
Low-Symmetry Phthalocyanines Bearing Carboxy-Groups: Synthesis, Spectroscopic and Quantum-Chemical Characterization

 , , ,
, , ,  and
and
Abstract
:1. Introduction
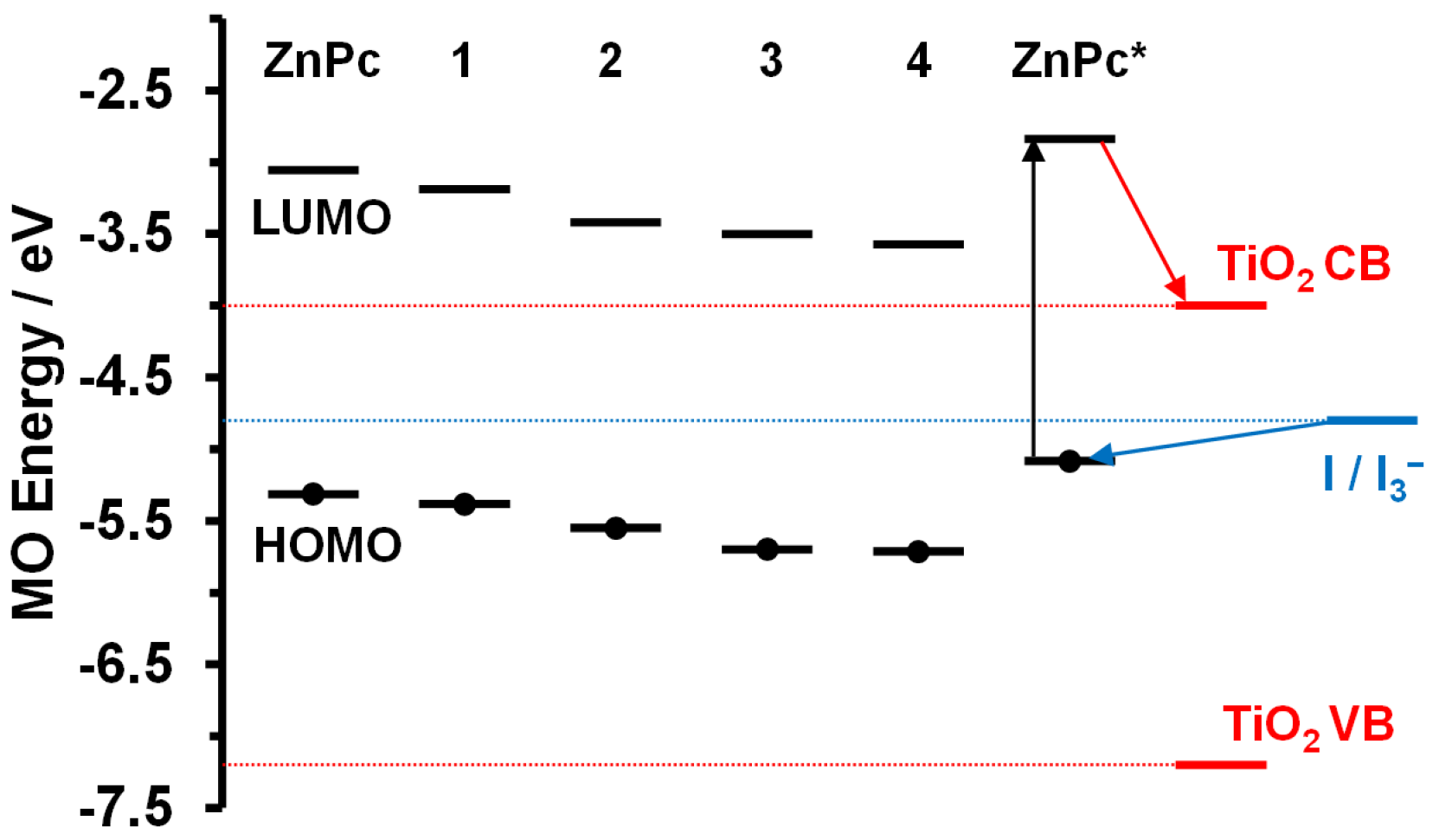
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.3. Computational Details
4.4. Synthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability: Samples of ZnPc1−4 are available from the authors. |
References
- Lukyanets, E.A.; Nemykin, V.N. The key role of peripheral substituents in the chemistry of phthalocyanines and their analogs. J. Porphyr. Phthalocyanines 2010, 14, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Nyokong, T.; Ahsen, V. (Eds.) Photosensitizers in Medicine, Environment, and Security; Springer: Dordrecht, The Netherlands, 2012; ISBN 978-90-481-3870-8. [Google Scholar]
- Koifman, O.I.; Ageeva, T.A.; Beletskaya, I.P.; Averin, A.D.; Yakushev, A.A.; Tomilova, L.G.; Dubinina, T.V.; Tsivadze, A.Y.; Gorbunova, Y.G.; Martynov, A.G.; et al. Macroheterocyclic Compound-a Key Building Block in New Functional Materials and Molecular Devices. Macroheterocycles 2020, 13, 311–467. [Google Scholar] [CrossRef]
- Mack, J.; Kobayashi, N. Low symmetry phthalocyanines and their analogues. Chem. Rev. 2011, 111, 281–321. [Google Scholar] [CrossRef] [PubMed]
- Urbani, M.; Ragoussi, M.E.; Nazeeruddin, M.K.; Torres, T.; Khaja, M. Phthalocyanines for dye-sensitized solar cells. Coord. Chem. Rev. 2019, 381, 1–64. [Google Scholar] [CrossRef]
- Cid, J.J.; Yum, J.H.; Jang, S.R.; Nazeeruddin, M.K.; Martínez-Ferrero, E.; Palomares, E.; Ko, J.; Grätzel, M.; Torres, T. Molecular cosensitization for efficient panchromatic dye-sensitized solar cells. Angew. Chem. Int. Ed. 2007, 46, 8358–8362. [Google Scholar] [CrossRef]
- Tortelli, S.; Cavazzini, M.; Orlandi, S.; Pozzi, G.; Pecnikaj, I.; Caramori, S.; Boaretto, R. Property tuning in unsymmetrical alkoxy zinc phthalocyanines by introduction of perfluoro-tert-butoxy end groups. J. Fluor. Chem. 2016, 188, 110–116. [Google Scholar] [CrossRef]
- Ikeuchi, T.; Mori, S.; Kobayashi, N.; Kimura, M. Low-Symmetrical Zinc(II) Benzonaphthoporphyrazine Sensitizers for Light-Harvesting in Near-IR Region of Dye-Sensitized Solar Cells. Inorg. Chem. 2016, 55, 5014–5018. [Google Scholar] [CrossRef]
- Ikeuchi, T.; Nomoto, H.; Masaki, N.; Griffith, M.J.; Mori, S.; Kimura, M. Molecular engineering of zinc phthalocyanine sensitizers for efficient dye-sensitized solar cells. Chem. Commun. 2014, 50, 1941. [Google Scholar] [CrossRef]
- Kimura, M.; Nomoto, H.; Suzuki, H.; Ikeuchi, T.; Matsuzaki, H.; Murakami, T.N.; Furube, A.; Masaki, N.; Griffith, M.J.; Mori, S. Molecular Design Rule of Phthalocyanine Dyes for Highly Efficient Near-IR Performance in Dye-Sensitized Solar Cells. Chem.-A Eur. J. 2013, 19, 7496–7502. [Google Scholar] [CrossRef]
- Virkki, K.; Hakola, H.; Urbani, M.; Tejerina, L.; Ince, M.; Martínez-Díaz, M.V.; Torres, T.; Golovanova, V.; Golovanov, V.; Tkachenko, N.V. Photoinduced Electron Injection from Zinc Phthalocyanines into Zinc Oxide Nanorods: Aggregation Effects. J. Phys. Chem. C 2017, 121, 9594–9605. [Google Scholar] [CrossRef]
- Nombona, N.; Antunes, E.; Litwinski, C.; Nyokong, T. Synthesis and photophysical studies of phthalocyanine-gold nanoparticle conjugates. Dalton Trans. 2011, 40, 11876. [Google Scholar] [CrossRef]
- Ballesteros, B.; Campidelli, S.; de la Torre, G.; Ehli, C.; Guldi, D.M.; Prato, M.; Torres, T. Synthesis, characterization and photophysical properties of a SWNT-phthalocyanine hybrid. Chem. Commun. 2007, 2950. [Google Scholar] [CrossRef]
- Yang, L.; Guo, L.; Chen, Q.; Sun, H.; Liu, J.; Zhang, X.; Pan, X.; Dai, S. Theoretical design and screening of panchromatic phthalocyanine sensitizers derived from TT1 for dye-sensitized solar cells. J. Mol. Graph. Model. 2012, 34, 1–9. [Google Scholar] [CrossRef]
- Milan, R.; Selopal, G.S.; Cavazzini, M.; Orlandi, S.; Boaretto, R.; Caramori, S.; Concina, I.; Pozzi, G. Dye-sensitized solar cells based on a push-pull zinc phthalocyanine bearing diphenylamine donor groups: Computational predictions face experimental reality. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, R.; Singh Selopal, G.; Cavazzini, M.; Orlandi, S.; Boaretto, R.; Caramori, S.; Concina, I.; Pozzi, G. Zinc phthalocyanines as light harvesters for SnO2-based solar cells: A case study. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, J.; Bunya, M.; Shimizu, Y.; Uoyama, H.; Komobuchi, N.; Okujima, T.; Uno, H.; Ito, S.; Stillman, M.J.; Ono, N.; et al. Application of MCD spectroscopy and TD-DFT to nonplanar core-modified tetrabenzoporphyrins: Effect of reduced symmetry on nonplanar porphyrinoids. Chem. Eur. J. 2008, 14, 5001–5020. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, T.; Kobayashi, N. Spectroscopic and structural properties of phthalocyanines deduced from their frontier molecular orbitals (MOs) and MO calculations. Coord. Chem. Rev. 2017, 345, 31–41. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Hadt, R.G.; Belosludov, R.V.; Mizuseki, H.; Kawazoe, Y. Influence of Molecular Geometry, Exchange-Correlation Functional, and Solvent Effects in the Modeling of Vertical Excitation Energies in Phthalocyanines Using Time-Dependent Density Functional Theory (TDDFT) and Polarized Continuum Model TDDFT Methods: Ca. J. Phys. Chem. A 2007, 111, 12901–12913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludov, R.V.; Nevonen, D.; Rhoda, H.M.; Sabin, J.R.; Nemykin, V.N. Simultaneous Prediction of the Energies of Q x and Q y Bands and Intramolecular Charge-Transfer Transitions in Benzoannulated and Non-Peripherally Substituted Metal-Free Phthalocyanines and Their Analogues: No Standard TDDFT Silver Bullet Yet. J. Phys. Chem. A 2019, 123, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040–1041, 45–53. [Google Scholar] [CrossRef]
- Martynov, A.G.; Mack, J.; May, A.K.; Nyokong, T.; Gorbunova, Y.G.; Tsivadze, A.Y. Methodological Survey of Simplified TD-DFT Methods for Fast and Accurate Interpretation of UV-Vis-NIR Spectra of Phthalocyanines. ACS Omega 2019, 4, 7265–7284. [Google Scholar] [CrossRef] [PubMed]
- Safonova, E.A.; Martynov, A.G.; Polovkova, M.A.; Ugolkova, E.A.; Minin, V.V.; Gorbunova, Y.G.; Tsivadze, A.Y. 5,8-Disubstituted crown-naphthalonitriles as a platform for highly soluble naphthalocyanines. Dye. Pigment. 2020, 180, 108484. [Google Scholar] [CrossRef]
- Yagodin, A.V.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y. Synthesis, electronic structure and NH-tautomerism of novel mono- and dibenzoannelated phthalocyanines. Dye. Pigment. 2020, 181, 108564. [Google Scholar] [CrossRef]
- May, A.; Majumdar, P.; Martynov, A.G.; Lapkina, L.A.; Troyanov, S.I.; Gorbunova, Y.G.; Tsivadze, A.Y.; Mack, J.; Nyokong, T. Optical limiting properties, structure and simplified TD-DFT calculations of scandium tetra-15-crown-5 phthalocyaninates. J. Porphyr. Phthalocyanines 2020, 24, 589–601. [Google Scholar] [CrossRef]
- Tejerina, L.; Martínez-Díaz, M.V.; Torres, T. Convergent Strategy for the Regioselective Synthesis of Nonaggregated α-Triaryl-β-carboxy Zinc Phthalocyanines. Org. Lett. 2015, 17, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Listorti, A.; López-Duarte, I.; Martínez-Díaz, M.V.; Torres, T.; DosSantos, T.; Barnes, P.R.F.; Durrant, J.R. Zn(ii) versus Ru(ii) phthalocyanine-sensitised solar cells. A comparison between singlet and triplet electron injectors. Energy Environ. Sci. 2010, 3, 1573–1579. [Google Scholar] [CrossRef]
- Aktas, E.; Jiménez-López, J.; Azizi, K.; Torres, T.; Palomares, E. Self-assembled Zn phthalocyanine as a robust p-type selective contact in perovskite solar cells. Nanoscale Horiz. 2020, 5, 1415–1419. [Google Scholar] [CrossRef]
- Gouterman, M. Optical Spectra and Electronic Structure of Porphyrins and Related Rings. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume III, pp. 1–165. [Google Scholar]
- Michl, J. Magnetic circular dichroism of aromatic molecules. Tetrahedron 1984, 40, 3845–3934. [Google Scholar] [CrossRef]
- Schaffer, A.M.; Gouterman, M.; Davidson, E.R. Porphyrins XXVIII. Extended Hückel calculations on metal phthalocyanines and tetrazaporphins. Theor. Chim. Acta 1973, 30, 9–30. [Google Scholar] [CrossRef]
- McHugh, A.J.; Gouterman, M.; Weiss, C. Porphyrins XXIV. Energy, oscillator strength, and Zeeman splitting calculations (SCMO-CI) for phthalocyanine, porphyrins, and related ring systems. Theor. Chim. Acta 1972, 24, 346–370. [Google Scholar] [CrossRef] [Green Version]
- Mack, J.; Wildervanck, M.; Nyokong, T. TD-DFT calculations and MCD spectroscopy of porphyrin and phthalocyanine analogues: Rational design of photosensitizers for PDT and NIR region sensor applications. Turk. J. Chem. 2014, 38, 1013–1026. [Google Scholar] [CrossRef]
- Nyokong, T.; Gasyna, Z.; Stillman, M.J. Analysis of the absorption and magnetic circular dichroism spectra of zinc phthalocyanine and the. pi.-cation-radical species [ZnPc(1−)].cntdot.+. Inorg. Chem. 1987, 26, 1087–1095. [Google Scholar] [CrossRef]
- Ough, E.; Nyokong, T.; Creber, K.A.M.; Stillman, M.J. Electrochemistry and Spectroscopy of Magnesium Phthalocyanine. Analysis of the Absorption and Magnetic Circular Dichroism Spectra. Inorg. Chem. 1988, 27, 2724–2732. [Google Scholar] [CrossRef]
- Stillman, M.J.; Nyokong, T. Absorption and Magnetic Circular Dichroism Spectral Properties of Phthalocyanines. Part 1: Complexes of the Dianion, Pc(-2). In Phthalocyanines: Properties and Applications 1; Leznoff, C.C., Lever, A.B.P., Eds.; VCH: Weinheim, Germany, 1989; pp. 133–289. [Google Scholar]
- Mack, J.; Stillman, M.J. Electronic Structures of Metal Phthalocyanine and Porphyrin Complexes from Analysis of the UV–Visible Absorption and Magnetic Circular Dichroism Spectra and Molecular Orbital Calculations. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: New York, NY, USA, 2003; pp. 43–116. [Google Scholar]
- Mack, J.; Stillman, M.J.; Kobayashi, N. Application of MCD spectroscopy to porphyrinoids. Coord. Chem. Rev. 2007, 251, 429–453. [Google Scholar] [CrossRef]
- Kobayashi, N.; Nakai, K. Applications of magnetic circular dichroism spectroscopy to porphyrins and phthalocyanines. Chem. Commun. 2007, 4077. [Google Scholar] [CrossRef]
- Isago, H. Optical Spectra of Phthalocyanines and Related Compounds A Guide for Beginners; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 978-4-431-55101-0. [Google Scholar]
- Mack, J.; Asano, Y.; Kobayashi, N.; Stillman, M.J. Application of MCD spectroscopy and TD-DFT to a highly non-planar porphyrinoid ring system. New insights on red-shifted porphyrinoid spectral bands. J. Am. Chem. Soc. 2005, 127, 17697–17711. [Google Scholar] [CrossRef] [PubMed]
- Chemcraft-Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 12 December 2021).
- Chemissian 4.65 Program (by L. Skripnikov). Available online: https://www.chemissian.com/ (accessed on 12 December 2021).
- Mack, J. Expanded, Contracted, and Isomeric Porphyrins: Theoretical Aspects. Chem. Rev. 2017, 117, 3444–3478. [Google Scholar] [CrossRef]
- Lu, H.; Mack, J.; Yang, Y.; Shen, Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasseen, F.A.; Al-Temimei, F.A. Electronic structures and photovoltaic properties of a novel phthalocyanine and titanium dioxide phthalocyanine for dye sensitized-solar cells. GSC Adv. Res. Rev. 2021, 6, 107–115. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, J.; Zheng, S. Designing Potential Donor Materials Based on DRCN5T with Halogen Substitutions: A DFT/TDDFT Study. Int. J. Mol. Sci. 2021, 22, 13498. [Google Scholar] [CrossRef]
- Scharber, M.C.; Mühlbacher, D.; Koppe, M.; Denk, P.; Waldauf, C.; Heeger, A.J.; Brabec, C.J. Design Rules for Donors in Bulk-Heterojunction Solar Cells—Towards 10% Energy-Conversion Efficiency. Adv. Mater. 2006, 18, 789–794. [Google Scholar] [CrossRef]
- Bourass, M.; Benjelloun, A.T.; Benzakour, M.; Mcharfi, M.; Hamidi, M.; Bouzzine, S.M.; Bouachrine, M. DFT and TD-DFT calculation of new thienopyrazine-based small molecules for organic solar cells. Chem. Cent. J. 2016, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Wei, S.; Wu, C.-M.; Li, S.; Guo, W. Can Polypyridyl Cu(I)-based Complexes Provide Promising Sensitizers for Dye-Sensitized Solar Cells? A Theoretical Insight into Cu(I) versus Ru(II) Sensitizers. J. Phys. Chem. C 2011, 115, 3753–3761. [Google Scholar] [CrossRef]
- Juma, J.M.; Vuai, S.A.H.; Babu, N.S. TD-DFT Investigations on Optoelectronic Properties of Fluorescein Dye Derivatives in Dye-Sensitized Solar Cells (DSSCs). Int. J. Photoenergy 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Toriumi, N.; Muranaka, A.; Hirano, K.; Yoshida, K.; Hashizume, D.; Uchiyama, M. 18π-Electron Tautomeric Benziphthalocyanine: A Functional Near-Infrared Dye with Tunable Aromaticity. Angew. Chem. Int. Ed. 2014, 53, 7814–7818. [Google Scholar] [CrossRef]
- Anderson, D.R.; Solntsev, P.V.; Rhoda, H.M.; Nemykin, V.N. How big is big? Separation by conventional methods, X-ray and electronic structures of positional isomers of bis- tert -butylisocyano adduct of 2(3),9(10),16(17),23(24)-tetrachloro-3(2),10(9),17(16),24(23)-tetra(2,6-di- iso -propylphenoxy)-phthalocyaninat. J. Porphyr. Phthalocyanines 2016, 20, 337–351. [Google Scholar] [CrossRef]
- Pozzi, G.; Quici, S.; Raffo, M.C.; Bignozzi, C.A.; Caramori, S.; Orlandi, M. Fluorous molecules for dye-sensitized solar cells: Synthesis and photoelectrochemistry of unsymmetrical zinc phthalocyanine sensitizers with bulky fluorophilic donor groups. J. Phys. Chem. C 2011, 115, 3777–3788. [Google Scholar] [CrossRef]
- Shimizu, M.; Tamagawa, T. Design and Characterization of 4,5-Bis(diarylamino)phthalic Acid Diesters as a New Class of Fluorophores Exhibiting Efficient Blue Emission in the Solid State. Eur. J. Org. Chem. 2015, 2015, 291–295. [Google Scholar] [CrossRef]
- Tylleman, B.; Gómez-Aspe, R.; Gbabode, G.; Geerts, Y.H.; Sergeyev, S. Ester-functionalized phthalonitriles and zinc phthalocyanines via palladium-catalyzed cyanation of 4,5-dichlorophthalates. Tetrahedron 2008, 64, 4155–4161. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G * basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
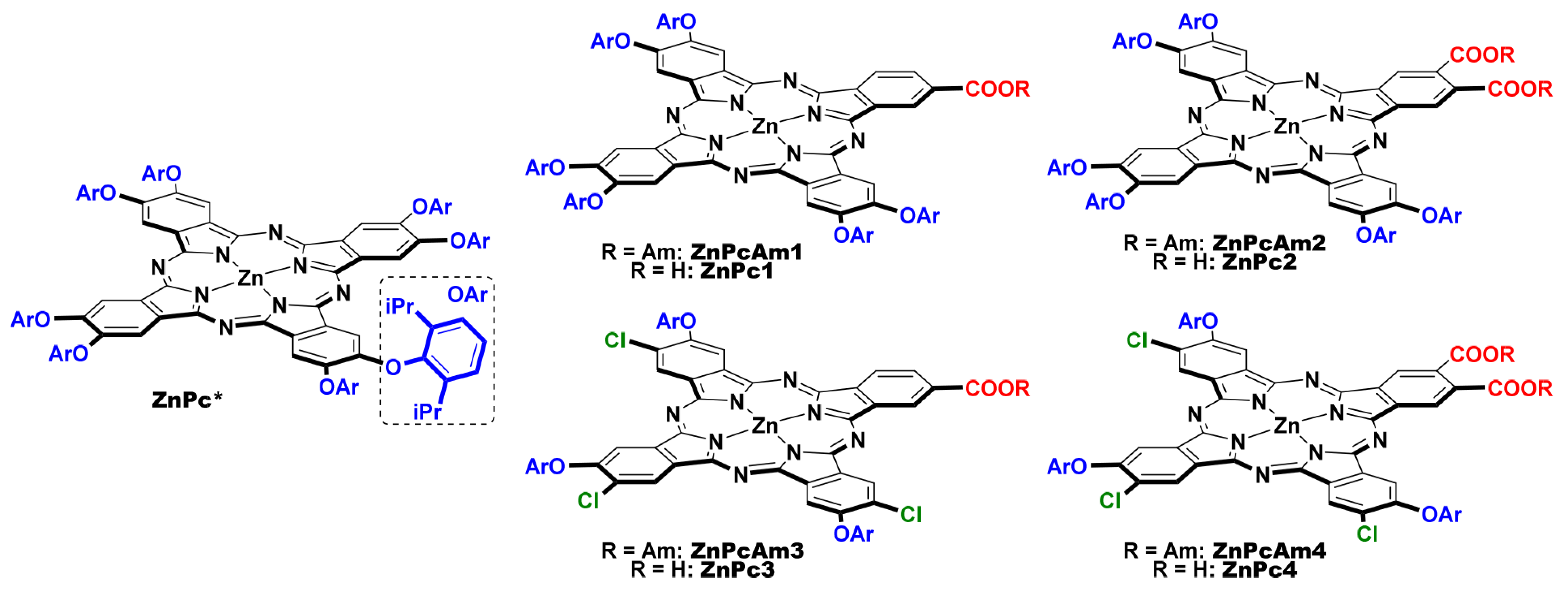
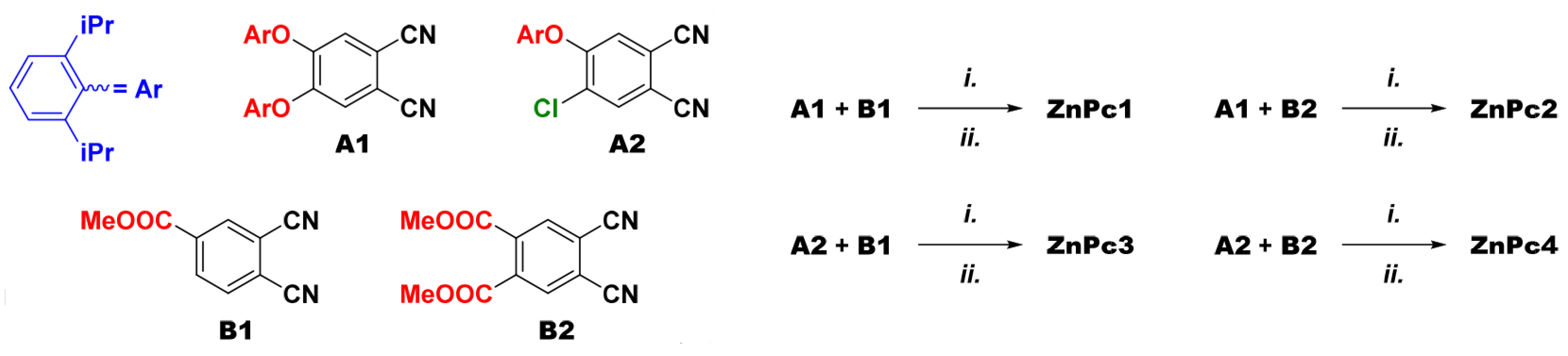
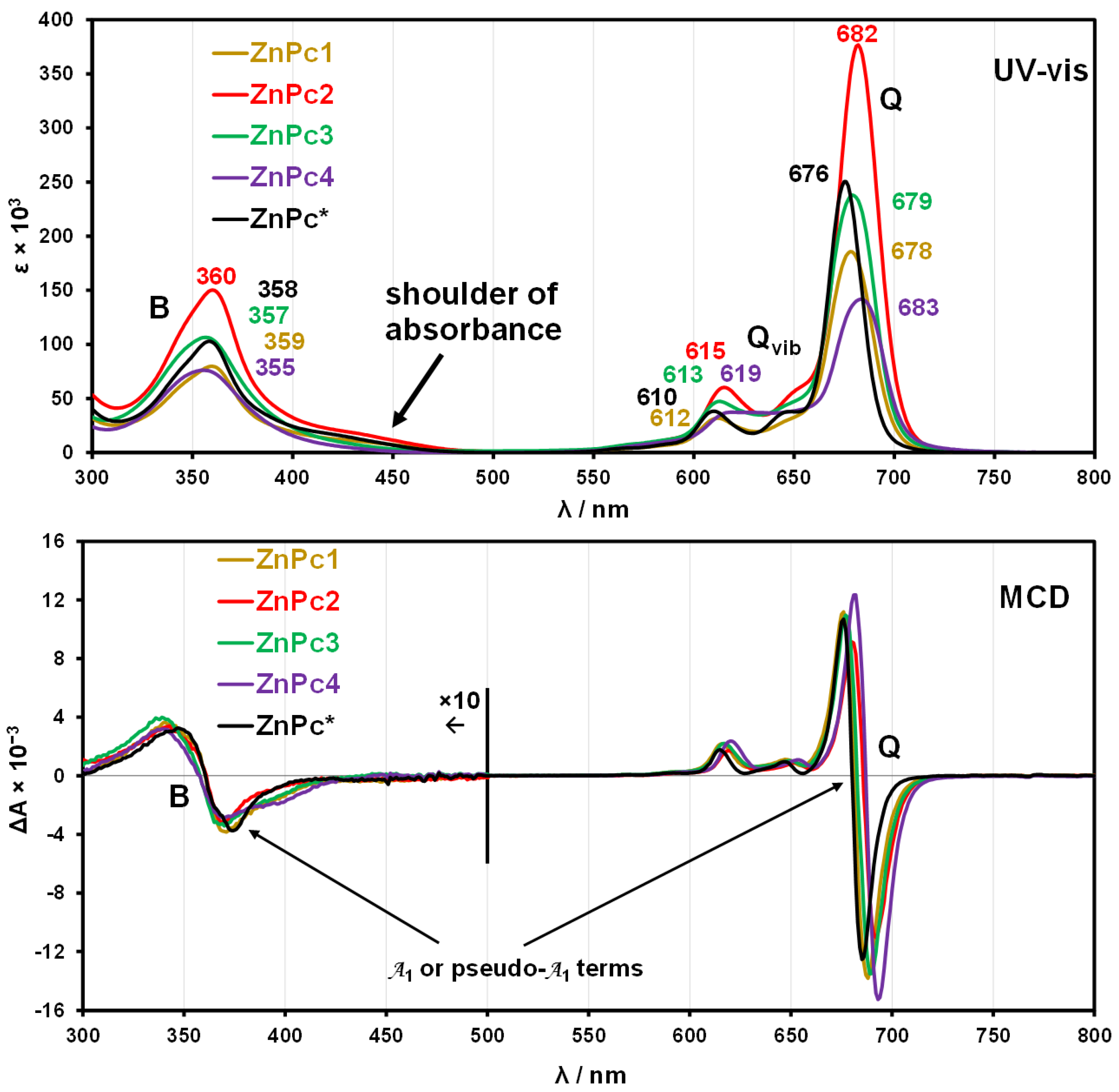

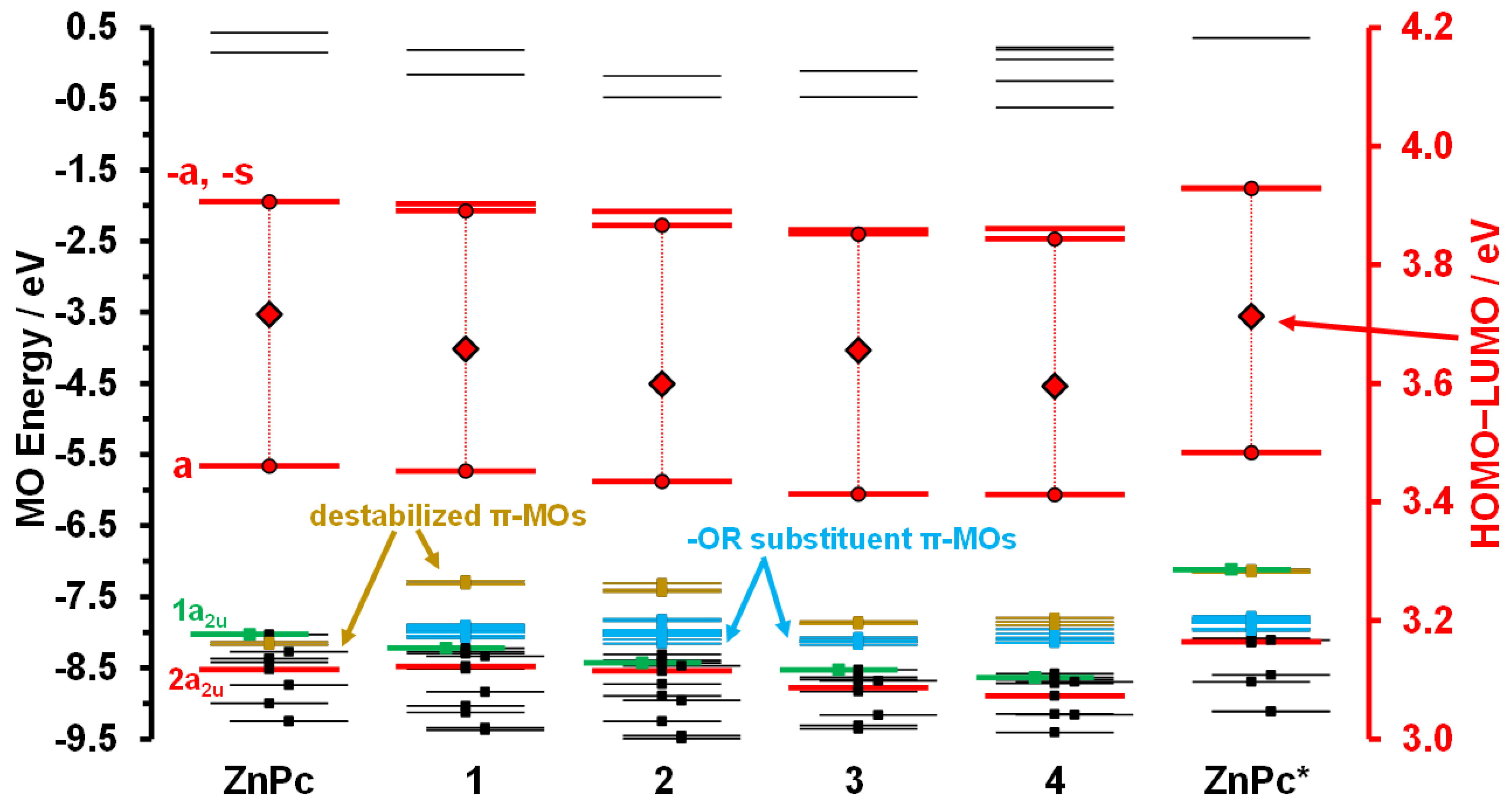


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λχαλχ, νμ (eV) | fcalc | Q Band Wavefunctions a | λχαλχ, νμ (eV) | fcalc | Wavefunctions of the Most Intense Bands Predicted in the B Band Region a | |
|---|---|---|---|---|---|---|
| ZnPc | 607 (2.04) 607 (2.04) | 0.48 0.48 | 94% a→-a; … 94% a → -s; … | 313 (3.96) | 0.02 | 70% H−2 (1b2u)Bz→-a; 20% 2a2u→-a; … |
| 313 (3.96) | 0.02 | 70% H−2 (1b2u)Bz→-s; 20% 2a2u→-s; … | ||||
| 295 (4.21) | 1.07 | 74% 1a2uBz →-a/-s; 9% 2a2u→-a/-s; 7% a→-a/-s; … | ||||
| 295 (4.21) | 1.07 | 74% 1a2uBz →-a/-s; 9% 2a2u→-a/-s; 7% a→-a/-s; … | ||||
| ZnPc* | 622 (1.99) 622 (1.99) | 0.56 0.56 | 92% a→-a; … 92% a → -s; … | 366 (3.39) | 0.04 | 73% H−1 (1b2u)Bz→-a; … |
| 366 (3.39) | 0.04 | 87% H−2 (1b2u)Bz→-a; … | ||||
| 332 (3.74) | 0.95 | 75% 1a2uBz→-a;18% H−2 (1b2u)Bz→-a; … | ||||
| 332 (3.74) | 0.95 | 72% 1a2uBz→-s;21% H−2 (1b2u)Bz→-s; … | ||||
| 295 (4.20) | 0.6 | 66% 2a2u→-s; 10% H−25 (1b1u) → -a; … | ||||
| 295 (4.20) | 0.6 | 66% 2a2u→-a; 10% H−25 (1b1u) → -s; … | ||||
| ZnPc1 | 626 (1.98) 613 (2.02) | 0.53 0.58 | 93% a→-s; … 92% a → -a; … | 372 (3.33) | 0.08 | 83% H−1Bz →-s; … |
| 346 (3.58) | 0.54 | 83% H−1Bz →-a; … | ||||
| 342 (3.63) | 0.16 | 73% H−2Bz →-a; … | ||||
| 333 (3.72) | 0.34 | 57% H−3Bz →-s; 19% a→L+2 (1b1u*); … | ||||
| 327 (3.79) | 0.11 | 69% a→L+2 (1b1u*); 20% H−3Bz→-s; … | ||||
| 311 (3.88) | 0.17 | 63% a→L+3 (1b2u*); 14% 1a2uBz→-a/-s; … | ||||
| 304 (4.08) | 0.3 | 51% 1a2uBz→-a/-s; 14% a → L+3 (1b2u*); … | ||||
| 299 (4.15) | 0.49 | 38% 2a2u→-a;15% H−17 (2eg)→-a/-s; 8% 1a2uBz→-a/-s; … | ||||
| 298 (4.17) | 0.58 | 27% 2a2u→-a/-s; 18% H−18 (1b1u) → -a/-s; 11% 1a2uBz → -s; … | ||||
| 286 (4.33) | 0.13 | 35% H−18 (1b1u)→-a; 14% H−17 (2eg)→-a/-s; 11% 1a2uBz→-s; | ||||
| ZnPc2 | 636 (1.95) 607 (2.04) | 0.57 0.56 | 94% a→-a; … 93% a → -s; … | 378 (3.28) | 0.09 | 67% H−2Bz →-a/-s; 19% H−1Bz→-s; … |
| 357 (3.47) | 0.4 | 88% H−1Bz →-a/-s; … | ||||
| 348 (3.56) | 0.17 | 85% H−2Bz →-a/-s; … | ||||
| 345 (3.62) | 0.15 | 76% a→L+3 (1b2u*); 10% H−3Bz→-s; … | ||||
| 331 (3.74) | 0.32 | 60% H−3Bz →-s; 20% a→L+3 (1b2u*); … | ||||
| 306 (4.05) | 0.71 | 34% 1a2uBz→-a; 31% H−16 (2eg) → -a/-s; … | ||||
| 303 (4.09) | 0.59 | 40% H−17 (1b1u) → -a/-s; 19% 1a2uBz → -s; … | ||||
| 288 (4.30) | 0.22 | 38% 1a2uBz→-a/-s; 17% H−17 (1b1u) → -a/-s; 7% 2a2u → -a/-s; … | ||||
| 286 (4.34) | 0.13 | 44% 1a2uBz→-a/-s; … | ||||
| ZnPc3 | 624 (1.99) 616 (2.01) | 0.53 0.61 | 93% a→-s; … 92% a → -a; … | 350 (3.54) | 0.06 | 49% H−1Bz →-s; 25% H−2Bz→-a/-s; … |
| 326 (3.80) | 0.51 | 34% H−1Bz →-a; 19% H−3Bz→-a; 12% H−2Bz→-a; … | ||||
| 315 (3.93) | 0.48 | 79% H−3Bz →-s; … | ||||
| 309 (4.01) | 0.5 | 38% 1a2uBz→-a; 26% a → L+3 (1b2u*); … | ||||
| 303 (4.09) | 0.14 | 53% a→L+3 (1b2u*); 26% 1a2uBz→-a/-s; … | ||||
| 298 (4.16) | 0.7 | 31% 1a2uBz→-a/-s; 20% 2a2u→-a/-s; 10% H−12 (1b1u*) → -a; … | ||||
| 297 (4.19) | 0.45 | 46% 2a2u→-a/-s;12% H−12 (1b1u*)→-a/-s; 11% 1a2uBz→-a/-s;… | ||||
| ZnPc4 | 635 (1.95) 612 (2.03) | 0.61 0.58 | 94% a→-s; … 94% a → -a; … | 353 (3.51) | 0.08 | 40% H−2Bz→-s; 30% H−1Bz→-s; … |
| 337 (3.67) | 0.07 | 72% a→L+2 (1b1u*); … | ||||
| 332 (3.74) | 0.48 | 80% H−1Bz→-a; … | ||||
| 328 (3.78) | 0.1 | 65% H−2Bz→-a; … | ||||
| 316 (3.93) | 0.17 | 81% a→L+3 (1b2u*); … | ||||
| 314 (3.95) | 0.46 | 80% H−3Bz→-s; … | ||||
| 302 (4.11) | 0.64 | 33% H−10 (2eg) → -a; 31% 1a2uBz → -a; … | ||||
| 299 (4.14) | 0.56 | 21% 2a2u→-a/-s; 19% 1a2uBz→-s; 13% H−10 (2eg) → -a/-s; 12% H−12 (1b1u) → -a/-s; … | ||||
| 298 (4.16) | 0.24 | 25% 2a2u→-a/-s;16% H−12 (1b1u)→-a/-s; 13% 1a2uBz→-s; … |
| λχαλχ, νμ (eV) | fcalc | Q Band Wavefunctions a | λχαλχ, νμ (eV) | fcalc | Wavefunctions of the Most Intense Bands Predicted in the B Band Region a | |
|---|---|---|---|---|---|---|
| ZnPc | 676 (1.84) 675 (1.84) | 0.57 0.57 | 97% a → -a/-s; … 97% a → -s/-a; … | 325 (3.81) | 0.28 | 74% H−2 (1b2u) → -a; 14% s → -a; … |
| 325 (3.81) | 0.28 | 74% H−2 (1b2u) → -s; 14% s → -s; … | ||||
| 308 (4.03) | 1.16 | 79% 1a2uBz → -a; 9% H−2 (1b2u) → -a; … | ||||
| 308 (4.03) | 1.16 | 74% 1a2uBz → -s; 9% H−2 (1b2u) → -s; … | ||||
| ZnPc* | 689 (1.80) 688 (1.80) | 0.59 0.59 | 95% a → -a/-s; … 95% a → -s/-a; … | 364 (3.41) | 0.63 | 70% 1a2uBz → -a/-s; H−2 (1b2u) → -a; … |
| 364 (3.41) | 0.64 | 71% 1a2uBz → -a/-s; 9% H−2 (1b2u) → -s; … | ||||
| 363 (3.41) | 0.18 | 58% H−18 (1b2gN) → -s; 18% 1a2uBz → -s; 13% H−1OAr → -s; … | ||||
| 363 (3.41) | 0.12 | 61% H−18 (1b2gN) → -s; 14% H−14OAr → -s; 13% 1a2uBz → -s; … | ||||
| 307 (4.04) | 0.85 | 62% 2a2u → -a; 16% H−25 (1b1u) → -s; … | ||||
| 307 (4.04) | 0.85 | 61% 2a2u → -s; 16% H−25 (1b1u) → -a; … | ||||
| ZnPc1 | 703 (1.76) 673 (1.84) | 0.6 0.59 | 97% a → -a; … 96% a → -s; … | 382 (3.25) | 0.42 | 80% H−1Bz → -a; … |
| 377 (3.29) | 0.17 | 82% H−2Bz → -a/-s; … | ||||
| 365 (3.39) | 0.39 | 75% H−3Bz → -s; … | ||||
| 320 (3.88) | 0.16 | 57% 1a2uBz → -a; 9% 2a2u → -a; 15% a → L+3 (1b2u*); … | ||||
| 312 (3.98) | 0.8 | 26% H−18 (1b1u) → -a; 25% 1a2uBz → -s; … | ||||
| 310 (4.00) | 0.51 | 58% 2a2u → -a;12% H−18 (1b1u) → -s; … | ||||
| 304 (4.08) | 0.16 | 31% H−17 (2eg) → -a; 27% a → L+4 (1a2u*); … | ||||
| 303 (4.09) | 0.15 | 45% H−17 (2eg) → -s; 24% a → L+4 (1a2u*); … | ||||
| 301 (4.12) | 0.16 | 40% H−17 (2eg) → -a/-s; 37% a → L+4 (1a2u*); … | ||||
| 300 (4.14) | 0.13 | 33% 1a2uBz → -s;29% H−18 (1b1u) → -a; … | ||||
| 292 (4.25) | 0.13 | 40% H−20 (2eg) → -a; 30% 2a2u → -s; … | ||||
| ZnPc2 | 723 (1.72) 663 (1.87) | 0.64 0.57 | 97% a → -a; … 97% a → -s; … | 395 (3.14) | 0.35 | 86% H−1Bz → -a/-s; … |
| 384 (3.23) | 0.14 | 84% H−2Bz → -a/-s; … | ||||
| 369 (3.36) | 0.27 | 52% a → L+2 (1b1u*); 38% H−3Bz → -s; … | ||||
| 361 (3.43) | 0.17 | 45% H−3Bz → -s; 36% a → L+2 (1b1u*); … | ||||
| 324 (3.82) | 0.27 | 57% H−16 (2eg) → -a; … | ||||
| 317 (3.91) | 0.53 | 51% H−11 (1b1u) → -a; 51% H−10OAr → -a; 9% 1a2uBz → -s; … | ||||
| 314 (3.95) | 0.6 | 42% 1a2uBz → -a; 14% 2a2u → -a; 10% H−21 (2eg) → -a; … | ||||
| 307 (4.04) | 0.15 | 54% H−4OAr → -a/-s; 18% 1a2uBz → -s; … | ||||
| 302 (4.11) | 0.38 | 40% 2a2u → -s; 10% H−4OAr → -s; … | ||||
| 296 (4.18) | 0.1 | 20% H−4OAr → -s; 15% H−10OAr → -a; 12% H-17 (1b1u) → -s; … | ||||
| ZnPc3 | 703 (1.76) 685 (1.81) | 0.6 0.62 | 97% a → -s; … 97% a → -a; … | 358 (3.46) | 0.13 | 42% H−1Bz → -a; 23% H−2Bz → -a; 20% a → L+2 (1b1u*); … |
| 355 (3.49) | 0.48 | 35% H−3Bz → -a; 21% H−1Bz → -a; 19% H−2Bz → -a; … | ||||
| 344 (3.61) | 0.38 | 75% H−3Bz → -s; 15% a → L+2 (1b1u*); … | ||||
| 325 (3.81) | 0.11 | 76% a → L+3 (1b2u*);7% 1a2uBz → -a; … | ||||
| 320 (3.88) | 0.16 | 49% 1a2uBz → -a; 16% 2a2u → -a; 15% a → L+3 (1b2u*); … | ||||
| 312 (3.97) | 0.69 | 36% 1a2uBz → -s; 23% a → L+4 (1a2u*); 14% H−12 (1b1u) → -a; … | ||||
| 310 (4.00) | 0.36 | 33% a → L+4 (1a2u*); 26% 2a2u → -a; 10% 1a2uBz → -s; … | ||||
| 309 (4.01) | 0.49 | 30% a → L+4 (1a2u*); 21% 2a2u → -a; … | ||||
| 302 (4.11) | 0.16 | 39% H−2OAr → -a; 12% 1a2uBz → -s;12% H−8OAr → -a; … | ||||
| ZnPc4 | 723 (1.71) 678 (1.83) | 0.64 0.6 | 97% a → -s; … 97% a → -a; … | 364 (3.41) | 0.16 | 35% H−1Bz → -a; 27% a → L+2 (1b1u*); 10% H−3Bz → -a; … |
| 361 (3.44) | 0.42 | 31% H−2Bz → -a; 21% a → L+2 (1b1u*); 15% H−1Bz → -a; … | ||||
| 341 (3.63) | 0.4 | 84% H−3Bz → -s; … | ||||
| 318 (3.90) | 0.14 | 47% H−10 (2eg) → -a; 16% H−4OAr → -a; … | ||||
| 314 (3.95) | 0.62 | 23% H−12 (1b1u) → -a; 14% 2a2u → -a; 10% H−7OAr → -a; … | ||||
| 312 (3.97) | 0.23 | 28% H−4OAr → -s; 15% 1a2uBz → -a;14% 2a2u → -a;… | ||||
| 310 (3.99) | 0.41 | 17% H−4OAr → -a; 12% H−5OAr → -s; 10% 1a2uBz → -s;… | ||||
| 305 (4.06) | 0.35 | 48% H−7OAr → -a; 27% 2a2u → -s; … | ||||
| 304 (4.08) | 0.09 | 30% 1a2uBz → -s;29% H−6OAr → -a; 19% H−14 (2eg) → -a; … | ||||
| 299 (4.15) | 0.07 | 23% H−12 (1b1u) → -a; 14% 2a2u → -a; … | ||||
| 297 (4.18) | 0.08 | 29% H−6OAr → -s; 18% 2a2u → -a;12% 1a2uBz → -s; … |
| HOMO (eV) | LUMO (eV) | Band gap (eV) | E00 a (eV) | EOXdye b (eV) | EOXdye* c (eV) | Voc d (eV) | ΔGinj e (eV) | ΔGregen f (eV) | f g | LHE h | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ZnPc | −5.31 | −3.05 | 2.26 | 1.86 | 5.31 | 3.46 | 1.01 | −0.54 | −0.51 | 0.48 | 0.67 |
| ZnPc1 | −5.38 | −3.19 | 2.19 | 1.84 | 5.38 | 3.55 | 1.08 | −0.45 | −0.58 | 0.56 | 0.72 |
| ZnPc2 | −5.55 | −3.42 | 2.13 | 1.83 | 5.55 | 3.72 | 1.25 | −0.28 | −0.75 | 0.56 | 0.72 |
| ZnPc3 | −5.70 | −3.50 | 2.19 | 1.83 | 5.70 | 3.86 | 1.40 | −0.14 | −0.90 | 0.56 | 0.73 |
| ZnPc4 | −5.71 | −3.58 | 2.14 | 1.82 | 5.71 | 3.89 | 1.41 | −0.11 | −0.91 | 0.57 | 0.73 |
| ZnPc* | −5.08 | −2.84 | 2.24 | 1.84 | 5.08 | 3.24 | 0.78 | −0.76 | −0.28 | 0.60 | 0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunin, D.A.; Ndebele, N.; Martynov, A.G.; Mack, J.; Gorbunova, Y.G.; Nyokong, T. Low-Symmetry Phthalocyanines Bearing Carboxy-Groups: Synthesis, Spectroscopic and Quantum-Chemical Characterization. Molecules 2022, 27, 524. https://doi.org/10.3390/molecules27020524
Bunin DA, Ndebele N, Martynov AG, Mack J, Gorbunova YG, Nyokong T. Low-Symmetry Phthalocyanines Bearing Carboxy-Groups: Synthesis, Spectroscopic and Quantum-Chemical Characterization. Molecules. 2022; 27(2):524. https://doi.org/10.3390/molecules27020524
Chicago/Turabian StyleBunin, Dmitry A., Nobuhle Ndebele, Alexander G. Martynov, John Mack, Yulia G. Gorbunova, and Tebello Nyokong. 2022. "Low-Symmetry Phthalocyanines Bearing Carboxy-Groups: Synthesis, Spectroscopic and Quantum-Chemical Characterization" Molecules 27, no. 2: 524. https://doi.org/10.3390/molecules27020524
APA StyleBunin, D. A., Ndebele, N., Martynov, A. G., Mack, J., Gorbunova, Y. G., & Nyokong, T. (2022). Low-Symmetry Phthalocyanines Bearing Carboxy-Groups: Synthesis, Spectroscopic and Quantum-Chemical Characterization. Molecules, 27(2), 524. https://doi.org/10.3390/molecules27020524