, 4164–4168. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, S.; Delpy, L.; Fleury, L.; Dougier, H.-L.; Sirac, C.; Cogné, M. Interallelic Class Switch Recombination Contributes Significantly to Class Switching in Mouse B Cells. J. Immunol. 2005, 174, 6176–6183. [Google Scholar] [CrossRef]
- Dougier, H.-L.; Reynaud, S.; Pinaud, E.; Carrion, C.; Delpy, L.; Cogné, M. Interallelic class switch recombination can reverse allelic exclusion and allowtrans-complementation of an IgH locus switching defect. Eur. J. Immunol. 2006, 36, 2181–2191. [Google Scholar] [CrossRef]
- Laffleur, B.; Bardet, S.M.; Garot, A.; Brousse, M.; Baylet, A.; Cogné, M. Immunoglobulin genes undergo legitimate repair in human B cells not only after cis-but also frequent trans-class switch recombination. Genes Immun. 2014, 15, 341–346. [Google Scholar] [CrossRef]
- Pinaud, E.; Aupetit, C.; Chauveau, C.; Cogne, M. Identification of a homolog of the Cα3′/hs3 enhancer and of an allelic variant of the 3′IgH/hs1,2 enhancer downstream the human immunoglobulin α1 gene. Eur. J. Immunol. 1997, 27, 2981–2985. [Google Scholar] [CrossRef]
- Garot, A.; Marquet, M.; Saintamand, A.; Bender, S.; Le Noir, S.; Rouaud, P.; Carrion, C.; Oruc, Z.; Bébin, A.-G.; Moreau, J.; et al. Sequential activation and distinct functions for distal and proximal modules within the IgH 3′ regulatory region. Proc. Natl. Acad. Sci. USA 2016, 113, 1618–1623. [Google Scholar] [CrossRef]
- Vincent-Fabert, C.; Fiancette, R.; Pinaud, E.; Truffinet, V.; Cogné, N.; Cogné, M.; Denizot, Y. Genomic deletion of the whole IgH 3′ regulatory region (hs3a, hs1,2, hs3b, and hs4) dramatically affects class switch recombination and Ig secretion to all isotypes. Blood 2010, 116, 1895–1898. [Google Scholar] [CrossRef]
- Rouaud-Tinguely, P.; Vincent-Fabert, C.; Saintamand, A.; Fiancette, R.; Marquet, M.; Robert, I.; Reina-San-Martin, B.; Pinaud, E.; Cogné, M.; Denizot, Y. The IgH 3′ regulatory region controls somatic hypermutation in germinal center B cells. J. Exp. Med. 2013, 210, 1501–1507. [Google Scholar] [CrossRef]
- Saintamand, A.; Vincent-Fabert, C.; Marquet, M.; Ghazzaui, N.; Magnone, V.; Pinaud, E.; Cogné, M.; Denizot, Y. Eμ and 3′RR IgH enhancers show hierarchic unilateral dependence in mature B-cells. Sci. Rep. 2017, 7, 442. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer-Nordgren, J.; Sirlin, S. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. EMBO J. 1986, 5, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Radcliffe, G.; Lin, Y.C.; Nietupski, J.; Berggren, L.; Sitia, R.; Severinson, E. Immunoglobulin heavy-chain switching may be directed by prior induction of transcripts from constant-region genes. Proc. Natl. Acad. Sci. USA 1988, 85, 7704–7708. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Gorham, B.; Li, S.C.; Bottaro, A.; Alt, F.W.; Rothman, P. Replacement of germ-line epsilon promoter by gene targeting alters control of immunoglobulin heavy chain class switching. Proc. Natl. Acad. Sci. USA 1993, 90, 3705–3709. [Google Scholar] [CrossRef]
- Bottaro, A.; Lansford, R.; Xu, L.; Zhang, J.; Rothman, P.; Alt, F.W. S region transcription per se promotes basal IgE class switch recombination but additional factors regulate the efficiency of the process. EMBO J. 1994, 13, 665–674. [Google Scholar] [CrossRef]
- Yewdell, W.T.; Chaudhuri, J. A transcriptional serenAID: The role of noncoding RNAs in class switch recombination. Int. Immunol. 2017, 29, 183–196. [Google Scholar] [CrossRef]
- Jung, S.; Rajewsky, K.; Radbruch, A. Shutdown of Class Switch Recombination by Deletion of a Switch Region Control Element. Science 1993, 259, 984–987. [Google Scholar] [CrossRef]
- Marchalot, A.; Ashi, M.O.; Lambert, J.-M.; Carrion, C.; Lecardeur, S.; Srour, N.; Delpy, L.; Le Pennec, S. Uncoupling Splicing from Transcription Using Antisense Oligonucleotides Reveals a Dual Role for I Exon Donor Splice Sites in Antibody Class Switching. Front. Immunol. 2020, 11, 780. [Google Scholar] [CrossRef]
- Kuzin, I.I.; Ugine, G.D.; Wu, D.; Young, F.; Chen, J.; Bottaro, A. Normal Isotype Switching in B Cells Lacking the Iμ Exon Splice Donor Site: Evidence for Multiple Iμ-Like Germline Transcripts. J. Immunol. 2000, 164, 1451–1457. [Google Scholar] [CrossRef]
- Yu, K.; Lieber, M.R. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair 2003, 2, 1163–1174. [Google Scholar] [CrossRef]
- Bottaro, A.; Young, F.; Chen, J.; Serwe, M.; Sablitzky, F.; Alt, F.W. Deletion of the IgH intronic enhancer and associated matrix-attachment regions decreases, but does not abolish, class switching at the mu locus. Int. Immunol. 1998, 10, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zou, Y.-R.; Rajewsky, K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 1993, 73, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Seidl, K.J.; Bottaro, A.; Vo, A.; Zhang, J.; Davidson, L.; Alt, F.W. An expressed neo(r) cassette provides required functions of the 1gamma2b exon for class switching. Int. Immunol. 1998, 10, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Harriman, G.R.; Bradley, A.; Das, S.; Rogers-Fani, P.; Davis, A.C. IgA class switch in I alpha exon-deficient mice. Role of germline transcription in class switch recombination. J. Clin. Investig. 1996, 97, 477–485. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Alt, F.W. Class-switch recombination: Interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004, 4, 541–552. [Google Scholar] [CrossRef]
- Yu, K.; Chedin, F.; Hsieh, C.-L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef]
- Basu, U.; Meng, F.-L.; Keim, C.; Grinstein, V.; Pefanis, E.; Eccleston, J.; Zhang, T.; Myers, D.; Wasserman, C.R.; Wesemann, D.R.; et al. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell 2011, 144, 353–363. [Google Scholar] [CrossRef]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Chao, J.; Rabadan, R.; Economides, A.N.; Basu, U. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature 2014, 514, 389–393. [Google Scholar] [CrossRef]
- Laffleur, B.; Lim, J.; Zhang, W.; Chen, Y.; Pefanis, E.; Bizarro, J.; Batista, C.R.; Wu, L.; Economides, A.N.; Wang, J.; et al. Noncoding RNA processing by DIS3 regulates chromosomal architecture and somatic hypermutation in B cells. Nat. Genet. 2021, 53, 230–242. [Google Scholar] [CrossRef]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Kazadi, D.; Sun, J.; Federation, A.; Chao, J.; Elliott, O.; Liu, Z.-P.; et al. RNA Exosome-Regulated Long Non-Coding RNA Transcription Controls Super-Enhancer Activity. Cell 2015, 161, 774–789. [Google Scholar] [CrossRef]
- Kilchert, C.; Wittmann, S.; Vasiljeva, L. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016, 17, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.; Zhang, W.; Laffleur, B.; Jha, M.K.; Lim, J.; Lee, H.; Wu, L.; Alvarez, N.S.; Liu, Z.-P.; Munteanu, E.L.; et al. Mechanism of noncoding RNA-associated N6-methyladenosine recognition by an RNA processing complex during IgH DNA recombination. Mol. Cell 2021, 81, 3949–3964. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, Z.; Chenuet, P.; Dalloul, I.; Boyer, F.; Aldigier, J.-C.; Laffleur, B.; El Makhour, Y.; Ryffel, B.; Quesniaux, V.F.; Togbé, D.; et al. G-quadruplex DNA targeting alters class-switch recombination in B cells and attenuates allergic inflammation. J. Allergy Clin. Immunol. 2018, 142, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Wang, L.; Meng, F.-L.; Hwang, J.K.; Alt, F.W.; Wu, H. AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell 2017, 67, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Vallur, A.C.; Maizels, N. Activities of human exonuclease 1 that promote cleavage of transcribed immunoglobulin switch regions. Proc. Natl. Acad. Sci. USA 2008, 105, 16508–16512. [Google Scholar] [CrossRef]
- Collier, D.A.; Griffin, J.A.; Wells, R.D. Non-B right-handed DNA conformations of homopurine.homopyrimidine sequences in the murine immunoglobulin C alpha switch region. J. Biol. Chem. 1988, 263, 7397–7405. [Google Scholar] [CrossRef]
- Chi, X.; Li, Y.; Qiu, X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 2020, 160, 233–247. [Google Scholar] [CrossRef]
- Abbott, R.K.; Thayer, M.; Labuda, J.; Silva, M.; Philbrook, P.; Cain, D.W.; Kojima, H.; Hatfield, S.; Sethumadhavan, S.; Ohta, A.; et al. Germinal Center Hypoxia Potentiates Immunoglobulin Class Switch Recombination. J. Immunol. 2016, 197, 4014–4020. [Google Scholar] [CrossRef]
- Cho, S.H.; Raybuck, A.L.; Stengel, K.; Wei, M.; Beck, T.C.; Volanakis, E.; Thomas, J.W.; Hiebert, S.; Haase, V.H.; Boothby, M.R. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016, 537, 234–238. [Google Scholar] [CrossRef]
- Tashiro, J.; Kinoshita, K.; Honjo, T. Palindromic but not G-rich sequences are targets of class switch recombination. Int. Immunol. 2001, 13, 495–505. [Google Scholar] [CrossRef]
- Zheng, S.; Kusnadi, A.; Choi, J.E.; Vuong, B.Q.; Rhodes, D.; Chaudhuri, J. NME proteins regulate class switch recombination. FEBS Lett. 2019, 593, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Monchaud, D.; Wong, J.M.Y. Global mapping of RNA G-quadruplexes (G4-RNAs) using G4RP-seq. Nat. Protoc. 2022, 17, 870–889. [Google Scholar] [CrossRef]
- Duquette, M.L.; Handa, P.; Vincent, J.A.; Taylor, A.F.; Maizels, N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004, 18, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Nguyen, N.L.B.; Burrows, C.J. Colocalization of m6A and G-Quadruplex-Forming Sequences in Viral RNA (HIV, Zika, Hepatitis B, and SV40) Suggests Topological Control of Adenosine N6-Methylation. ACS Central Sci. 2019, 5, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Ookuro, Y.; Iida, K.; Nagasawa, K.; Yoshida, W. Destabilization of DNA and RNA G-quadruplex structures formed by GGA repeat due to N6-methyladenine modification. Biochem. Biophys. Res. Commun. 2022, 597, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Czerniak, T.; Saenz, J.P. Lipid membranes modulate the activity of RNA through sequence-dependent interactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2119235119. [Google Scholar] [CrossRef]
- Zheng, S.; Vuong, B.Q.; Vaidyanathan, B.; Lin, J.-Y.; Huang, F.-T.; Chaudhuri, J. Non-coding RNA Generated following Lariat Debranching Mediates Targeting of AID to DNA. Cell 2015, 161, 762–773. [Google Scholar] [CrossRef]
- De Almeida, C.R.; Dhir, S.; Dhir, A.; Moghaddam, A.E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N.J. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol. Cell 2018, 70, 650–662. [Google Scholar] [CrossRef]
- Schrader, C.E.; Guikema, J.E.J.; Linehan, E.K.; Selsing, E.; Stavnezer, J. Activation-Induced Cytidine Deaminase-Dependent DNA Breaks in Class Switch Recombination Occur during G1 Phase of the Cell Cycle and Depend upon Mismatch Repair. J. Immunol. 2007, 179, 6064–6071. [Google Scholar] [CrossRef]
- Wiedemann, E.-M.; Peycheva, M.; Pavri, R. DNA Replication Origins in Immunoglobulin Switch Regions Regulate Class Switch Recombination in an R-Loop-Dependent Manner. Cell Rep. 2016, 17, 2927–2942. [Google Scholar] [CrossRef]
- Bochman, M.L.; Schwacha, A. The Mcm Complex: Unwinding the Mechanism of a Replicative Helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652–683. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Ba, Z.; Liang, Z.; Dring, E.W.; Hu, H.; Lou, J.; Kyritsis, N.; Zurita, J.; Shamim, M.S.; et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 2019, 573, 600–604. [Google Scholar] [CrossRef]
- Li, L.; Williams, P.; Ren, W.; Wang, M.Y.; Gao, Z.; Miao, W.; Huang, M.; Song, J.; Wang, Y. YY1 interacts with guanine quadruplexes to regulate DNA looping and gene expression. Nat. Chem. Biol. 2021, 17, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.A.; Garrett, F.E.; Price-Whelan, A.; Birshtein, B.K. Comparative analysis of human and mouse 3′ Igh regulatory regions identifies distinctive structural features. Mol. Immunol. 2005, 42, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Le Noir, S.; Laffleur, B.; Carrion, C.; Garot, A.; Lecardeur, S.; Pinaud, E.; Denizot, Y.; Skok, J.; Cogné, M. The IgH locus 3′ cis-regulatory super-enhancer co-opts AID for allelic transvection. Oncotarget 2017, 8, 12929–12940. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, I.; Boyer, F.; Dalloul, Z.; Pignarre, A.; Caron, G.; Fest, T.; Chatonnet, F.; Delaloy, C.; Durandy, A.; Jeannet, R.; et al. Locus suicide recombination actively occurs on the functionally rearranged IgH allele in B-cells from inflamed human lymphoid tissues. PLoS Genet. 2019, 15, e1007721. [Google Scholar] [CrossRef] [PubMed]
- Péron, S.; Laffleur, B.; Denis-Lagache, N.; Cook-Moreau, J.; Filloux, M.; Cogne, M. IgH locus suicide recombination, or when B cells surrender! Med. Sci. MS 2012, 28, 551–554. [Google Scholar] [CrossRef]
- Xu, Y.-Z.; Jenjaroenpun, P.; Wongsurawat, T.; Byrum, S.D.; Shponka, V.; Tannahill, D.; Chavez, E.A.; Hung, S.S.; Steidl, C.; Balasubramanian, S.; et al. Activation-induced cytidine deaminase localizes to G-quadruplex motifs at mutation hotspots in lymphoma. NAR Cancer 2020, 2, zcaa029. [Google Scholar] [CrossRef] [PubMed]
- Duquette, M.L.; Pham, P.; Goodman, M.F.; Maizels, N. AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation. Oncogene 2005, 24, 5791–5798. [Google Scholar] [CrossRef]
- Yewdell, W.T.; Kim, Y.; Chowdhury, P.; Lau, C.M.; Smolkin, R.M.; Belcheva, K.T.; Fernandez, K.C.; Cols, M.; Yen, W.-F.; Vaidyanathan, B.; et al. A Hyper-IgM Syndrome Mutation in Activation-Induced Cytidine Deaminase Disrupts G-Quadruplex Binding and Genome-wide Chromatin Localization. Immunity 2020, 53, 952–970. [Google Scholar] [CrossRef]
- Durandy, A.; Peron, S.; Taubenheim, N.; Fischer, A. Activation-induced cytidine deaminase: Structure–function relationship as based on the study of mutants. Hum. Mutat. 2006, 27, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Revy, P.; Muto, T.; Levy, Y.; Geissmann, F.; Plebani, A.; Sanal, O.; Catalan, N.; Forveille, M.; Dufourcq-Lagelouse, R.; Gennery, A.; et al. Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell 2000, 102, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Samaniego-Castruita, D.; Dong, Z.; González-Avalos, E.; Yan, Q.; Sarma, K.; Rao, A. TET deficiency perturbs mature B cell homeostasis and promotes oncogenesis associated with accumulation of G-quadruplex and R-loop structures. Nat. Immunol. 2022, 23, 99–108. [Google Scholar] [CrossRef]
- Orlanski, S.; Labi, V.; Reizel, Y.; Spiro, A.; Lichtenstein, M.; Levin-Klein, R.; Koralov, S.B.; Skversky, Y.; Rajewsky, K.; Cedar, H.; et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc. Natl. Acad. Sci. USA 2016, 113, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Béguelin, W.; Fontán, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov. 2018, 8, 1632–1653. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.; Love, C.L.; Waldrop, A.; Leppä, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kumari, S.; Rodriguez, R.; Balasubramanian, S. Small-molecule-mediated G-quadruplex isolation from human cells. Nat. Chem. 2010, 2, 1095–1098. [Google Scholar] [CrossRef]
- Brunori, M.; Gilson, E. Télomère et cancer: Quoi de plus à la fin ? Med. Sci. MS 2005, 21, 37–42. [Google Scholar] [CrossRef]
- Groelly, F.J.; Porru, M.; Zimmer, J.; Benainous, H.; De Visser, Y.; Kosova, A.A.; Di Vito, S.; Serra, V.; Ryan, A.; Leonetti, C.; et al. Anti-tumoural activity of the G-quadruplex ligand pyridostatin against BRCA1/2-deficient tumours. EMBO Mol. Med. 2022, 14, e14501. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Zheng, B.-X.; Li, Y.; Huang, X.-H.; Lin, D.-M.; Chen, C.-C.; Hou, J.-Q.; Ou, T.-M.; Wong, W.-L.; Zhang, K.; et al. Rational design of small-molecules to recognize G-quadruplexes of c-MYC promoter and telomere and the evaluation of their in vivo antitumor activity against breast cancer. Nucleic Acids Res. 2022, 50, 1829–1848. [Google Scholar] [CrossRef] [PubMed]
- Debbarma, S.; Acharya, P.C. Targeting G-Quadruplex Dna for Cancer Chemotherapy. Curr. Cancer Drug Targets 2022, 19, 13–25. [Google Scholar] [CrossRef]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [PubMed]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Bardin, C.; Leroy, J.L. The formation pathway of tetramolecular G-quadruplexes. Nucleic Acids Res. 2008, 36, 477–488. [Google Scholar] [CrossRef]
- Bödör, C.; Bognár, Á.; Reiniger, L.; Szepesi, A.; Toth, E.; Kopper, L.; Matolcsy, A. Aberrant somatic hypermutation and expression of activation-induced cytidine deaminase mRNA in mediastinal large B-cell lymphoma. Br. J. Haematol. 2005, 129, 373–376. [Google Scholar] [CrossRef]
- Nagaoka, H.; Tran, T.H.; Kobayashi, M.; Aida, M.; Honjo, T. Preventing AID, a physiological mutator, from deleterious activation: Regulation of the genomic instability that is associated with antibody diversity. Int. Immunol. 2010, 22, 227–235. [Google Scholar] [CrossRef]
- Takizawa, M.; Tolarová, H.; Li, Z.; Dubois, W.; Lim, S.; Callen, E.; Franco, S.; Mosaico, M.; Feigenbaum, L.; Alt, F.W.; et al. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J. Exp. Med. 2008, 205, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Wang, J.; Nuccio, S.P.; Mao, H.; Di Antonio, M. Short LNA-modified oligonucleotide probes as efficient disruptors of DNA G-quadruplexes. Nucleic Acids Res. 2022, 50, 7247–7259. [Google Scholar] [CrossRef] [PubMed]
- Bossaert, M.; Pipier, A.; Riou, J.-F.; Noirot, C.; Nguyên, L.-T.; Serre, R.-F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2α (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. Elife 2021, 10, e65184. [Google Scholar] [CrossRef] [PubMed]
- Zyner, K.G.; Mulhearn, D.S.; Adhikari, S.; Cuesta, S.M.; Di Antonio, M.; Erard, N.; Hannon, G.J.; Tannahill, D.; Balasubramanian, S. Genetic interactions of G-quadruplexes in humans. Elife 2019, 8, e46793. [Google Scholar] [CrossRef] [PubMed]
- McLuckie, K.I.E.; Di Antonio, M.; Zecchini, H.; Xian, J.; Caldas, C.; Krippendorff, B.-F.; Tannahill, D.; Lowe, C.; Balasubramanian, S. G-Quadruplex DNA as a Molecular Target for Induced Synthetic Lethality in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 9640–9643. [Google Scholar] [CrossRef] [PubMed]
- Pipier, A.; Devaux, A.; Lavergne, T.; Adrait, A.; Couté, Y.; Britton, S.; Calsou, P.; Riou, J.F.; Defrancq, E.; Gomez, D. Constrained G4 structures unveil topology specificity of known and new G4 binding proteins. Sci. Rep. 2021, 11, 13469. [Google Scholar] [CrossRef]
- Calderwood, S.K. A critical role for topoisomerase IIb and DNA double strand breaks in transcription. Transcription 2016, 7, 75–83. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.-R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521. [Google Scholar] [CrossRef]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Fridman, W.H.; Petitprez, F.; Meylan, M.; Chen, T.W.-W.; Sun, C.-M.; Roumenina, L.T.; Sautès-Fridman, C. B cells and cancer: To B or not to B? J. Exp. Med. 2020, 218, e20200851. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougoüin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.-M.; Italiano, A.; Sautès-Fridman, C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dézé, O.; Laffleur, B.; Cogné, M. Roles of G4-DNA and G4-RNA in Class Switch Recombination and Additional Regulations in B-Lymphocytes. Molecules 2023, 28, 1159. https://doi.org/10.3390/molecules28031159
Dézé O, Laffleur B, Cogné M. Roles of G4-DNA and G4-RNA in Class Switch Recombination and Additional Regulations in B-Lymphocytes. Molecules. 2023; 28(3):1159. https://doi.org/10.3390/molecules28031159
Chicago/Turabian StyleDézé, Ophélie, Brice Laffleur, and Michel Cogné. 2023. "Roles of G4-DNA and G4-RNA in Class Switch Recombination and Additional Regulations in B-Lymphocytes" Molecules 28, no. 3: 1159. https://doi.org/10.3390/molecules28031159
APA StyleDézé, O., Laffleur, B., & Cogné, M. (2023). Roles of G4-DNA and G4-RNA in Class Switch Recombination and Additional Regulations in B-Lymphocytes. Molecules, 28(3), 1159. https://doi.org/10.3390/molecules28031159