1. Introduction
Since the synthesis of urea in 1828, humans have opened the door to organic synthesis. With the development of isolation and identification techniques and organic synthesis methods, people are pushing the limits of complex molecular synthesis, especially complex natural products, such as vitamin B12 and anemone toxin. Natural products are the main source of innovative medicines, such as artemisinin and paclitaxel. From 1981 to 2020, over 50% of the small molecule drugs approved by the FDA were derived from natural products or the parent molecular structure of natural products [
1]. It can be seen that innovative drug development based on active natural products has played a crucial role in the field of original drug research and development. However, active natural products are often very scarce in nature and have low direct druggability. Therefore, research on the total synthesis of active natural products can not only solve the problem of limited natural resources and prepare a large amount of active natural products but also enable the rapid synthesis of natural product analogues with diverse skeletons and functional groups, building a natural product-like compound library to conduct extensive biological activity screening and drug development research. It has important scientific significance and potential application value in the field of natural product synthesis and new drug development. Thus, the development of practical synthetic methods and efficient synthetic strategies in natural product total synthesis has always been an active field in organic synthesis and has attracted great attention from synthetic chemists.
Deconstructive synthesis, namely building new (and often more challenging) structures through bond cleavage and the formation of easily accessible moieties, has emerged as a useful design principle in preparing complex bioactive natural products and other target molecules [
2]. The basic logic of deconstructive synthesis is to construct a “hardly accessible” skeleton from an “easily accessible” skeleton through skeletal reorganization. Meanwhile, the rapid construction of polycyclic molecular frameworks, the precise instillation of multi-stereocenters bearing crowed quaternary carbon centers, and the efficient induction of versatile functional groups should also be highlighted. It also facilitates the continuous modification of key advanced intermediates to improve the overall efficiency of the synthetic route. The subtlety of deconstructive synthesis often lies in its creative approach of building intricate molecular frameworks, resulting in the design of easily accessible intermediates that could significantly reduce the challenge of total synthesis. The development of deconstructive synthesis strategies is often inspired by the molecular structural characteristics of natural products, which also tests the creativity and personalized perspective of synthetic chemists.
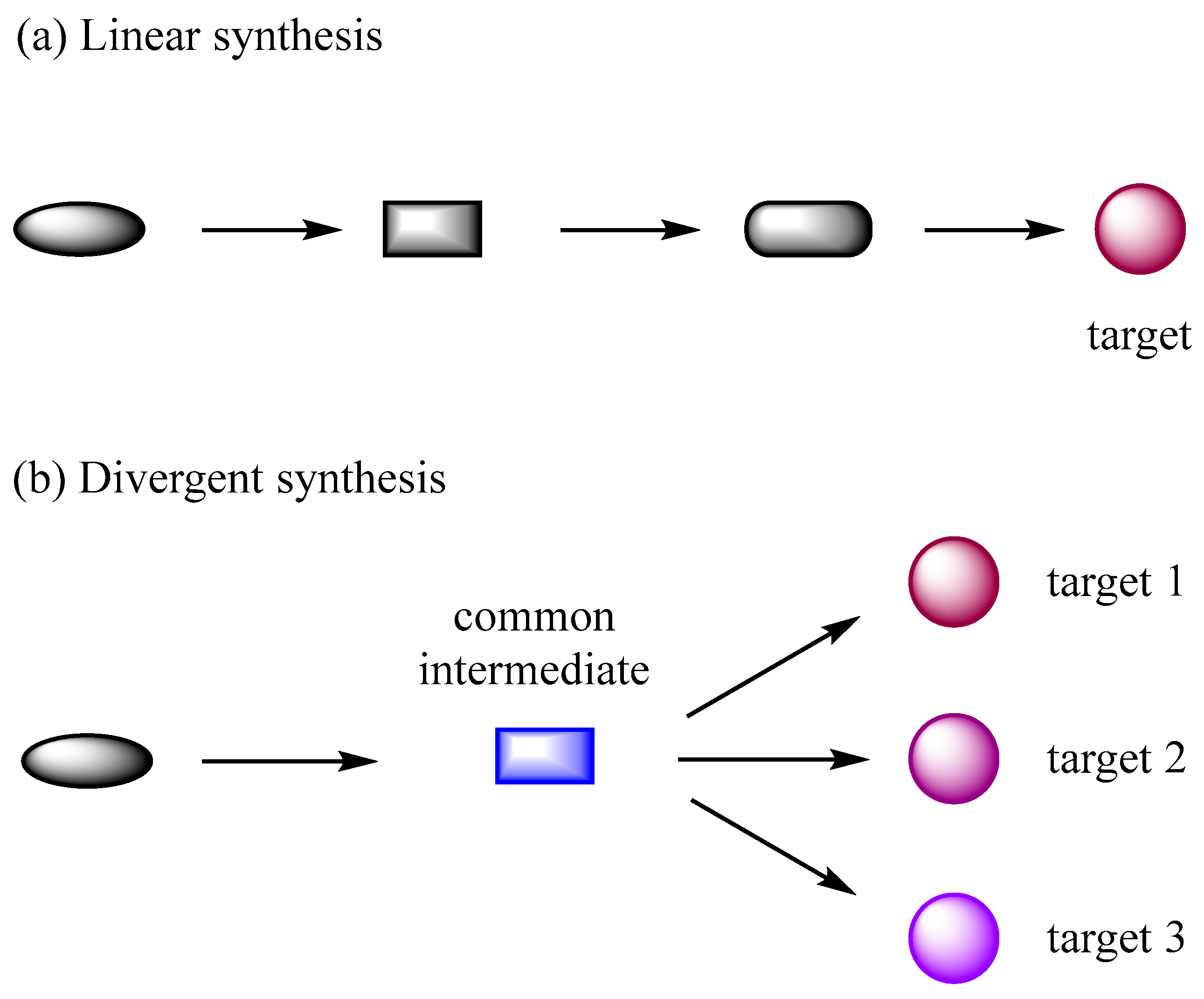
Divergent synthesis, in short, involves working from one common intermediate to many natural products (
Figure 1). In general, a class of natural products often shares a similar chemical structure with various functional groups or different molecular frameworks that can be constructed from a common intermediate in biosynthesis. The original definition of divergent synthesis was proposed and demonstrated by Boger in 1984 (
Scheme 1) [
3]. “Divergent” is defined as a common intermediate (preferably an advanced intermediate) being converted, separately, to at least two natural products. Applications of divergent synthesis include not only preparing molecules in the same family but also accessing natural products that have the same molecular skeletons from different families. Divergent total synthesis, also defined as “collective total synthesis” by MacMillan, is when multiple skeletons of natural products are prepared from a versatile common intermediate [
4].
Compared with linear synthesis that can only achieve one natural product at a time, the divergent strategy provides a more efficient approach to access many natural products with similar or different molecular skeletons from a versatile molecule through a number of routine chemical operations. In addition, the divergent synthesis of natural products is also more conducive to building a natural-product-like compound library to conduct more biological activity screening and innovative drug development based on the bioactive natural products.
The application of both strategies requires organic chemists to be familiar with the skeleton, functional group, oxidation state, and other characteristics of the target molecules. One of the most challenging steps is to design a suitable advanced intermediate that can be easily prepared and quickly converted to as many target molecules as possible. Meanwhile, the common intermediate should also be close to the target molecule to reduce the total number of synthesis steps.
During the past decade, many research groups have developed diverse, novel synthetic methodologies to realize a lot of impressive approaches for the divergent and deconstructive synthesis of natural products. Previous literature on the divergent strategy in natural product total syntheses from 2013 to June 2017 has been summarized in
Chemical Reviews [
5]. Excellent reviews focusing on deconstructive synthesis through rearrangement reactions already exist, providing more comprehensive perspectives for authors who have interests in this field [
6,
7,
8,
9,
10,
11,
12].
In the last three years, many research groups have accomplished a lot of creative synthetic routes to syntheses of natural products with complex structures and broad biological activities. In this review, we will first introduce five excellent examples to demonstrate the synthetic utilities of the deconstructive and divergent strategies in natural product total synthesis. Then, we will summarize our previous work on the deconstructive and divergent synthesis of bioactive natural products to demonstrate the high efficiency and simplicity of these two strategies in natural product total synthesis. After a brief introduction of the respective bioactive natural products, we will discuss the details of the synthetic routes and how to combine the deconstructive and divergent strategies in target molecules.
2. Selected Deconstructive and Divergent Syntheses of Natural Product (2021–2023)
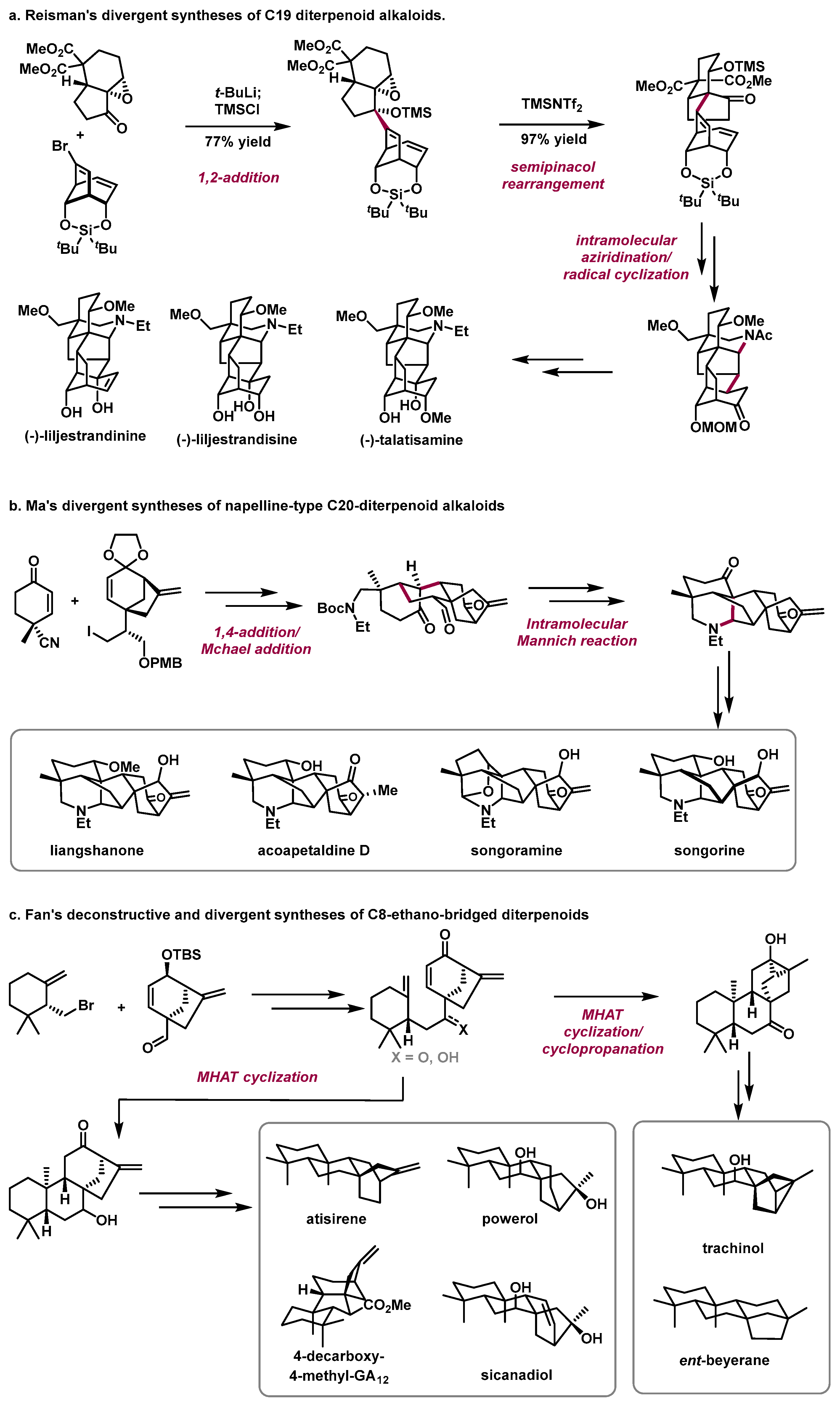
In 2021, the Reisman group from the California Institute of Technology reported the divergent total syntheses of three C19 diterpenoid alkaloids: (−)-talatisamine, (−)-liljestrandisine, and (−)-liljestrandinine from phenol (
Scheme 2a) [
13]. The highlights of this work include (1) a 1,2-addition/semipinacol rearrangement sequence to efficiently couple two complex fragments and construct the quaternary carbon center and (2) an intramolecular aziridination and radical cyclization to assemble the pentacyclic skeleton of the target alkaloids.
In 2022, the Ma group from the Shanghai Institute of Organic Chemistry reported an asymmetric divergent approach to the total synthesis of six napelline-type C20-diterpenoid alkaloids in a convergent manner (
Scheme 2b) [
14]. The highlights of this work include (1) a diastereoselective intermolecular Cu-mediated conjugate addition to couple the two fragments; (2) an intramolecular Michael addition reaction to construct the tetracyclic skeleton; and (3) an intramolecular Mannich cyclization to rapidly construct the azabicyclo [3.2.1] octane motif of the target molecules.
In 2023, the Fan group from Lanzhou University published a deconstructive and divergent synthesis of nine C8-ethano-bridged diterpenoids based on late-stage skeletal diversification in the longest linear 8 to 11 steps starting from readily available chiral materials (
Scheme 2c) [
15]. Crucial advanced intermediates with different structural skeletons were rapidly constructed through regioselective and diastereoselective metal-hydride hydrogen atom transfer (MHAT) cyclization from the
multi-reactivity of densely functionalized polycyclic substrates.
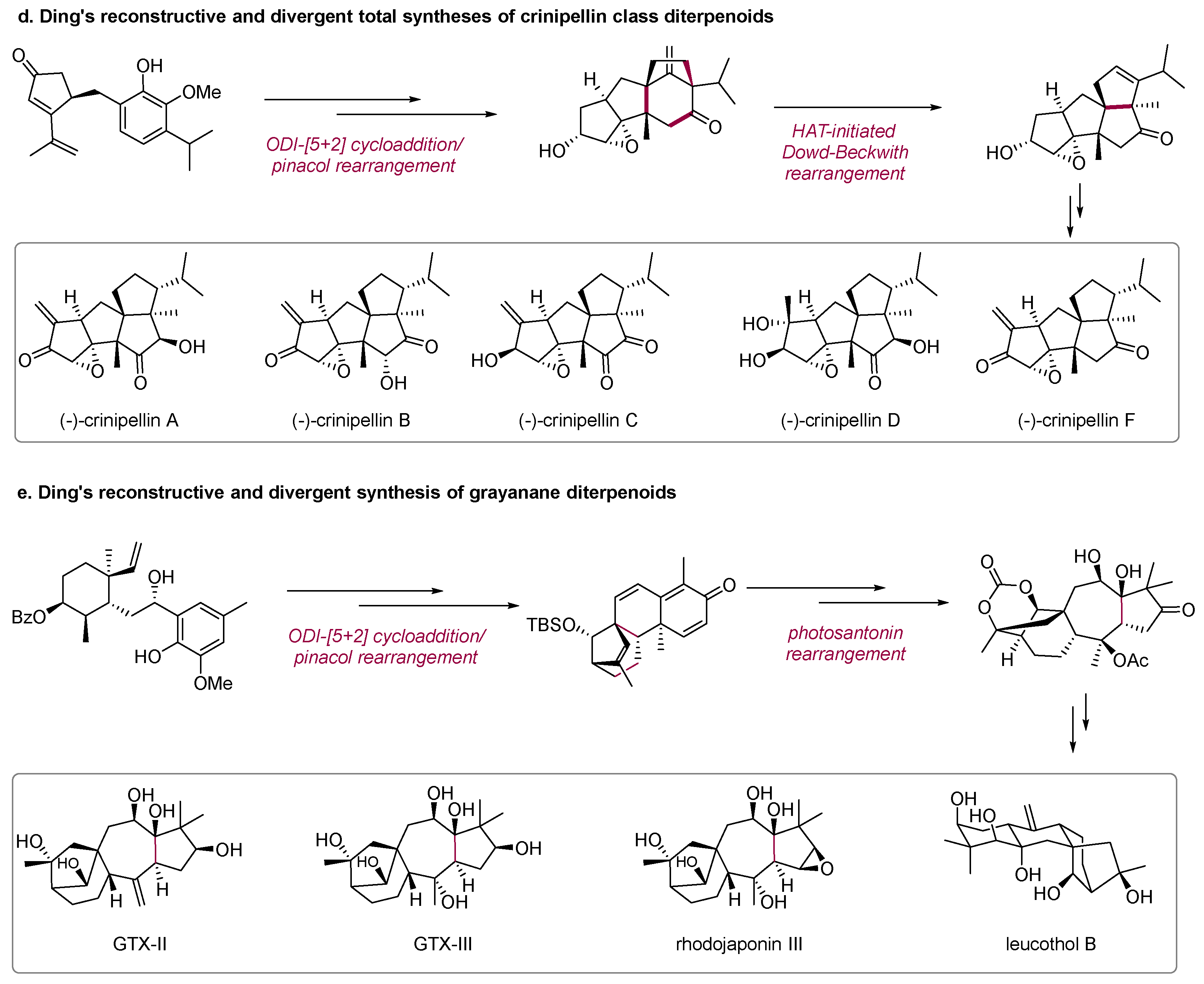
In 2022, the Ding group from Zhejiang University developed deconstructive and divergent syntheses of eight tetraquinane diterpenoids through a HAT-initiated Dowd−Beckwith rearrangement reaction for the efficient assembly of diversely functionalized polyquinane frameworks (
Scheme 2d) [
16].
In 2023, the Ding group finished the deconstructive and divergent syntheses of nine grayanane diterpenoids that belong to five distinct subtypes from a common advanced tetracyclic intermediate which was prepared through a tandem intramolecular oxidative dearomatization-induced (ODI) [5 + 2] cycloaddition/pinacol rearrangement to construct [3.2.1]-bicyclic skeleton and a photosantonin rearrangement to assemble the 5/7 bicyclic framework (
Scheme 2e) [
17].
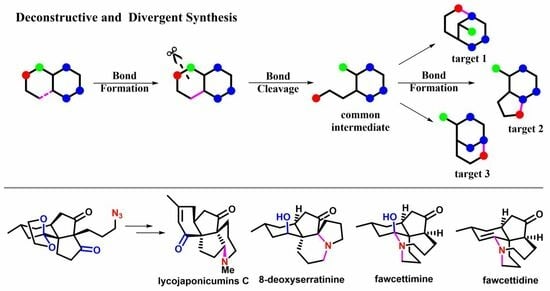
3. Divergent Syntheses of Fawcettimine-Class Lycopodium Alkaloids
Lycopodium alkaloids are structurally complex natural products with quinolizine, pyridine, or
α-pyridone, originally identified in the
Lycopodium genus.
Lycopodium alkaloids exhibit important biological activities. For example, Huperzine A is a potent inhibitor of acetylcholinesterase (AChE) and shows promise in the treatment of Alzheimer’s disease (AD). The fawcettimine-class
Lycopodium alkaloids are a class of structurally unique alkaloids with complex and unique skeletons bearing quaternary carbon centers, such as fawcettimine (
1); fawcettidine (
2); lycojaponicumins C (
3); and 8-deoxyserratinine (
4) (
Scheme 3) [
18]. In particular, lycojaponicumins C (
3), isolated by Yu and co-workers in 2012 from the traditional Chinese medicine
Lycopodium japonicum, exhibits lipopolysaccharide (LPS)-induced pro-inflammatory factors in BV2 macrophages [
19]. The fawcettimine-class
Lycopodium alkaloids feature fused tetracyclic skeletons, including two common
cis-hydroindene (6/5 bicycle) motifs and two diverse ring systems bearing quaternary carbon centers. The
Lycopodium alkaloids have attracted great attention from synthetic chemists and medicinal chemists for their unique chemical structures and broad biological activities. During the past decade, several elegant approaches for the total synthesis of
Lycopodium alkaloids have been reported [
20].
In 2013, Tu’s group reported a creative approach to divergent synthesis of four fawcettimine-class
Lycopodium alkaloids, namely, fawcettimine (
1), fawcettidine (
2), lycojaponicumins C (
3), and 8-deoxyserratinine (
4) [
21]. In this work, the authors designed a common intermediate which can transform to alkaloids
1–
4 in a divergent manner. Meanwhile, a practical methodology to quickly construct the
cis-hydroindene skeleton was designed and developed to demonstrate that natural product total synthesis can promote the development of novel synthetic methodologies and synthetic strategies.
After optimization of the Mukaiyama–Michael addition conditions, coupling of two simple building blocks
5 and
6 was realized in the presence of triflimide to afford silyl ether
7 with excellent yield and high diastereoselectivity (
Scheme 4). Then, Cu(tbs)
2-catcalyzed carbene cyclization designed by our group was successfully conducted, and decarboxylation of the resulting cyclization product provided the desired ketone
9 with a
cis-6/5 bicyclic skeleton and a quaternary carbon center with a 55% yield [
22]. The bicycle
9 could also be scalable prepared with good yield by using a one-pot operation. Notably, this novel methodology offers an efficient way to construct the
cis-6/5 bicyclic skeleton from simple building blocks which can apply to the total synthesis of other complex natural products. Then, Dickmann condensation of the ketone ester
9 gave an enolate of tricyclic trione
10, which in situ reacted with π-allylpalladium complex to deliver the tricyclic compound
11 with angular 6/5/5 tricyclic molecular framework bearing two quaternary carbon centers. Selective protection of the ketone on the six-membered ring and hydroboration of the terminal double bond of
11 gave the desired alcohol
12, which could readily be converted into the common intermediate, the tricyclic azide
13, through the Mitsunobu reaction.
The tricyclic azide
13 possesses an angular 6/5/5 tricyclic framework, two contiguous quaternary carbon centers, two carbonyl groups, and an azide chain and can serve as a versatile common intermediate to divergent synthesis alkaloids, namely, fawcettimine (
1), fawcettidine (
2), lycojaponicumins C (
3), and 8-deoxyserratinine (
4), via the deconstructive strategy. On the one hand, the regioselective
aza-Wittig reaction and reduction of
13 yielded tetracyclic amine
14, which was transformed to (−)-lycojaponicumins C (
1) through several chemical operations to install the double bond and N-methyl group of the target molecule (
Scheme 5). On the other hand, the regioselective Schmidt rearrangement of
15, obtained from deprotection of
14, was realized and provided the desired tetracyclic lactam
16 in a useful synthetic yield. Treatment of
16 with Lawesson’s reagent followed by a reduction of the resulting sulfo-lactam with Raney Ni gave tetracyclic amine
17 with a 66% yield. Then, a late-stage skeletal diversification of tetracyclie
17 was conducted to synthesize three alkaloids with different skeletons. Thus, compound
17 could divergently transform to the following: (−)-8-deoxyserratinine (
4) via selective NaBH
4-reduction; (+)-fawcettimine (
1) under an SmI
2/THF reductive condition via selective C4-N cleavage/
aza-ketalization; and (+)-fawcettidine (
2) under a Zn /HOAc harsh reductive condition via C4-N cleavage/
aza-ketalization/dehydration.
In this work, the Tu group have explored a divergent and deconstructive strategy for the total syntheses of four fawcettimine-class Lycopodium alkaloids, namely, fawcettimine (1, 10 steps), fawcettidine (2, 12 steps), lycojaponicumins C (3, 12 steps), and 8-deoxyserratinine (4, 12 steps) from simple building blocks 5 and 6. A major innovation of this strategy involved the design of a versatile common intermediate 13, which can synthesize not only the fawcettimine-class Lycopodium alkaloids but also other complex natural products. Other highlights of this work include (i) two consecutive one-pot procedures to rapidly assemble the angular 6/5/5 tricyclic framework bearing two contiguous quaternary carbon centers at the early stage; (ii) a highly regioselective aza-Wittig reaction, Schmidt rearrangement, and selective C4-N cleavage to construct the differently sized rings of target natural products for late-stage skeletal diversification.
4. Deconstructive Syntheses of Cyclopiane Class Tetracyclic Diterpenes
The
Cyclopiane class tetracyclic diterpenes are a class of unique bioactive natural products isolated from fermentation of marine-derived entophytic fungi of the
Penicillium genus [
23,
24,
25]. Conidiogenone (
19) and conidiogenol (
20) exhibit potent conidiation-inducing activity, while conidiogenone B (
21) shows high antibacterial activities (
Figure 2). The structures of the
Cyclopiane class tetracyclic diterpenes feature a highly fused and strained tetracyclic (6/5/5/5) skeleton, 6–8 consecutive chiral centers, and four sterically hindered quaternary carbon centers. The
Cyclopiane-class tetracyclic diterpenes have attracted great attention for their unprecedented chemical structures and important biological activities. During the past decade, several elegant approaches for the total syntheses of
Cyclopiane class tetracyclic diterpenes have been reported [
26,
27,
28,
29]. The first total syntheses of three
Cyclopiane class tetracyclic diterpenes, namely, conidiogenone (
19), conidiogenol (
20), and conidiogenone B (
21), was accomplished by the Tu group in 2016 through the use of a well-designed semipinacol rearrangement as a key step to construct the requisite spirocyclic (6/5) skeleton and sterically hindered quaternary carbon center of the target molecules [
26].
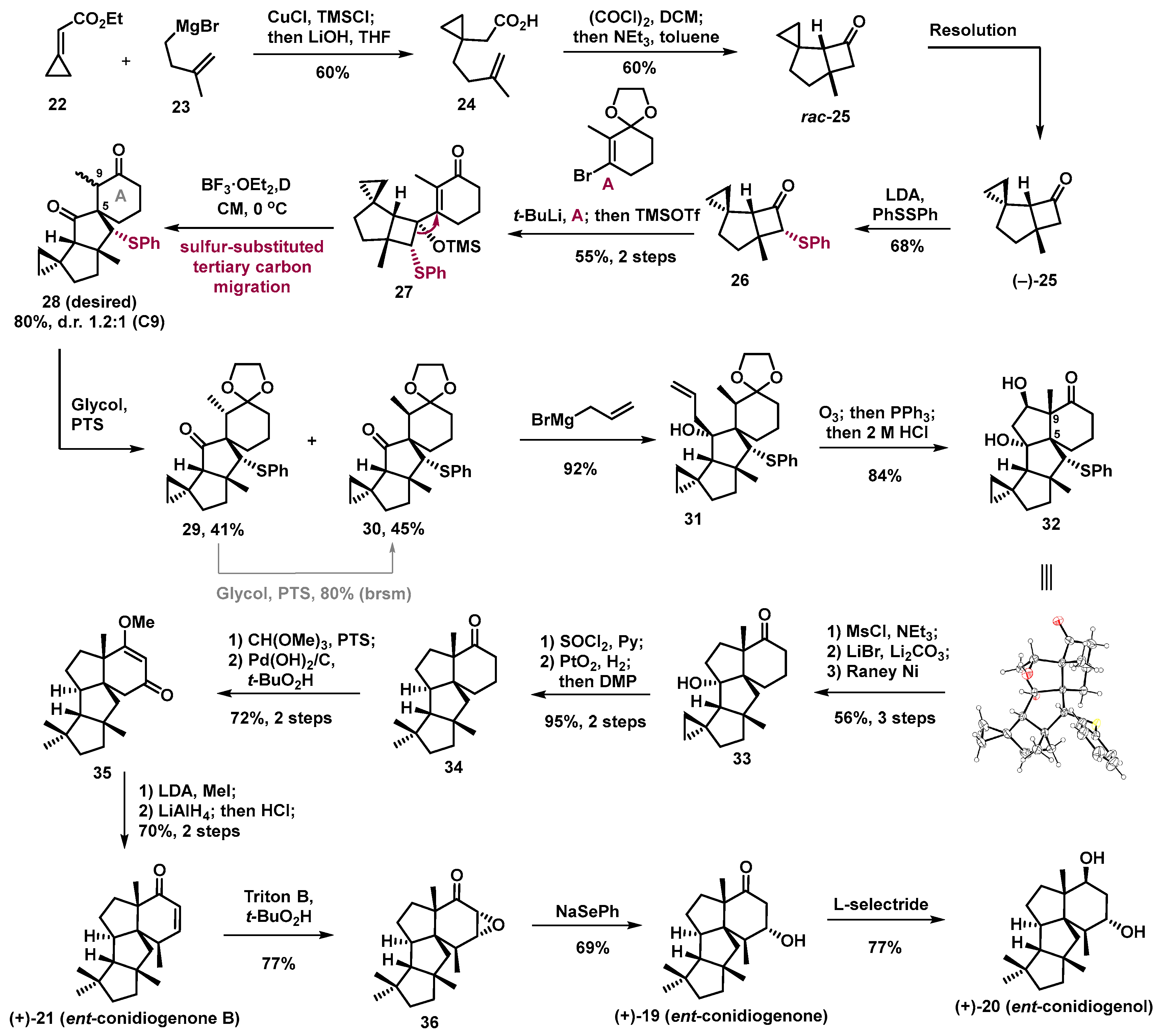
This work started with the preparation of enantioenriched cyclobutanone
25 that was obtained via chiral resolution of racemic
25, which was prepared in two steps from simple building blocks
22 and
23 via 1, 4-addition, and [2 + 2] cyclization (
Scheme 6). The well-designed vinyl cyclobutnol silylether precursor
27 was successfully synthesized by coupling known vinyl bromide
A and phenylthiol-cyclcobutanone
26, which was obtained from enantioenriched
25 and phenyl disulphide in the presence of LDA. The phenylthioether group was firstly introduced to adjust the electron density of expected carbon in semipinacol rearrangement, aiming to enhance a dominated migration of the expected carbon of the rearrangement precursor
27. Fortunately, under the BF
3·OEt
2/DCM condition, the expected semipinacol rearrangement occurred and provided the desired spirocyclic product
28 bearing the correct quaternary carbon center with an 80% yield and with a ratio of 1.2:1 at the C9 position. The structure and stereocenters of
28 were confirmed via X-ray crystallographic analysis of its derivative
32 (
vide infra). After serval chemical operations, the last five-membered ring of the target molecule was successfully constructed via acid-promoted Aldol cyclization of aldehyde, which was obtained from terminal olefin
31 through Ozonization. After removal of the unnecessary functional group on the triquinane (5/5/5 tricyclic) motif, tetracycle
35 was obtained with a high yield. Then, stereoselective installation of a methyl group in the presence of LDA and LiAlH
4 reduction of the resulting ketone, followed by quenching with HCl aqueous solution to install the enone motif, provided conidiogenone B (
21) with a 70% yield. However, the positive optical rotation and CD spectrum data of synthetic conidiogenone B were opposite to those of the natural occurring one. Thus, the correct absolute configuration of naturally occurring conidiogenone B (
21) is, in fact, the enantiomer of the originally assigned absolute configuration. Meanwhile, stereoselective epoxidation of conidiogenone B, followed by reduction of the resulting epoxide with NaSePh, afforded conidiogenone (
19) with a 54% yield over two steps. Finally, diastereoselective reduction of conidiogenone (
19) with L-selectride gave another diterpene conidiogenol (
20) with a 77% yield.
In this work, the Tu group achieved the first total synthesis of the cyclopiane class tetracyclic diterpene conidiogenone B and its transformation into conidiogenone and conidiogenol by installing a SPh “directing” group on the expected migratory carbon of the precursor to perform regio- and diastereo-selective semipinacol-type rearrangement. The absolute configuration of naturally occurring conidiogenone B (21) is also corrected through this synthesis. More importantly, the 6/5/5 tricyclic ring system of the target molecule was rapidly constructed via well-designed semipinacol rearrangement in one step, which not only constructed a crowded ring system bearing a quaternary carbon center but also reserved versatile functional groups for the introduction of the last ring and vicinal quaternary carbon center (C9), making the synthetic route more pretrial and efficient.
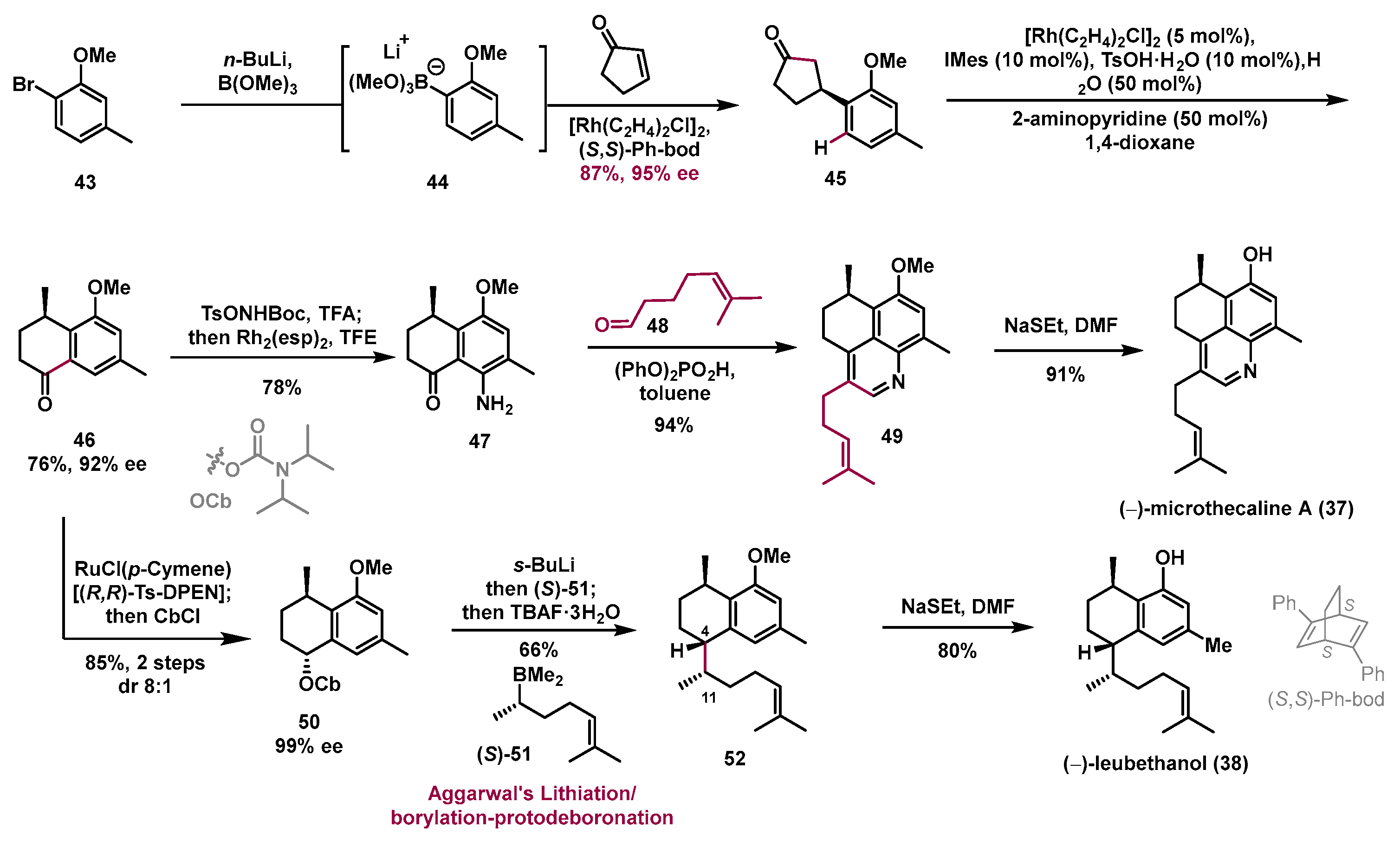
5. Asymmetric Total Syntheses of Serrulatane and Amphilectane Diterpenoids
Serrulatane and amphilectane diterpenoids are a class of polycyclic natural products that exhibit broad biological activities, such as
anti-malarial,
anti-tuberculosis, and
anti-bacterial properties. (
Figure 3). (−)-Microthecaline A (
37) is a unique nitrogen-containing serrulatane-type diterpenoid that exhibits
anti-malarial activity against
Plasmodium falciparum (IC
50 = 7.7 μM) [
30]. (−)-Leubethanol (
38) is a serrulatane diterpene isolated from the roots of
Leucophyllum frutescens with important
anti-tuberculosis activity (MIC 6.25–12.50 μg/mL) [
31]. Because of their wide-ranging biological activities and challenging structures, synthesis of serrulatane and amphilectane diterpenoids have attracted great attention from synthetic chemists, and several total syntheses have been accomplished in the past decade. In 2020, the Dong group developed a divergent and innovative approach to quickly access diverse serrulatane diterpenoids, namely, (−)-microthecaline A (
37), (−)-leubethanol (
38), (+)-
seco-pseudopteroxazole (
39), amphilectane diterpenoids (+)-pseudopteroxazole (
40), (+)-pseudopterosin G–J aglycone (
41), and (−)-pseudopterosin A–F aglycone (
42), by using a Rhodium catalytic C–C/C–H cascade reaction to construct the common skeleton, α-tetralone [
32].
In this work, asymmetric Rhodium-catalyzed 1,4-addition was applied to the preparation of the enantioenriched 3-arylcyclopentanone
45, which was the precursor of the C–C activation reaction (
Scheme 7). Compound
45 could be conveniently accessed with a 87% yield with 95% ee through asymmetric 1,4-addition of 2-cyclopentenone with fresh prepared arylborate salt
44, which was obtained from commercially available 1-bromo-2-methoxy-4-methylbenzene
43 and B(OMe)
3 in the presence of
n-BuLi. Under the previously reported C–C/C–H activation conditions, the desired α-tetralone
46 was obtained with a 76% yield and 92% ee. Aniline
47 was isolated with a 79% yield from
46 through Kurti/Falck’s Rhodium-catalyzed amination. The quinoline
49 was prepared via Friedländer condensation of aniline
47 and aldehyde
48 in the presence of (PhO)
2PO
2H with an excellent yield. Finally, removal of the methyl group by NaSEt provided (−)-microthecaline A (
37) with a 91% yield. Meanwhile, α-tetralone
46 could also convert into carbamate
50 through Noyori asymmetric reduction and carbamoylation of the resulting alcohol. Notably, under Aggarwal’s lithiation-borylation reaction condition, compound
52 was obtained with a 66% yield by coupling the freshly prepared chiral organoborane
51 with carbamate
50. Finally, removal of the methyl group with NaSEt gave (−)-leubethanol (
38) with an 80% yield.
Using the synthetic route of preparation of compound
52, the common advanced intermediate
58 was obtained from aryl boronic acid
53, 2-cyclopentenone, and chiral organoborane
51 in five steps with a 30% overall yield (
Scheme 8). Then, a direct cross-dehydrogenative-coupling (CDC) of
58 was realized under
o-chloranil/MeCN conditions to afford the desired tricyclic cyclization products
59 and
60 with a 40% combined yield with a ratio of 1.4:1. Finally, (+)-
seco-pseudopteroxazole (
39) and (+)-pseudopteroxazole (
40) were synthesized from
58 and
59 via deprotection and one-pot oxidative oxazole formation, respectively. Pseudopterosin G–J aglycone (
41) and (−)-pseudopterosin A–F aglycone (
42) were prepared from
59 and
60 through deprotection, respectively.
In this work, the use of catalytic C–C/C–H activation of 3-arylcyclopentanones as a key step has been illustrated in the enantioselective total synthesis of a range of diterpenoids, namely, (−)-microthecaline A (37, five steps), (−)-leubethanol (38, six steps), (+)-seco-pseudopteroxazole (39, seven steps), (+)-pseudopteroxazole (40, eight steps), (+)-pseudopterosin G–J aglycone (41, eight steps), and (−)-pseudopterosin A–F aglycone (42, eight steps). This strategy can accelerate asymmetric construction of the poly-substituted tetrahydronaphthalene cores, therefore significantly simplifying the overall synthesis. This is a nice example of the design of common advanced intermediates for divergent synthesis of two classes of bioactive natural products through a deconstruction strategy. With the power of the new synthetic methodology and strategy, the synthetic approach to these diterpenoids is significantly shorter than that in previous work, which could remarkably accelerate the investigation of their potential as drug candidates in drug discovery and development.
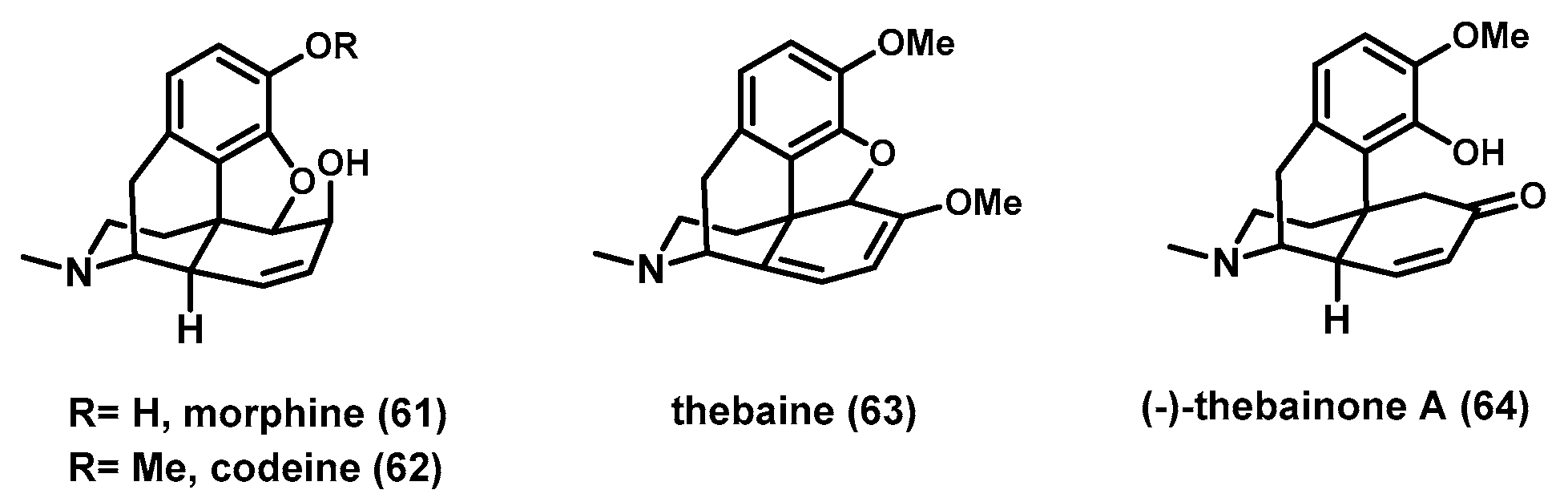
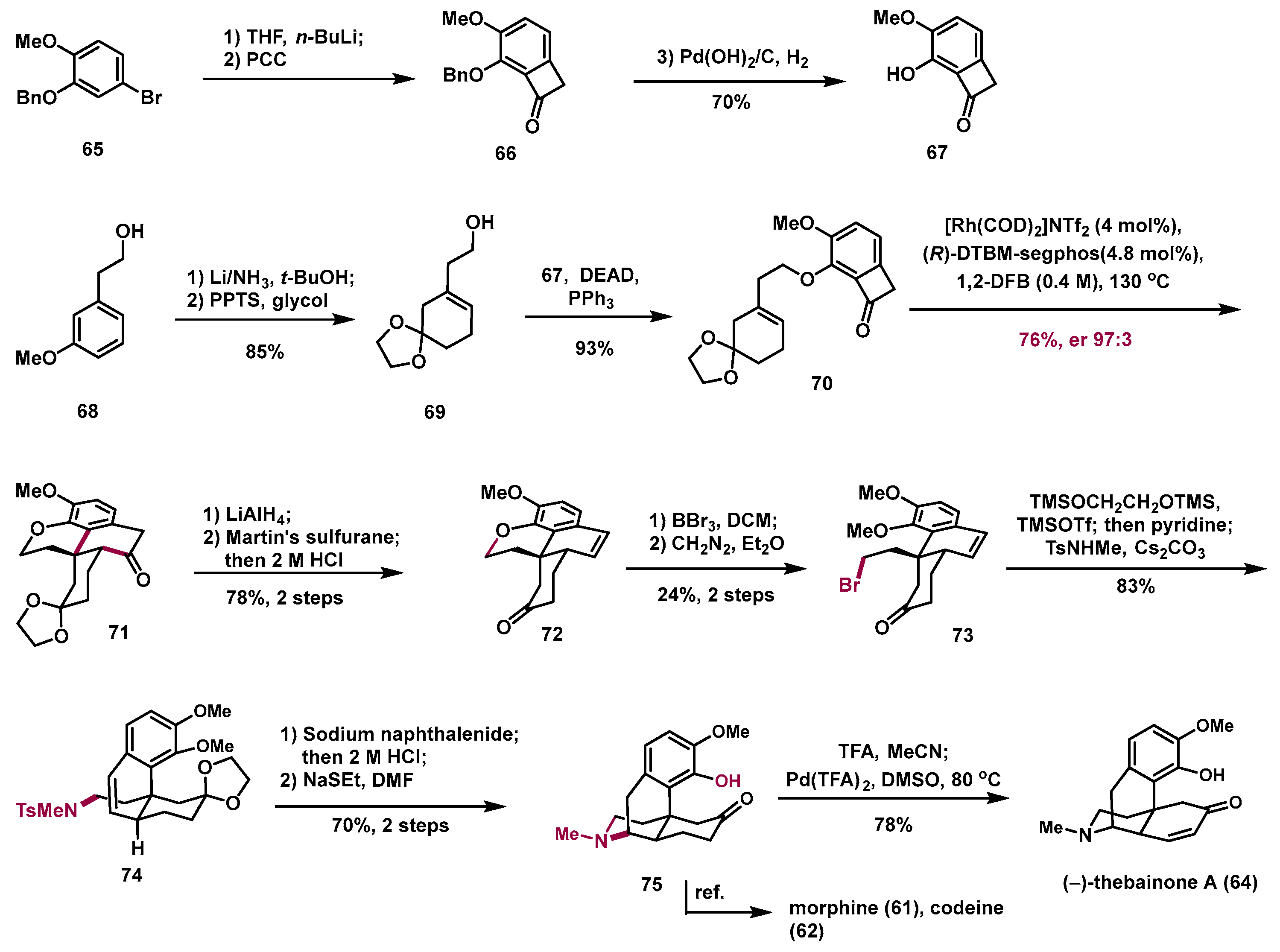
6. Deconstructive Synthesis of Morphine Alkaloid (−)-Thebainone A
Morphine and codeine have attracted great attention for their powerful biological activity and medical applications (
Figure 4). Modification of the morphine alkaloids is still an active field in drug discovery. Therefore, developing a new asymmetric synthetic route of morphine alkaloids and their analogues is highly desirable for exploring their potential utilities in drug discovery and development. In 2021, the Dong group reported a concise enantioselective deconstructive synthesis of the morphine alkaloid thebainone A for the first time, as well as formal synthesis of codeine and morphine from commercially available materials [
2,
33]. The high efficiency of the synthetic strategy is enabled by an asymmetric Rh-catalyzed C–C activation reaction (cut-and-sew) to access the all-carbon fused-rings structure and quaternary carbon center.
According to the previously synthetic route, benzocyclobutenone
67 was prepared from commercially available compound
65 in three steps on a decagram scale with a 70% overall yield, while alcohol
69 was accessed through Birch reduction and ketal formation of commercially available anisole
68 (
Scheme 9). Mitsunobu coupling of phenol
67 and alcohol
69 delivered the desired precursor
70 with a 93% yield. Because of the acid-sensitive ketal, sterically hindered trisubstituted olefin, and relatively long linker of compound
70, the efficiency of C–C activation is a challenge. After screening a series of C–C activation reaction conditions, we successfully realized Rh-catalyzed enantioselective C–C activation and obtained the desired tetracyclic compound
71 with a 76% yield with 97:3 er on a gram scale. Notably, compound
71 contains the all-carbon fused 6/6/6 rings bearing the quaternary carbon centers of the target molecules. This step not only sets the requested stereochemistry at the C13 and C14 positions but also forms all the C−C bonds present in the natural products. Compound
71 transformed into styrene
72 with a 78% yield via LAH-reduction, elimination, and deprotection. Cleavage of the C−O bond with BBr
3, followed by methylation of the resulting diphenol provided the desired alkyl bromide
73. Then, a one-pot sequence of ketal installation and SN2 amination of alkyl bromide smoothly delivered sulfonamide
78, which could provide the common intermediate
75 through formal hydroamination under a sodium naphthalenide condition to construct the piperidine and selectively deprotect the more sterically hindered methyl ether in the presence of NaSEt. The common intermediate
75 could not only transform into morphine alkaloid (−)-thebainone A (
64) through desaturation under Stahl’s condition but also serve as a known precursor for the syntheses of codeine (
61) and morphine (
62) [
13].
In this work, the Dong group developed a novel strategy for the synthesis of the morphine alkaloid (−)-thebainone A (67). Key steps included (i) construction of the all-carbon 6/6/6 tricyclic skeleton bearing a quaternary carbon center through an asymmetric Rh-catalyzed C–C bond activation reaction from easily accessible benzocyclobutenone; (ii) construction of a piperidine ring from dihydropyran through C–O bond cleavage with BBr3 and C-N bond formation in the presence of sodium naphthalenide. This creative approach is an excellent example of the application of the deconstructive strategy in the total synthesis of natural products. Furthermore, a reoptimized catalytic C–C bond activation condition was also discovered, with good substrate scope and potential application in the synthesis of other polycyclic natural products.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}