
Trans-(±)-TTPG-B Attenuates Cell Cycle Progression and Inhibits Cell Proliferation on Cholangiocarcinoma Cells
 , and
, and
Abstract
1. Introduction
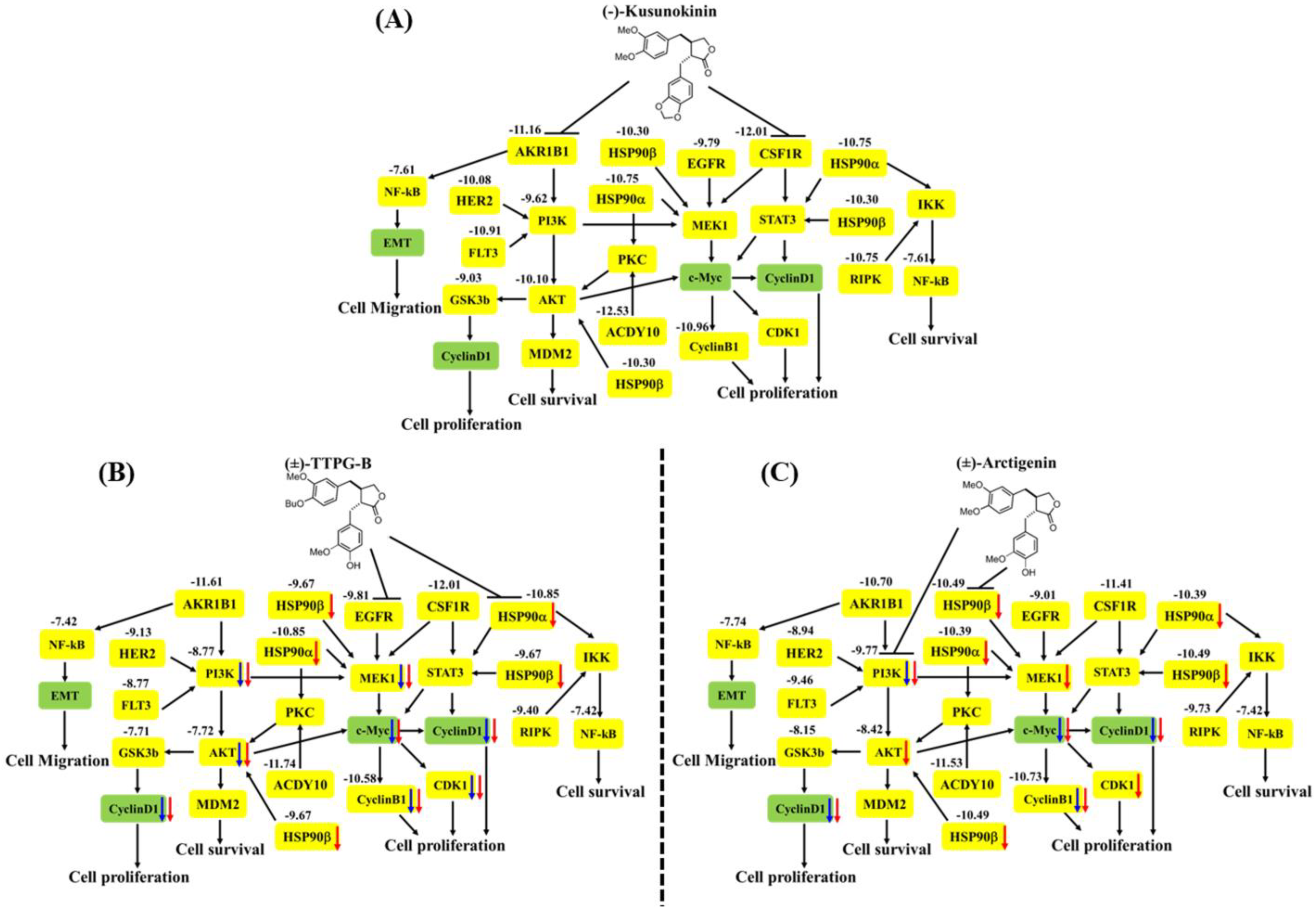
2. Results
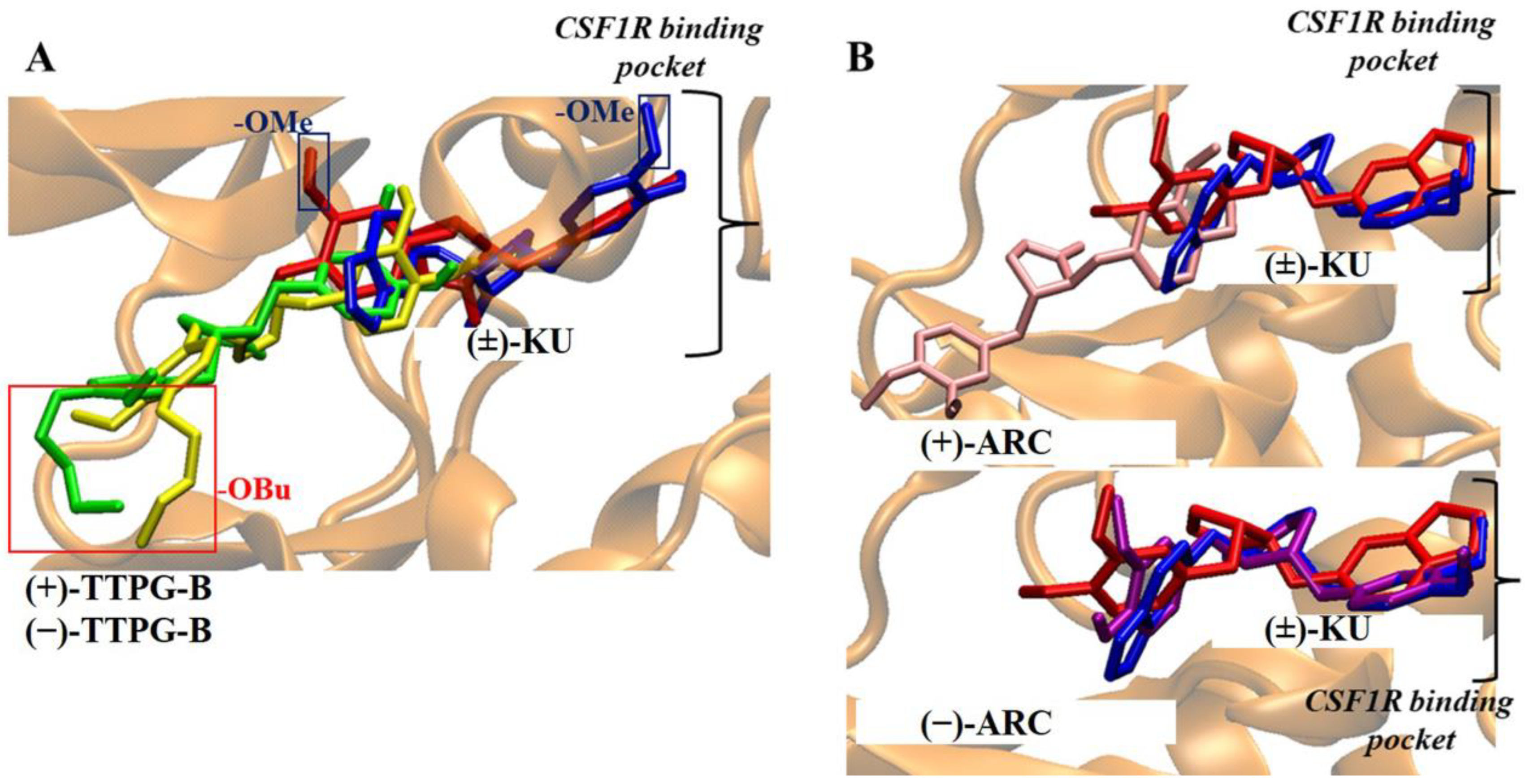
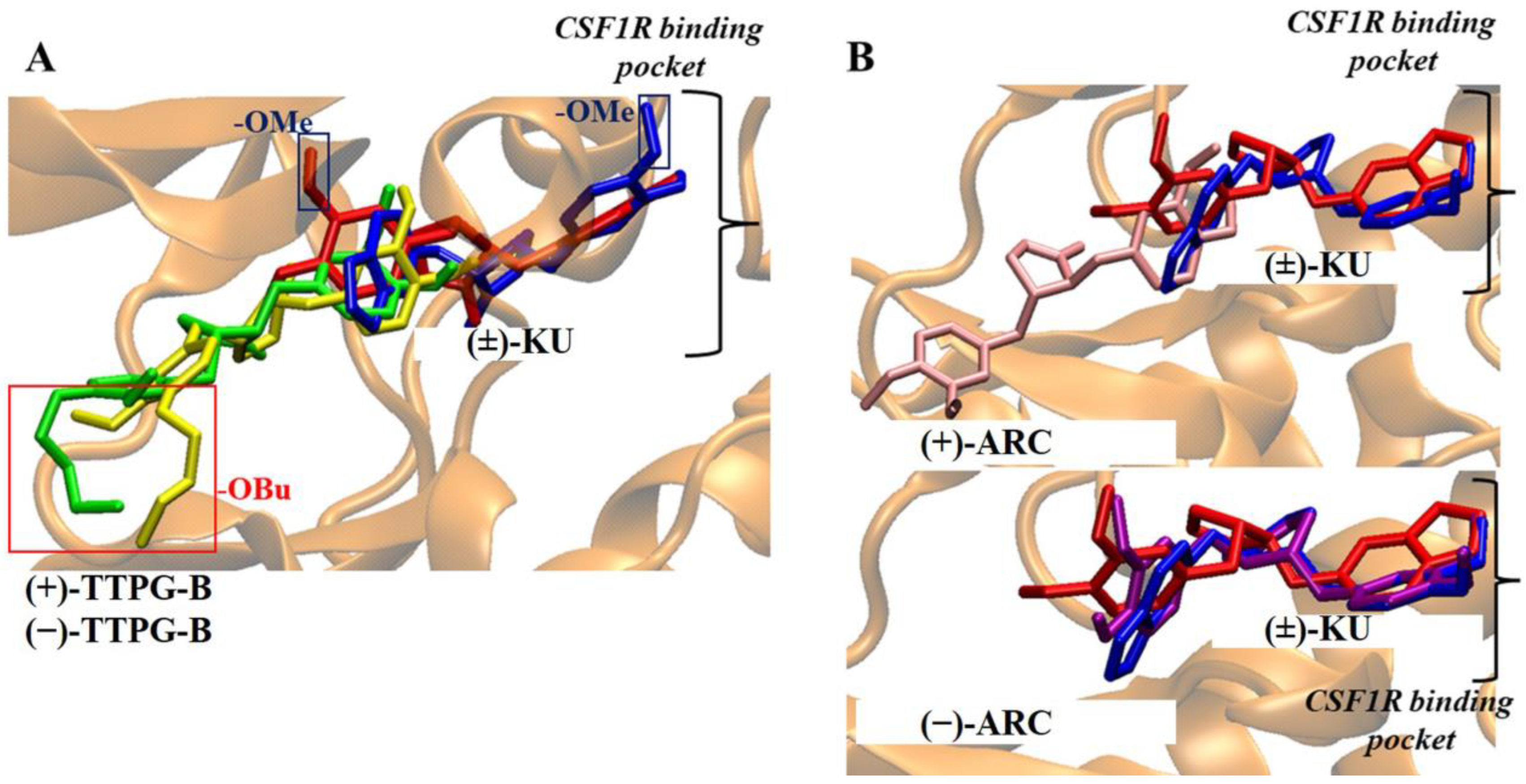
2.1. Molecular Docking of (±)-TTPG-B and (±)-ARC
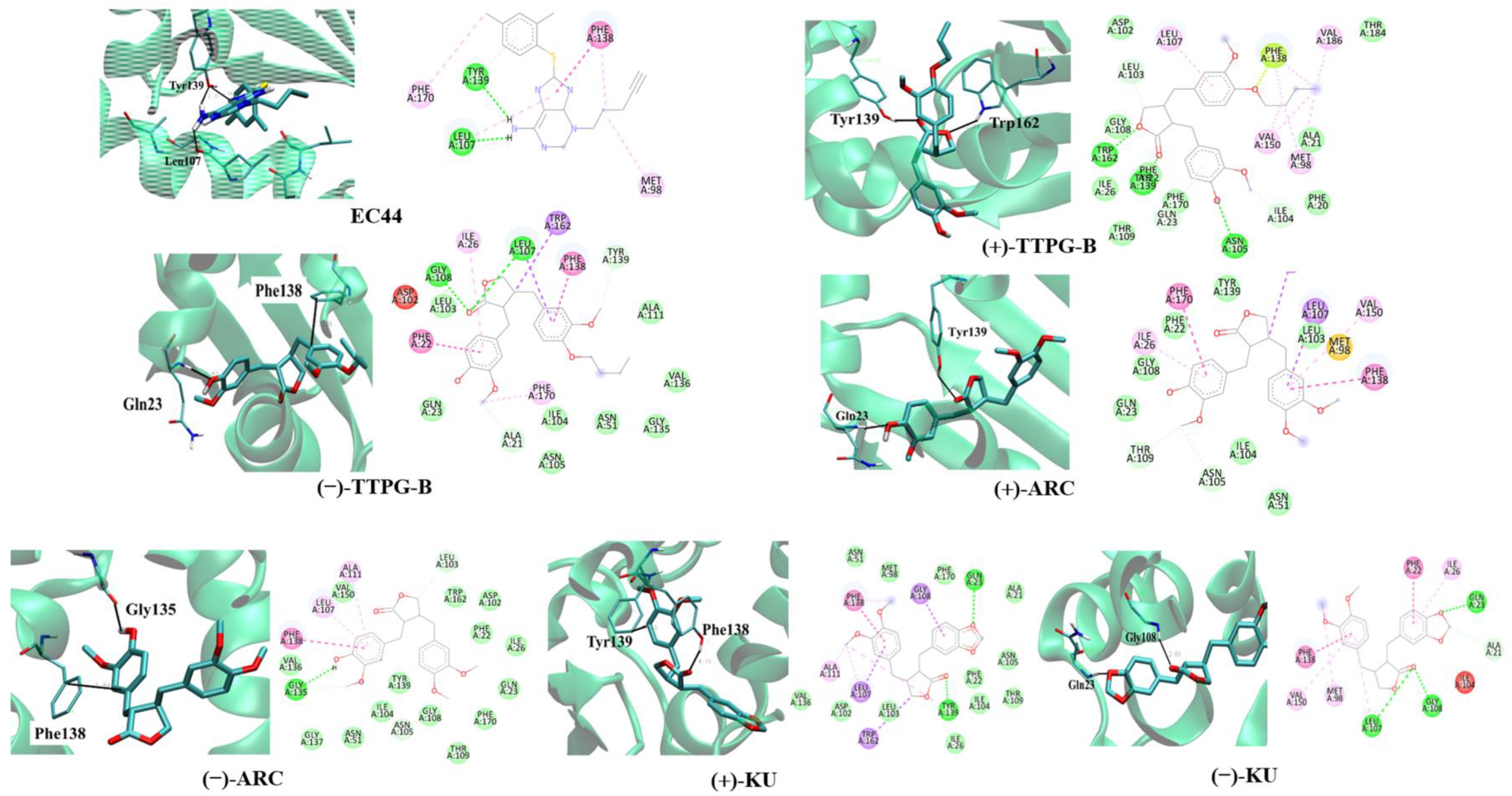
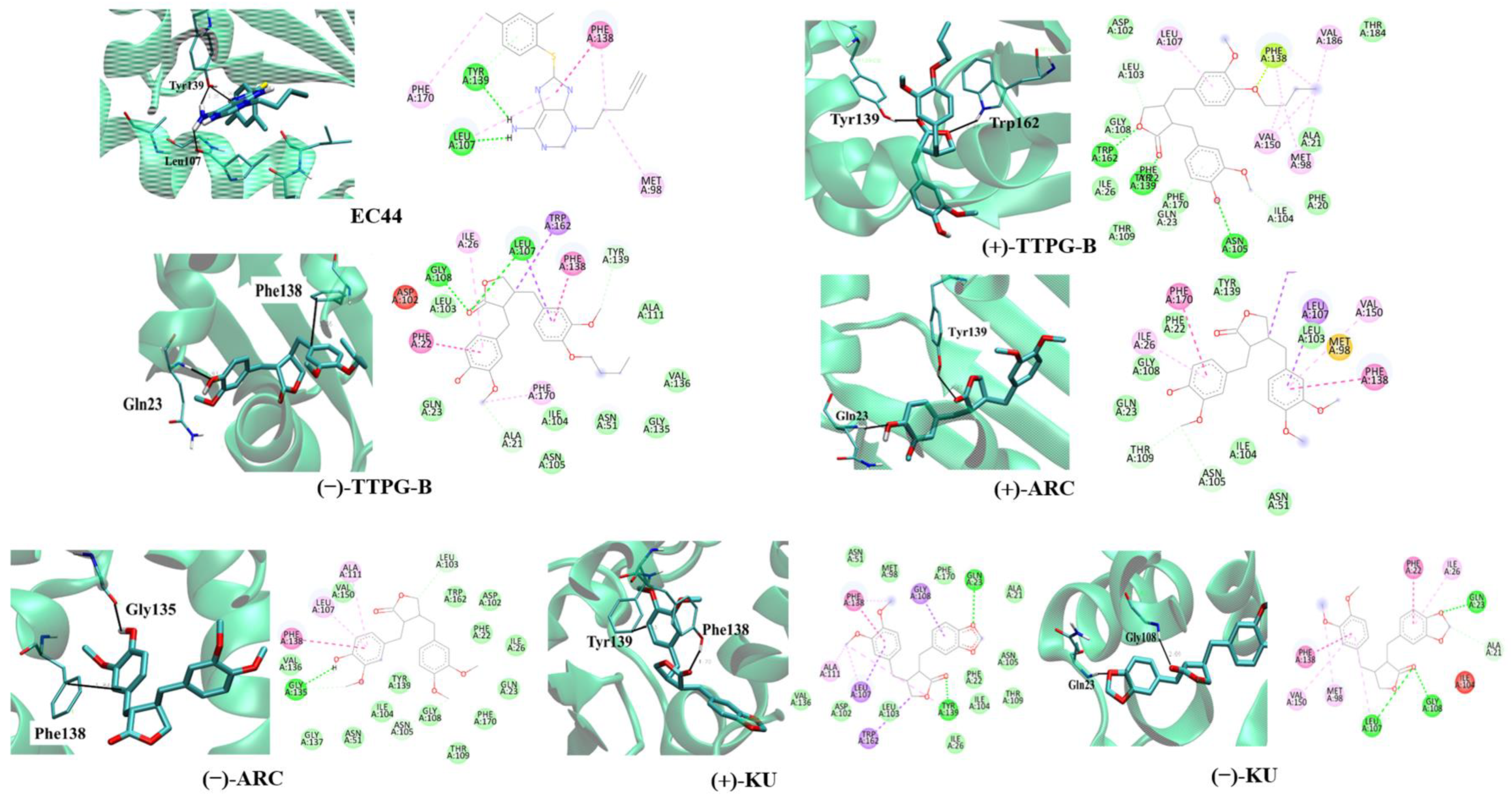
2.2. Binding Position of (±)-TTPG-B and (±)-ARC and on HSP90α
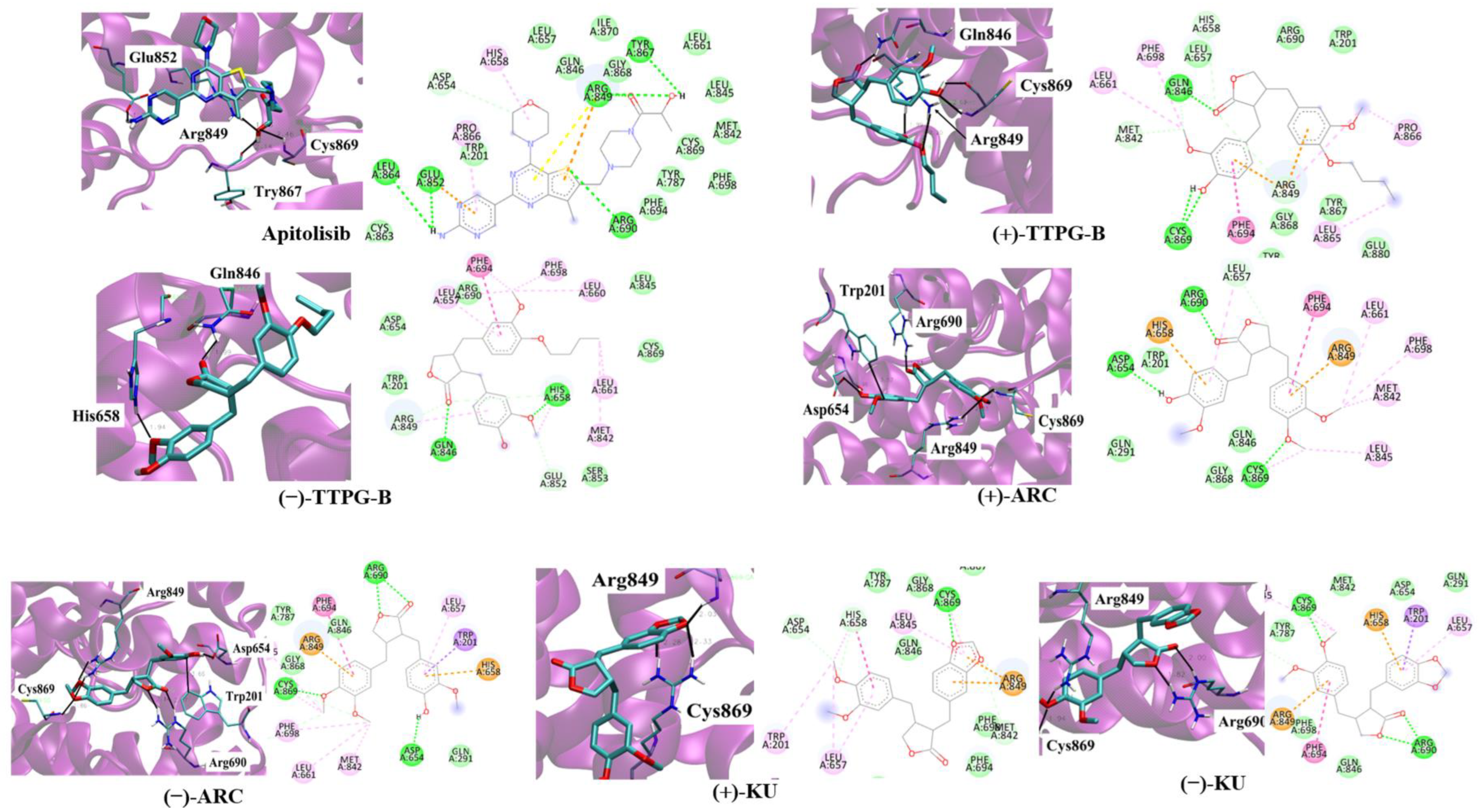
2.3. Binding Position of (±)-TTPG-B and (±)-ARC on PI3K
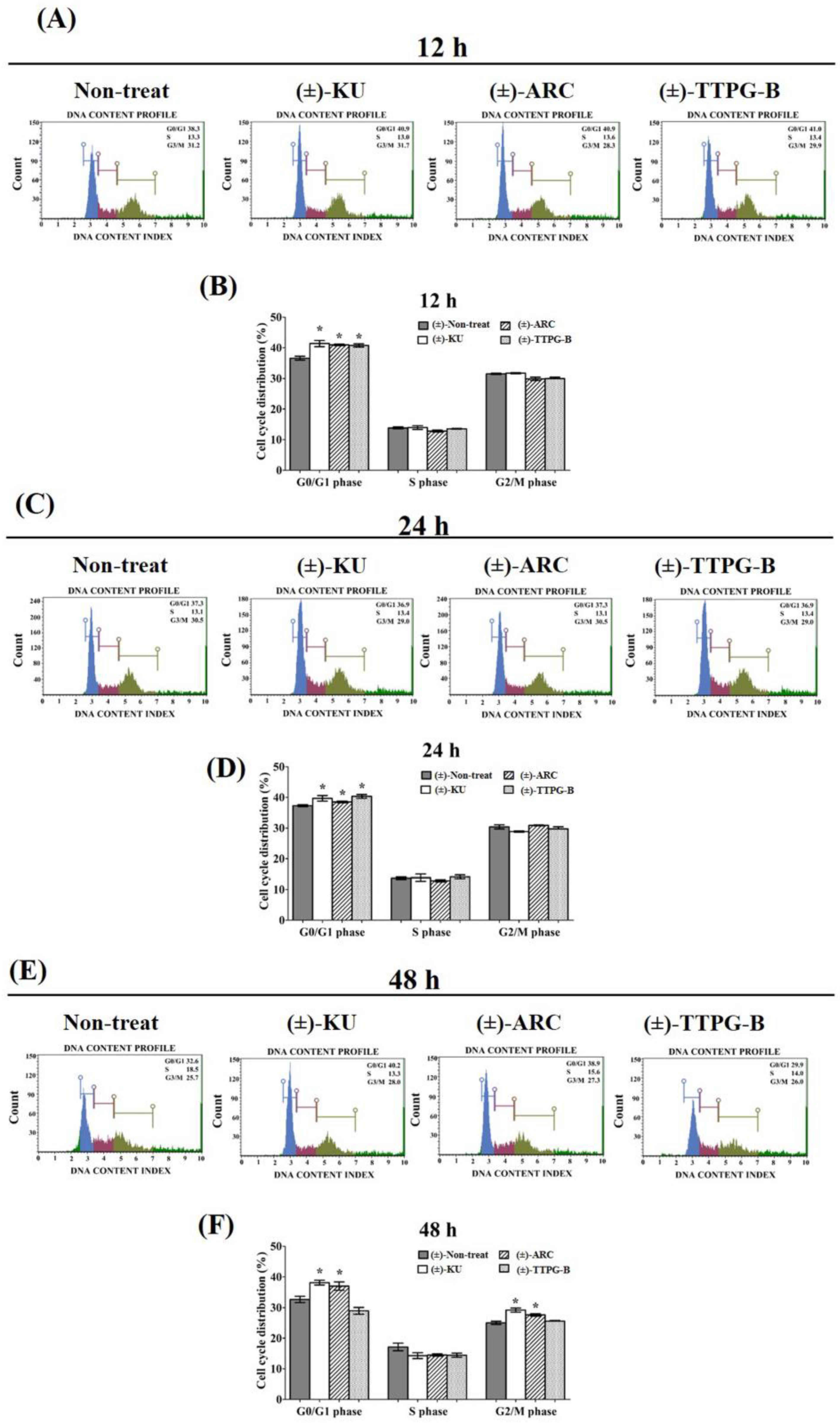
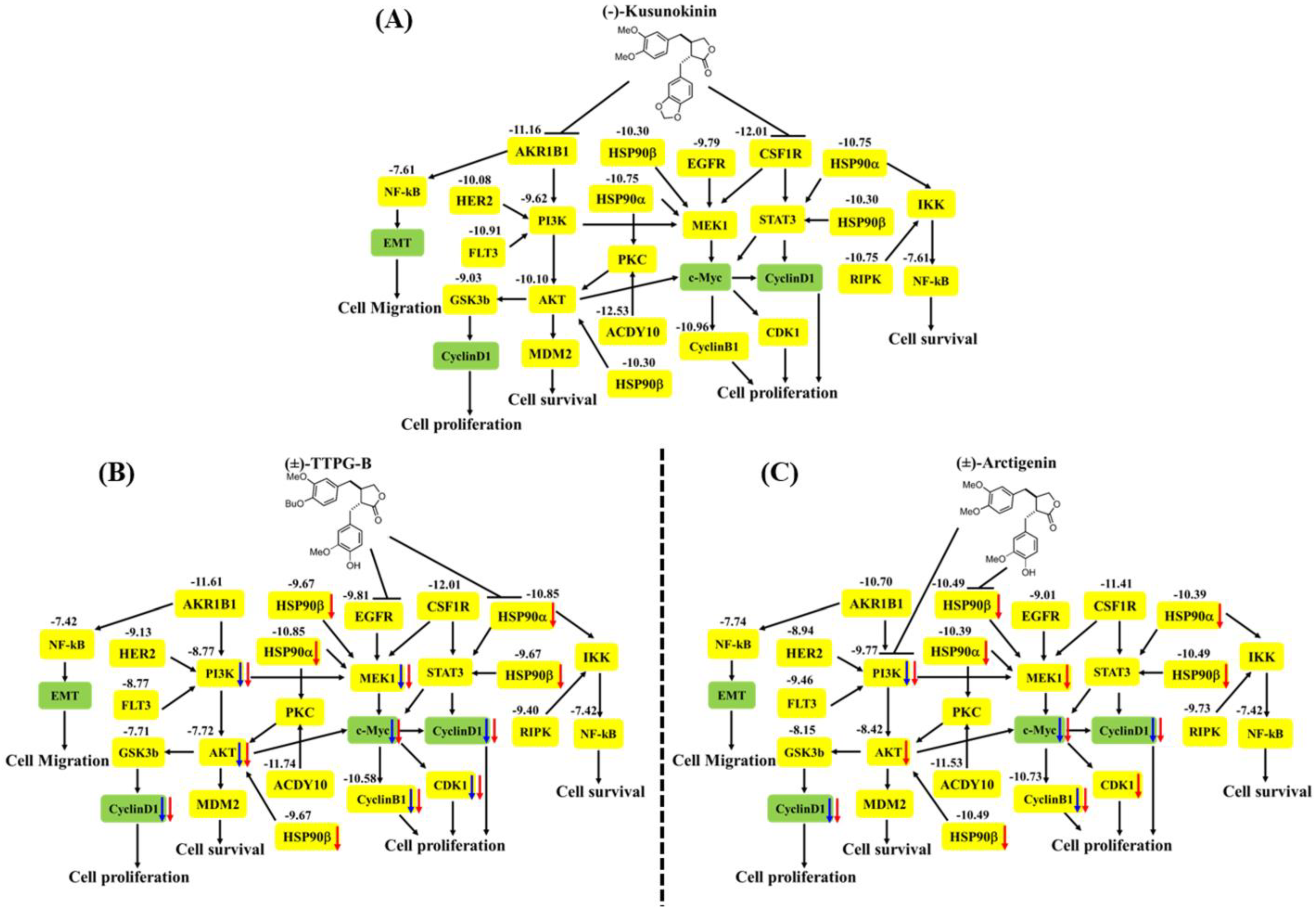
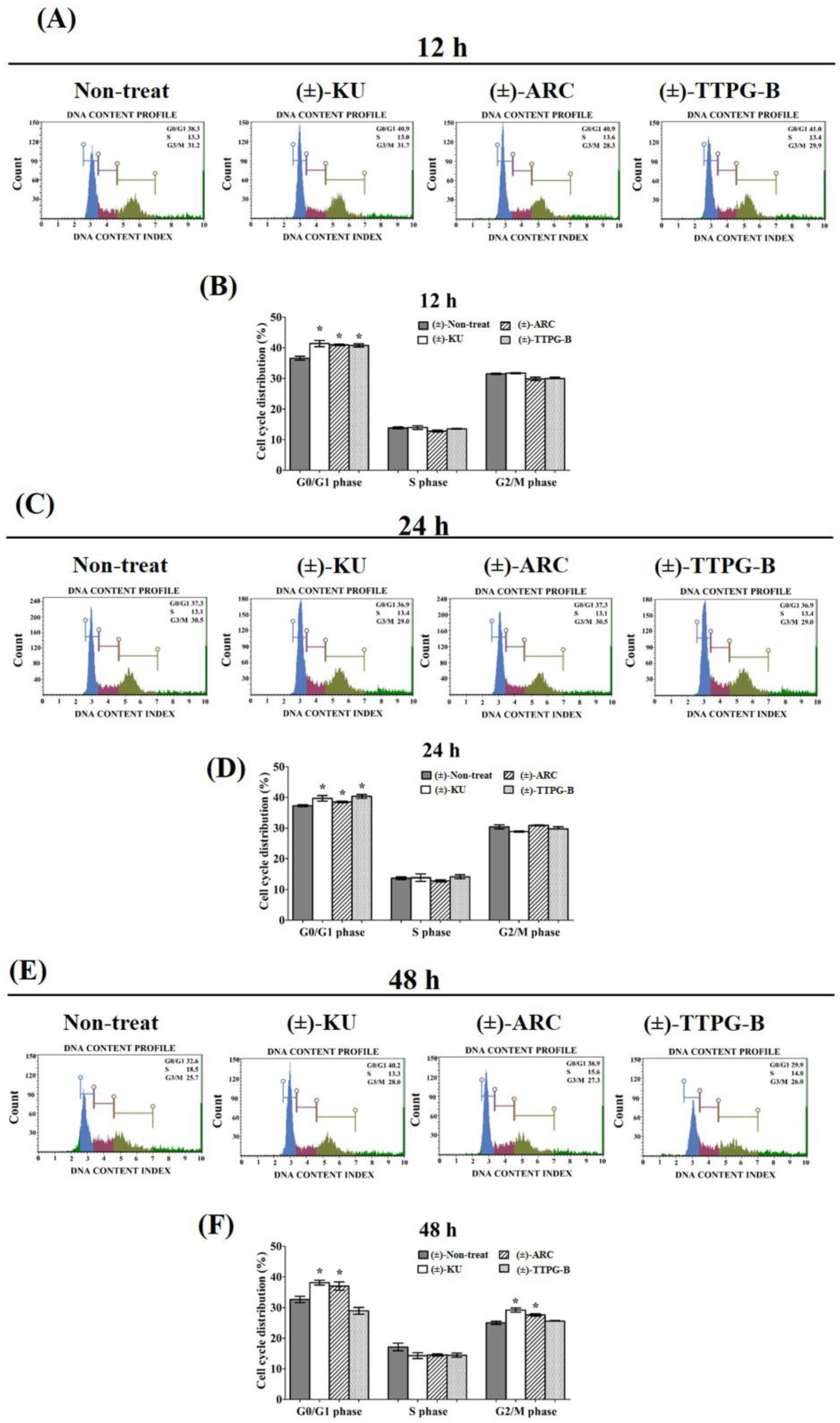
2.4. (±)-TTPG-B and (±)-ARC Dominated Cell-Cycle Arrest
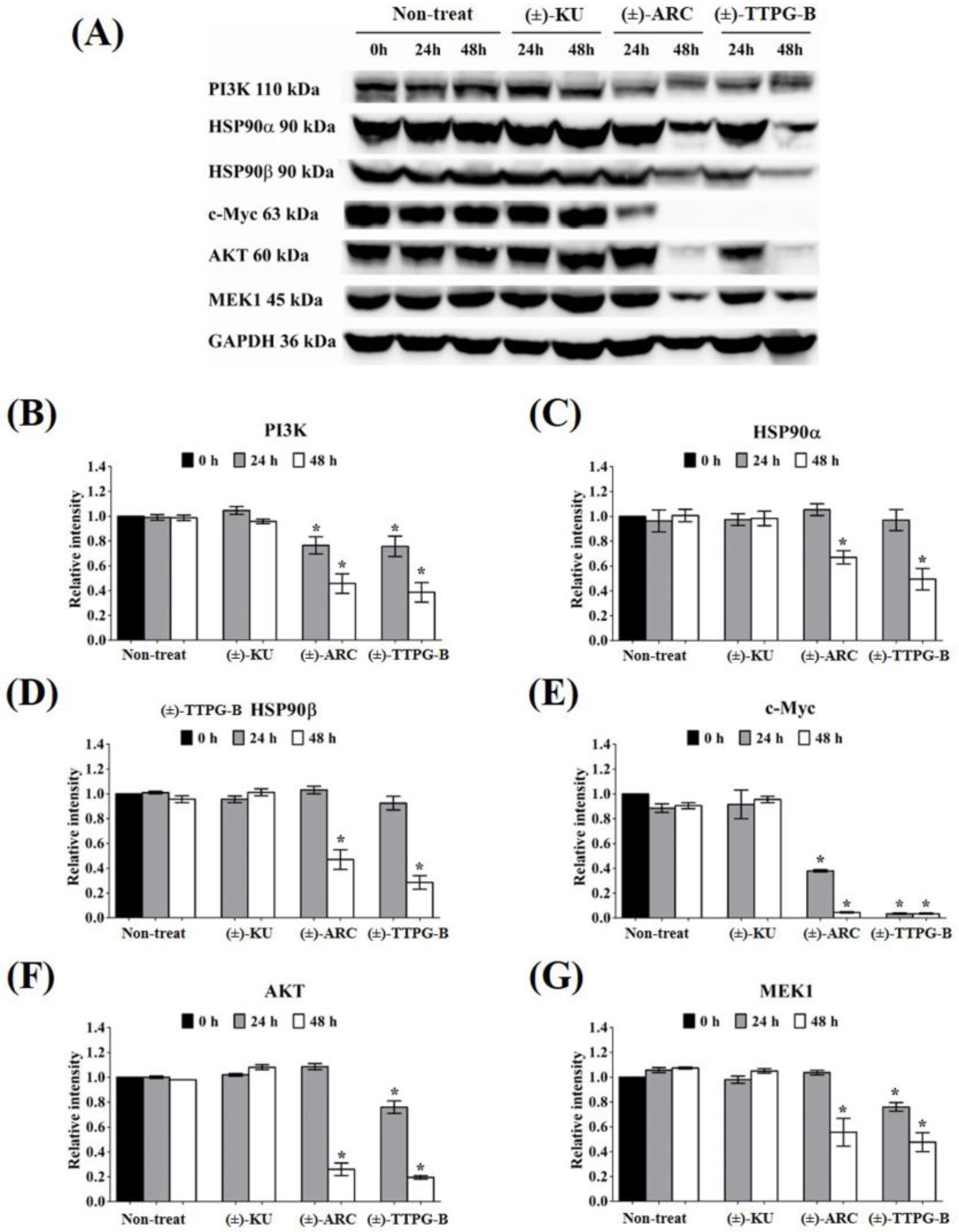
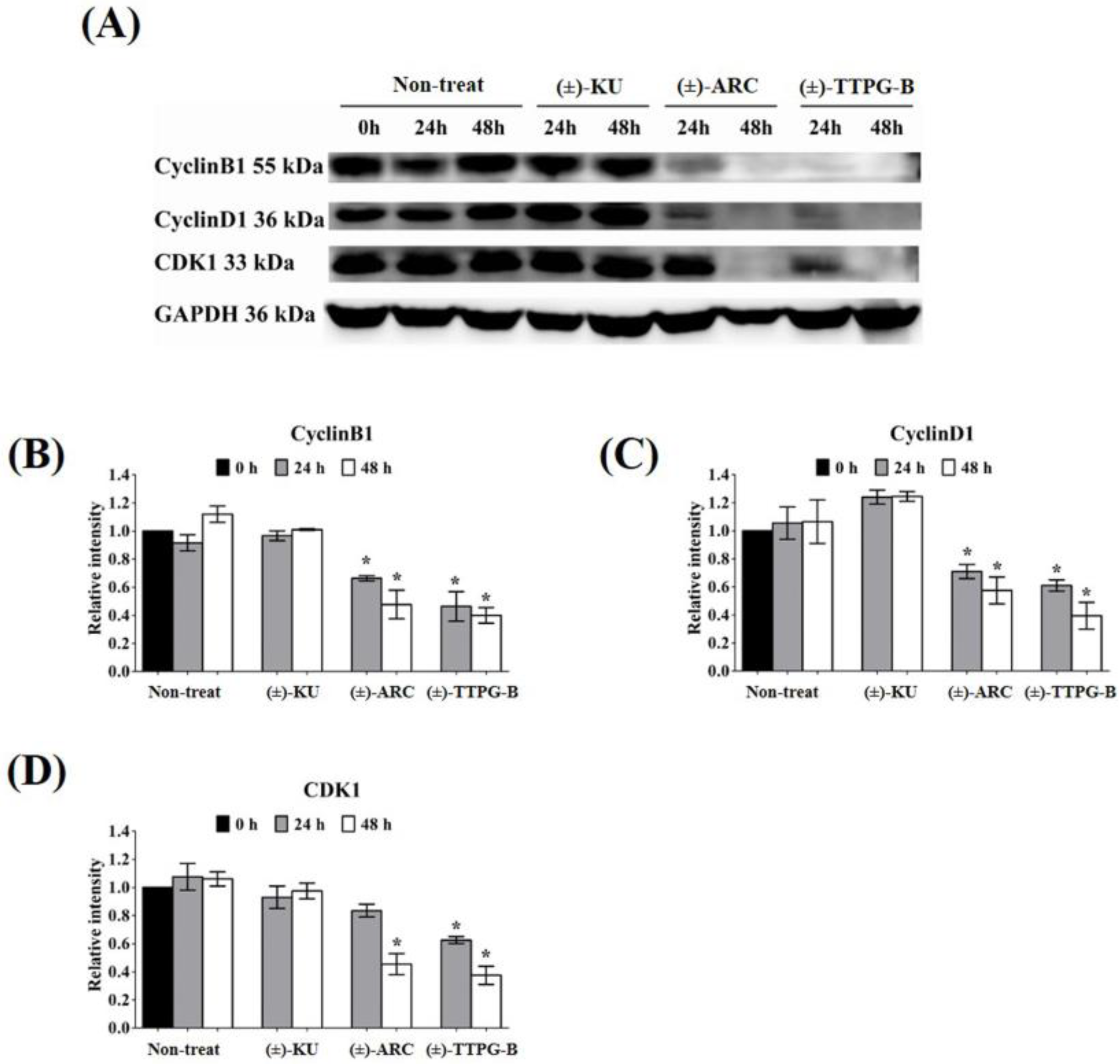
2.5. (±)-TTPG-B and (±)-ARC Suppressed HSP90α and PI3K, and Their Downstream Proteins
3. Discussion
4. Materials and Methods
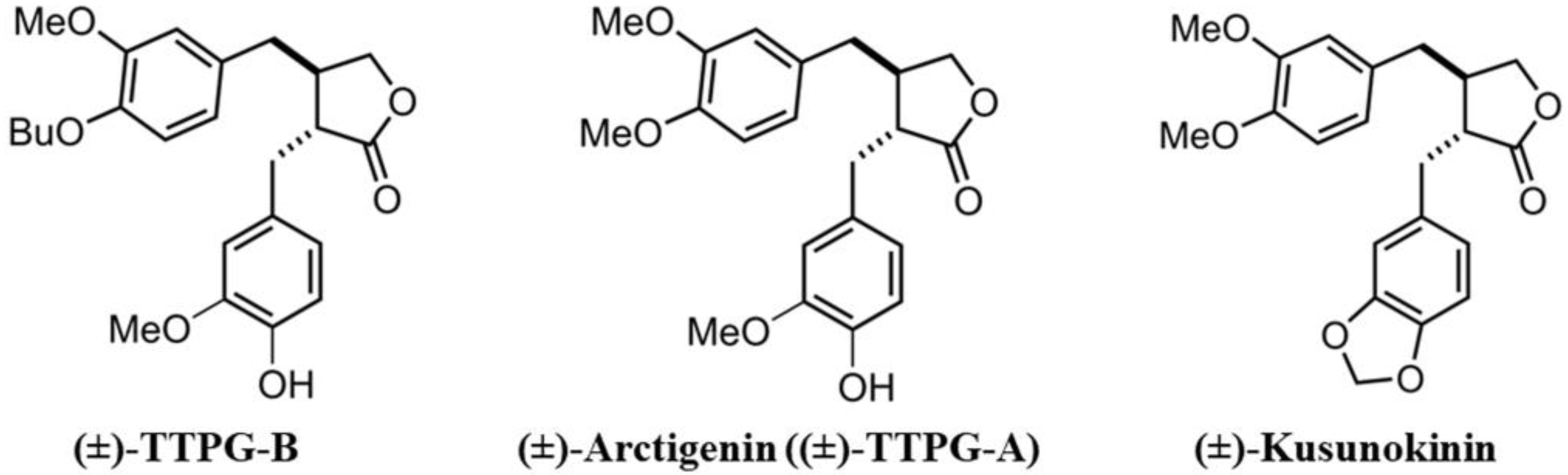
4.1. Compound Acquisition
4.2. Molecular Docking
4.3. Cell Culture
4.4. Cell Cycle Analysis Assay
4.5. Western Blot Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef]
- Cho, S.M.; Esmail, A.; Raza, A.; Dacha, S.; Abdelrahim, M. Timeline of FDA-Approved Targeted Therapy for Cholangiocarcinoma. Cancers 2022, 14, 2641. [Google Scholar] [CrossRef]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Fouassier, L.; Marzioni, M.; Afonso, M.B.; Dooley, S.; Gaston, K.; Giannelli, G.; Rodrigues, C.M.P.; Lozano, E.; Mancarella, S.; Segatto, O.; et al. Signalling networks in cholangiocarcinoma: Molecular pathogenesis, targeted therapies and drug resistance. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39 (Suppl. S1), 43–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, X.; Fu, J.; Xu, W.; Yuan, J. The Role of Tumour Metabolism in Cisplatin Resistance. Front. Mol. Biosci. 2021, 8, 691795. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Lee, K.H.; Kim, J.W.; Suh, K.J.; Nam, A.R.; Bang, J.H.; Bang, Y.J.; Oh, D.Y. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: Phase Ib study. Br. J. Cancer 2019, 121, 332–339. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Thongpanchang, T.; Wongma, K.; Tedasen, A.; Sukpondma, Y.; Graidist, P. Anticancer activity of synthetic (±)-kusunokinin and its derivative (±)-bursehernin on human cancer cell lines. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 117, 109115. [Google Scholar] [CrossRef]
- Tedasen, A.; Dokduang, S.; Sukpondma, Y.; Lailerd, N.; Madla, S.; Sriwiriyajan, S.; Rattanaburee, T.; Tipmanee, V.; Graidist, P. (-)-Kusunokinin inhibits breast cancer in N-nitrosomethylurea-induced mammary tumor rats. Eur. J. Pharmacol. 2020, 882, 173311. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Tipmanee, V.; Tedasen, A.; Thongpanchang, T.; Graidist, P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 129, 110361. [Google Scholar] [CrossRef]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (-)-Kusunokinin as a Potential Aldose Reductase Inhibitor: Equivalency Observed via AKR1B1 Dynamics Simulation. ACS Omega 2021, 6, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Tanawattanasuntorn, T.; Rattanaburee, T.; Thongpanchang, T.; Graidist, P. Trans-(±)-Kusunokinin Binding to AKR1B1 Inhibits Oxidative Stress and Proteins Involved in Migration in Aggressive Breast Cancer. Antioxid. 2022, 11, 2347. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Sermmai, P.; Tangthana-Umrung, K.; Thongpanchang, T.; Graidist, P. Anticancer Activity of (±)-Kusunokinin Derivatives towards Cholangiocarcinoma Cells. Molecules 2022, 27, 8291. [Google Scholar] [CrossRef] [PubMed]
- Basset, C.A.; Conway de Macario, E.; Leone, L.G.; Macario, A.J.L.; Leone, A. The chaperone system in cancer therapies: Hsp90. J. Mol. Histol. 2023, 54, 105–118. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhu, H.P.; Xie, X.; Mao, Q.; Liu, Y.Q.; He, X.H.; Peng, C.; Jiang, Q.L.; Huang, W. Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 1845. [Google Scholar] [CrossRef]
- El-Shafey, H.W.; Gomaa, R.M.; El-Messery, S.M.; Goda, F.E. Synthetic approaches, anticancer potential, HSP90 inhibition, multitarget evaluation, molecular modeling and apoptosis mechanistic study of thioquinazolinone skeleton: Promising antibreast cancer agent. Bioorganic Chem. 2020, 101, 103987. [Google Scholar] [CrossRef] [PubMed]
- Lawal, B.; Lo, W.C.; Mokgautsi, N.; Sumitra, M.R.; Khedkar, H.; Wu, A.T.; Huang, H.S. A preclinical report of a cobimetinib-inspired novel anticancer small-molecule scaffold of isoflavones, NSC777213, for targeting PI3K/AKT/mTOR/MEK in multiple cancers. Am. J. Cancer Res. 2021, 11, 2590–2617. [Google Scholar]
- Arthur, D.E.; Uzairu, A. Molecular docking studies on the interaction of NCI anticancer analogues with human Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit. J. King Saud. Univ. Sci. 2019, 31, 1151–1166. [Google Scholar] [CrossRef]
- Wang, T.; Qi, D.; Hu, X.; Li, N.; Zhang, X.; Liu, H.; Zhong, C.; Zhang, J. A novel evodiamine amino derivative as a PI3K/AKT signaling pathway modulator that induces apoptosis in small cell lung cancer cells. Eur. J. Pharmacol. 2021, 906, 174215. [Google Scholar] [CrossRef]
- Proskuriakova, E.; Khedr, A. Current Targeted Therapy Options in the Treatment of Cholangiocarcinoma: A Literature Review. Cureus 2022, 14, e26233. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Schwartz, S.J.; Sun, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2009, 12, 17–27. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Protein | Docking Score (kcal/mol−1) | Name | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (−)-KU | (+)-KU | (−)-ARC | (+)-ARC | (−)-TTPG-B | (+)-TTPG-B | Known Inhibitor | |||
| Cell signaling | |||||||||
| 1. | ACDY10 | −12.53 | −11.80 | −11.41 | −11.53 | −11.74 | −11.43 | −8.23 | SQ-22536 |
| 2. | AKT | −10.10 | −10.26 | −8.42 | −8.30 | −7.72 | −7.09 | −9.50 | Capivasertib |
| 3. | CSF1R | −12.01 | −10.58 | −11.41 | −9.29 | −8.96 | −9.35 | −12.29 | Pexidartinib |
| 4. | EGFR | −9.79 | −9.55 | −9.09 | −9.01 | −8.71 | −9.81 | −9.41 | Erlotinib |
| 5. | FLT3 | −10.91 | −9.42 | −9.46 | −8.53 | −8.77 | −8.58 | −10.56 | Gilteritinib |
| 6. | GSK3b | −8.24 | −9.03 | −7.83 | −8.15 | −7.71 | −7.60 | −5.37 | Tideglusib |
| 7. | HER2 | −9.25 | −10.08 | −8.94 | −8.37 | −9.13 | −8.96 | −9.03 | SCHEMBL3505765 |
| 8. | Hsp90α | −10.75 | −10.52 | −10.39 | −9.66 | −10.12 | −10.85 | −9.59 | EC44 |
| 9. | Hsp90β | −10.30 | −10.57 | −10.49 | −8.83 | −9.30 | −9.67 | −10.45 | Sunitinib |
| 10. | JAK1 | −8.29 | −8.46 | −8.27 | −9.01 | −7.98 | −7.62 | −10.54 | CHEMBL3779913 |
| 11. | MDM2 | −7.32 | −7.72 | −6.69 | −7.78 | −7.02 | −7.04 | −9.11 | CHEMBL1234332 |
| 12. | MEK1 | −10.38 | −10.72 | −10.10 | −8.99 | −9.80 | −9.40 | −13.09 | Trametinib |
| 13. | mTOR | −6.91 | −7.10 | −6.70 | −7.11 | −6.61 | −6.90 | −7.48 | AZD8055 |
| 14. | NF-kB | −7.61 | −7.80 | −7.74 | −7.22 | −7.42 | −6.88 | −7.16 | BAY11-7085 |
| 15. | PI3K | −9.62 | −8.89 | −9.77 | −9.13 | −8.17 | −8.77 | −9.54 | Apitolisib |
| 16. | PKC | −9.89 | −9.74 | −9.01 | −8.36 | −8.65 | −8.27 | −13.33 | Sotrastaurin |
| 17. | RIPK | −10.41 | −10.75 | −9.73 | −9.89 | −9.37 | −9.40 | −9.00 | SCHEMBL4397912 |
| 18. | STAT3 | −7.68 | −7.51 | −7.31 | −7.20 | −6.66 | −6.94 | −9.90 | STAT5-IN-2 |
| Cell invasion | |||||||||
| 1. | AKR1B1 | −11.16 | −11.80 | −10.60 | −10.70 | −11.61 | −11.40 | −10.18 | Epalrestat |
| 2. | VEGFR | −8.93 | −9.53 | −9.10 | −9.48 | −8.72 | −8.66 | −11.93 | Tivazanib |
| Cell cycle | |||||||||
| 1. | CDK1 | −10.42 | −10.06 | −10.06 | −9.51 | −10.15 | −9.63 | −11.32 | CGP74514A |
| 2. | CyclinB1 | −10.96 | −10.41 | −10.52 | −10.73 | −10.58 | −10.35 | −9.40 | Q27097368 |
| HSP90α | ||||||
|---|---|---|---|---|---|---|
| EC44 (Inhibitor) | (+)-TTPG-B | (−)-TTPG-B | (+)-ARC | (−)-ARC | (+)-KU | (−)-KU |
| Leu107 | Leu107 | Leu107 | ||||
| Tyr139 | Tyr139 | Tyr139 | Tyr139 | |||
| Gln23 | Gln23 | Gln23 | ||||
| Phe138 | Phe138 | Phe138 | ||||
| Trp162 | Gly108 | Gly135 | Gly108 | |||
| PI3K | ||||||
|---|---|---|---|---|---|---|
| Apitolisib (Inhibitor) | (+)-TTPG-B | (−)-TTPG-B | (+)-ARC | (−)-ARC | (+)-KU | (−)-KU |
| Arg849 | Arg849 | Arg849 | Arg849 | Arg849 | Arg849 | |
| Gln852 | ||||||
| Tyr867 | ||||||
| Cys869 | Cys869 | Cys869 | Cys869 | Cys869 | Cys869 | |
| Trp201 | Trp201 | |||||
| Arg649 | Arg649 | |||||
| Arg690 | Arg690 | Arg690 | ||||
| Gln846 | Gln846 | |||||
| His658 | Asp654 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rattanaburee, T.; Chompunud Na Ayudhya, C.; Thongpanchang, T.; Tipmanee, V.; Graidist, P. Trans-(±)-TTPG-B Attenuates Cell Cycle Progression and Inhibits Cell Proliferation on Cholangiocarcinoma Cells. Molecules 2023, 28, 7342. https://doi.org/10.3390/molecules28217342
Rattanaburee T, Chompunud Na Ayudhya C, Thongpanchang T, Tipmanee V, Graidist P. Trans-(±)-TTPG-B Attenuates Cell Cycle Progression and Inhibits Cell Proliferation on Cholangiocarcinoma Cells. Molecules. 2023; 28(21):7342. https://doi.org/10.3390/molecules28217342
Chicago/Turabian StyleRattanaburee, Thidarath, Chompunud Chompunud Na Ayudhya, Tienthong Thongpanchang, Varomyalin Tipmanee, and Potchanapond Graidist. 2023. "Trans-(±)-TTPG-B Attenuates Cell Cycle Progression and Inhibits Cell Proliferation on Cholangiocarcinoma Cells" Molecules 28, no. 21: 7342. https://doi.org/10.3390/molecules28217342
APA StyleRattanaburee, T., Chompunud Na Ayudhya, C., Thongpanchang, T., Tipmanee, V., & Graidist, P. (2023). Trans-(±)-TTPG-B Attenuates Cell Cycle Progression and Inhibits Cell Proliferation on Cholangiocarcinoma Cells. Molecules, 28(21), 7342. https://doi.org/10.3390/molecules28217342