

The Bioavailability of Drugs—The Current State of Knowledge

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
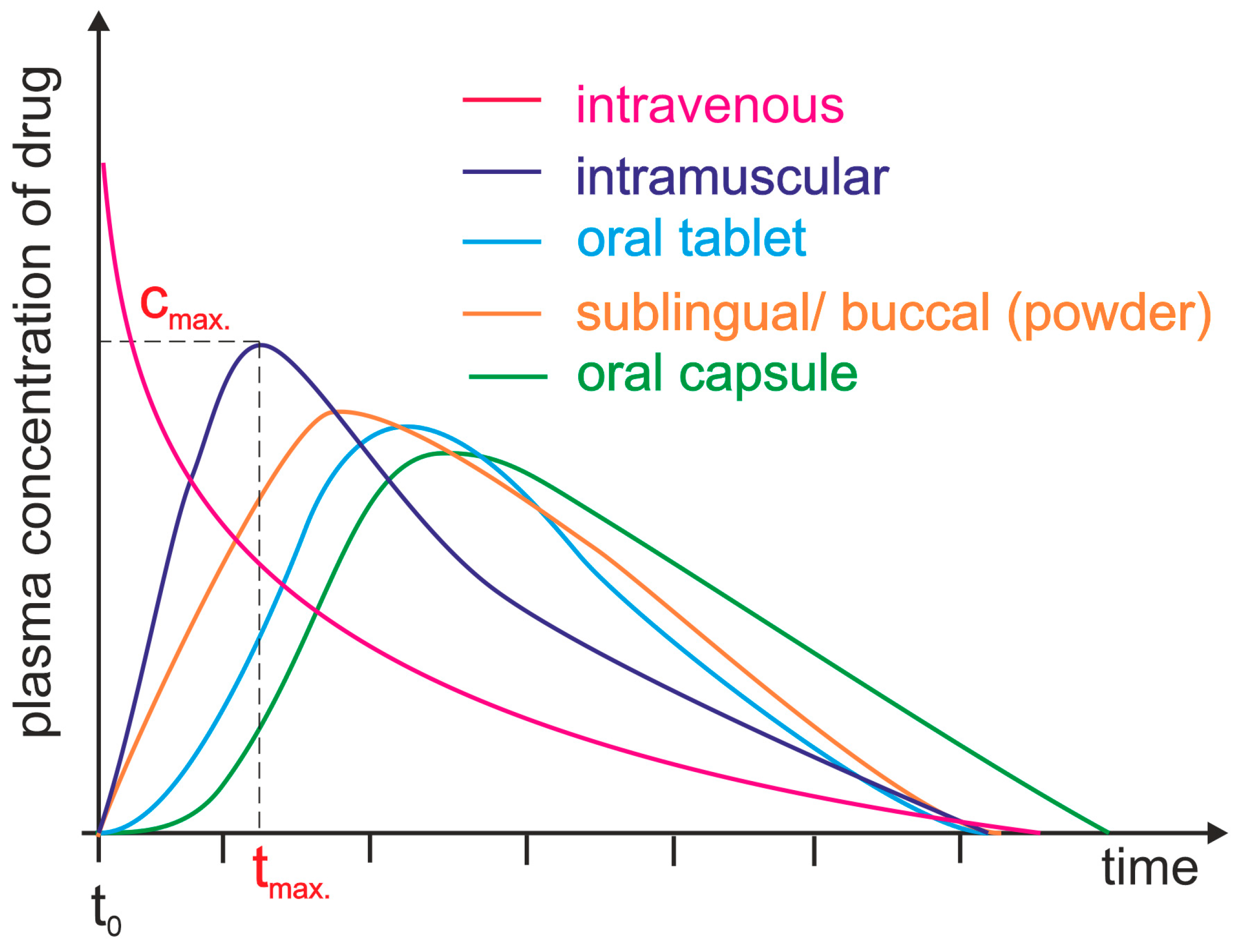
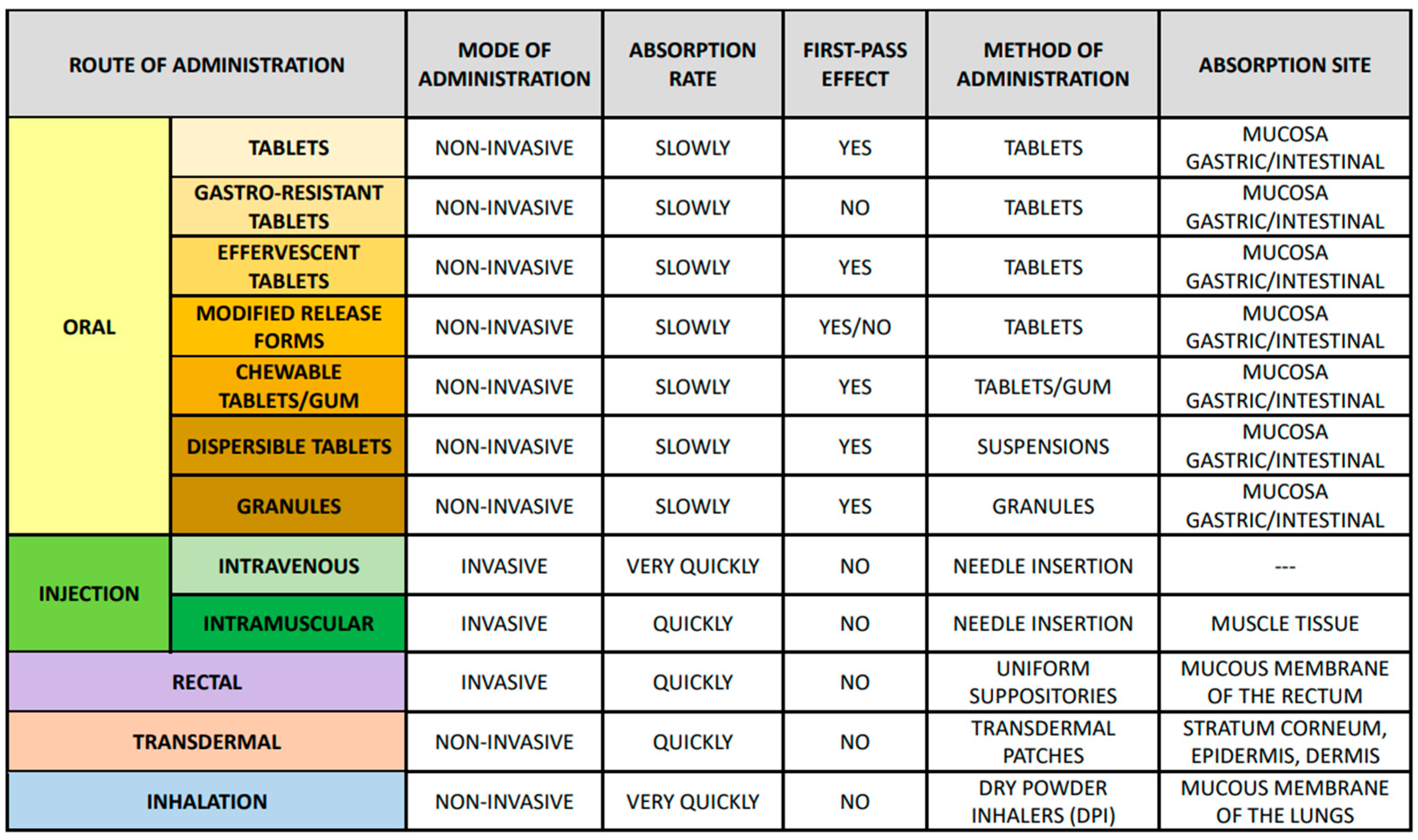
3. Bioavailability of Drugs: Basic Concepts and Controlling Factors
4. Methods for Assessing Drug Bioavailability
5. Drugs with Poorly Described Bioavailability
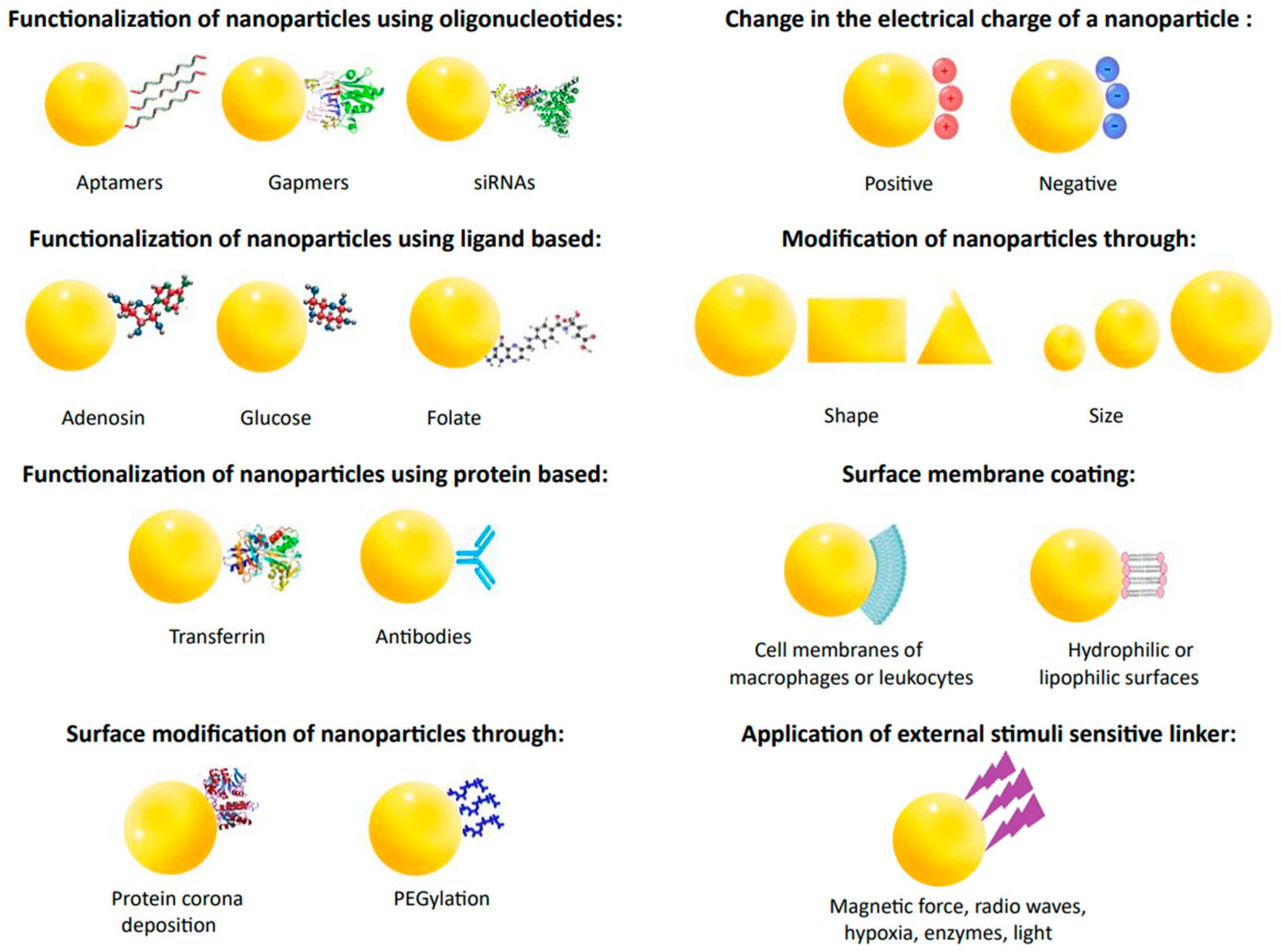
6. Challenges and Prospects in Drug Bioavailability Research
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herkenne, C.; Alberti, I.; Naik, A.; Kalia, Y.N.; Mathy, F.X.; Preat, V.; Guy, R.H. In vivo methods for the assessment of topical drug bioavailability. Pharm. Res. 2008, 25, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Morales, A.; Hatley, O.J.; Turner, D.; Galetin, A.; Aarons, L.; Rostami-Hodjegan, A. The use of ROC analysis for the qualitative prediction of human oral bioavailability from animal data. Pharm. Res. 2014, 31, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.; Gardner, I.; Swales, N. An introduction to drug disposition: The basic principles of absorption, distribution, metabolism, and excretion. Toxicol. Pathol. 1995, 23, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.N.; Amidon, G.L. A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals. J. Clin. Pharmacol. 2002, 42, 620–643. [Google Scholar] [CrossRef] [PubMed]
- Doogue, M.P.; Polasek, T.M. The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Kumar, R.; Kishor, K.; Mlsna, T.; Pittman, C.U., Jr.; Mohan, D. Pharmaceuticals of emerging concern in aquatic systems: Chemistry, occurrence, effects, and removal methods. Chem. Rev. 2019, 119, 3510–3673. [Google Scholar] [CrossRef]
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. Gut reaction: Impact of systemic diseases on gastrointestinal physiology and drug absorption. Drug Discov. Today 2019, 24, 417–427. [Google Scholar] [CrossRef]
- Wagner, J.G. History of pharmacokinetics. Pharmacol. Therapeut. 1981, 12, 537–562. [Google Scholar] [CrossRef]
- Lin, L.; Wong, H. Predicting oral drug absorption: Mini review on physiologically-based pharmacokinetic models. Pharmaceutics 2017, 9, 41. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Pi, C.; Feng, X.; Hou, Y.; Zhao, L.; Wei, Y. The influence of nanoparticle properties on oral bioavailability of drugs. Int. J. Nanomed. 2020, 15, 6295–6310. [Google Scholar] [CrossRef]
- Graveno, M.; Stratford, R.E. Absorption, Distribution, Metabolism, and Excretion of Biopharmaceutical Drug Products. In ADME Processes in Pharmaceutical Sciences: Dosage, Design and Pharmacotherapy Success; Talevi, A., Quiroga, P.A.M., Eds.; Springer Nature: Cham, Switzerland, 2018; pp. 241–270. [Google Scholar]
- Zucker, I.; Prendergast, B.J. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol. Sex Differ. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Moher, D. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Int. J. Sur. 2021, 88, 105906. [Google Scholar] [CrossRef] [PubMed]
- Alston, A.B.; Digigow, R.; Fluhmann, B.; Wacker, M.G. Putting square pegs in round holes: Why traditional pharmacokinetic principles cannot universally be applied to iron-carbohydrate complexes. Eur. J. Pharm. Biopharm. 2023, 188, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Hedaya, M.A. Basic Pharmacokinetics, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Wang, F.; Yang, G.; Zhou, Y.; Song, H.; Xiong, L.; Wang, L.; Shen, X. Pharmacokinetics of niazirin from Moringa oleifera Lam in rats by UPLC-MS/MS: Absolute bioavailability and dose proportionality. eFood 2022, 3, e39. [Google Scholar] [CrossRef]
- EFSA Panel on Nutrition, Novel Foods and Food Allergens (NDA); Turck, D.; Bohn, T.; Castenmiller, J.; De Henauw, S.; Hirsch-Ernst, K.I.; Pentieva, K. Conversion of calcium-l-methylfolate and (6S)-5-methyltetrahydrofolic acid glucosamine salt into dietary folate equivalents. EFSA J. 2022, 20, e07452. [Google Scholar] [CrossRef] [PubMed]
- Currie, G.M. Pharmacology, part 2: Introduction to pharmacokinetics. J. Nucl. Med. Technol. 2018, 46, 221–230. [Google Scholar] [CrossRef]
- Tuntland, T.; Ethell, B.; Kosaka, T.; Blasco, F.; Zang, R.X.; Jain, M.; Hoffmaster, K. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol. 2014, 5, 174. [Google Scholar] [CrossRef]
- Wei, M.; Zhang, X.; Pan, X.; Wang, B.; Ji, C.; Qi, Y.; Zhang, J.Z. HobPre: Accurate prediction of human oral bioavailability for small molecules. J. Cheminform. 2022, 14, 1–10. [Google Scholar] [CrossRef]
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Kalaimathi, K.; Shine, K.; Gandhi, G.R.; Vijayakumar, S.; Ayyanar, M.; Amalraj, S.; Jose, J. Cyanobacterial metabolites as novel potential suppressors of breast cancer: A comparative in silico pharmacological assessment. Intell. Pharm. 2023, 1, 133–144. [Google Scholar] [CrossRef]
- Stillhart, C.; Vučićević, K.; Augustijns, P.; Basit, A.W.; Batchelor, H.; Flanagan, T.R.; Müllertz, A. Impact of gastrointestinal physiology on drug absorption in special populations—An UNGAP review. Eur. J. Pharm. Sci. 2020, 147, 105280. [Google Scholar] [CrossRef] [PubMed]
- Belayneh, A.; Molla, F. The effect of coffee on pharmacokinetic properties of drugs: A review. Biomed. Res. Int. 2020, 2020, 7909703. [Google Scholar] [CrossRef] [PubMed]
- Sochacka, J.; Lipska, I. Rola α1-kwaśnej glikoproteiny surowicy krwi ludzkiej w procesie wiązania leków, sytuacja w Polsce i na świecie. Farmacja Polska 2014, 70, 55–62. [Google Scholar]
- Jahanban-Esfahlan, A.; Ostadrahimi, A.; Jahanban-Esfahlan, R.; Roufegarinejad, L.; Tabibiazar, M.; Amarowicz, R. Recent developments in the detection of bovine serum albumin. Int. J. Biol. Macromol. 2019, 138, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M. Into the labyrinth of the lipocalin α1-acid glycoprotein. Front. Physiol. 2021, 847, 1–10. [Google Scholar] [CrossRef]
- Zsila, F.; Iwao, Y. The drug binding site of human α1-acid glycoprotein: Insight from induced circular dichroism and electronic absorption spectra. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2007, 1770, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Bian, C.; Zhu, L.; Zhao, G.; Huang, Z.; Huang, M. Effect of human serum albumin on drug metabolism: Structural evidence of esterase activity of human serum albumin. J. Struct. Biol. 2007, 157, 348–355. [Google Scholar] [CrossRef]
- Peters, T.; Stewart, A.J. Albumin research in the 21st century. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 12, 5351–5353. [Google Scholar] [CrossRef]
- Rabbani, G.; Ahn, S.N. Structure, enzymatic activities, glycation and therapeutic potential of human serum albumin: A natural cargo. Int. J. Biol. Macromol. 2019, 123, 979–990. [Google Scholar] [CrossRef]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Bi, H. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Settimo, L.; Bellman, K.; Knegtel, R.M. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm. Res. 2014, 31, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Gaohua, L.; Miao, X.; Dou, L. Crosstalk of physiological pH and chemical pKa under the umbrella of physiologically based pharmacokinetic modeling of drug absorption, distribution, metabolism, excretion, and toxicity. Expert Opin. Drug Met. 2021, 17, 1103–1124. [Google Scholar] [CrossRef]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Augustijns, P. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Yang, Z. Review of current strategies for delivering Alzheimer’s disease drugs across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.O.; He, M. Unlocking the Power of Exosomes for Crossing Biological Barriers in Drug Delivery. Pharmaceutics 2021, 13, 122. [Google Scholar] [CrossRef]
- Kratzer, I.; Ek, J.; Stolp, H. The molecular anatomy and functions of the choroid plexus in healthy and diseased brain. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183430. [Google Scholar] [CrossRef]
- Uchida, Y.; Goto, R.; Usui, T.; Tachikawa, M.; Terasaki, T. Blood-Arachnoid Barrier as a Dynamic Physiological and Pharmacological Interface between Cerebrospinal Fluid and Blood. In Drug Delivery to the Brain: Physiological Concepts, Methodologies and Approaches; Springer International Publishing: Cham, Germany, 2022; pp. 93–121. [Google Scholar] [CrossRef]
- Kiecker, C. The origins of the circumventricular organs. J. Anat. 2018, 232, 540–553. [Google Scholar] [CrossRef]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliver. Rev. 2020, 165, 1–14. [Google Scholar] [CrossRef]
- Villaseñor, R.; Lampe, J.; Schwaninger, M.; Collin, L. Intracellular transport and regulation of transcytosis across the blood–brain barrier. Cell. Mol. Life Sci. 2019, 76, 1081–1092. [Google Scholar] [CrossRef]
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Félix, M.; Burke, M.; Chen, H.H.; Patterson, C.; Mittal, S. Predicting bioavailability of monoclonal antibodies after subcutaneous administration: Open innovation challenge. Adv. Drug Deliver. Rev. 2020, 167, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Jin, Y. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Bannigan, P.; Aldeghi, M.; Bao, Z.; Häse, F.; Aspuru-Guzik, A.; Allen, C. Machine learning directed drug formulation development. Adv. Drug Deliver. Rev. 2021, 175, 113806. [Google Scholar] [CrossRef] [PubMed]
- Tanguay, M.; Girard, J.; Scarsi, C.; Mautone, G.; Larouche, R. Pharmacokinetics and comparative bioavailability of a levothyroxine sodium oral solution and soft capsule. Clin. Pharm. Dug Dev. 2019, 8, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Shariare, M.H.; Altamimi, M.A.; Marzan, A.L.; Tabassum, R.; Jahan, B.; Reza, H.M.; Kazi, M. In vitro dissolution and bioavailability study of furosemide nanosuspension prepared using design of experiment (DoE). Saudi Pharm. J. 2019, 27, 96–105. [Google Scholar] [CrossRef]
- Fijałkowski, Ł.; Skubiszewska, M.; Grześk, G.; Koech, F.K.; Nowaczyk, A. Acetylsalicylic acid–primus inter pares in Pharmacology. Molecules 2022, 27, 8412. [Google Scholar] [CrossRef]
- Pitiot, A.; Heuzé-Vourc’h, N.; Sécher, T. Alternative Routes of Administration for Therapeutic Antibodies. Antibodies 2022, 11, 56. [Google Scholar] [CrossRef]
- Alghamdi, M.A.; Fallica, A.N.; Virzì, N.; Kesharwani, P.; Pittalà, V.; Greish, K. The promise of nanotechnology in personalized medicine. J. Pers. Med. 2022, 12, 673. [Google Scholar] [CrossRef]
- Gröber, U. Magnesium and drugs. Int. J. Mol. Sci. 2019, 20, 2094. [Google Scholar] [CrossRef] [PubMed]
- Moyer, A.M.; Matey, E.T.; Miller, V.M. Individualized medicine: Sex, hormones, genetics, and adverse drug reactions. Pharmacol. Res. Perspect. 2019, 7, e00541. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Ashiru-Oredope, D.A.; Yao, Z.; Dou, L.; Madla, C.M.; Taherali, F.; Basit, A.W. Boosting drug bioavailability in men but not women through the action of an excipient. Int. J. Pharm. 2020, 587, 119678. [Google Scholar] [CrossRef] [PubMed]
- Angeles, P.C.; Robertsen, I.; Seeberg, L.T.; Krogstad, V.; Skattebu, J.; Sandbu, R.; Hjelmesæth, J. The influence of bariatric surgery on oral drug bioavailability in patients with obesity: A systematic review. Obes. Rev. 2019, 20, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Minuesa, G.; Arimany-Nardi, C.; Erkizia, I.; Cedeno, S.; Moltó, J.; Clotet, B.; Martinez-Picado, J. P-glycoprotein (ABCB1) activity decreases raltegravir disposition in primary CD4+ P-gphigh cells and correlates with HIV-1 viral load. J. Antimicrob. Chemoth. 2016, 71, 2782–2792. [Google Scholar] [CrossRef]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef]
- Australian Product Information Tivicay (dolutegravir) Film-Coated Tablets and TIVICAY PD (dolutegravir) Dispersible Tablets. Available online: https://www.tga.gov.au/sites/default/files/2022-08/auspar-tivicay-tivicay-pd-220705-pi.pdf (accessed on 10 October 2023).
- Shultz, M.D. Two decades under the influence of the rule of five and the changing properties of approved oral drugs: Miniperspective. J. Med. Chem. 2018, 62, 1701–1714. [Google Scholar] [CrossRef]
- U.S. FDA Approves GlaxoSmithKline’s HIV Drug Tivicay. Available online: https://www.reuters.com/article/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 (accessed on 5 October 2023).
- Chen, M.L.; Sun, A.; Cao, W.; Eliason, A.; Mendez, K.M.; Getzler, A.J.; Sundrud, M.S. Physiological expression and function of the MDR1 transporter in cytotoxic T lymphocytes. J. Exp. Med. 2020, 217, 5. [Google Scholar] [CrossRef]
- Cheng, L.; Wong, H. Food effects on oral drug absorption: Application of physiologically-based pharmacokinetic modeling as a predictive tool. Pharmaceutics 2020, 12, 672. [Google Scholar] [CrossRef]
- Selyutina, O.Y.; Polyakov, N.E. Glycyrrhizic acid as a multifunctional drug carrier–From physicochemical properties to biomedical applications: A modern insight on the ancient drug. Int. J. Pharm. 2019, 559, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Dev. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Liu, M.; Hong, C.; Li, G.; Sun, J.; Wang, J.; Xie, Y. The effects of pH, surfactant, ion concentration, coformer, and molecular arrangement on the solubility behavior of myricetin cocrystals. Acta Pharm. Sin. B 2019, 9, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Z.; Zhou, W.; Liang, X.; Zhou, G.; Han, C.C.; Liu, Y. Mechanism of a long-term controlled drug release system based on simple blended electrospun fibers. J. Control. Release 2020, 320, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Juvale, I.I.A.; Hamid, A.A.A.; Abd Halim, K.B.; Has, A.T.C. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Santbergen, M.J.; Van der Zande, M.; Gerssen, A.; Bouwmeester, H.; Nielen, M.W. Dynamic in vitro intestinal barrier model coupled to chip-based liquid chromatography mass spectrometry for oral bioavailability studies. Anal. Bioanal. Chem. 2020, 412, 1111–1122. [Google Scholar] [CrossRef]
- Attwa, M.W.; AlRabiah, H.; Mostafa, G.A.; Kadi, A.A. Development of an LC-MS/MS method for quantification of sapitinib in human liver microsomes: In silico and in vitro metabolic stability evaluation. Molecules 2023, 28, 2322. [Google Scholar] [CrossRef]
- Shinha, K.; Nihei, W.; Ono, T.; Nakazato, R.; Kimura, H. A pharmacokinetic–pharmacodynamic model based on multi-organ-on-a-chip for drug–drug interaction studies. Biomicrofluidics 2020, 14, 044108. [Google Scholar] [CrossRef]
- Fu, C.; Shi, H.; Chen, H.; Zhang, K.; Wang, M.; Qiu, F. Oral bioavailability comparison of artemisinin, deoxyartemisinin, and 10-deoxoartemisinin based on computer simulations and pharmacokinetics in rats. ACS Omega 2020, 6, 889–899. [Google Scholar] [CrossRef]
- Hashemzadeh, N.; Rahimpour, E.; Jouyban, A. Applications of Exhaled Breath Condensate Analysis for Drug Monitoring and Bioequivalence Study of Inhaled Drugs. J. Pharm. Pharm. Sci. 2022, 25, 391–401. [Google Scholar] [CrossRef]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A clinical pharmacokinetic and pharmacodynamic review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, Y.; Min, M.; Li, Y. Effect of dietary supplements on Se bioavailability: A comprehensive in vitro and in vivo study. Ecotox. Environ. Saf. 2022, 231, 113193. [Google Scholar] [CrossRef] [PubMed]
- Perez-Medina, C.; Teunissen, A.J.; Kluza, E.; Mulder, W.J.; Van der Meel, R. Nuclear imaging approaches facilitating nanomedicine translation. Adv. Drug Deliver. Rev. 2020, 154, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Dałek, P.; Drabik, D.; Wołczańska, H.; Foryś, A.; Jagas, M.; Jędruchniewicz, N.; Langner, M. Bioavailability by design—Vitamin D3 liposomal delivery vehicles. Nanomed. Nanotechnol. Biol. Med. 2022, 43, 102552. [Google Scholar] [CrossRef]
- Leite, P.M.; Martins, M.A.P.; das Graças Carvalho, M.; Castilho, R.O. Mechanisms and interactions in concomitant use of herbs and warfarin therapy: An updated review. Biomed. Pharmacother. 2021, 143, 112103. [Google Scholar] [CrossRef]
- Lewis, R.J.; Trager, W.F. Warfarin metabolism in man: Identification of metabolites in urine. J. Clin. Investig. 1970, 49, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, M.; Xiong, L.; Feng, W.; Williams, R.O., III. Bioavailability improvement of carbamazepine via oral administration of modified-release amorphous solid dispersions in rats. Pharmaceutics 2020, 12, 11–1023. [Google Scholar] [CrossRef]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 inducer carbamazepine and its drug–drug interaction potential: A physiologically based pharmacokinetic modeling approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef]
- Hsin, C.H.; Stoffel, M.S.; Gazzaz, M.; Schaeffeler, E.; Schwab, M.; Fuhr, U.; Taubert, M. Combinations of common SNPs of the transporter gene ABCB1 influence apparent bioavailability, but not renal elimination of oral digoxin. Sci. Rep. 2020, 10, 12457. [Google Scholar] [CrossRef]
- Ibrahim, N.A.M. An up-to-date review of digoxin toxicity and its management. Int. J. Res. Pharm. Pharm. Sci. 2019, 4, 59–64. [Google Scholar]
- Pawar, S.R.; Barhate, S.D. Solubility enhancement (Solid Dispersions) novel boon to increase bioavailability. J. Drug Deliv. Ther. 2019, 9, 583–590. [Google Scholar] [CrossRef]
- Kareem, S.H.K.A. Quality by Design Approach for Bioavailability Enhancement of Some Hydrophobic Drugs. Available online: https://shodhgangotri.inflibnet.ac.in/bitstream/20.500.14146/13393/1/final%20synopsis%20corrected.pdf (accessed on 5 October 2023).
- Pireddu, R.; Schlich, M.; Marceddu, S.; Valenti, D.; Pini, E.; Fadda, A.M.; Sinico, C. Nanosuspensions and microneedles roller as a combined approach to enhance diclofenac topical bioavailability. Pharmaceutics 2020, 12, 1140. [Google Scholar] [CrossRef] [PubMed]
- Sardana, K.; Mathachan, S.R. Super bioavailable itraconazole and its place and relevance in recalcitrant dermatophytosis: Revisiting skin levels of itraconazole and minimum inhibitory concentration data. Indian Dermatol. Online J. 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Bardelmeijer, H.A.; Beijnen, J.H.; Brouwer, K.R.; Rosing, H.; Nooijen, W.J.; Schellens, J.H.; van Tellingen, O. Increased oral bioavailability of paclitaxel by GF120918 in mice through selective modulation of P-glycoprotein. Clin. Cancer Res. 2000, 6, 4416–4421. [Google Scholar] [PubMed]
- Jirofti, N.; Poorsargol, M.; Sarhaddi, F.; Jahani, A.; Kadkhoda, J.; Kalalinia, F.; Taboada, P. Polymer stabilized, phenytoin-loaded nanomicelles as promising nanocarriers: In silico and in vitro evaluations. Eur. Polym. J. 2023, 196, 112228. [Google Scholar] [CrossRef]
- Markovic, M.; Zur, M.; Ragatsky, I.; Cvijić, S.; Dahan, A. BCS Class IV oral drugs and absorption windows: Regional-dependent intestinal permeability of furosemide. Pharmaceutics 2020, 12, 1175. [Google Scholar] [CrossRef]
- Virili, C.; Brusca, N.; Capriello, S.; Centanni, M. Levothyroxine therapy in gastric malabsorptive disorders. Front. Endocrinol. 2021, 11, 621616. [Google Scholar] [CrossRef]
- Bailey, D.G. Fruit juice inhibition of uptake transport: A new type of food–drug interaction. Brit. J. Clin. Pharmacol. 2010, 70, 645–655. [Google Scholar] [CrossRef]
- Park, J.; Agyemang, A.; Chow, M.S. Can currently available drugs for erectile dysfunction be re-formulated to achieve rapid effect? J. Asian Assoc. Sch. Pharm. 2019, 8, 58–63. [Google Scholar]
- Ansari, M.J.; Aldawsari, M.F.; Zafar, A.; Soltani, A.; Yasir, M.; Jahangir, M.A.; Sarjadi, M.S. In vitro release and cytotoxicity study of encapsulated sulfasalazine within LTSP micellar/liposomal and TSP micellar/niosomal nano-formulations. Alex. Eng. J. 2022, 61, 9749–9756. [Google Scholar] [CrossRef]
- Oglah, M.K.; Bashir, M.K.; Mustafa, Y.F. Hypericin and its analogues: A review of their biological activities. Turk. J. Field Crops 2021, 26, 259–269. [Google Scholar]
- Lin, Y.; Li, Y.; Zeng, Y.; Tian, B.; Qu, X.; Yuan, Q.; Song, Y. Pharmacology, toxicity, bioavailability, and formulation of magnolol: An update. Front. Pharmacol. 2021, 12, 632767. [Google Scholar] [CrossRef] [PubMed]
- Podoll, T.; Pearson, P.G.; Evarts, J.; Ingallinera, T.; Bibikova, E.; Sun, H.; Slatter, J.G. Bioavailability, biotransformation, and excretion of the covalent bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab. Dispos. 2019, 47, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Smidova, V.; Michalek, P.; Goliasova, Z.; Eckschlager, T.; Hodek, P.; Adam, V.; Heger, Z. Nanomedicine of tyrosine kinase inhibitors. Theranostics 2021, 11, 1546. [Google Scholar] [CrossRef]
- O’Laughlin, K.; Tobolowsky, F.A.; Elmor, R.; Overton, R.; O’Connor, S.M.; Damon, I.K.; Veillard, M. Clinical use of tecovirimat (Tpoxx) for treatment of monkeypox under an investigational new drug protocol—United States, May–August 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1190. [Google Scholar] [CrossRef] [PubMed]
- Wanjari, A.S.; Pathak, S.S.; Rajput, D.; Wanjari, D.S.; Jadhao, S.; Gokarn, R. Effect of Piper longum Linn on the oral bioavailability of Phenytoin. Int. J. Bot. Stud. 2020, 5, 119–122. [Google Scholar]
- Eedara, B.B.; Veerareddy, P.R.; Jukanti, R.; Bandari, S. Improved oral bioavailability of fexofenadine hydrochloride using lipid surfactants: Ex vivo, in situ and in vivo studies. Drug Dev. Ind. Pharm. 2014, 40, 1030–1043. [Google Scholar] [CrossRef]
- Deore, A.B.; Dhumane, J.R.; Wagh, R.; Sonawane, R. The stages of drug discovery and development process. Asian J. Pharm. Res. Dev. 2019, 7, 62–67. [Google Scholar] [CrossRef]
- Cruz-Burgos, M.; Losada-Garcia, A.; Cruz-Hernández, C.D.; Cortés-Ramírez, S.A.; Camacho-Arroyo, I.; Gonzalez-Covarrubias, V.; Rodríguez-Dorantes, M. New approaches in oncology for repositioning drugs: The case of PDE5 inhibitor sildenafil. Front. Oncol. 2021, 11, 627229. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, Y.; Sun, Q.; Zhang, Z.; Zhao, M.; Peng, C.; Shi, S. Ginsenosides emerging as both bifunctional drugs and nanocarriers for enhanced antitumor therapies. J. Nanobiotechnol. 2021, 19, 1–40. [Google Scholar] [CrossRef]
- Hedaya, M.; Bandarkar, F.; Nada, A. In vitro and in vivo evaluation of ibuprofen nanosuspensions for enhanced oral bioavailability. Med. Prin. Pract. 2021, 30, 361–368. [Google Scholar] [CrossRef]
- Lizoń, A.; Tisończyk, J.; Gajewska, M.; Drożdż, R. Silver nanoparticles as a tool for the study of interactions between proteins. Int. J. Mol. Sci. 2021, 22, 9703. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Porat, D.; Markovic, M.; Zur, M.; Kister, O.; Langguth, P. Optimized in silico modeling of drug absorption after gastric bypass: The case of metformin. Pharmaceutics 2021, 13, 1873. [Google Scholar] [CrossRef] [PubMed]
- Drozd, K.V.; Boycov, D.E.; Manin, A.N.; Churakov, A.V.; Perlovich, G.L. Two Novel Drug–Drug Cocrystals of Antifungal Clotrimazole with Nonsteroidal Anti-Inflammatory Drugs. Cryst. Growth Des. 2023, 23, 6597–6608. [Google Scholar] [CrossRef]
- Batisai, E. Multicomponent crystals of anti-tuberculosis drugs: A mini-review. RSC Adv. 2020, 10, 37134–37141. [Google Scholar] [CrossRef]
- Sulttan, S.; Rohani, S. Controlled drug release of smart magnetic self-assembled micelle, kinetics and transport mechanisms. J. Pharm. Sci. 2022, 111, 2378–2388. [Google Scholar] [CrossRef]
- Voronin, A.P.; Ramazanova, A.G.; Surov, A.O.; Churakov, A.V.; Perlovich, G.L. Multicomponent Crystals of Amitriptyline as Potential Controlled-Release Systems: Synthesis, Crystal Packing Analysis, and Dissolution Study. Cryst. Growth Des. 2023, 23, 6926–6943. [Google Scholar] [CrossRef]
- Khodov, I.A.; Belov, K.V.; Krestyaninov, M.A.; Sobornova, V.V.; Dyshin, A.A.; Kiselev, M.G. Does DMSO Affect the Conformational Changes of Drug Molecules in Supercritical CO2 Media? J. Mol. Liq. 2023, 384, 122230. [Google Scholar] [CrossRef]
- Khodov, I.A.; Belov, K.V.; Sobornova, V.V.; Dyshin, A.A.; Kiselev, M.G. Exploring the temperature-dependent proportions of lidocaine conformers equilibria in supercritical carbon dioxide via NOESY. J. Mol. Liq. 2023, 387, 122620. [Google Scholar] [CrossRef]
- Zavalishin, M.N.; Pimenov, O.A.; Belov, K.V.; Khodov, I.A.; Gamov, G.A. Chemical equilibria in aqueous solutions of H [AuCl4] and bovine or human serum albumin. J. Mol. Liq. 2023, 389, 122914. [Google Scholar] [CrossRef]
- Bohn, T.; Desmarchelier, C.; Dragsted, L.O.; Nielsen, C.S.; Stahl, W.; Rühl, R.; Borel, P. Host-related factors explaining interindividual variability of carotenoid bioavailability and tissue concentrations in humans. Mol. Nutr. Food Res. 2017, 61, 1600685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, Y.; Huang, W.; Jin, M.; Gao, Z. The influence of the gut microbiota on the bioavailability of oral drugs. Acta Pharm. Sin. B 2021, 11, 1789–1812. [Google Scholar] [CrossRef]
- Trucillo, P. Drug carriers: Classification, administration, release profiles, and industrial approach. Processes 2021, 9, 470. [Google Scholar] [CrossRef]
- Landersdorfer, C.B.; Gwee, A.; Nation, R.L. Clinical pharmacological considerations in an early intravenous to oral antibiotic switch: Are barriers real or simply perceived? Clin. Microbiol. Infec. 2023, 29, 1120–1125. [Google Scholar] [CrossRef]
- Koziolek, M.; Alcaro, S.; Augustijns, P.; Basit, A.W.; Grimm, M.; Hens, B.; Corsetti, M. The mechanisms of pharmacokinetic food-drug interactions–A perspective from the UNGAP group. Eur. J. Pharm. Sci. 2019, 134, 31–59. [Google Scholar] [CrossRef]
- Drenth-van Maanen, A.C.; Wilting, I.; Jansen, P.A. Prescribing medicines to older people—How to consider the impact of ageing on human organ and body functions. Brit. J. Clin. Pharmacol. 2020, 86, 1921–1930. [Google Scholar] [CrossRef]
- Baillie, T.A.; Cayen, M.N.; Fouda, H.; Gerson, R.J.; Green, J.D.; Grossman, S.J.; Shipley, L.A. Drug metabolites in safety testing. Toxicol. Appl. Pharm. 2002, 182, 188–196. [Google Scholar] [CrossRef]
- May, M.; Schindler, C.; Engeli, S. Modern pharmacological treatment of obese patients. Ther. Adv. Endocrinol. Metab. 2020, 11, 1–19. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stielow, M.; Witczyńska, A.; Kubryń, N.; Fijałkowski, Ł.; Nowaczyk, J.; Nowaczyk, A. The Bioavailability of Drugs—The Current State of Knowledge. Molecules 2023, 28, 8038. https://doi.org/10.3390/molecules28248038
Stielow M, Witczyńska A, Kubryń N, Fijałkowski Ł, Nowaczyk J, Nowaczyk A. The Bioavailability of Drugs—The Current State of Knowledge. Molecules. 2023; 28(24):8038. https://doi.org/10.3390/molecules28248038
Chicago/Turabian StyleStielow, Marlena, Adrianna Witczyńska, Natalia Kubryń, Łukasz Fijałkowski, Jacek Nowaczyk, and Alicja Nowaczyk. 2023. "The Bioavailability of Drugs—The Current State of Knowledge" Molecules 28, no. 24: 8038. https://doi.org/10.3390/molecules28248038
APA StyleStielow, M., Witczyńska, A., Kubryń, N., Fijałkowski, Ł., Nowaczyk, J., & Nowaczyk, A. (2023). The Bioavailability of Drugs—The Current State of Knowledge. Molecules, 28(24), 8038. https://doi.org/10.3390/molecules28248038