

Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds

Abstract
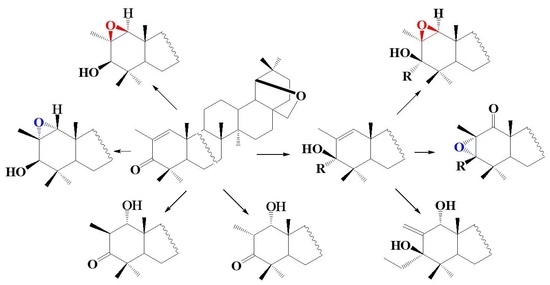
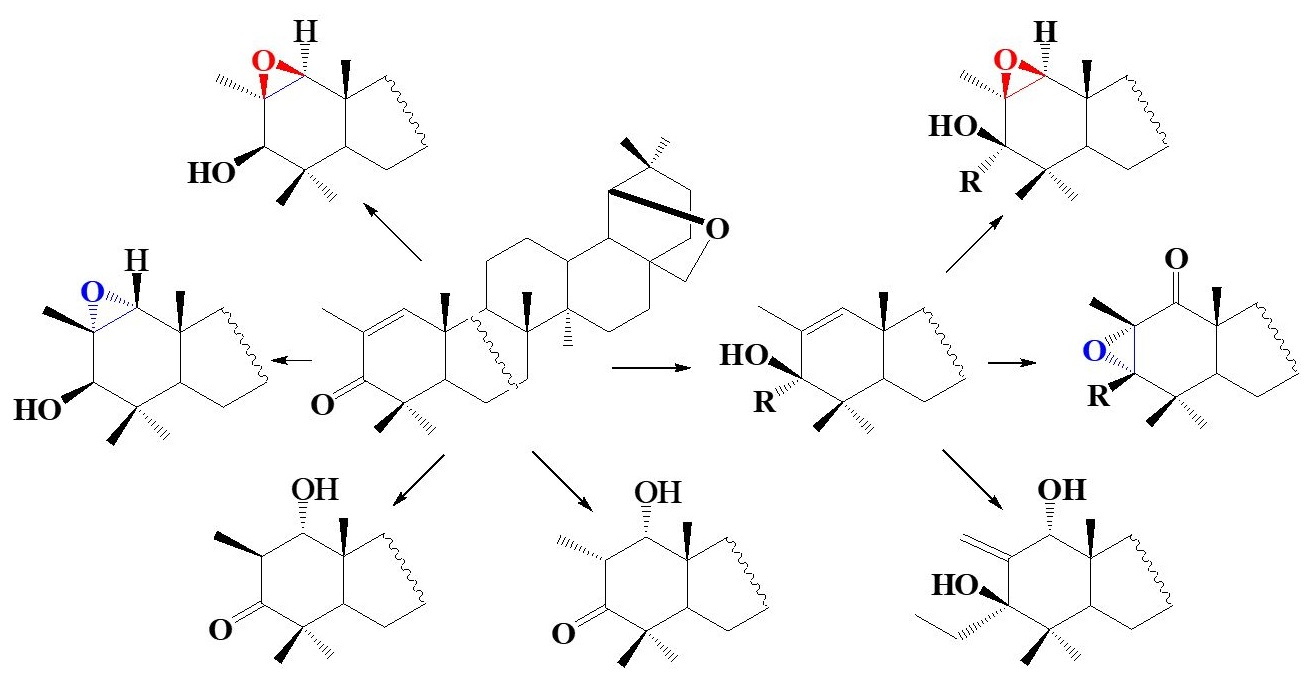
1. Introduction
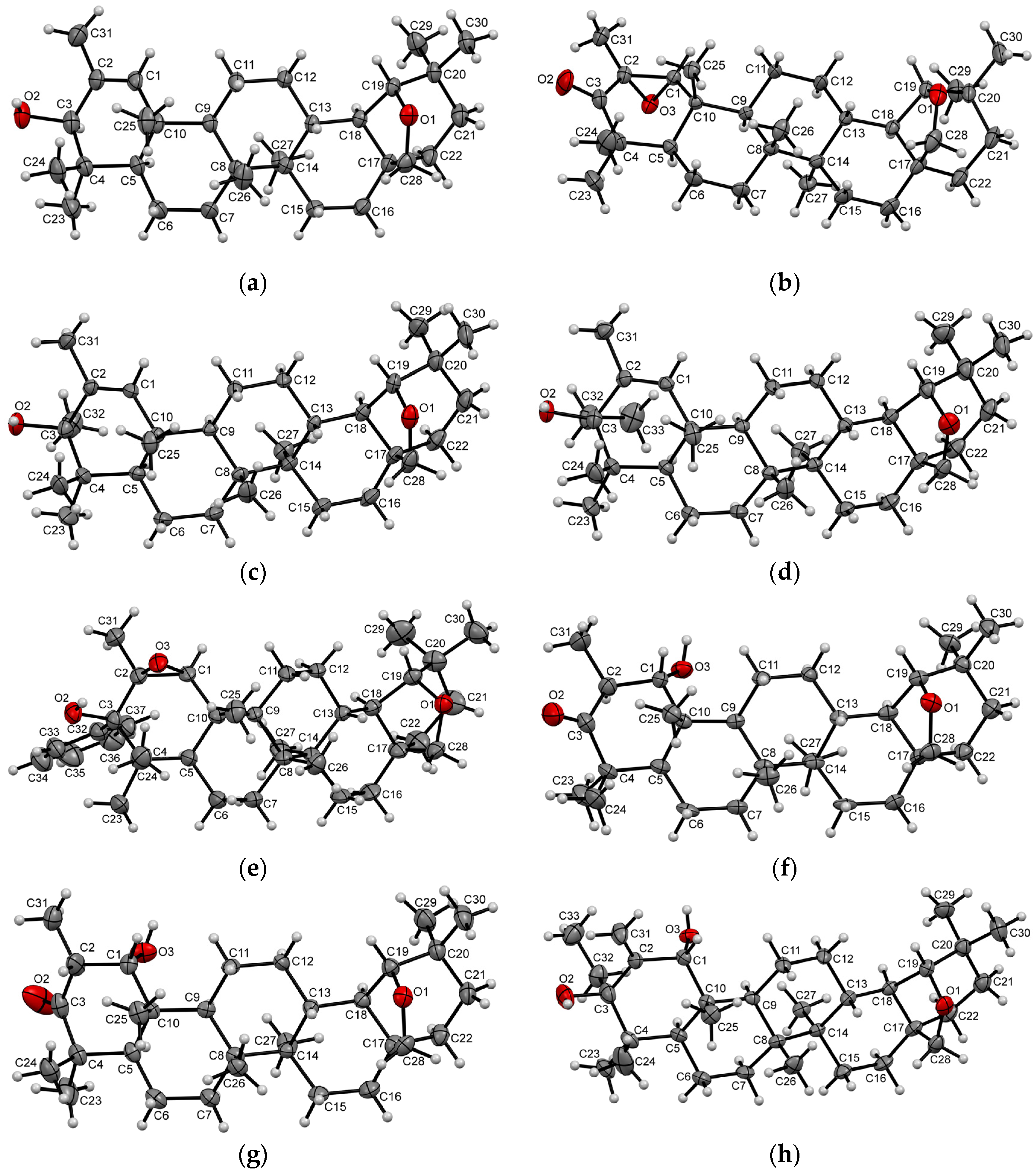
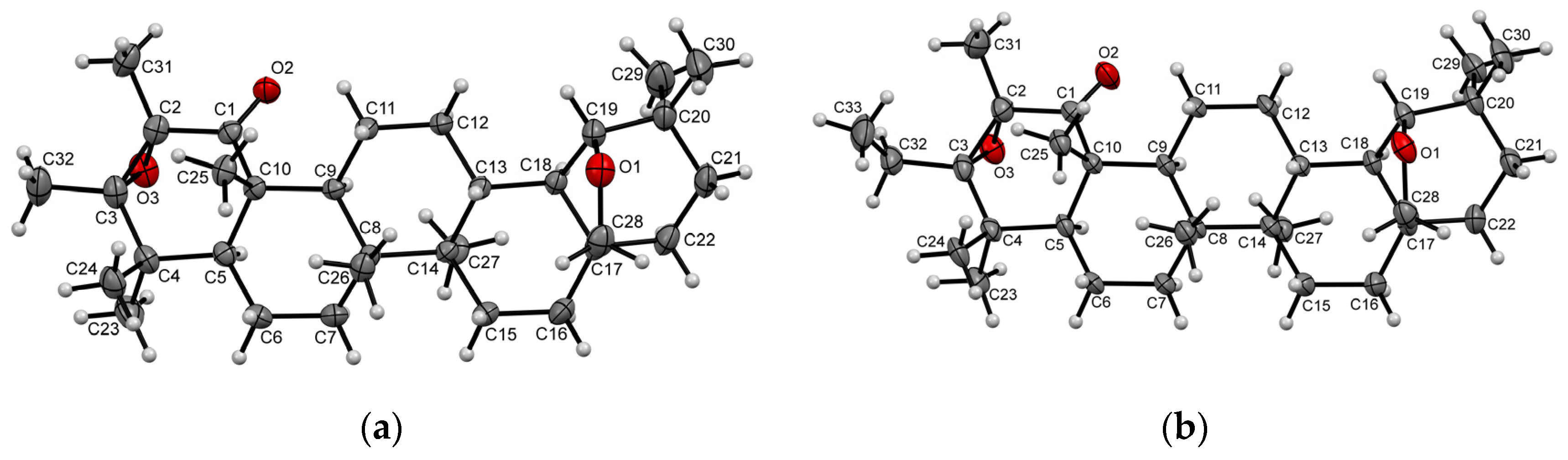
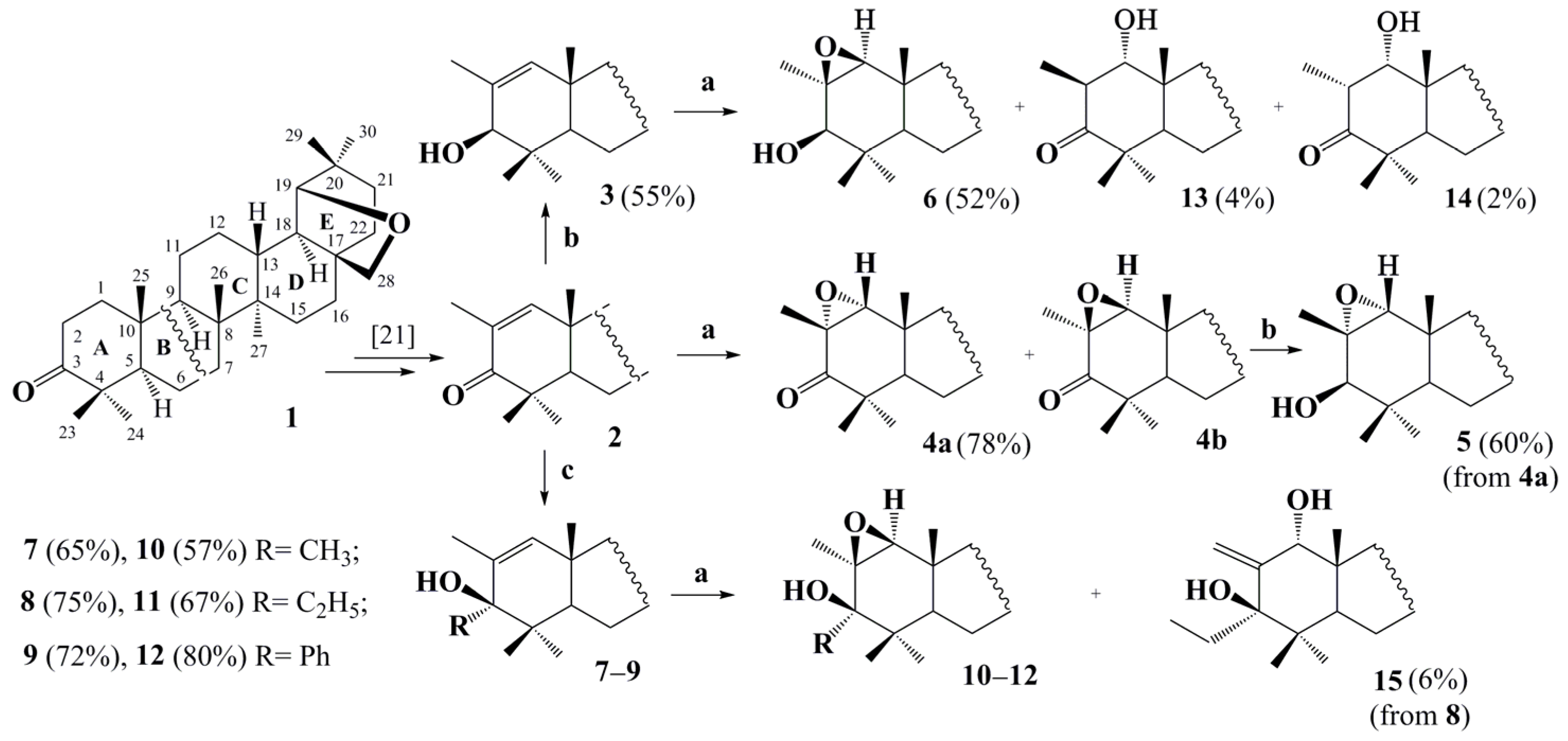
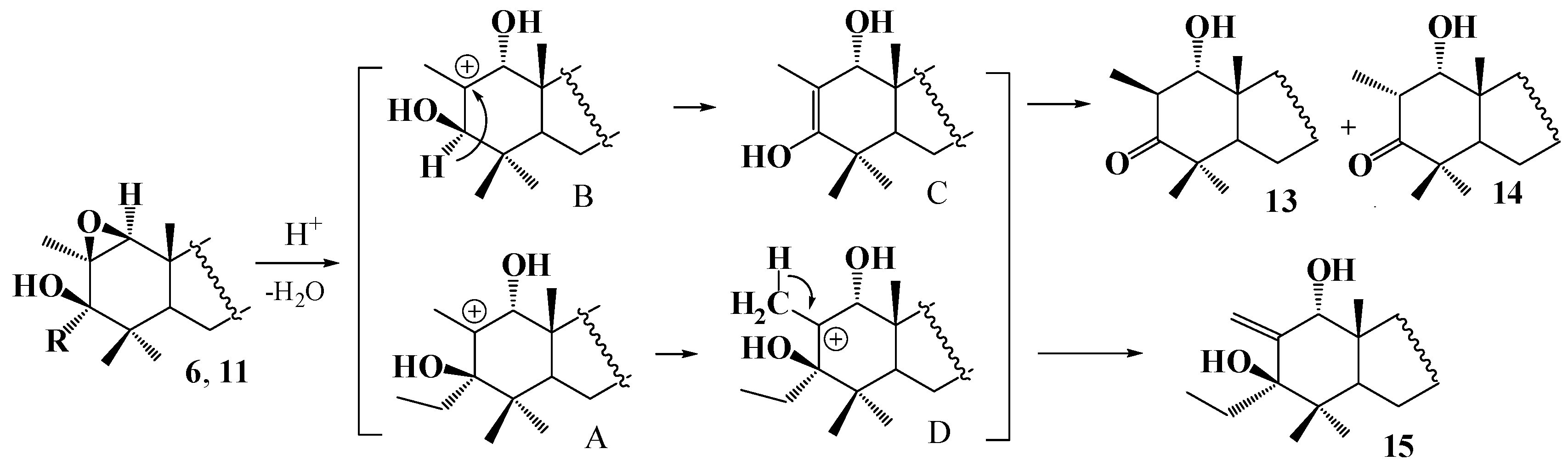
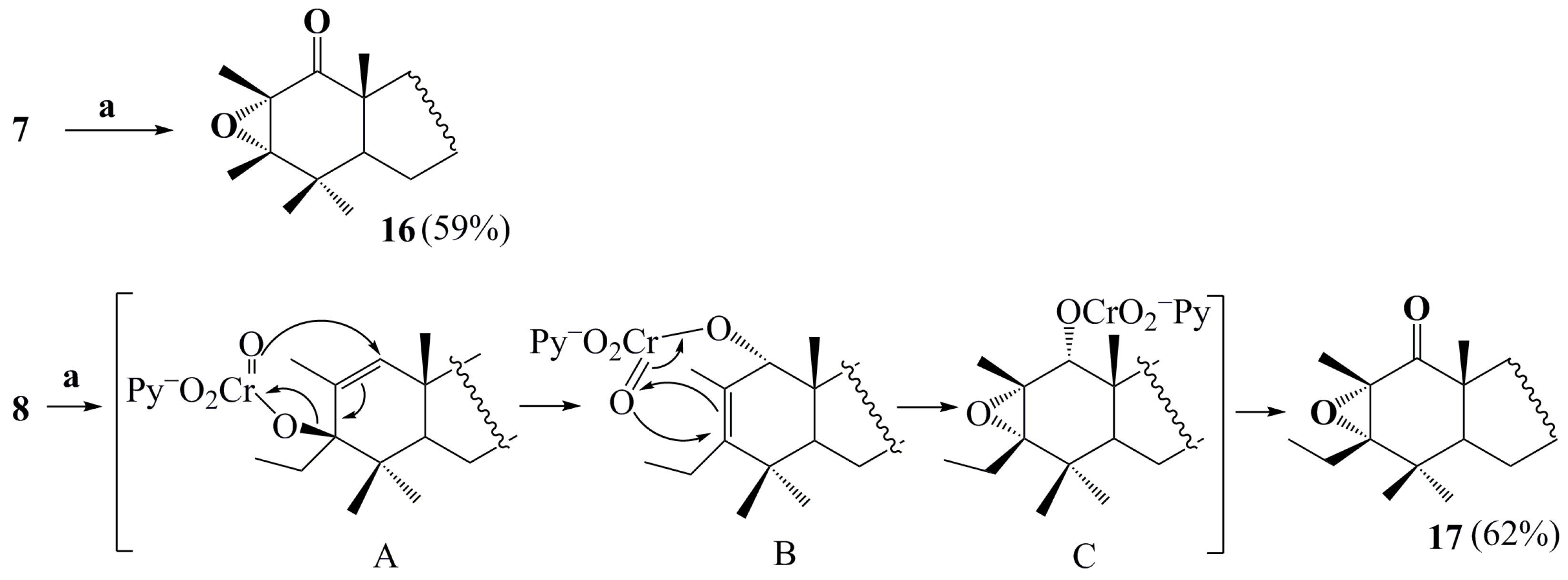
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Procedure for Preparing Compounds , , –, –
4.2. General Procedure for Preparing Compounds and
4.3. General Procedure for Preparing Compounds –
4.4. General Procedure for Preparing Compounds and
4.5. Screening for Cytotoxic Activity of Compounds , , , –, , and
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Markov, A.V.; Zenkova, M.A.; Logashenko, E.B. Modulation of tumour-related signaling pathways by natural pentacyclic triterpenoids and their semisynthetic derivatives. Curr. Med. Chem. 2017, 24, 1277–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, R.H.; Wang, M.; Xu, G.B.; Liao, S.G. Prodrugs of triterpenoids and their derivatives. Eur. J. Med. Chem. 2017, 131, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Xu, J.; Lin, A.; Wu, X.; Wu, L.; Xie, W. Recent advances for the synthesis of selenium-containing small molecules as potent antitumor agents. Curr. Med. Chem. 2018, 25, 2009–2033. [Google Scholar] [CrossRef]
- Wu, H.F.; Morris-Natschke, S.L.; Xu, X.D.; Yang, M.H.; Cheng, Y.Y.; Yu, S.S.; Lee, K.H. Recent advances in natural anti-HIV triterpenoids and analogs. Med. Res. Rev. 2020, 40, 2339–2385. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Şoica, C.; Voicu, M.; Ghiulai, R.; Dehelean, C.; Racoviceanu, R.; Trandafirescu, C.; Roșca, O.-J.; Nistor, G.; Mioc, M.; Mioc, A. Natural compounds in sex hormone-dependent cancers: The role of triterpenes as therapeutic agents. Front. Endocrinol. 2021, 11, 612396. [Google Scholar] [CrossRef]
- Nadendla, R.R. Principles of Organic Medicinal Chemistry; New Age Publishers: New Delhi, India, 2005; 322p. [Google Scholar]
- Urban, M.; Klinot, J.; Tislerova, I.; Biedermann, D.; Hajduch, M.; Cisarova, I.; Sarek, J. Reactions of activated lupane oxo-compounds with diazomethane: An approach to new derivatives of cytotoxic triterpenes. Synthesis 2006, 23, 3979–3986. [Google Scholar] [CrossRef]
- Michaudel, Q.; Journot, G.; Regueiro-Ren, A.; Goswami, A.; Guo, Z.; Tully, T.P.; Zou, L.; Ramabhadran, R.O.; Houk, K.N.; Baran, P.S. Improving physical properties via C-H oxidation: Chemical and enzymatic approaches. Angew. Chem. Int. Ed. 2014, 53, 12091–12096. [Google Scholar] [CrossRef]
- Huang, R.Z.; Jin, L.; Wang, C.G.; Xu, X.J.; Du, Y.; Liao, N.; Ji, M.; Liao, Z.X.; Wang, H.S. A pentacyclic triterpene derivative possessing polyhydroxyl ring A suppresses growth of HeLa cells by reactive oxygen species-dependent NF-κB pathway. Eur. J. Pharmacol. 2018, 838, 157–169. [Google Scholar] [CrossRef]
- Guo, H.; Wang, H.; Huo, Y.X. Engineering critical enzymes and pathways for improved triterpenoid biosynthesis in yeast. ACS Synth. Biol. 2020, 9, 2214–2227. [Google Scholar] [CrossRef]
- Shah, S.; Tan, H.; Sultan, S.; Faridz, M.; Shah, M.; Nurfazilah, S.; Hussain, M. Microbial-catalyzed biotransformation of multifunctional triterpenoids derived from phytonutrients. Int. J. Mol. Sci. 2014, 15, 12027–12060. [Google Scholar] [CrossRef]
- Luchnikova, N.A.; Grishko, V.V.; Ivshina, I.B. Biotransformation of oleanane and ursane triterpenic acids. Molecules 2020, 25, 5526. [Google Scholar] [CrossRef]
- Berger, M.; Knittl-Frank, C.; Bauer, S.; Winter, G.; Maulide, N. Application of relay C−H oxidation logic to polyhydroxylated oleanane triterpenoids. Chem 2020, 6, 1183–1189. [Google Scholar] [CrossRef]
- Mu, T.; Wei, B.; Zhu, D.; Yu, B. Site-selective C-H hydroxylation of pentacyclic triterpenoids directed by transient chiral pyridine-imino groups. Nat. Commun. 2020, 11, 4371. [Google Scholar] [CrossRef]
- Borkova, L.; Hodon, J.; Urban, M. Synthesis of betulinic acid derivatives with modified A-ring and their application as potential drug candidates. Asian J. Org. Chem. 2018, 7, 1542–1560. [Google Scholar] [CrossRef]
- Huang, L.; Luo, H.; Li, Q.; Wang, D.; Zhang, J.; Hao, X.; Yang, X. Pentacyclic triterpene derivatives possessing polyhydroxyl ring A inhibit Gram-positive bacteria growth by regulating metabolism and virulence genes expression. Eur. J. Med. Chem. 2015, 95, 64–75. [Google Scholar] [CrossRef]
- Huang, L.R.; Hao, X.J.; Li, Q.J.; Wang, D.P.; Zhang, J.X.; Luo, H.; Yang, X.S. 18β-glycyrrhetinic acid derivatives possessing a trihydroxylated a ring are potent gram-positive antibacterial agents. J. Nat. Prod. 2016, 79, 721–731. [Google Scholar] [CrossRef]
- Motlhanka, D.; Houghton, P.; Miljkovic-Brake, A.; Habtemariam, S. A novel pentacyclic triterpene glycoside from a resin of Commiphora glandulosa from Botswana. Afr. J. Pharm. Pharmacol. 2010, 4, 549–554. [Google Scholar]
- Liang, Z.M.; Wang, X.H.; Huang, L.R.; Li, Q.J.; Guan, T.Q.; Hao, X.J.; Luo, H.; Yang, X.S. 1α,2α-Epoxy-3β-hydroxy oleanolic acid derivatives regulation of the metabolism, haemolysis and β-lactamase gene expression in vitro and their structure–microbicidal activity relationship. Bioorganic Med. Chem. Lett. 2016, 26, 3870–3875. [Google Scholar] [CrossRef]
- Konysheva, A.V.; Zhukova, A.E.; Dmitriev, M.V.; Grishko, V.V. Synthesis and intramolecular cyclization of a 2,3-seco-oleanane triterpenoid with an ethylketone fragment. Chem. Nat. Compd. 2018, 54, 1094–1099. [Google Scholar] [CrossRef]
- Hanson, J.R.; Hitchcock, P.B.; Kiran, I. The stereochemistry of epoxidation of steroidal 4,6-dienes. J. Chem. Res.—Part S 1999, 3, 198–199. [Google Scholar] [CrossRef]
- Kinot, J.; Krumpolc, M.; Vystrčil, A. Triterpenes. IX. Reaction of isomeric 2,3-epoxides with Grignard reagent. Collect. Czechoslov. Chem. Commun. 1966, 31, 3174–3181. [Google Scholar] [CrossRef]
- Kehrli, A.R.H.; Taylor, D.A.H.; Niven, M. The synthesis of a 1α,2α,3α-triacetoxy limonoid. J. Chem. Soc. Perkin Trans. 1990, 7, 2057–2065. [Google Scholar] [CrossRef]
- García-Granados, A.; López, P.E.; Melguizo, E.; Parra, A.; Simeó, Y. Oxidation of several triterpenic diene and triene systems. Oxidative cleavage to obtain chiral intermediates for drimane and phenanthrene semi-synthesis. Tetrahedron 2004, 60, 3831–3845. [Google Scholar] [CrossRef]
- Amer, H.; Mereiter, K.; Stanetty, C.; Hofinger, A.; Czollner, L.; Beseda, I.; Jordis, U.; Kueenburg, B.; Claßen-Houben, D.; Kosma, P. Synthesis and crystal structures of ring A modified glycyrrhetinic acid derivatives derived from 2,3-oxirane and 2,3-thiirane intermediates. Tetrahedron 2010, 66, 4390–4402. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Khusnutdinova, E.F.; Korlyukov, A.A. Stereospecific epoxidation of an olean-18(19)-ene-type triterpenoid. Chem. Nat. Compd. 2011, 46, 900–901. [Google Scholar] [CrossRef]
- Mikhailova, L.R.; Budaev, A.S.; Spirikhin, L.V.; Baltina, L.A. Oxidation of licorice-root triterpene-acid derivatives by m-chloroperbenzoic acid. Chem. Nat. Compd. 2019, 55, 88–91. [Google Scholar] [CrossRef]
- Pakulski, Z.; Cmoch, P.; Korda, A.; Luboradzki, R.; Gwardiak, K.; Karczewski, R. Rearrangements of the betulin core. Synthesis of terpenoids possessing the bicyclo[3.3.1]nonane fragment by rearrangement of lupane-type epoxides. J. Org. Chem. 2021, 86, 1084–1095. [Google Scholar] [CrossRef]
- Paryzek, Z. Epoxidation of lanost-9(11)-enes. The effect of a β-carbonyl group upon the stereochemistry of epoxidation. J. Chem. Soc. Perkin Trans. 1978, 1, 329–3369. [Google Scholar] [CrossRef]
- Kvasnica, M.; Tislerova, I.; Sarek, J.; Sejbal, J.; Cisarova, I. Preparation of new oxidized 18-α-oleanane derivatives. Collect. Czech. Chem. Commun. 2005, 70, 1447–1464. [Google Scholar] [CrossRef]
- Tolmacheva, I.A.; Shelepen’kina, L.N.; Shashkov, A.S.; Grishko, V.V.; Glushkov, V.A.; Tolstikov, A.G. Reaction of 3-acetoxy-(2,3),(19β,28)-diepoxyoleanane with cyclic and linear amines. Chem. Nat. Compd. 2007, 43, 153–158. [Google Scholar] [CrossRef]
- Csuk, R.; Nitsche, C.; Sczepek, R.; Schwarz, S.; Siewert, B. Synthesis of antitumor-active betulinic acid-derived hydroxypropargylamines by copper-catalyzend mannich reactions. Arch. Pharm. 2013, 346, 232–246. [Google Scholar] [CrossRef]
- Pereslavtseva, A.V.; Tolmacheva, I.A.; Slepukhin, P.A.; El’tsov, O.S.; Kucherov, I.I.; Eremin, V.F.; Grishko, V.V. Synthesis of A-pentacyclic triterpene α,β-alkenenitriles. Chem. Nat. Compd. 2014, 49, 1059–1066. [Google Scholar] [CrossRef]
- Konysheva, A.V.; Nebogatikov, V.O.; Tolmacheva, I.A.; Dmitriev, M.V.; Grishko, V.V. Synthesis of cytotoxically active derivatives based on alkylated 2,3-seco-triterpenoids. Eur. J. Med. Chem. 2017, 140, 74–83. [Google Scholar] [CrossRef]
- Konysheva, A.V.; Eroshenko, D.V.; Grishko, V.V. Synthesis, Cyclization, and cytotoxic activity of 2,3-secolupane triterpenoids with an ethylketone fragment. Nat. Prod. Commun. 2019, 14, 1–7. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry Part A: Structure and Mechanisms, 5th ed.; Springer: New York, NY, USA, 2007; 1199p. [Google Scholar]
- Kočovský, P. Stereochemistry of epoxidation of allylic and homoallylic cyclohexene alcohols. J. Chem. Soc. Perkin Trans. 1994, 13, 1759–1763. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Afonso, C.A.M. Brønsted acid-catalyzed dihydroxylation of olefins in aqueous medium. Adv. Synth. Catal. 2011, 353, 2920–2926. [Google Scholar] [CrossRef]
- Jat, J.L.; Kumar, G. Isomerization of Epoxides. Adv. Synth. Catal. 2019, 361, 4426–4441. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Ganesh, V. Oxidation adjacent to oxygen of alcohols by chromium reagents. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 7, pp. 277–294. [Google Scholar]
- Luzzio, F.A. 1,3-Oxidative transpositions of allylic alcohols in organic synthesis. Tetrahedron 2012, 68, 5323–5339. [Google Scholar] [CrossRef]
- Papillaud, B.; Tiffon, F.; Taran, M.; Miguel, B.A.S.; Delmond, B. Part I. epoxydes diterpeniques: Synthese et reactivite d’epoxydes derives d’acides resiniques. Tetrahedron 1985, 41, 1845–1857. [Google Scholar] [CrossRef]
- Ley, S.V.; Madin, A. Oxidation adjacent to oxygen of alcohols by chromium reagents. In Comprehensive Organic Synthesis; Trost, B.M., Ed.; Plenum: New York, NY, USA, 1991; Volume 7, 251p. [Google Scholar]
- Kharitonov, Y.V.; Shul’ts, E.E.; Shakirov, M.M. Synthetic transformations of higher terpenoids. XXXIII.*Preparation of 15,16-dihydroisopimaric acid and methyl dihydroisopimarate and their transformations. Chem. Nat. Compd. 2014, 49, 2857–2899. [Google Scholar] [CrossRef]
- Singha, R.; Ghosh, P. Phytochemical investigation of Sapium baccatum: Identification of 3α-hydroxy-1α, 2α-epoxy lupan. J. Indian Chem. Soc. 2018, 95, 549–552. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Sen’kova, A.V.; Moralev, A.D.; Savin, I.A.; Komarova, N.I.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. Novel Epoxides of Soloxolone Methyl: An Effect of the Formation of Oxirane Ring and Stereoisomerism on Cytotoxic Profile, Anti-Metastatic and Anti-Inflammatory Activities In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 6214. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Molina, M.T.; Anjum, S. Naturally occurring cyclohexane epoxides: Sources, biological activities and synthesis. Chem. Rev. 2004, 104, 2857–2899. [Google Scholar] [CrossRef] [PubMed]
- Way2Drug Predictive Services. PASS Online. Available online: http://www.pharmaexpert.ru/passonline/index.php (accessed on 6 April 2022).
- Poroikov, V.V. Computer-aided drug design: From discovery of novel pharmaceutical agents to systems pharmacology. Biomed. Chem. 2020, 66, 30–41. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Keil, B. Laboratory Technika Organicke Chemie; Nakladatelstvi ČSAV: Praha, Czech Republic, 1963; 751p. [Google Scholar]
- CrysAlisPro, Version 1.171.37.33 (Release 27-03-2014 CrysAlis171.NET); Agilent Technologies: Yarnton, UK, 2014.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP - A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Compound | Estimated Activity, Pa | ||||||
|---|---|---|---|---|---|---|---|
| Antineoplastic | Colorectal Cancer | Colon Cancer | Lung Cancer | Ovarian Cancer | Carcinoma | Thyroid Cancer | |
| 2 | 0.940 | 0.914 | 0.911 | 0.786 | 0.780 | - | - |
| 3 | 0.961 | 0.900 | 0.898 | 0.819 | 0.774 | - | 0.703 |
| 4a, 4b | 0.983 | 0.902 | 0.900 | 0.819 | 0.758 | 0.742 | - |
| 5 | 0.967 | 0.898 | 0.898 | 0.845 | 0.820 | 0.778 | 0.710 |
| 6 | 0.967 | 0.898 | 0.895 | 0.845 | 0.820 | 0.778 | 0.778 |
| 7 | 0.911 | 0.886 | 0.884 | 0.786 | 0.710 | - | - |
| 8 | 0.911 | 0.886 | 0.884 | 0.769 | - | - | - |
| 9 | 0.911 | 0.870 | 0.868 | 0.747 | 0.744 | - | - |
| 10 | 0.959 | 0.899 | 0.896 | 0.827 | 0.748 | 0.736 | - |
| 11 | 0.960 | 0.901 | 0.898 | 0.826 | 0.755 | 0.726 | - |
| 12 | 0.954 | 0.837 | 0.833 | 0.756 | - | - | - |
| 13, 14 | 0.959 | 0.888 | 0.881 | 0.826 | 0.822 | - | - |
| 15 | 0.934 | 0.890 | 0.887 | 0.832 | 0.701 | - | - |
| 16 | 0.938 | 0.898 | 0.895 | 0.852 | 0.735 | - | 0.716 |
| 17 | 0.950 | 0.856 | 0.852 | 0.810 | - | - | - |
| Tested Compound | IC50 (Mean ± SD), μM | |||||
|---|---|---|---|---|---|---|
| HEpG2 | HCT116 | MS | RD TE32 | A549 | MCF-7 | |
| 2 ** | 132.1 ± 15.61 | >200 | 156.1 ± 24.83 | >200 | 136.8 ± 48.1 | 60.94 ± 2.32 |
| 3 | 135.6 ± 13.21 | 128.2 ± 15.66 | 52.56 ± 4.58 | 145.3 ± 35.3 | 96.42 ± 12.96 | 115.8 ± 6.14 |
| 4a | 129.7 ± 13.52 | 129.1 ± 15.16 | 44.02 ± 6.80 | 142.3 ± 24.8 | 39.17 ± 5.11 | 97.27 ± 10.93 |
| 5 | >200 | >200 | >200 | >200 | >200 | 78.72 ± 13.55 |
| 9 | >200 | >200 | >200 | >200 | >200 | 45.27 ± 4.18 |
| 10 | >200 | >200 | >200 | >200 | >200 | >200 |
| 11 | >200 | >200 | >200 | >200 | >200 | 45.88 ± 7.43 |
| 12 | >200 | >200 | >200 | >200 | >200 | 37.08 ± 5.05 |
| 17 | >200 | >200 | >200 | >200 | >200 | 117.00 ± 4.24 |
| DOX * | 1.78 ± 0.31 | 1.96 ± 0.19 | 1.29 ± 0.16 | 1.27 ± 0.03 | 2.04 ± 0.22 | 0.14 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krainova, G.; Beloglazova, Y.; Dmitriev, M.; Grishko, V. Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds. Molecules 2023, 28, 550. https://doi.org/10.3390/molecules28020550
Krainova G, Beloglazova Y, Dmitriev M, Grishko V. Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds. Molecules. 2023; 28(2):550. https://doi.org/10.3390/molecules28020550
Chicago/Turabian StyleKrainova, Gulnaz, Yulia Beloglazova, Maksim Dmitriev, and Victoria Grishko. 2023. "Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds" Molecules 28, no. 2: 550. https://doi.org/10.3390/molecules28020550
APA StyleKrainova, G., Beloglazova, Y., Dmitriev, M., & Grishko, V. (2023). Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds. Molecules, 28(2), 550. https://doi.org/10.3390/molecules28020550