
Emission Quenching in Tetraphenylfuran Crystal: Why This Propeller-Shaped Molecule Does Not Emit in the Condensed Phase


Abstract
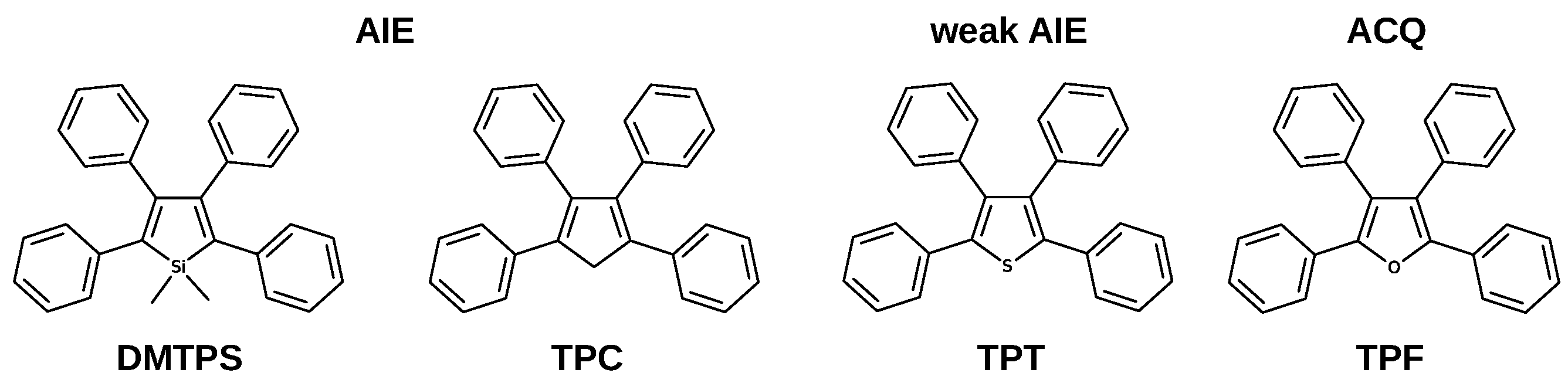
:1. Introduction
2. Computational Details
3. Results and Discussion
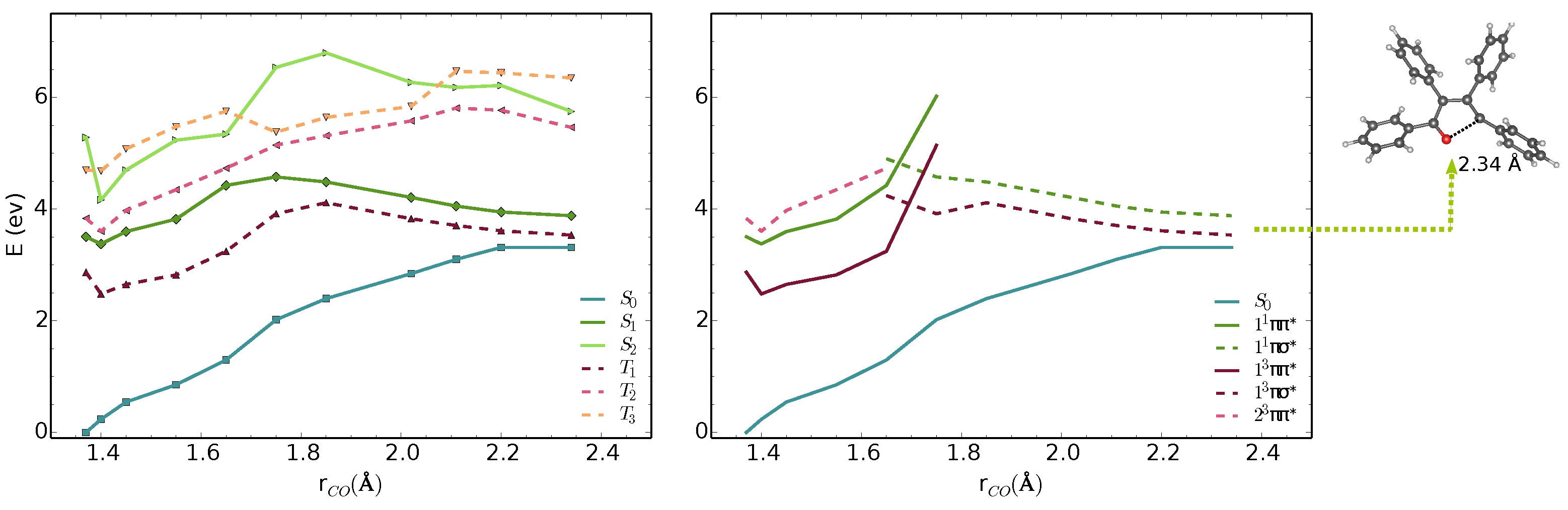
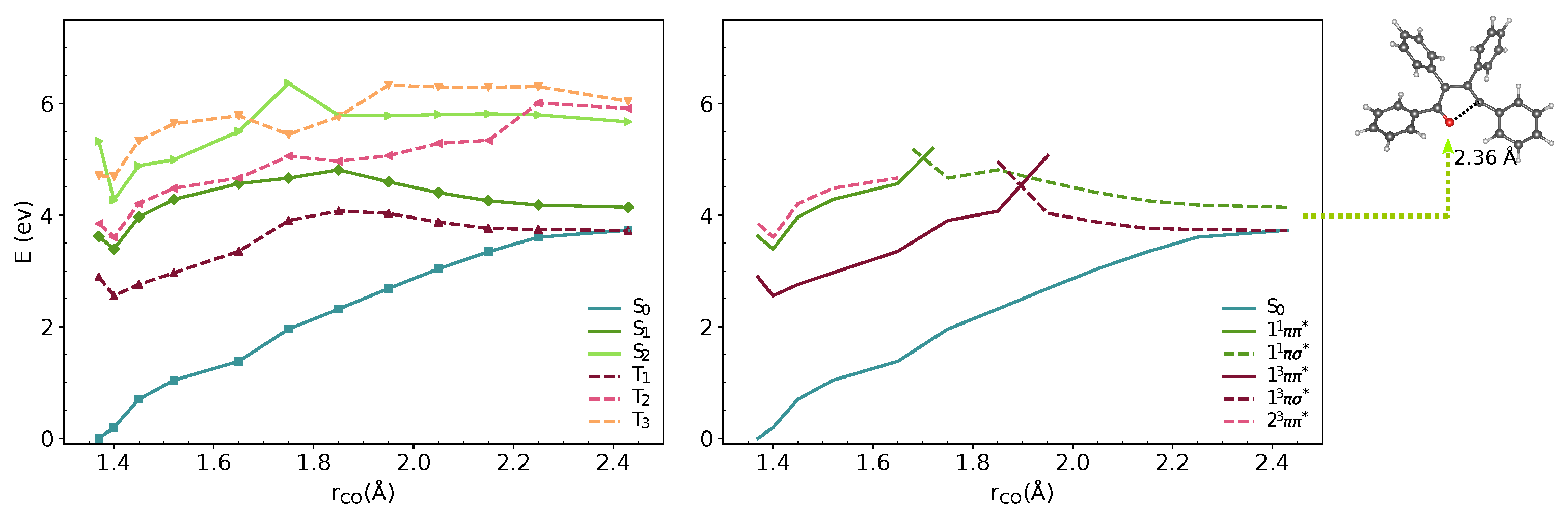
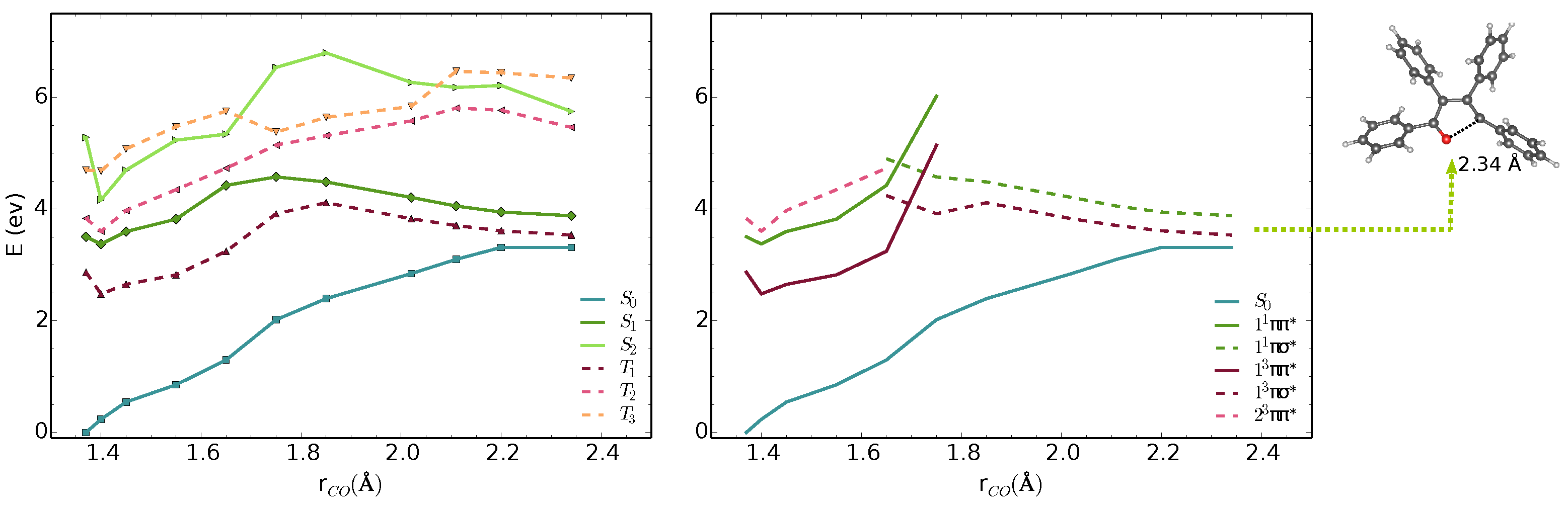
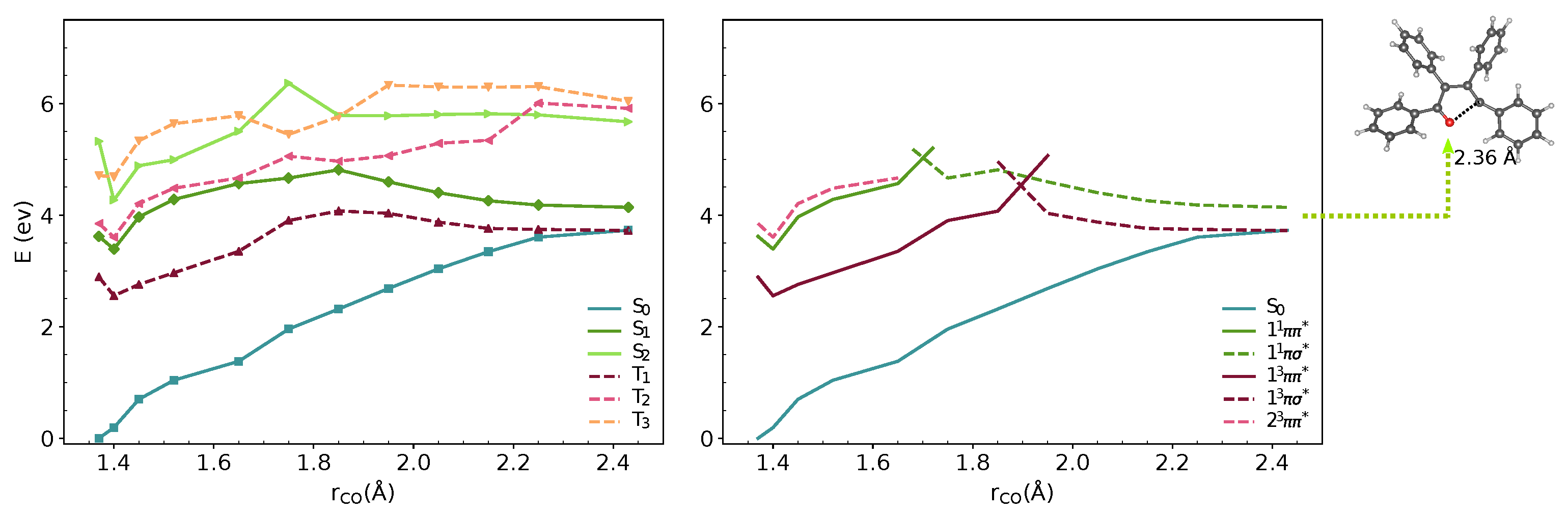
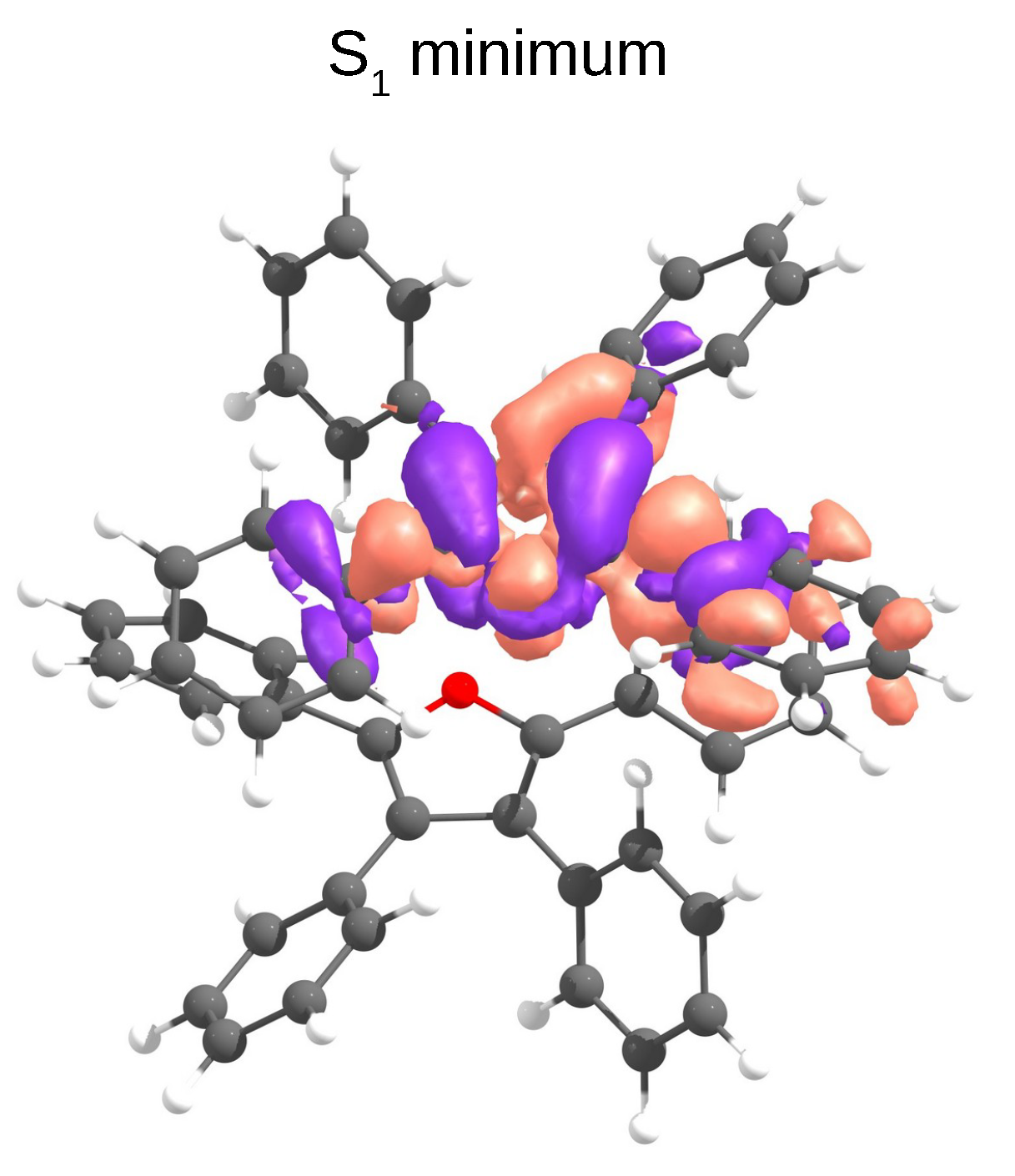
3.1. Vertical Excitations and Radiative Mechanisms
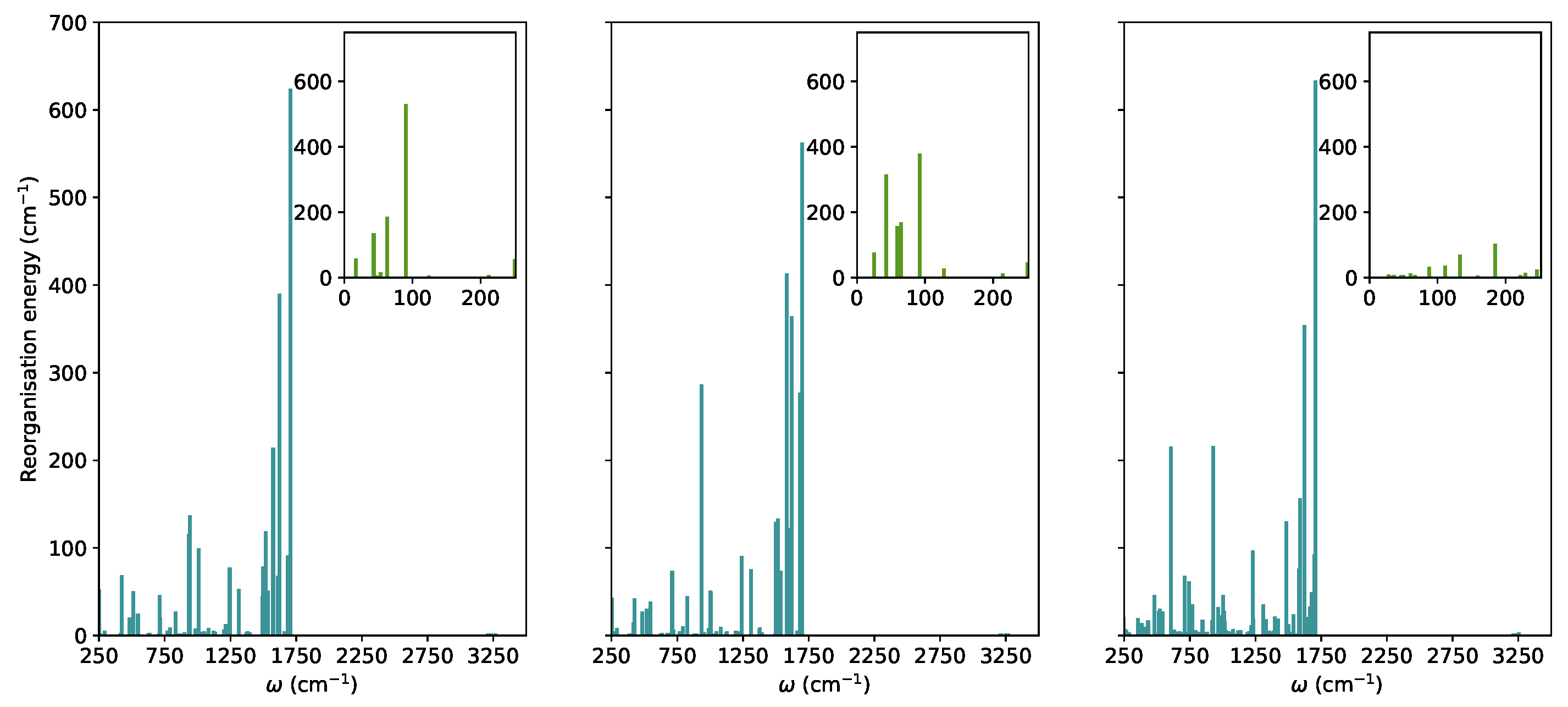
3.2. Nonradiative Relaxation Mechanisms
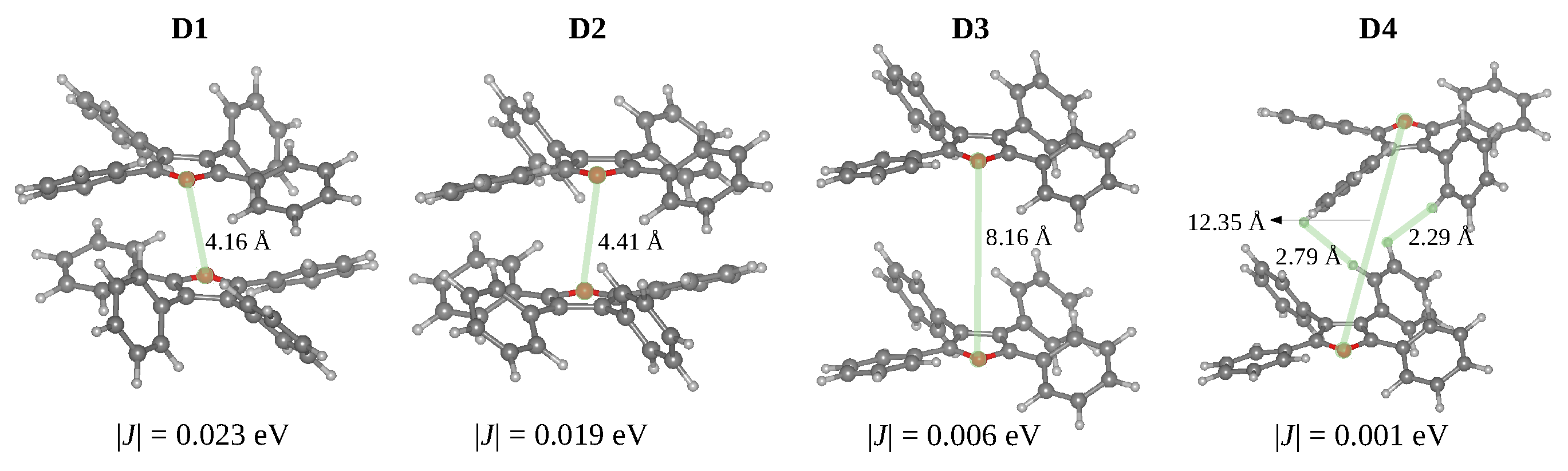
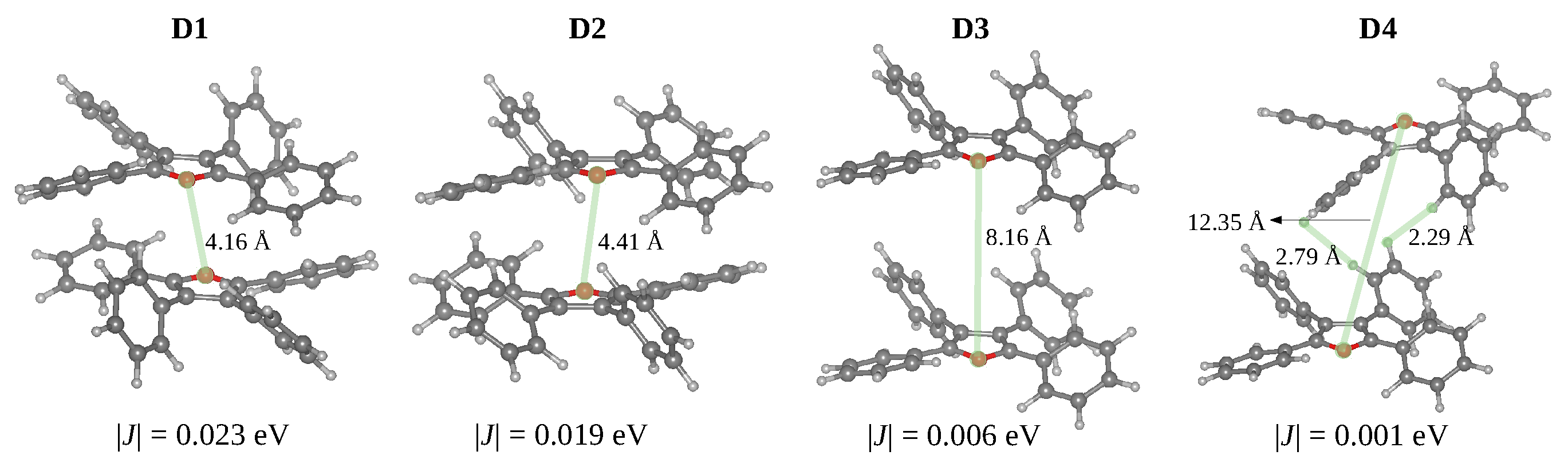
3.3. Crystal Structure: Intermolecular Interactions and Exciton Transport
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, J.; Aguilar Suarez, L.E.; Yoon, S.J.; Varghese, S.; Serpa, C.; Park, S.Y.; Lüer, L.; Roca-Sanjuán, D.; Milián-Medina, B.; Gierschner, J. Solid State Luminescence Enhancement in π-Conjugated Materials: Unraveling the Mechanism beyond the Framework of AIE/AIEE. J. Phys. Chem. C 2017, 121, 23166–23183. [Google Scholar] [CrossRef]
- Peng, X.L.; Ruiz-Barragan, S.; Li, Z.S.; Li, Q.S.; Blancafort, L. Restricted Access to a Conical Intersection to Explain Aggregation Induced Emission in Dimethyl Tetraphenylsilole. J. Mater. Chem. C 2016, 4, 2802–2810. [Google Scholar] [CrossRef]
- Crespo-Otero, R.; Li, Q.; Blancafort, L. Exploring Potential Energy Surfaces for Aggregation-Induced Emission—From Solution to Crystal. Chem. Asian J. 2019, 14, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Zhuang, Z.; Zhao, Z.; Tang, B.Z. AIEgens based on main group heterocycles. J. Mater. Chem. C 2018, 6, 11835–11852. [Google Scholar] [CrossRef]
- Dommett, M.; Rivera, M.; Crespo-Otero, R. How Inter- and Intramolecular Processes Dictate Aggregation-Induced Emission in Crystals Undergoing Excited-State Proton Transfer. J. Phys. Chem. Lett. 2017, 8, 6148–6153. [Google Scholar] [CrossRef] [PubMed]
- Dommett, M.; Rivera, M.; Smith, M.T.H.; Crespo-Otero, R. Molecular and crystalline requirements for solid state fluorescence exploiting excited state intramolecular proton transfer. J. Mater. Chem. C 2020, 8, 2558–2568. [Google Scholar] [CrossRef]
- Li, W.; Zhu, L.; Shi, Q.; Ren, J.; Peng, Q.; Shuai, Z. Excitonic coupling effect on the nonradiative decay rate in molecular aggregates: Formalism and application. Chem. Phys. Lett. 2017, 683, 507–514. [Google Scholar] [CrossRef]
- Yu, G.; Yin, S.; Liu, Y.; Chen, J.; Xu, X.; Sun, X.; Ma, D.; Zhan, X.; Peng, Q.; Shuai, Z.; et al. Structures, Electronic States, Photoluminescence, and Carrier Transport Properties of 1,1-Disubstituted 2,3,4,5-Tetraphenylsiloles. J. Am. Chem. Soc. 2005, 127, 6335–6346. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, L.; Crespo-Otero, R. Understanding Aggregation Induced Emission in a Propeller-Shaped Blue Emitter. ChemPhotoChem 2019, 3, 907–915. [Google Scholar] [CrossRef]
- Stojanović, L.; Crespo-Otero, R. Aggregation-Induced Emission in the Tetraphenylthiophene Crystal: The Role of Triplet States. J. Phys. Chem. C 2020, 124, 17752–17761. [Google Scholar] [CrossRef]
- Nie, H.; Hu, K.; Cai, Y.; Peng, Q.; Zhao, Z.; Hu, R.; Chen, J.; Su, S.J.; Qin, A.; Tang, B.Z. Tetraphenylfuran: Aggregation-induced emission or aggregation-caused quenching? Mater. Chem. Front. 2017, 1, 1125–1129. [Google Scholar] [CrossRef]
- Gu, Y.; Li, N.; Shao, G.; Wang, K.; Zou, B. Mechanism of Different Piezoresponsive Luminescence of 2,3,4,5-Tetraphenylthiophene and 2,3,4,5-Tetraphenylfuran: A Strategy for Designing Pressure-Induced Emission Enhancement Materials. J. Phys. Chem. Lett. 2020, 11, 678–682. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Liu, M.; Yang, Q.; Chen, Y. From Tetraphenylfurans to Ring-Opened (Z)-1,4-Enediones: ACQ Fluorophores versus AIEgens with Distinct Responses to Mechanical Force and Light. Chem. Eur. J. 2018, 24, 13197–13204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, X.; Zhang, P.; Huang, X.; Zheng, X.; Chen, M.; Feng, H.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Ionization and Anion-π+ Interaction: A New Strategy for Structural Design of Aggregation-Induced Emission Luminogens. J. Am. Chem. Soc. 2017, 139, 16974–16979. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Stratmann, E.R.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Caillie, C.V.; Amos, R.D. Geometric derivatives of excitation energies using SCF and DFT. Chem. Phys. Lett. 1999, 308, 249–255. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Christiansen, O.; Koch, H.; Jørgensen, P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. [Google Scholar] [CrossRef]
- Hättig, C.; Köhn, A. Transition moments and excited-state first-order properties in the coupled-cluster model CC2 using the resolution-of-the-identity approximation. J. Chem. Phys. 2002, 117, 6939–6951. [Google Scholar] [CrossRef]
- Hättig, C. Geometry optimizations with the coupled-cluster model CC2 using the resolution-of-the-identity approximation. J. Chem. Phys. 2003, 118, 7751–7761. [Google Scholar] [CrossRef]
- Köhn, A.; Hättig, C. Analytic gradients for excited states in the coupled-cluster model CC2 employing the resolution-of-the-identity approximation. J. Chem. Phys. 2003, 119, 5021–5036. [Google Scholar] [CrossRef]
- Roos, B.O.; Sadlej, A.J.; Malmqvist, P.Å.; Andersson, K.; Wolinski, K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Vancoillie, S.; Delcey, M.G.; Lindh, R.; Vysotskiy, V.; Malmqvist, P.Å.; Veryazov, V. Parallelization of a multiconfigurational perturbation theory. J. Comput. Chem. 2013, 34, 1937–1948. [Google Scholar] [CrossRef]
- Roos, B.O.; Taylor, P.R.; Siegbahn, P.E.M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K.; Morokuma, K. Updated Branching Plane for Finding Conical Intersections without Coupling Derivative Vectors. J. Chem. Theory Comput. 2010, 6, 1538–1545. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fdez Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [Green Version]
- TURBOMOLE V7.0 2015, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 10 December 2021).
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395–502. [Google Scholar] [CrossRef]
- Dapprich, S.; Komaromi, I.; Suzie Byun, K.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. Theochem 1999, 461–462, 121. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, K.M.; Cornell, W.D.; Kollman, P.A.; Ferguson, D.M.; Cieplak, P.; Caldwell, J.W.; Bayly, C.I.; Gould, I.R.; Spellmeyer, D.C.; Fox, T. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 2005, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Rivera, M.; Dommett, M.; Crespo-Otero, R. ONIOM(QM:QM’) Electrostatic Embedding Schemes for Photochemistry in Molecular Crystals. J. Chem. Theory Comput. 2019, 15, 2504–2516. [Google Scholar] [CrossRef]
- Rivera, M.; Dommett, M.; Sidat, A.; Rahim, W.; Crespo-Otero, R. fromage: A library for the study of molecular crystal excited states at the aggregate scale. J. Comput. Chem. 2020. [Google Scholar] [CrossRef]
- Aradi, B.; Hourahine, B.; Frauenheim, T. DFTB+, a Sparse Matrix-Based Implementation of the DFTB Method. J. Phys. Chem. A 2007, 111, 5678–5684. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Levich, V.G.; Dogonadze, R.R. Theory of non-radiation electron transitions from ion to ion in solutions. Dokl. Akad. Nauk SSSR 1959, 124, 123–126. [Google Scholar]
- Levich, V.G. Present state of the theory of oxidation-reduction in solution (bulk and electrode reactions). Adv. Electrochem. Electrochem. Eng. 1966, 4, 249–371. [Google Scholar]
- Jortner, J. Temperature dependent activation energy for electron transfer between biological molecules. J. Chem. Phys. 1976, 64, 4860–4867. [Google Scholar] [CrossRef]
- Reimers, J.R. A practical method for the use of curvilinear coordinates in calculations of normal-mode-projected displacements and duschinsky rotation matrices for large molecules. J. Chem. Phys. 2001, 115, 9103–9109. [Google Scholar] [CrossRef]
- Aragó, J.; Troisi, A. Dynamics of the excitonic coupling in organic crystals. Phys. Rev. Lett. 2015, 114, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Fornari, R.P.; Aragó, J.; Troisi, A. Exciton Dynamics in Phthalocyanine Molecular Crystals. J. Phys. Chem. C 2016, 120, 7987–7996. [Google Scholar] [CrossRef] [Green Version]
- Aragó, J.; Troisi, A. Regimes of Exciton Transport in Molecular Crystals in the Presence of Dynamic Disorder. Adv. Funct. Mater. 2015, 26, 2316–2325. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy (eV) | ||||
|---|---|---|---|---|
| Vacuum/Solution | Crystal | |||
| Absorption | Emission | Absorption | Emission | |
| RI-CC2/aug-cc-pVDZ | 4.10 (0.58) | 3.45 (0.71) | - | - |
| TD-B3LYP/6-31G(d) | 3.72 (0.47) | 3.16 (0.50) | 3.72 (0.41) | 3.19 (0.43) |
| TD-B97X-D/6-31G(d) | 4.26 (0.51) | 3.47 (0.60) | 4.26 (0.49) | 3.49 (0.52) |
| TD-B97X-D/6-31G(d)/PCM | 4.19 (0.63) | 3.19 (0.89) | - | - |
| MS-2-CASPT2/6-31G(d) | 3.50 | 3.14 | 3.61 | 3.20 |
| Experimental [11] | 3.79 | 3.24 | - | 3.24 |
| Solution | Crystal | |
|---|---|---|
| Experimental [11] | ||
| 0.40 | 0.01 | |
| 0.68 | 0.06 | |
| 5.88 | 0.67 | |
| 8.84 | 1.66 | |
| Predicted | ||
| 3.93 | 3.89 | |
| 1.1 | 0.7 |
| Structure | State | E | f | |J| |
|---|---|---|---|---|
| Monomer | () | 3.9434 | 0.72 | - |
| D1 | () | 3.9077 | 0.00 | 0.023 |
| () | 3.9544 | 1.30 | ||
| D2 | () | 3.9209 | 0.00 | 0.019 |
| () | 3.9605 | 1.31 | ||
| D3 | () | 3.9377 | 0.00 | 0.006 |
| () | 3.9495 | 1.40 | ||
| D4 | () | 3.9422 | 0.00 | 0.001 |
| () | 3.9437 | 1.42 |
| Crystal | ||||
|---|---|---|---|---|
| TPF | 0.70 | 0.023 | 1.42 | 1.17 |
| TPT | 1.00 | 0.015 | 1.35 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojanović, L.; Crespo-Otero, R. Emission Quenching in Tetraphenylfuran Crystal: Why This Propeller-Shaped Molecule Does Not Emit in the Condensed Phase. Molecules 2022, 27, 522. https://doi.org/10.3390/molecules27020522
Stojanović L, Crespo-Otero R. Emission Quenching in Tetraphenylfuran Crystal: Why This Propeller-Shaped Molecule Does Not Emit in the Condensed Phase. Molecules. 2022; 27(2):522. https://doi.org/10.3390/molecules27020522
Chicago/Turabian StyleStojanović, Ljiljana, and Rachel Crespo-Otero. 2022. "Emission Quenching in Tetraphenylfuran Crystal: Why This Propeller-Shaped Molecule Does Not Emit in the Condensed Phase" Molecules 27, no. 2: 522. https://doi.org/10.3390/molecules27020522
APA StyleStojanović, L., & Crespo-Otero, R. (2022). Emission Quenching in Tetraphenylfuran Crystal: Why This Propeller-Shaped Molecule Does Not Emit in the Condensed Phase. Molecules, 27(2), 522. https://doi.org/10.3390/molecules27020522