
Essential Rule Derived from Thermodynamics and Kinetics Studies of Benzopyran Compounds


Abstract
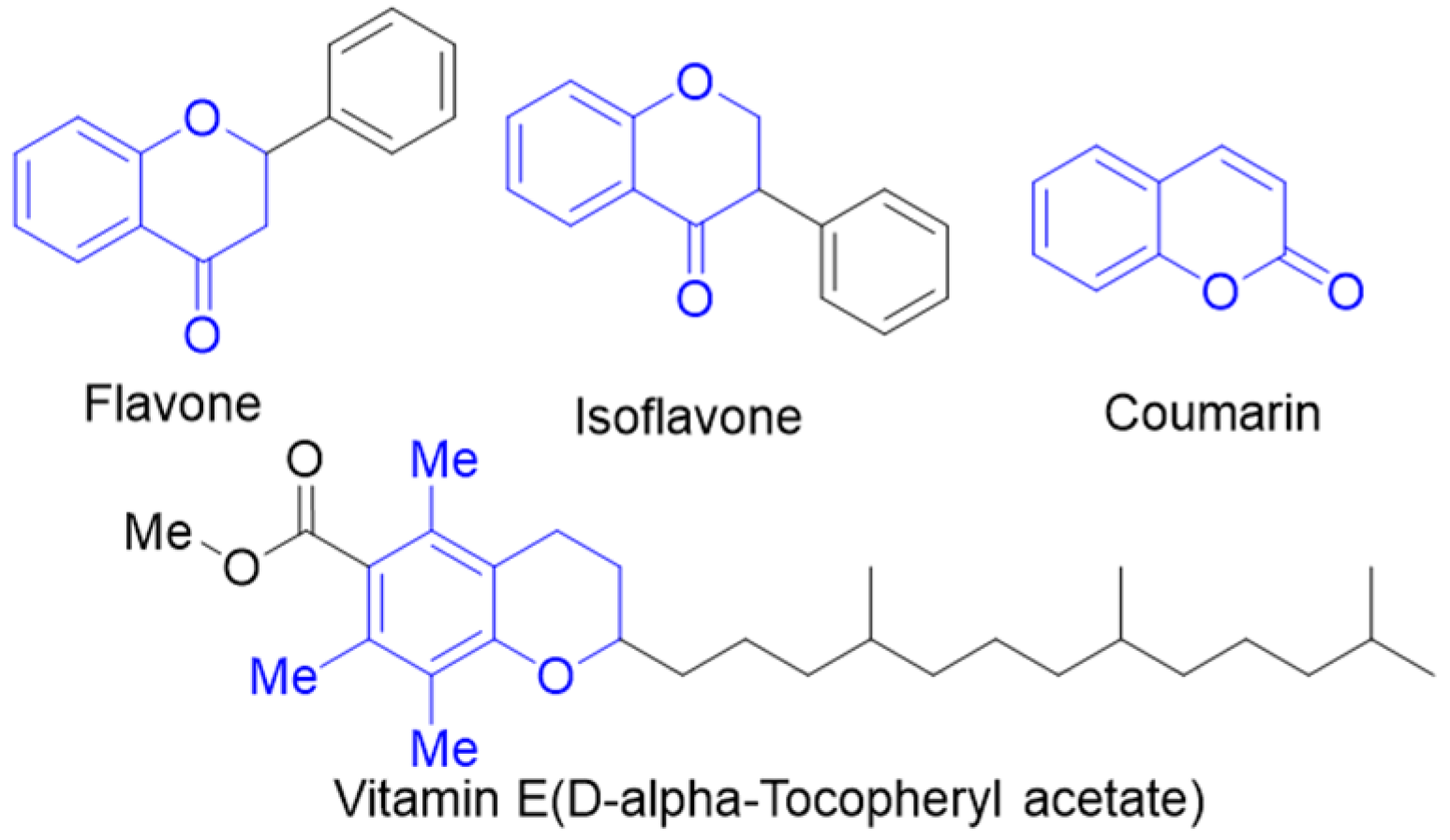
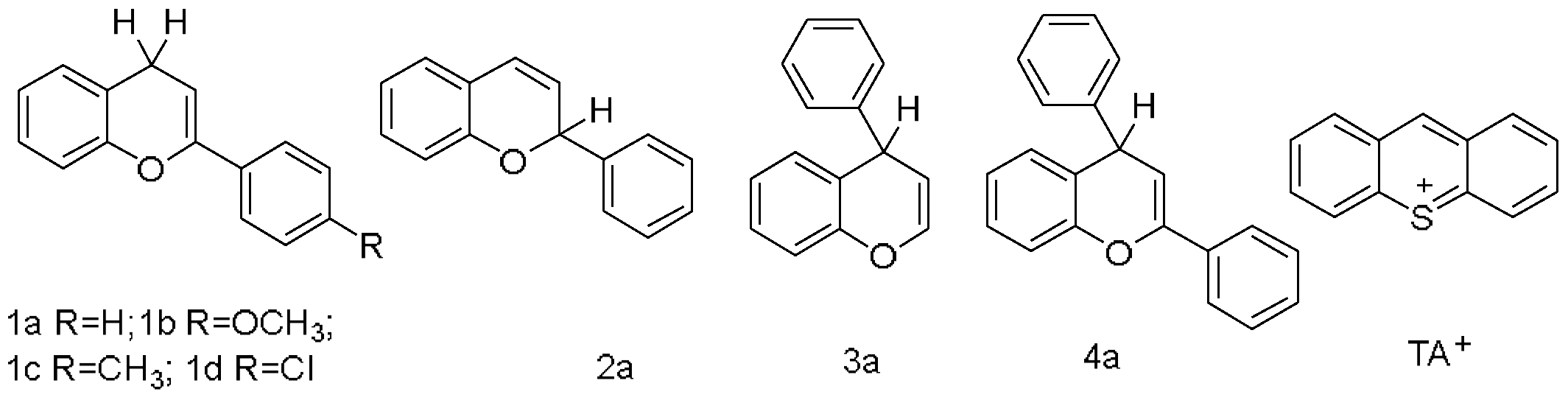
:1. Introduction
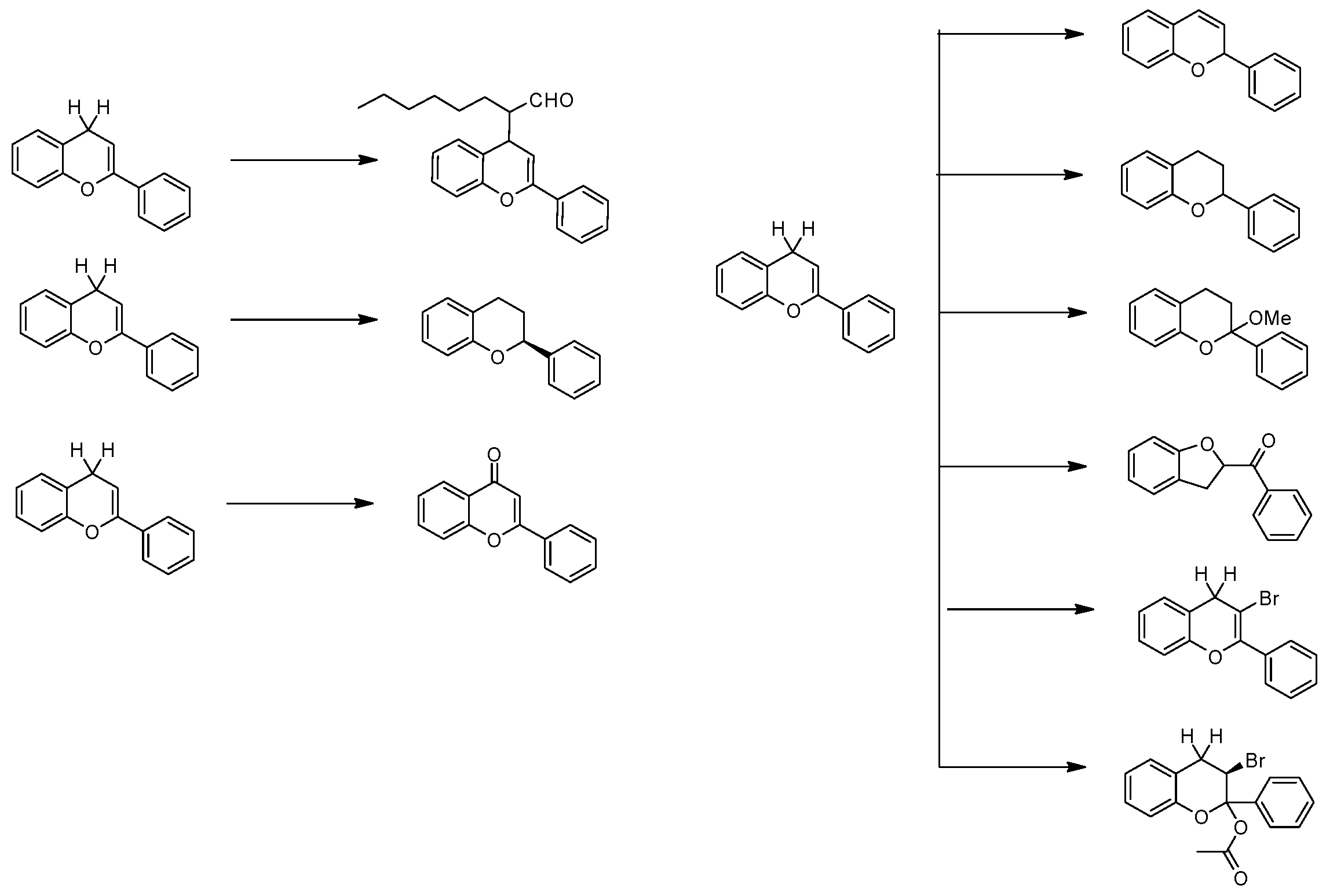
2. Results
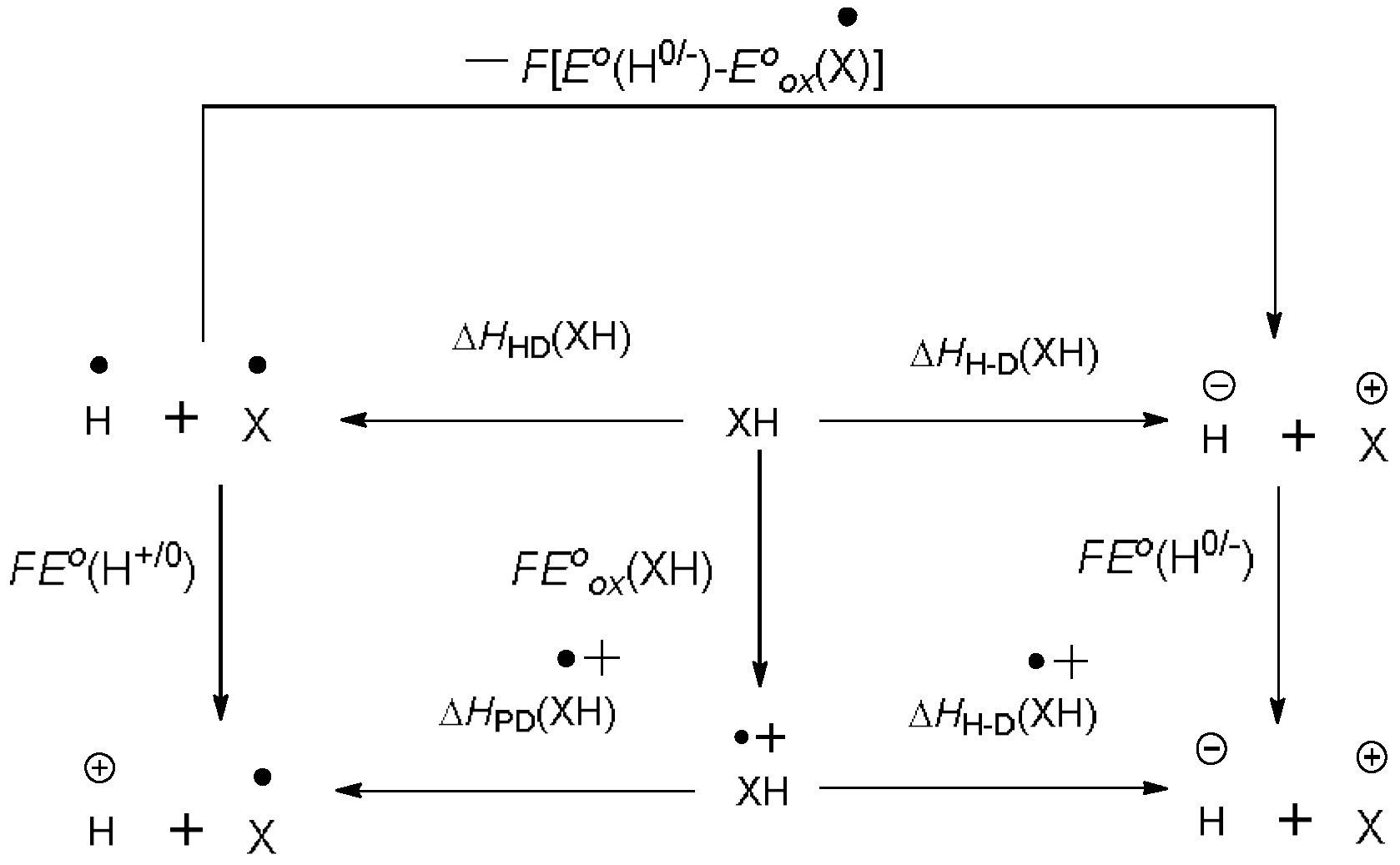
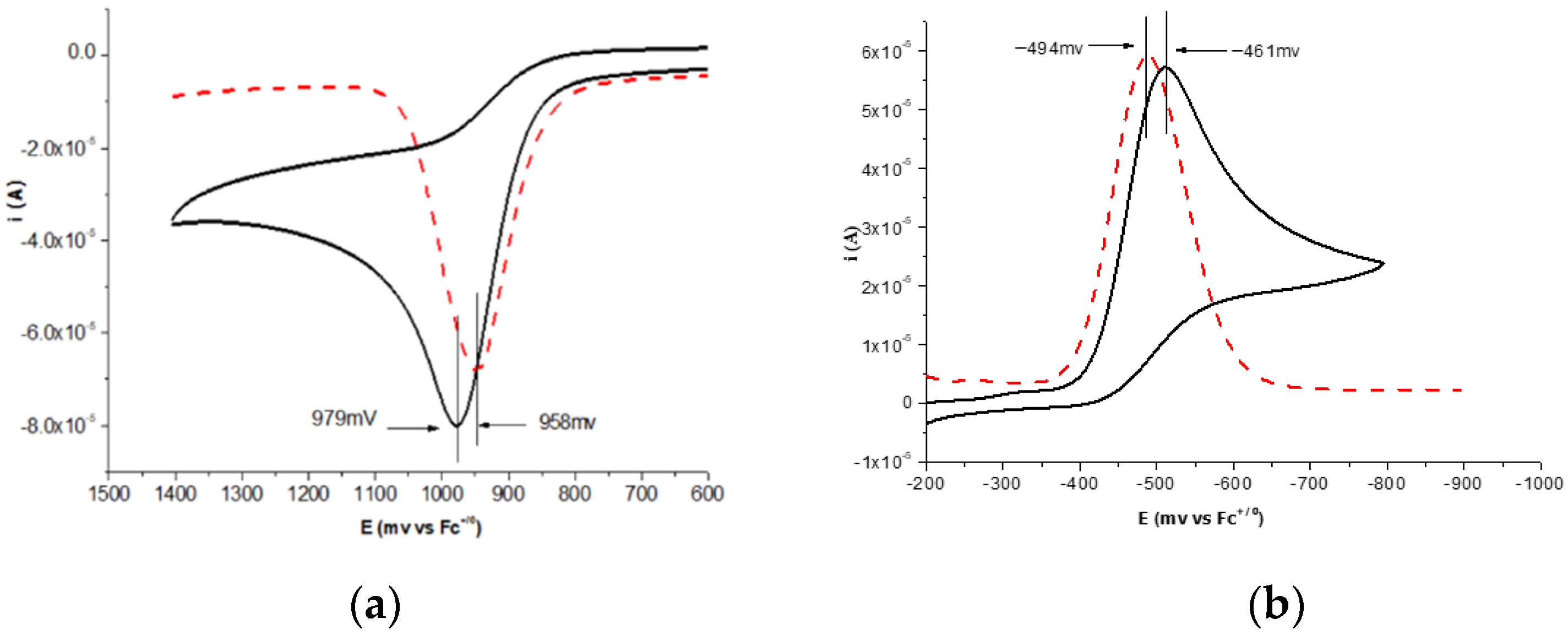
2.1. The Statement of Measurement Methods
2.2. Data Collection Method
2.3. Data Analysis Process
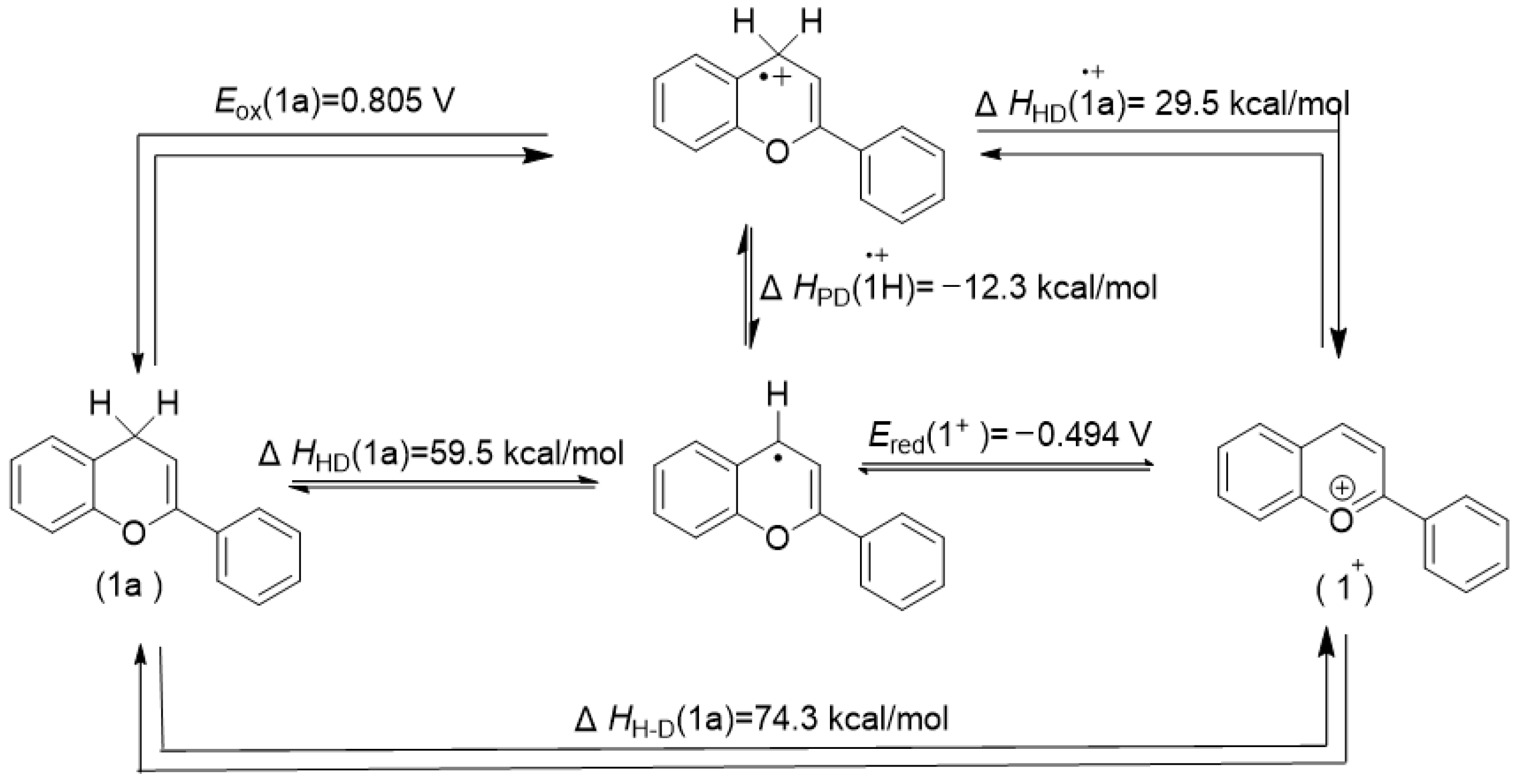
3. Discussion
3.1. The Statement of Novel Findings
3.2. Accuracy and Reliability of the Novel Findings
3.3. The Application Value of Novel Findings
3.4. Assessment of Novel Findings
4. Experimental Procedures
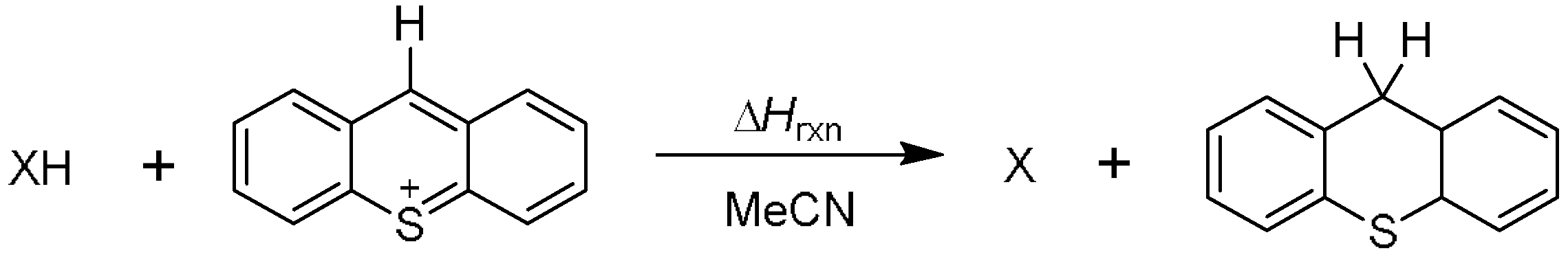
4.1. Measurment of Redox Potentials
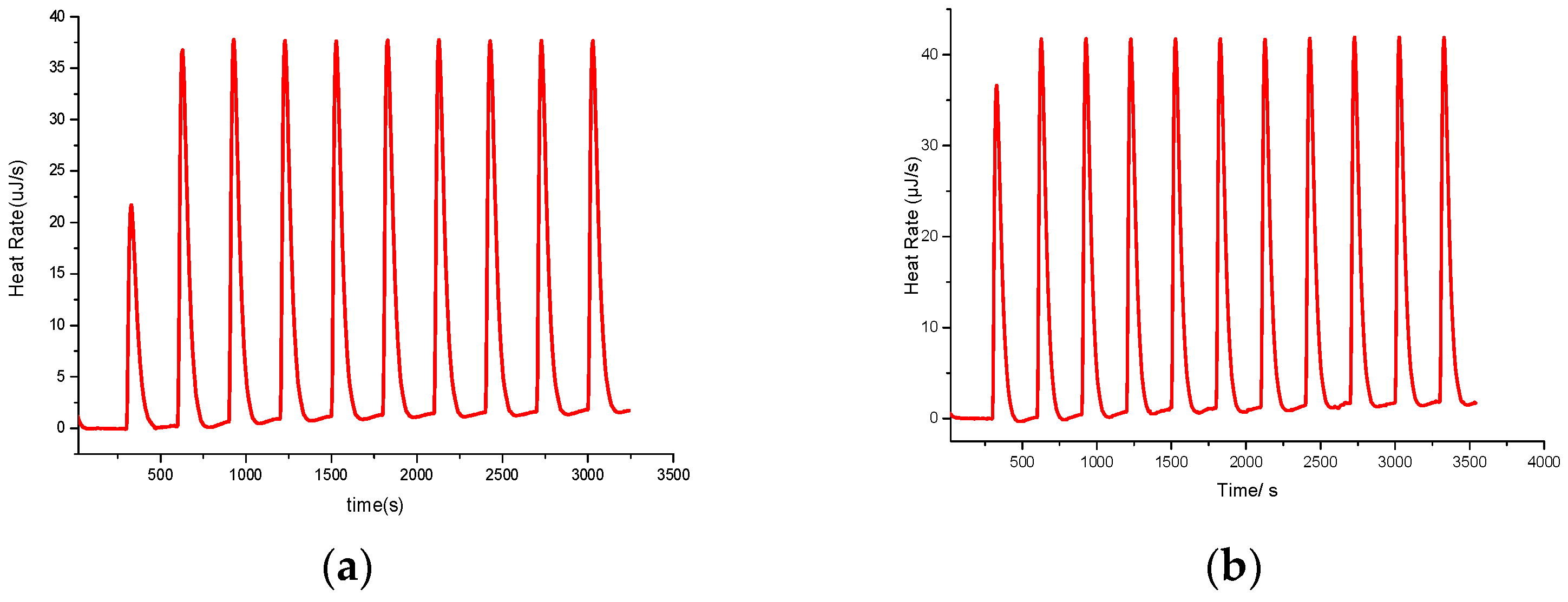
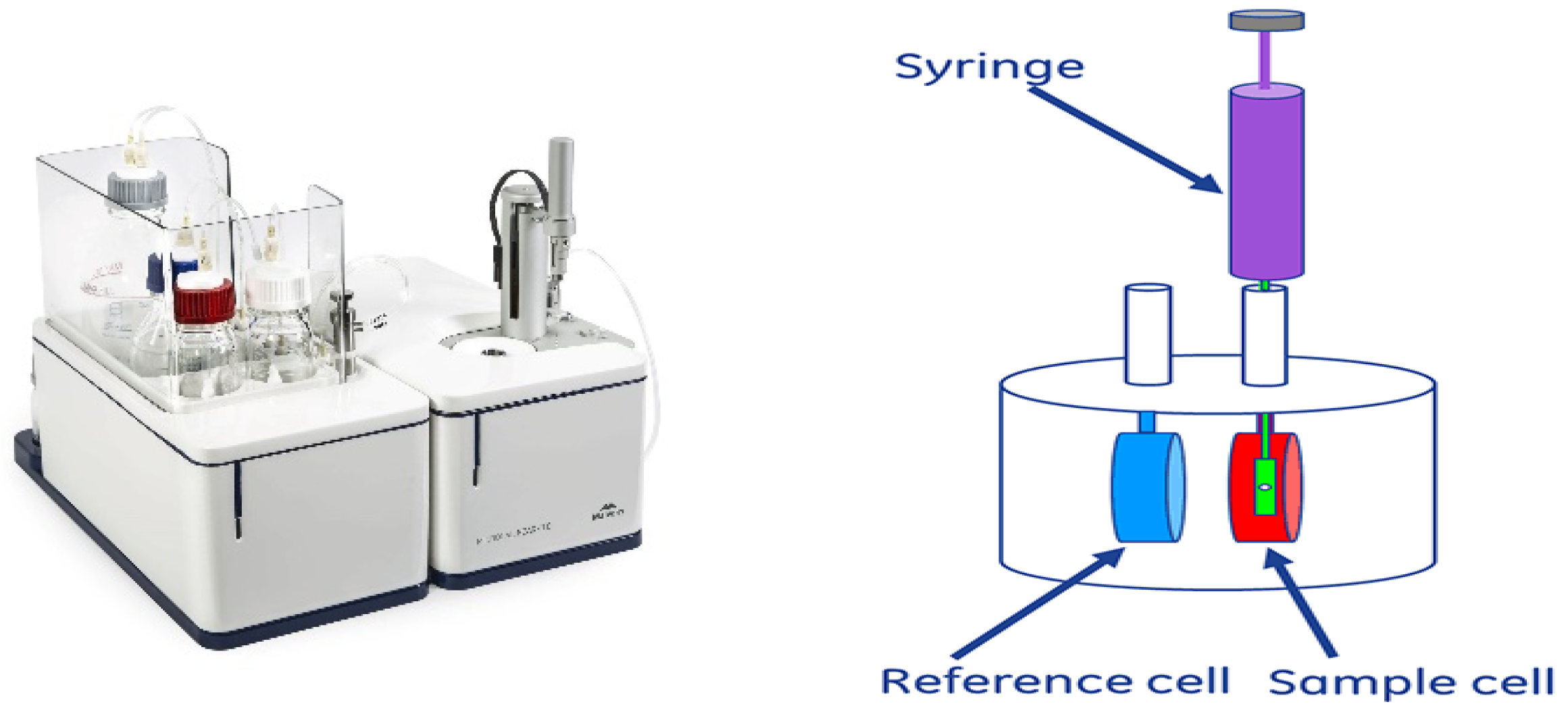
4.2. Isothermal Titration Calorimetry (ITC)
4.3. Kinetics Measurment and Calculation Process
5. Conclusions
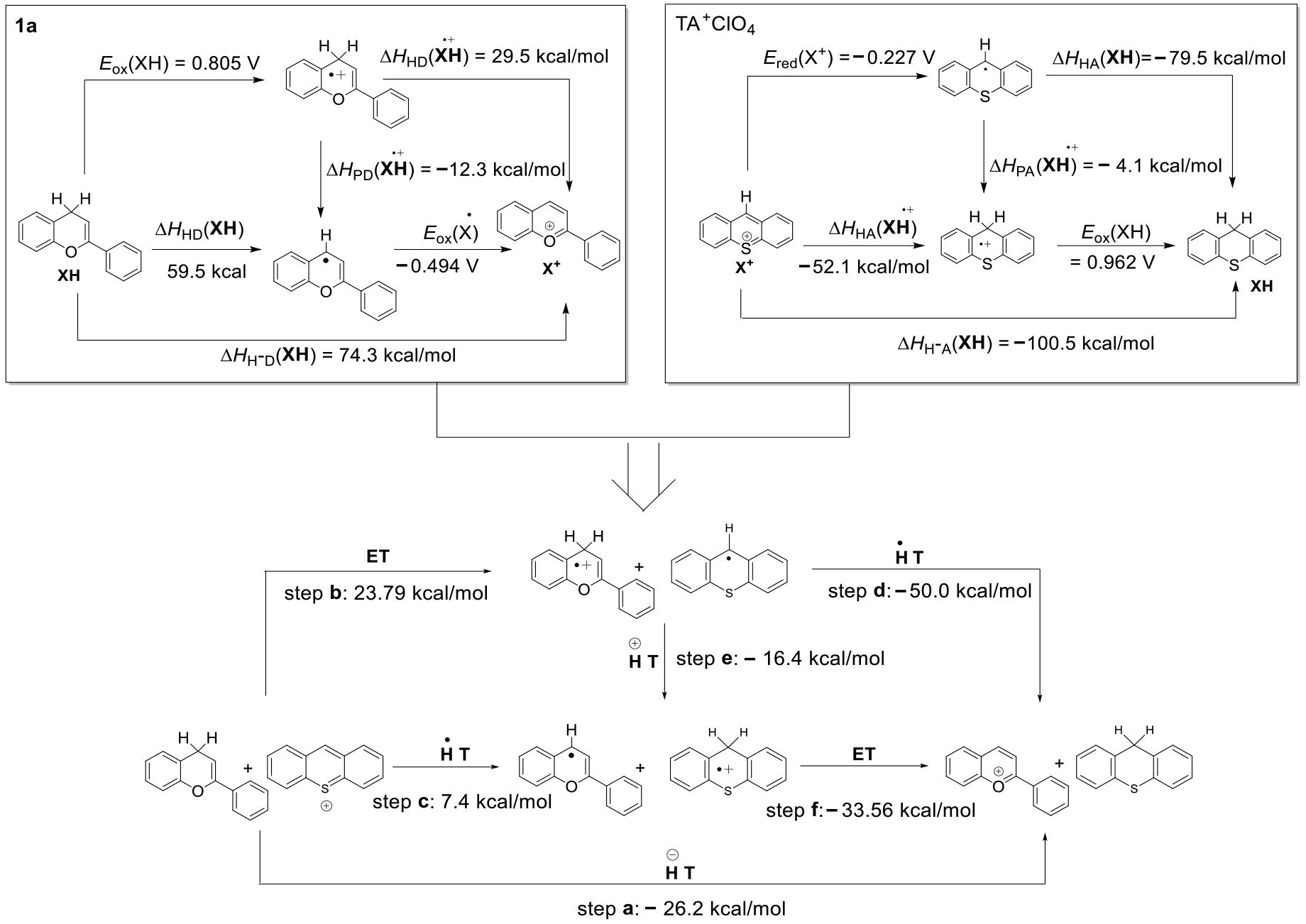
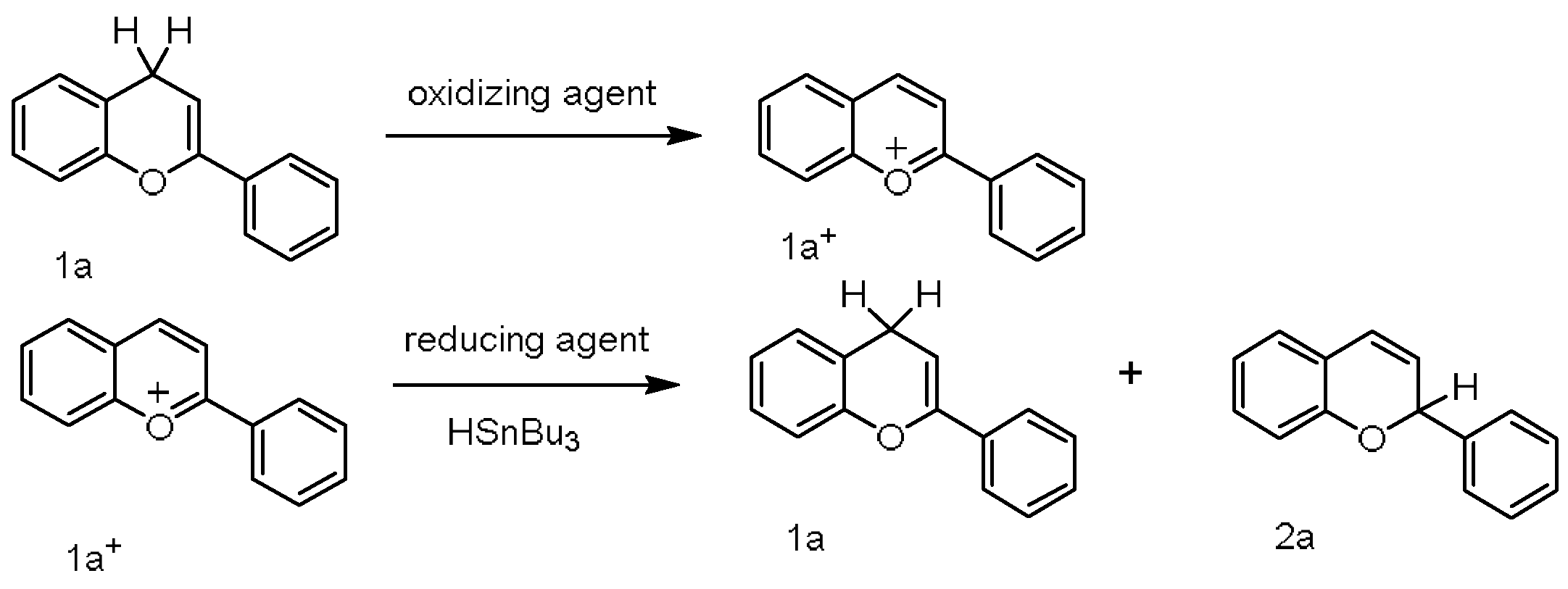
- According to the analysis of kinetics and thermodynamics data for the reactions between four types of benzofuran compounds with TA+ClO4−, along with the Molecular ID and reaction thermodynamic platforms, were obtained, proving that these reactions follow a one-step hydride transfer mechanism.
- For the hydride transfer reaction, the hydride affinity of the C–H bond at the reaction site and the activation energy of the reaction exhibited excellent linear dependence on the benzene ring para substituent constants. A simultaneous correlation between thermodynamics and kinetics was predicted to appear in the hydride transfer reactions of the 2a, 3a, and 4a compounds.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Teng, H.; Zheng, Y.-M.; Cao, H.; Huang, Q.; Xiao, J.-B.; Chen, L. Enhancement of bioavailability and bioactivity of diet-derived flavonoids by application of nanotechnology: A review. Crit. Rev. Food. Sci. 2023, 63, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, R.; Williamson, G. Citrus polyphenols and risk of type 2 diabetes: Evidence from mechanistic studies. Crit. Rev. Food. Sci. 2023, 63, 2178–2202. [Google Scholar] [CrossRef] [PubMed]
- Cardoso Reis, A.C.; Valente, G.M.; Silva, B.M.; Brito Magalhães, C.L.; Kohlhoff, M.; Brandão, G.C. Anti-arboviral activity and chemical characterization of hispidulin and ethanolic extracts from Millingtonia hortensis L.f. and Oroxylum indicum (L.) Kurz (Bignoniaceae). Nat. Prod. Res. 2023, 37, 613–617. [Google Scholar] [CrossRef]
- Tian, M.-R.; Du, K.; Zhi, Y.-L.; Xue, G.-M.; Zhao, Z.-Z.; Si, Y.-Y.; Chen, H.; Sun, Y.-J.; Feng, W.-S.; Ma, J.-L. LSD1 inhibitors from the roots of Pueraria lobate. J. Asian. Nat. Prod. Res. 2023, 25, 44–52. [Google Scholar] [CrossRef]
- Aboshabana, R.; Elmansi, H.; El-Enany, N. Investigation of facile spectroscopic approaches for rapid calycosin determination in invitro biological samples and pharmaceuticals; application to the content uniformity of capsules. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 286, 121904. [Google Scholar] [CrossRef]
- Nezamivand-Chegini, M.; Metzger, S.; Moghadam, A.; Tahmasebi, A.; Koprivova, A.; Eshghi, S.; Mohammadi-Dehchesmeh, M.; Kopriva, S.; Niazi, A.; Ebrahimie, E. Integration of transcriptomic and metabolomic analyses provides insights into response mechanisms to nitrogen and phosphorus deficiencies in soybean. Plant. Sci. 2023, 326, 111498. [Google Scholar] [CrossRef]
- Lopes, M.; Sanches-Silva, A.; Castilho, M.; Cavaleiro, C.; Ramos, F. Halophytes as source of bioactive phenolic compounds and their potential applications. Crit. Rev. Food. Sci. 2023, 63, 1078–1101. [Google Scholar] [CrossRef]
- Arya, A.; Kumar, S.; Paul, R.; Suryavanshi, A.; Kain, D.; Sahoo, R.N. Ethnopharmacological survey of indigenous medicinal plants of Palampur, Himachal Pradesh in north-western Himalaya, India. Adv. Tradit. Med. 2023, 23, 169–212. [Google Scholar] [CrossRef]
- Yuan, Z.-H.; Yang, Y.-S.; Lv, P.-C.; Zhu, H.-L. Recent Progress in Small-Molecule Fluorescent Probes for Detecting Mercury Ions. Critical Reviews in Analytical Chemistry. Crit. Rev. Anal. Chem. 2022, 52, 250–274. [Google Scholar] [CrossRef]
- Nguemo, R.T.; Mbouangouere, R.; Bitchagno, G.T.M.; Tchuenguem, R.; Temgoua, E.V.N.; Ndontsa, B.L.; Mpetga, J.S.; Opatz, T.; Ngouela, A.S.; Tane, P. A new ceramide from the leaves of Lannea schimperi (Hochst. ex A. Rich.) Engl. Nat. Prod. Res. 2022, 36, 515–522. [Google Scholar] [CrossRef]
- Motkowski, R.; Maciejczyk, M.; Hryniewicka, M.; Karpińska, J.; Mikołuć, B. Effect of Statin Therapy on the Plasma Concentrations of Retinol, Alpha-Tocopherol and Coenzyme Q10 in Children with Familial Hypercholesterolemia. Cardiovasc Drug. Ther. 2022, 36, 75–84. [Google Scholar] [CrossRef]
- Abbas-Mohammadi, M.; Moridi, F.M.; Salehi, P.; Ebrahimi, S.N.; Sonboli, A.; Kelso, C.; Skropeta, D. Molecular networking based dereplication of AChE inhibitory compounds from the medicinal plant Vincetoxicum funebre (Boiss. & Kotschy). J. Biomol. Struct. Dyn. 2022, 40, 1942–1951. [Google Scholar] [PubMed]
- Fides, B.; Montse, G.C.; Luca, Z.; Elena, B.P.G.C. Catalytic stereoselective benzylic C–H functionalizations by oxidative C–H activation and organocatalysis. Chem. Commun. 2009, 39, 5919–5921. [Google Scholar]
- Carine, V.; Alejandro, B.; Frederik, M.; Andreas, P. Enantioselective Synthesis of Chromanes by Iridium-Catalyzed Asymmetric Hydrogenation of 4H-Chromenes. Synlett 2008, 20, 3167–3171. [Google Scholar]
- Hatnm, N.A.R.; Nacy, W.G. Oxythallation of flavenes. I: Direct conversion of flavenes to flavenes with thallium (III) nitrate. Tetrahedron Lett. 1983, 24, 4455–4456. [Google Scholar] [CrossRef]
- Geoffrey, C.B.; Ben, R.B.; Ian, A.S.; William, R.T. Reactions of Flav-2-enes and Flav-2-en-4-ones (Flavones). J. Chem. Soc. Perkin Trans. I 1983, 8, 1831–1846. [Google Scholar]
- Wayner, D.D.M.; Parker, V.D. Bond energies in solution from electrode potentials and thermochemical cycles. A simplified and general approach. Acc. Chem. Res. 1993, 26, 287–294. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Li, X.-T.; Han, S.-H.; Mei, L.-R. Conversion and Origin of Normal and Abnormal Temperature Dependences of Kinetic Isotope Effect in Hydride Transfer Reactions. J. Org. Chem. 2012, 77, 4774–4783. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Zhang, M.-T.; Yu, A.; Wang, C.-H.; Cheng, J.-P. Hydride, Hydrogen Atom, Proton, and Electron Transfer Driving Forces of Various Five-Membered Heterocyclic Organic Hydrides and Their Reaction Intermediates in Acetonitrile. J. Am. Chem. Soc. 2008, 130, 2501–2516. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Deng, F.-H.; Yang, J.-D.; Li, X.-T.; Chen, Q.; Lei, N.-P.; Meng, F.-K.; Zhao, X.-P.; Han, S.-H.; Hao, E.-J.; et al. A Classical but New Kinetic Equation for Hydride Transfer Reactions. Org. Biomol. Chem. 2013, 11, 6071–6089. [Google Scholar] [CrossRef]
- O’Dea, J.J.; Osteryoung, J.; Osteryoung, R.A. Theory of square wave voltammetry for kinetic systems. Anal. Chem. 1981, 53, 695–701. [Google Scholar] [CrossRef]
- Shen, G.-B.; Xia, K.; Li, X.-T.; Li, J.-L.; Fu, Y.-H.; Yuan, L.; Zhu, X.-Q. Prediction of Kinetic Isotope Effects for Various Hydride Transfer Reactions Using a New Kinetic Model. J. Phys. Chem. A 2016, 120, 1779–1799. [Google Scholar] [CrossRef] [PubMed]
- Claudia, F.; Grigoriy, R.; Herbert, M. Kinetics of the Reactions of Flavylium Ions with π-Nucleophiles. Eur. J. Org. Chem. 2001, 2001, 4451–4456. [Google Scholar]
- Li, Y.; Zhu, X.-Q. Theoretical Prediction of Activation Free Energies of Various Hydride Self-Exchange Reactions in Acetonitrile at 298 K. ACS Omega 2018, 3, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, U.M.; Amina, S.; Bilal, A.K.; Muhammad, N.Z.; Ishtiaq, A.; Muhammad, Z. Synthesis, Molecular Docking Studies and Biological Evaluation of 3-Iminoaurones as Acetylcholinesterase and Butyrylcholinesterase Inhibitors. Lett. Drug. Des. Discov. 2017, 14, 1035–1041. [Google Scholar]
- Nuno, R.C.; Pedro, M.C.; Vania, A.; Teresa, D.; Luis, F.V.; Pedro, M.P. Water as the reaction medium for multicomponent reactions basedon boronic acids. Tetrahedron 2010, 66, 2736–2745. [Google Scholar]
- Stan, S.H.; Sami, E.F. Tandem Arylation-Reduction of Acyl Heterocycles Convenient Synthesis of Benzyl [1,2]. J. Heterocyclic. Chem. 1987, 24, 1205–1213. [Google Scholar]
- Wu, Y.-C.; Liu, L.; Liu, Y.-L.; Wang, D.; Chen, Y.-J. TFA-Mediated Tandem Friedel-Crafts Alkylation/Cyclization/Hydrogen Transfer Process for the Synthesis of Flavylium Compounds. J. Org. Chem. 2007, 72, 9383–9386. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydride (XH) | ΔHrxn a | Eox (XH) b | Eox(X•) b | ||
|---|---|---|---|---|---|
| CV | OSWV | CV | OSWV | ||
| 1b | 27.2 | 0.711 | 0.680 | −0.556 | −0.584 |
| 1c | 26.8 | 0.754 | 0.725 | −0.502 | −0.530 |
| 1a | 26.2 | 0.835 | 0.805 | −0.461 | −0.494 |
| 1d | 25.4 | 0.902 | 0.870 | −0.412 | −0.440 |
| 2a | 26.8 | 0.979 | 0.958 | −0.461 | −0.494 |
| 3a | 9.8 | 0.940 | 0.925 | −0.505 | −0.528 |
| 4a | 14.3 | 1.012 | 0.997 | −0.549 | −0.570 |
| TA+ClO4− | 0.990 | 0.962 | −0.238 | −0.227 | |
| XH | ΔHH-D(XH) a | ΔHHD(XH) b | ΔHPD(XH•+) b | ΔHHD(XH•+) b | k2 (M−1S−1) c |
|---|---|---|---|---|---|
| 1b | 73.3 | 60.6 | −8.3 | 31.4 | 306.52 |
| 1c | 73.7 | 59.7 | −10.2 | 30.8 | 290.83 |
| 1a | 74.3 | 59.5 | −12.3 | 29.5 | 161.93 |
| 1d | 75.1 | 59.0 | −14.2 | 28.8 | 90.44 |
| 2a | 83.7 | 58.9 | −16.4 | 25.4 | 72.26 |
| 3a | 90.7 | 76.7 | −2.2 | 43.2 | 2.76 |
| 4a | 86.2 | 73.1 | −3.0 | 37.0 | 11.73 |
| Species | Thermodynamic Parameters | Diagnoses of the Characteristic Properties |
|---|---|---|
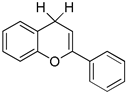 | ΔHH-D(1a) = 74.3 kcal/mol ΔHHD(1a) = 59.5 kcal/mol Eox(1a) = 0.805 V | moderately strong hydride donor strong hydrogen donor and antioxidant weak one-e− reductant |
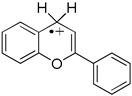 | ΔHPD(1a•+) = −12.3kcal/mol ΔHHD(1a•+) = 29.5 kcal/mol Ered(1a•+) = 0.805 V | very strong organic acid good hydrogen donor and antioxidant strong one-e− oxidant |
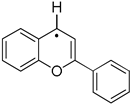 | ΔHPA(1a•) = 12.3 kcal/mol ΔHHA(a1•) = −59.5 kcal/mol Eox(1a•) = −0.494V | weak base mildly strong hydrogen acceptor strong one-e− reductant |
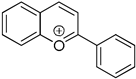 | ΔHH-A(1a+) = −74.3 kcal/mol ΔHHA(1a+) = −29.5 kcal/mol Ered(1a+) = −0.494 V | strong hydride acceptor strong hydrogen acceptor weak one-e− oxidant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Hu, X.; Zhu, X. Essential Rule Derived from Thermodynamics and Kinetics Studies of Benzopyran Compounds. Molecules 2023, 28, 8039. https://doi.org/10.3390/molecules28248039
Chen B, Hu X, Zhu X. Essential Rule Derived from Thermodynamics and Kinetics Studies of Benzopyran Compounds. Molecules. 2023; 28(24):8039. https://doi.org/10.3390/molecules28248039
Chicago/Turabian StyleChen, Baolong, Xin Hu, and Xiaoqing Zhu. 2023. "Essential Rule Derived from Thermodynamics and Kinetics Studies of Benzopyran Compounds" Molecules 28, no. 24: 8039. https://doi.org/10.3390/molecules28248039
APA StyleChen, B., Hu, X., & Zhu, X. (2023). Essential Rule Derived from Thermodynamics and Kinetics Studies of Benzopyran Compounds. Molecules, 28(24), 8039. https://doi.org/10.3390/molecules28248039