
A New Perspective on SPME and SPME Arrow: Formaldehyde Determination by On-Sample Derivatization Coupled with Multiple and Cooling-Assisted Extractions

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Method Optimization
2.2. SPME Absorption by PDMS Coating
2.3. Cooling-Assisted SPME
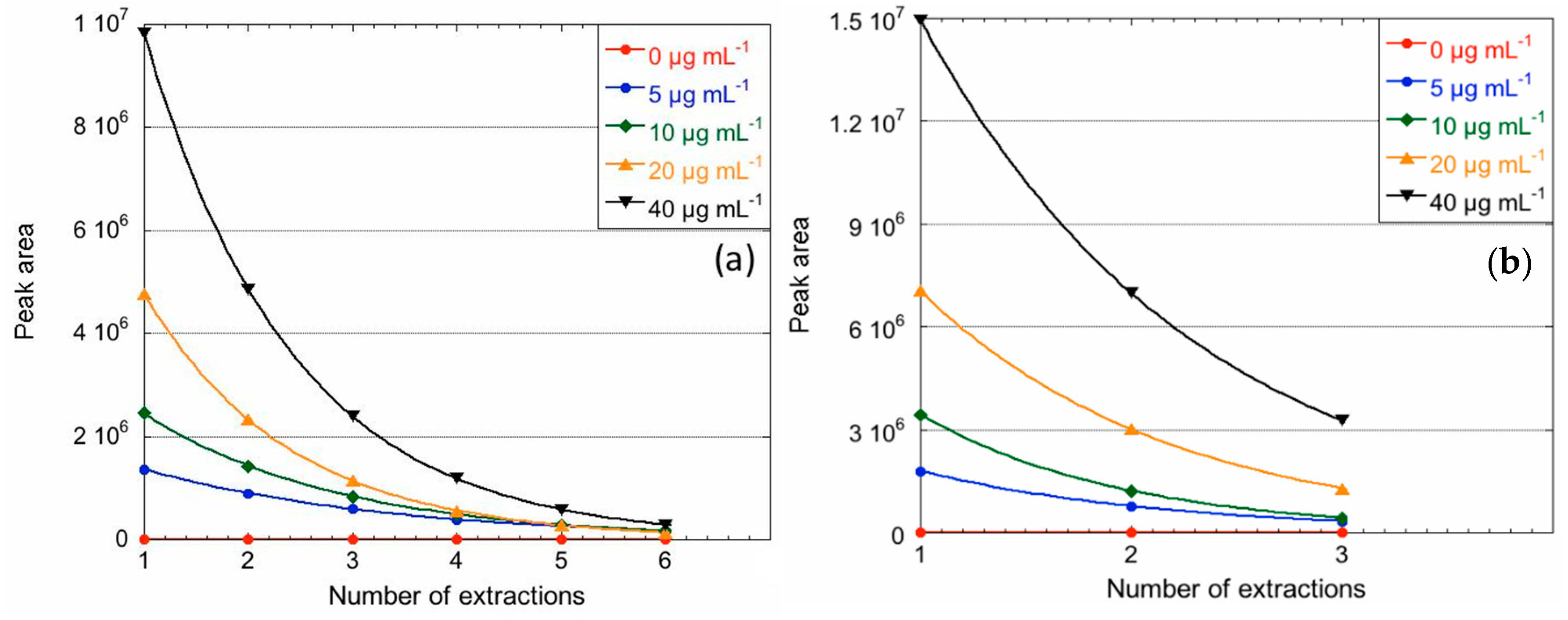
2.4. MSPME Extraction
2.5. Real Samples Analysis
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Samples
3.3. PFBHA On-Sample Derivatization Routine and Online SPME Sampling
3.4. Online Robotic System
3.5. GC-MS Operating Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Formacare. Available online: https://www.formacare.eu/ (accessed on 22 March 2023).
- Kong, L.; Li, B.; Zhao, L.; Zhang, R.; Wang, C. Density, Viscosity, Surface Tension, Excess Property and Alkyl Chain Length for 1, 4-Butanediol (1)+ 1, 2-Propanediamine (2) Mixtures. J. Mol. Liq. 2021, 326, 115107. [Google Scholar] [CrossRef]
- Dreyfors, J.M.; Jones, S.B.; Sayed, Y. Hexamethylenetetramine: A Review. Am. Ind. Hyg. Assoc. J. 1989, 50, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Ince, M.; Ince, O.K.; Ondrasek, G.; Ince, M.; Ince, O.K.; Ondrasek, G. Biochemical Toxicology: Heavy Metals and Nanomaterials; IntechOpen: London, UK, 2020; ISBN 978-1-78984-697-3. [Google Scholar]
- Dugheri, S.; Massi, D.; Mucci, N.; Berti, N.; Cappelli, G.; Arcangeli, G. Formalin Safety in Anatomic Pathology Workflow and Integrated Air Monitoring Systems for the Formaldehyde Occupational Exposure Assessment. Int. J. Occup. Med. Environ. Health 2021, 34, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Special Rapporteur on Toxics and Human Rights. Available online: https://www.ohchr.org/en/special-procedures/sr-toxics-and-human-rights (accessed on 24 March 2023).
- Nielsen, G.D.; Larsen, S.T.; Wolkoff, P. Re-Evaluation of the WHO (2010) Formaldehyde Indoor Air Quality Guideline for Cancer Risk Assessment. Arch. Toxicol. 2017, 91, 35–61. [Google Scholar] [CrossRef]
- Yu, L.; Wang, B.; Cheng, M.; Yang, M.; Gan, S.; Fan, L.; Wang, D.; Chen, W. Association between Indoor Formaldehyde Exposure and Asthma: A Systematic Review and Meta-Analysis of Observational Studies. Indoor Air 2020, 30, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, W.; Zhang, X.; Dong, T.; Zeng, H.; Fan, Q. Association between Formaldehyde Exposure and Miscarriage in Chinese Women. Medicine 2017, 96, e7146. [Google Scholar] [CrossRef]
- A Review of Human Carcinogens. Available online: https://monographs.iarc.who.int/wp-content/uploads/2018/06/mono100F.pdf (accessed on 15 April 2023).
- Formaldehyde Can Cause Rare Cancers, New EPA Analysis Finds (3). Available online: https://news.bloomberglaw.com/environment-and-energy/formaldehyde-can-cause-rare-cancers-new-epa-analysis-finds (accessed on 24 March 2023).
- 12460-3:2020(En); Wood-Based Panels—Determination of Formaldehyde Release—Part 3: Gas Analysis Method. ISO: Geneva, Switzerland, 2020.
- Branch, L.S. Consolidated Federal Laws of Canada, Formaldehyde Emissions from Composite Wood Products Regulations. Available online: https://laws-lois.justice.gc.ca/eng/regulations/SOR-2021-148/FullText.html (accessed on 24 March 2023).
- European Commission. Directorate General for Health and Food Safety. Scientific Advice on the Threshold for the Warning ‘Contains Formaldehyde’ in Annex V, Preamble Point 2 for Formaldehyde-Releasing Substances. Available online: https://data.europa.eu/doi/10.2875/269855 (accessed on 24 March 2023).
- De Groot, A.C.; White, I.R.; Flyvholm, M.-A.; Lensen, G.; Coenraads, P.-J. Formaldehyde-Releasers in Cosmetics: Relationship to Formaldehyde Contact Allergy. Contact Dermat. 2010, 62, 2–17. [Google Scholar] [CrossRef]
- Circular No. 21/2017/TT-BCT Formaldehyde, Aromatic Amines in Textile Products. Available online: https://english.luatvietnam.vn/circular-no-21-2017-tt-bct-on-promulgating-national-technical-regulations-on-content-of-117773-doc1.html (accessed on 24 March 2023).
- Piccinini, P.; Senaldi, C.; Summa, C. European Survey on the Release of Formaldehyde from Textiles. Available online: https://publications.jrc.ec.europa.eu/repository/handle/JRC36150 (accessed on 24 March 2023).
- 14184-1:2011; Textiles—Determination of Formaldehyde—Part 1: Free and Hydrolised Formaldehyde (Water Extraction Methods). ISO: Geneva, Switzerland, 2011.
- Nowshad, F.; Islam, M.N.; Khan, M.S. Concentration and Formation Behavior of Naturally Occurring Formaldehyde in Foods. Agric. Food Secur. 2018, 7, 17. [Google Scholar] [CrossRef]
- European Food Safety Authority. Endogenous Formaldehyde Turnover in Humans Compared with Exogenous Contribution from Food Sources. EFSA J. 2014, 12, 3550. [Google Scholar] [CrossRef]
- Health, C. for D. and R. Code of Federal Regulations—Title 21—Food and Drugs. Available online: https://www.fda.gov/medical-devices/medical-device-databases/code-federal-regulations-title-21-food-and-drugs (accessed on 24 March 2023).
- Perna, R.B.; Bordini, E.J.; Deinzer-Lifrak, M. A Case of Claimed Persistent Neuropsychological Sequelae of Chronic Formaldehyde Exposure: Clinical, Psychometric, and Functional Findings. Arch. Clin. Neuropsychol. Off. J. Natl. Acad. Neuropsychol. 2001, 16, 33–44. [Google Scholar] [CrossRef]
- Chemicals of High Concern to Children—Washington State Department of Ecology. Available online: https://ecology.wa.gov/Regulations-Permits/Reporting-requirements/Childrens-Safe-Products-Act-Reporting/Chemicals-of-high-concern-to-children (accessed on 29 March 2023).
- Understanding Formaldehyde in Children’s and Consumer Products. Available online: https://www.health.state.mn.us/communities/environment/childenvhealth/docs/edu_formaldehyde.pdf (accessed on 24 March 2023).
- Mo, C. Formaldehyde Regulations in the United States: An Overview. Available online: https://www.compliancegate.com/formaldehyde-regulations-united-states/ (accessed on 24 March 2023).
- Gao, P. The Exposome in the Era of One Health. Environ. Sci. Technol. 2021, 55, 2790–2799. [Google Scholar] [CrossRef] [PubMed]
- Dugheri, S.; Bonari, A.; Gentili, M.; Cappelli, G.; Pompilio, I.; Bossi, C.; Arcangeli, G.; Campagna, M.; Mucci, N. High-Throughput Analysis of Selected Urinary Hydroxy Polycyclic Aromatic Hydrocarbons by an Innovative Automated Solid-Phase Microextraction. Molecules 2018, 23, 1869. [Google Scholar] [CrossRef]
- Lin, E.Z.; Esenther, S.; Mascelloni, M.; Irfan, F.; Godri Pollitt, K.J. The Fresh Air Wristband: A Wearable Air Pollutant Sampler. Environ. Sci. Technol. Lett. 2020, 7, 308–314. [Google Scholar] [CrossRef]
- Dugheri, S.; Mucci, N.; Cappelli, G.; Trevisani, L.; Bonari, A.; Bucaletti, E.; Squillaci, D.; Arcangeli, G. Advanced Solid-Phase Microextraction Techniques and Related Automation: A Review of Commercially Available Technologies. J. Anal. Methods Chem. 2022, 2022, e8690569. [Google Scholar] [CrossRef]
- Poole, C.; Mester, Z.; Miró, M.; Pedersen-Bjergaard, S.; Pawliszyn, J. Glossary of Terms Used in Extraction (IUPAC Recommendations 2016). Pure Appl. Chem. 2016, 88, 517–558. [Google Scholar] [CrossRef]
- Arthur, C.L.; Pawliszyn, J. Solid Phase Microextraction with Thermal Desorption Using Fused Silica Optical Fibers. Anal. Chem. 1990, 62, 2145–2148. [Google Scholar] [CrossRef]
- Herrington, J.S.; Gómez-Ríos, G.A.; Myers, C.; Stidsen, G.; Bell, D.S. Hunting Molecules in Complex Matrices with SPME Arrows: A Review. Separations 2020, 7, 12. [Google Scholar] [CrossRef]
- SPME Arrow Tip = Less Septa Piercing Force. Available online: https://www.restek.com/it/chromablography/chromablography/spme-arrow-tip--less-septa-piercing-force/ (accessed on 24 March 2023).
- Dennenlöhr, J.; Thörner, S.; Maxminer, J.; Rettberg, N. Analysis of Selected Staling Aldehydes in Wort and Beer by GC-EI-MS/MS Using HS-SPME with On-Fiber Derivatization. J. Am. Soc. Brew. Chem. 2020, 78, 284–298. [Google Scholar] [CrossRef]
- Reyes-Garcés, N.; Gionfriddo, E.; Gómez-Ríos, G.A.; Alam, M.N.; Boyacı, E.; Bojko, B.; Singh, V.; Grandy, J.; Pawliszyn, J. Advances in Solid Phase Microextraction and Perspective on Future Directions. Anal. Chem. 2018, 90, 302–360. [Google Scholar] [CrossRef]
- Crucello, J.; Sampaio, N.M.F.M.; Junior, I.M.; Carvalho, R.M.; Gionfriddo, E.; Marriott, P.J.; Hantao, L.W. Automated Method Using Direct-Immersion Solid-Phase Microextraction and on-Fiber Derivatization Coupled with Comprehensive Two-Dimensional Gas Chromatography High-Resolution Mass Spectrometry for Profiling Naphthenic Acids in Produced Water. J. Chromatogr. A 2023, 1692, 463844. [Google Scholar] [CrossRef]
- Martos, P.A.; Pawliszyn, J. Sampling and Determination of Formaldehyde Using Solid-Phase Microextraction with On-Fiber Derivatization. Anal. Chem. 1998, 70, 2311–2320. [Google Scholar] [CrossRef]
- Cancilla, D.A.; Hee, S.S.Q. O-(2,3,4,5,6-Pentafluorophenyl)Methylhydroxylamine Hydrochloride: A Versatile Reagent for the Determination of Carbonyl-Containing Compounds. J. Chromatogr. A 1992, 627, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Bellugi, I.; Gambi, D.; Pacenti, M.; Dugheri, S.; Focardi, L.; Tulli, G. Compound A, Formaldehyde and Methanol Concentrations during Low-Flow Sevoflurane Anaesthesia: Comparison of Three Carbon Dioxide Absorbers. Acta Anaesthesiol. Scand. 2007, 51, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Dugheri, S.; Cappelli, G.; Fanfani, N.; Ceccarelli, J.; Trevisani, L.; Sarti, M.; Squillaci, D.; Bucaletti, E.; Gori, R.; Mucci, N.; et al. Formaldehyde Analysis by On-Fiber Derivatization: A Study of the Kinetic Models of Adsorption for Divinylbenzene. Quím. Nova 2023, in press. [Google Scholar] [CrossRef]
- Dugheri, S.; Massi, D.; Mucci, N.; Marrubini, G.; Cappelli, G.; Speltini, A.; Bonferoni, M.C.; Arcangeli, G. Exposure to Airborne Formaldehyde: Sampling and Analytical Methods—A Review. Trends Environ. Anal. Chem. 2021, 29, e00116. [Google Scholar] [CrossRef]
- Dugheri, S.; Bonari, A.; Pompilio, I.; Colpo, M.; Mucci, N.; Montalti, M.; Arcangeli, G. Development of an Innovative Gas Chromatography-Mass Spectrometry Method for Assessment of Formaldehyde in the Workplace Atmosphere. Acta Chromatogr. 2017, 29, 511–514. [Google Scholar] [CrossRef]
- Vaz, E.B.; Santos, M.F.C.; de Jesus, E.G.; Vieira, K.M.; Osório, V.M.; Menini, L. Development of Methodology for Detection of Formaldehyde-DNPH in Milk Manager by Central Composite Rotational Design and GC/MS. Res. Soc. Dev. 2022, 11, e16411931575. [Google Scholar] [CrossRef]
- Shin, H.-S.; Lim, H.-H. Simple Determination of Formaldehyde in Fermented Foods by HS-SPME-GC/MS. Int. J. Food Sci. Technol. 2012, 47, 350–356. [Google Scholar] [CrossRef]
- Kim, H.-J.; Shin, H.-S. Simple and Automatic Determination of Aldehydes and Acetone in Water by Headspace Solid-Phase Microextraction and Gas Chromatography-Mass Spectrometry. J. Sep. Sci. 2011, 34, 693–699. [Google Scholar] [CrossRef]
- Bao, M.; Joza, P.J.; Masters, A.; Rickert, W.S. Analysis of Selected Carbonyl Compounds in Tobacco Samples by Using Pentafluorobenzylhydroxylamine Derivatization and Gas Chromatography-Mass Spectrometry. Contrib. Tob. Nicotine Res. 2014, 26, 86–97. [Google Scholar] [CrossRef]
- Wardencki, W.; Orlita, J.; Namieśnik, J. Comparison of Extraction Techniques for Gas Chromatographic Determination of Volatile Carbonyl Compounds in Alcohols. Fresenius J. Anal. Chem. 2001, 369, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Hudson, E.D.; Ariya, P.A.; Gélinas, Y.; Hudson, E.D.; Ariya, P.A.; Gélinas, Y. A Method for the Simultaneous Quantification of 23 C1–C9 Trace Aldehydes and Ketones in Seawater. Environ. Chem. 2011, 8, 441–449. [Google Scholar] [CrossRef]
- Bao, M.; Pantani, F.; Griffini, O.; Burrini, D.; Santianni, D.; Barbieri, K. Determination of Carbonyl Compounds in Water by Derivatization–Solid-Phase Microextraction and Gas Chromatographic Analysis. J. Chromatogr. A 1998, 809, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Tessini, C.; Müller, N.; Mardones, C.; Meier, D.; Berg, A.; von Baer, D. Chromatographic Approaches for Determination of Low-Molecular Mass Aldehydes in Bio-Oil. J. Chromatogr. A 2012, 1219, 154–160. [Google Scholar] [CrossRef]
- Chericoni, S.; Battistini, I.; Dugheri, S.; Pacenti, M.; Giusiani, M. Novel Method for Simultaneous Aqueous in Situ Derivatization of THC and THC-COOH in Human Urine Samples: Validation and Application to Real Samples. J. Anal. Toxicol. 2011, 35, 193–198. [Google Scholar] [CrossRef]
- Pacenti, M.; Dugheri, S.; Traldi, P.; Degli Esposti, F.; Perchiazzi, N.; Franchi, E.; Calamante, M.; Kikic, I.; Alessi, P.; Bonacchi, A.; et al. New Automated and High-Throughput Quantitative Analysis of Urinary Ketones by Multifiber Exchange-Solid Phase Microextraction Coupled to Fast Gas Chromatography/Negative Chemical-Electron Ionization/Mass Spectrometry. J. Autom. Methods Manag. Chem. 2010, 2010, 972926. [Google Scholar] [CrossRef]
- Ghiasvand, A.R.; Hajipour, S.; Heidari, N. Cooling-Assisted Microextraction: Comparison of Techniques and Applications. TrAC Trends Anal. Chem. 2016, 77, 54–65. [Google Scholar] [CrossRef]
- Ghiasvand, A.; Yazdankhah, F.; Paull, B. Heating-, Cooling- and Vacuum-Assisted Solid-Phase Microextraction (HCV-SPME) for Efficient Sampling of Environmental Pollutants in Complex Matrices. Chromatographia 2020, 83, 531–540. [Google Scholar] [CrossRef]
- Koster, E.H.M.; de Jong, G.J. Multiple Solid-Phase Microextraction. J. Chromatogr. A 2000, 878, 27–33. [Google Scholar] [CrossRef]
- Tena, M.T.; Carrillo, J.D. Multiple Solid-Phase Microextraction: Theory and Applications. TrAC Trends Anal. Chem. 2007, 26, 206–214. [Google Scholar] [CrossRef]
- Schmarr, H.-G.; Sang, W.; Ganß, S.; Fischer, U.; Köpp, B.; Schulz, C.; Potouridis, T. Analysis of Aldehydes via Headspace SPME with ON-Fiber Derivatization to Their O-(2,3,4,5,6-Pentafluorobenzyl)oxime Derivatives and Comprehensive 2D-GC-MS. J. Sep. Sci. 2008, 31, 3458–3465. [Google Scholar] [CrossRef] [PubMed]
- Hudson, E.D.; Okuda, K.; Ariya, P.A. Determination of Acetone in Seawater Using Derivatization Solid-Phase Microextraction. Anal. Bioanal. Chem. 2007, 388, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-M.; Senthurchelvan, R.; Munoz, J.; Basak, S.; Rajeshwar, K.; Benglas-Smith, G.; Howell, B.C. Photolytic and Photocatalytic Destruction of Formaldehyde in Aqueous Media. J. Electrochem. Soc. 1996, 143, 1562. [Google Scholar] [CrossRef]
- Dugheri, S.; Cappelli, G.; Ceccarelli, J.; Fanfani, N.; Trevisani, L.; Squillaci, D.; Bucaletti, E.; Gori, R.; Mucci, N.; Arcangeli, G. Innovative Gas Chromatographic Determination of Formaldehyde by Miniaturized Extraction and On-Fiber Derivatization, via SPME and SPME Arrow. Quím. Nova 2022, 45, 1236–1244. [Google Scholar] [CrossRef]
- Güneş, K.; Can, Z.; Arda, A. Determination of Formaldehyde in Textile Dye and Auxiliary Chemicals with Headspace Gas Chromatography-Flame Ionization Detector. Turk. J. Chem. 2022, 46, 575–581. [Google Scholar] [CrossRef]
- Tuduri, L.; Desauziers, V.; Fanlo, J.L. Potential of Solid-Phase Microextraction Fibers for the Analysis of Volatile Organic Compounds in Air. J. Chromatogr. Sci. 2001, 39, 521–529. [Google Scholar] [CrossRef]
- Louch, D.; Motlagh, S.; Pawliszyn, J. Dynamics of Organic Compound Extraction from Water Using Liquid-Coated Fused Silica Fibers. Anal. Chem. 1992, 64, 1187–1199. [Google Scholar] [CrossRef]
- Selecting the Appropriate SPME Fiber Coating—Effect of Analyte Molecular Weight and Polarity. Available online: https://www.sigmaaldrich.com/IT/en/technical-documents/technical-article/analytical-chemistry/solid-phase-microextraction/selecting-the-appropriate (accessed on 29 March 2023).
- Pawliszyn, J. Theory of Solid-Phase Microextraction. J. Chromatogr. Sci. 2000, 38, 270–278. [Google Scholar] [CrossRef]
- Pacenti, M.; Dugheri, S.; Villanelli, F.; Bartolucci, G.; Calamai, L.; Boccalon, P.; Arcangeli, G.; Vecchione, F.; Alessi, P.; Kikic, I.; et al. Determination of Organic Acids in Urine by Solid-Phase Microextraction and Gas Chromatography–Ion Trap Tandem Mass Spectrometry Previous ‘in Sample’ Derivatization with Trimethyloxonium Tetrafluoroborate. Biomed. Chromatogr. 2008, 22, 1155–1163. [Google Scholar] [CrossRef]
- Moreira, N.; Araújo, A.M.; Rogerson, F.; Vasconcelos, I.; Freitas, V.D.; Pinho, P.G. de Development and Optimization of a HS-SPME-GC-MS Methodology to Quantify Volatile Carbonyl Compounds in Port Wines. Food Chem. 2019, 270, 518–526. [Google Scholar] [CrossRef]
- Xu, S.; Li, H.; Wu, H.; Xiao, L.; Dong, P.; Feng, S.; Fan, J. A Facile Cooling-Assisted Solid-Phase Microextraction Device for Solvent-Free Sampling of Polycyclic Aromatic Hydrocarbons from Soil Based on Matrix Solid-Phase Dispersion Technique. Anal. Chim. Acta 2020, 1115, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ghiasvand, A.R.; Hosseinzadeh, S.; Pawliszyn, J. New Cold-Fiber Headspace Solid-Phase Microextraction Device for Quantitative Extraction of Polycyclic Aromatic Hydrocarbons in Sediment. J. Chromatogr. A 2006, 1124, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Jia, J.; Sun, T.; Wang, Y.; Liao, L. Application of a Novel Cold Activated Carbon Fiber-Solid Phase Microextraction for Analysis of Organochlorine Pesticides in Soil. J. Environ. Sci. Health Part B 2007, 42, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Carasek, E.; Pawliszyn, J. Screening of Tropical Fruit Volatile Compounds Using Solid-Phase Microextraction (SPME) Fibers and Internally Cooled SPME Fiber. J. Agric. Food Chem. 2006, 54, 8688–8696. [Google Scholar] [CrossRef]
- Ghiasvand, A.R.; Setkova, L.; Pawliszyn, J. Determination of Flavour Profile in Iranian Fragrant Rice Samples Using Cold-Fibre SPME–GC–TOF–MS. Flavour Fragr. J. 2007, 22, 377–391. [Google Scholar] [CrossRef]
- Haddadi, S.H.; Niri, V.H.; Pawliszyn, J. Study of Desorption Kinetics of Polycyclic Aromatic Hydrocarbons (PAHs) from Solid Matrices Using Internally Cooled Coated Fiber. Anal. Chim. Acta 2009, 652, 224–230. [Google Scholar] [CrossRef]
- Banitaba, M.H.; Hosseiny Davarani, S.S.; Kazemi Movahed, S. Comparison of Direct, Headspace and Headspace Cold Fiber Modes in Solid Phase Microextraction of Polycyclic Aromatic Hydrocarbons by a New Coating Based on Poly(3,4-Ethylenedioxythiophene)/Graphene Oxide Composite. J. Chromatogr. A 2014, 1325, 23–30. [Google Scholar] [CrossRef]
- Pati, S.; Tufariello, M.; Crupi, P.; Coletta, A.; Grieco, F.; Losito, I. Quantification of Volatile Compounds in Wines by HS-SPME-GC/MS: Critical Issues and Use of Multivariate Statistics in Method Optimization. Processes 2021, 9, 662. [Google Scholar] [CrossRef]
- Hakkarainen, M. Developments in Multiple Headspace Extraction. J. Biochem. Biophys. Methods 2007, 70, 229–233. [Google Scholar] [CrossRef]
- Wercinski, S.A. Solid Phase Microextraction: A Practical Guide; CRC Press: Boca Raton, FL, USA, 1999; ISBN 978-1-4398-3238-7. [Google Scholar]
- Wang, C.-H.; Su, H.; Chou, J.-H.; Lin, J.-Y.; Huang, M.-Z.; Lee, C.-W.; Shiea, J. Multiple Solid Phase Microextraction Combined with Ambient Mass Spectrometry for Rapid and Sensitive Detection of Trace Chemical Compounds in Aqueous Solution. Anal. Chim. Acta 2020, 1107, 101–106. [Google Scholar] [CrossRef]
- Pérez-Olivero, S.J.; Pérez-Pont, M.L.; Conde, J.E.; Pérez-Trujillo, J.P. Determination of Lactones in Wines by Headspace Solid-Phase Microextraction and Gas Chromatography Coupled with Mass Spectrometry. J. Anal. Methods Chem. 2014, 2014, e863019. [Google Scholar] [CrossRef] [PubMed]
- Ezquerro, Ó.; Pons, B.; Tena, M.T. Multiple Headspace Solid-Phase Microextraction for the Quantitative Determination of Volatile Organic Compounds in Multilayer Packagings. J. Chromatogr. A 2003, 999, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Cancilla, D.A.; Chou, C.-C.; Barthel, R.; Hee, S.S.Q. Characterization of the 0-(2,3,4,5,6-Pentafluorobenzyl)- Hydroxylamine Hydrochloride (PFBOA) Derivatives of Some Aliphatic Mono- and Dialdehydes and Quantitative Water Analysis of These Aldehydes. J. AOAC Int. 1992, 75, 842–853. [Google Scholar] [CrossRef]
- Ojala, M.; Kotiaho, T.; Siirilä, J.; Sihvonen, M.-L. Analysis of Aldehydes and Ketones from Beer as O-(2,3,4,5,6-Pentafluorobenzyl)hydroxylamine Derivatives. Talanta 1994, 41, 1297–1309. [Google Scholar] [CrossRef]
- Feher, I.; Schmutzer, G.; Voica, C.; Moldovan, Z. Determination of Formaldehyde in Romanian Cosmetic Products Using Coupled GC/MS System after SPME Extraction. AIP Conf. Proc. 2013, 1565, 294–297. [Google Scholar] [CrossRef]
- Tang, X.; Bai, Y.; Duong, A.; Smith, M.T.; Li, L.; Zhang, L. Formaldehyde in China: Production, Consumption, Exposure Levels, and Health Effects. Environ. Int. 2009, 35, 1210–1224. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PFBHA Solution Concentration (mg/mL) | PFBHA Solution Volume Used (µL) | Derivatization Target | Sample Volume (mL) | Sample Matrix | Reference |
|---|---|---|---|---|---|
| 12 | 50 | Acetone | 10 | Seawater | [58] |
| 12 | 100 | Aliphatic aldehydes C1 to C9 (from formaldehyde to nonanal), acetone, acrolein, butanone, furfural, benzaldehyde, methylglyoxal, glyoxal, 2,4-pentane dione | 20 | Seawater | [48] |
| 2 | 1000 | Formaldehyde, acetaldehyde, acetone, propionaldehyde, acrolein, isobutyraldehyde, butyraldehyde, pentanal, crotonaldehyde, isovaleraldehyde, hexanal | 9 | Alcoholic beverages | [47] |
| 6 | 40 | 23 carbonyl compounds, including C1–C10 saturated aliphatic and unsaturated aldehydes, ketones, and dialdehydes | 4.0–8.5 | Water | [49] |
| SPME | SPME Arrow | |||
|---|---|---|---|---|
| HS | DI | HS | DI | |
| Slope ± Standard Error | (8.7 ± 0.1) × 104 | (7.6 ± 0.1) × 104 | (2.37 ± 0.04) × 105 | (2.01 ± 0.08) × 105 |
| Intercept ± Standard Error | (1.0 ± 2.5) × 104 | (3.4 ± 3.0) × 104 | −(1.0 ± 0.9) × 104 | (1.7 ± 1.5) × 105 |
| Exposure time (min) | 30 | 20 | 30 | 20 |
| Mean recovery (%) | 14.8 | 14.2 | 38.3 | 37.4 |
| R2 | 0.9994 | 0.9989 | 0.9990 | 0.9962 |
| Average CV% | 9.1 | 10.3 | 8.9 | 9.7 |
| Cooling SPME | Cooling SPME Arrow | |
|---|---|---|
| Slope ± Standard Error | (2.44 ± 0.03) × 105 | (3.74 ± 0.07) × 105 |
| Intercept ± Standard Error | (2.7 ± 7.1) × 104 | −(1.5 ± 1.4) × 105 |
| Exposure temperature (°C) | 10 | 10 |
| Mean recovery (%) | 41.7 | 60.5 |
| R2 | 0.9994 | 0.9991 |
| Average CV% | 8.9 | 7.6 |
| Cooling MSPME | Cooling MSPME Arrow | |
|---|---|---|
| Slope ± Standard Error | (4.8 ± 0.1) × 105 | (6.32 ± 0.05) × 105 |
| Intercept ± Standard Error | (0.1 ± 2.0) × 105 | −(2.0 ± 1.0) × 105 |
| Mean recovery (%) | 97.4 | 96.3 |
| β | 0.56 | 0.41 |
| CV% range | 5.7–10.2 | 4.8–9.6 |
| R2 | 0.9990 | 0.9998 |
| Column | Technique | LOD | LOQ | |
|---|---|---|---|---|
| Cooling MSPME | Fused-silica 35% Ph (30 m × 0.25 mm, 0.25 μm film thickness) | GC-MS | 11 ng L−1 | 36 ng L−1 |
| Cooling MSPME Arrow | Fused-silica 35% Ph (30 m × 0.25 mm, 0.25 μm film thickness) | GC-MS | 8 ng L−1 | 26 ng L−1 |
| Bao et al. [49] | Fused-silica capillary column (30 m × 0.25 mm I.D., 0.25 μm film thickness) | SPME-GC-ECD | 20 ng L−1 | - |
| Gunes et al. [61] | Wax capillary column (30 m × 0.32 mm ID, 0.25 μm film thickness) | GC-FID | 50 µg L−1 | 167 µg L−1 |
| Hudson et al. [58] | 5%-phenyl-methylpolysi-loxane (30 m × 0.25 mm id × 0.25 μm film thickness) | SPME-GC-MS | 3.0 ng L−1 | - |
| Wardencki et al. [47] | Fused-silica containing Rtx-5 (30 m × 0.32 i.d., 3 μm film thickness) | SPME-GC-ECD | 5 ng L−1 | - |
| Matrix | Mean FA Content (mg/L) | R2 | Average CV% | CV% Range | ||||
|---|---|---|---|---|---|---|---|---|
| SPME | SPME Arrow | SPME | SPME Arrow | SPME | SPME Arrow | SPME | SPME Arrow | |
| Green apple | 11.8 | 11.2 | 0.9873 | 0.9990 | 7.5 | 6.6 | 6.8–9.8 | 5.3–8.8 |
| Plum | 9.16 | 11.4 | 0.9939 | 0.9969 | 6.2 | 5.9 | 5.3–8.5 | 5.2–9.2 |
| Tomato | 14.5 | 20.8 | 0.9841 | 0.9930 | 8.1 | 5.3 | 6.2–9.6 | 4.2–7.3 |
| Shampoo | 3.51 | 6.9 | 0.9984 | 0.9882 | 7.0 | 4.2 | 5.9–8.2 | 3.9–6.7 |
| Face-wash | 4.92 | 8.0 | 0.9901 | 0.9987 | 7.6 | 4.5 | 6.5–8.4 | 3.8–6.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugheri, S.; Cappelli, G.; Fanfani, N.; Ceccarelli, J.; Marrubini, G.; Squillaci, D.; Traversini, V.; Gori, R.; Mucci, N.; Arcangeli, G. A New Perspective on SPME and SPME Arrow: Formaldehyde Determination by On-Sample Derivatization Coupled with Multiple and Cooling-Assisted Extractions. Molecules 2023, 28, 5441. https://doi.org/10.3390/molecules28145441
Dugheri S, Cappelli G, Fanfani N, Ceccarelli J, Marrubini G, Squillaci D, Traversini V, Gori R, Mucci N, Arcangeli G. A New Perspective on SPME and SPME Arrow: Formaldehyde Determination by On-Sample Derivatization Coupled with Multiple and Cooling-Assisted Extractions. Molecules. 2023; 28(14):5441. https://doi.org/10.3390/molecules28145441
Chicago/Turabian StyleDugheri, Stefano, Giovanni Cappelli, Niccolò Fanfani, Jacopo Ceccarelli, Giorgio Marrubini, Donato Squillaci, Veronica Traversini, Riccardo Gori, Nicola Mucci, and Giulio Arcangeli. 2023. "A New Perspective on SPME and SPME Arrow: Formaldehyde Determination by On-Sample Derivatization Coupled with Multiple and Cooling-Assisted Extractions" Molecules 28, no. 14: 5441. https://doi.org/10.3390/molecules28145441
APA StyleDugheri, S., Cappelli, G., Fanfani, N., Ceccarelli, J., Marrubini, G., Squillaci, D., Traversini, V., Gori, R., Mucci, N., & Arcangeli, G. (2023). A New Perspective on SPME and SPME Arrow: Formaldehyde Determination by On-Sample Derivatization Coupled with Multiple and Cooling-Assisted Extractions. Molecules, 28(14), 5441. https://doi.org/10.3390/molecules28145441