
Symmetry Breaking Charge Transfer in DNA-Templated Perylene Dimer Aggregates
 , ,
, ,  ,
,  ,
,
Abstract
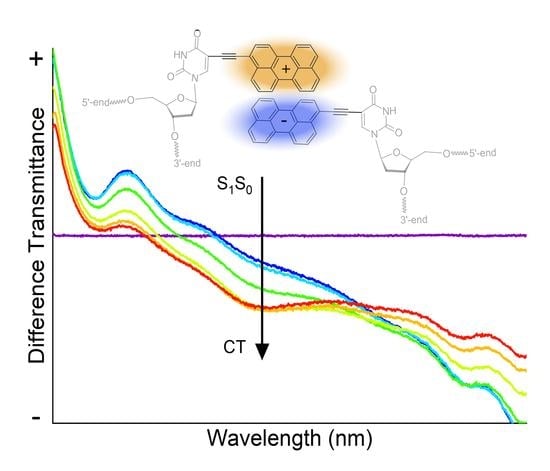
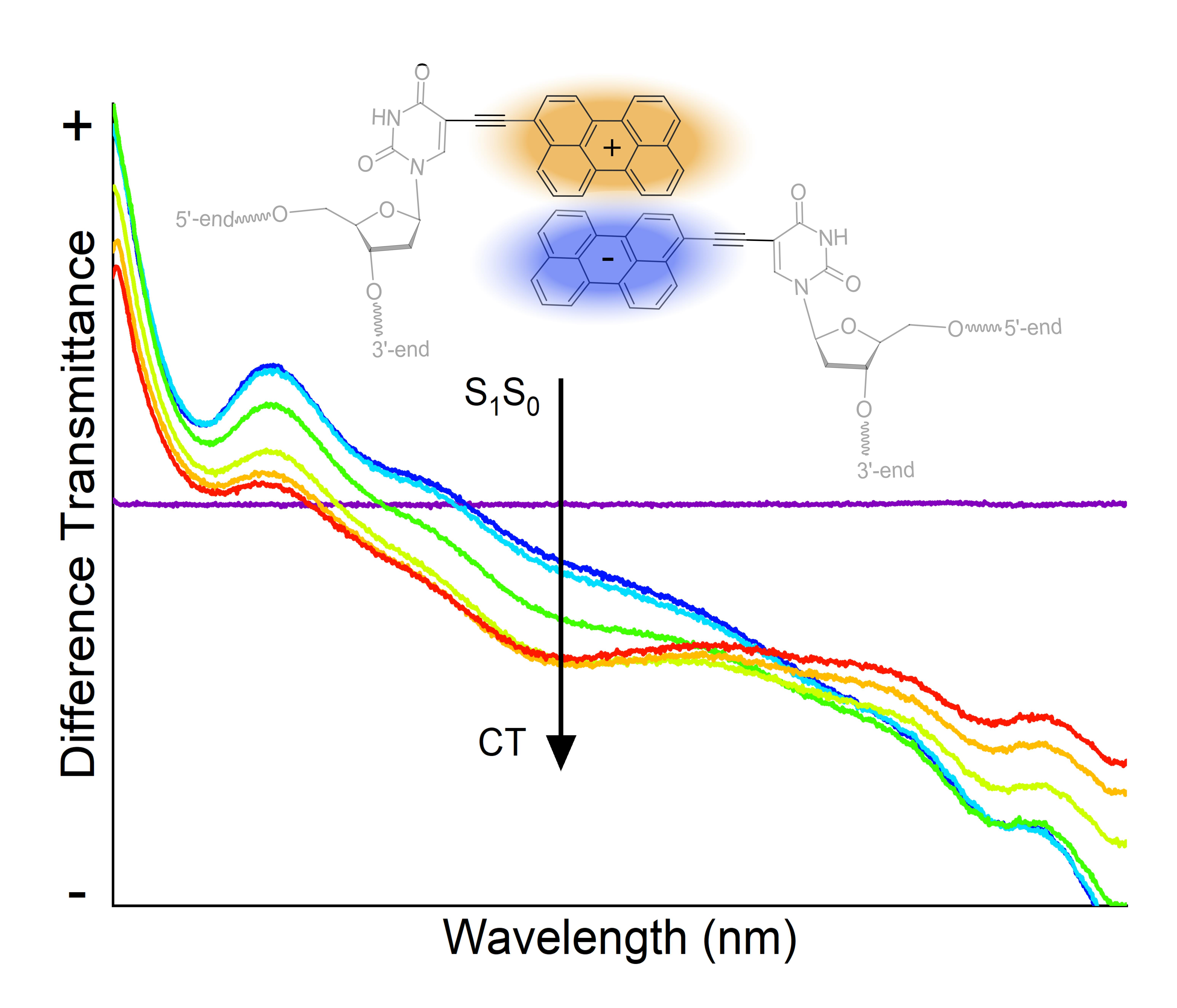
1. Introduction
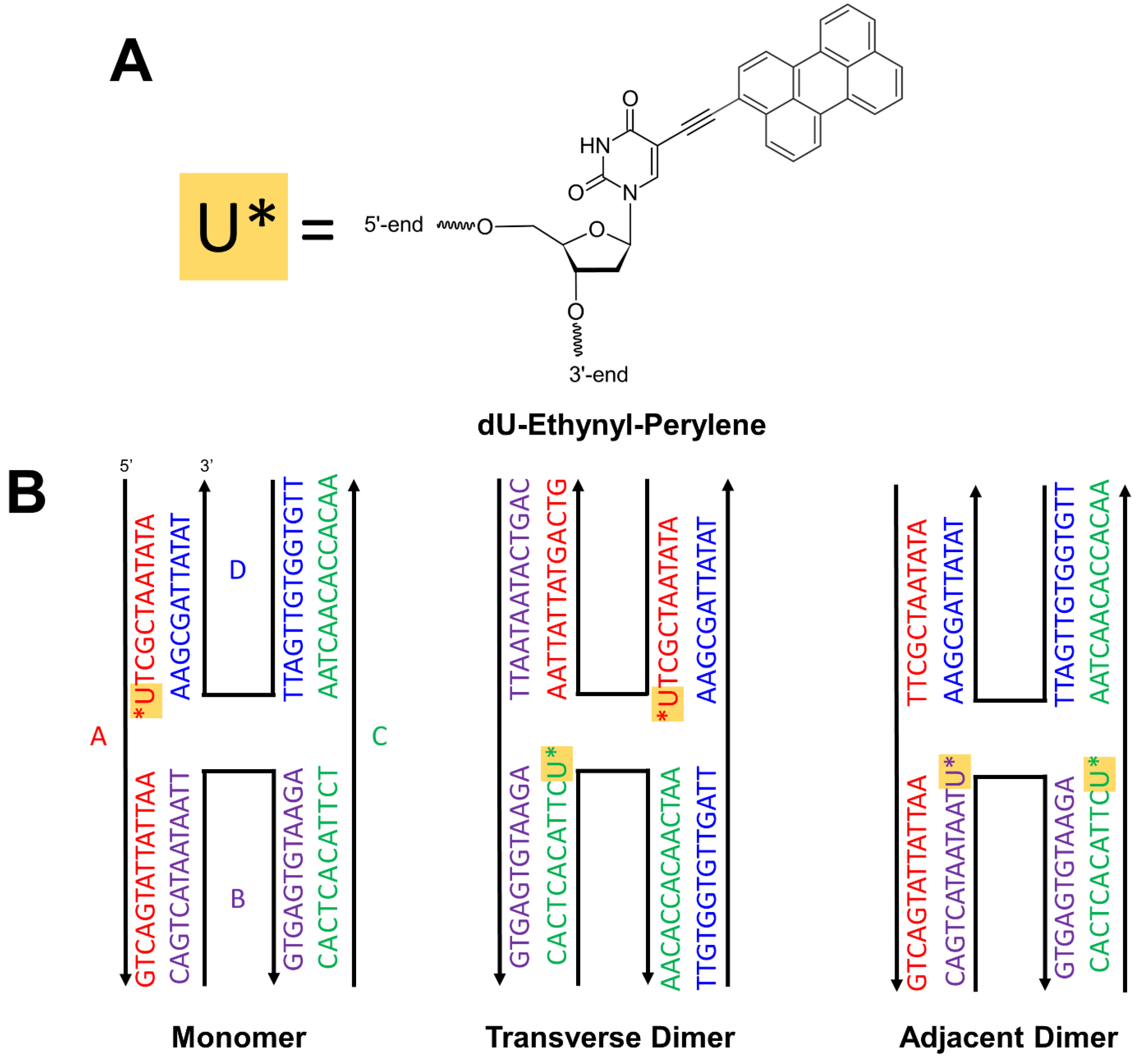
2. Materials and Methods
2.1. DNA-Dye Construction Preparation
2.2. Steady-State Absorption Spectroscopy
2.3. Steady-State Fluorescence Spectroscopy
2.4. Time-Correlated Single Photon Counting
2.5. Femtosecond Visible Transient Absorption Spectroscopy
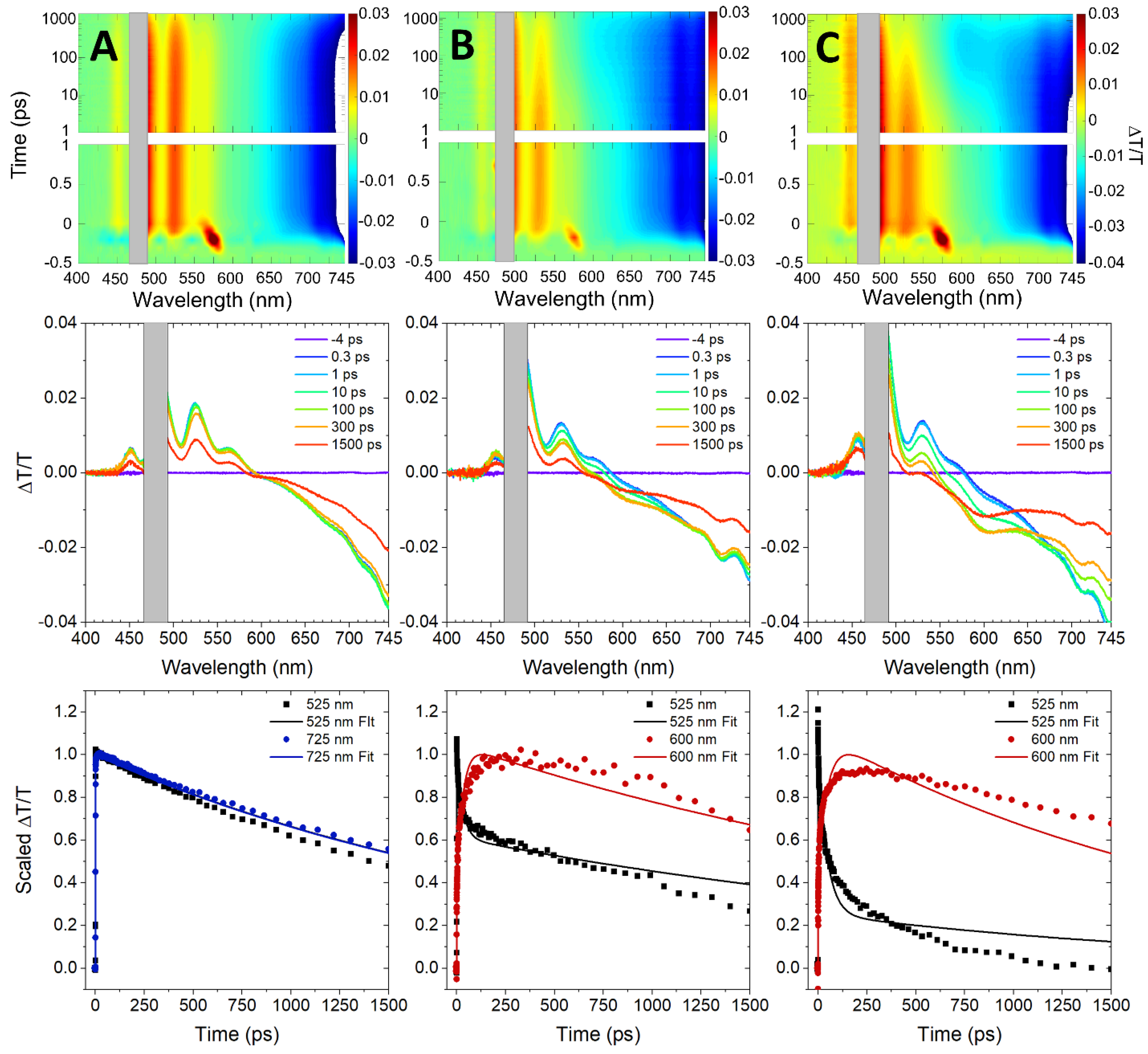
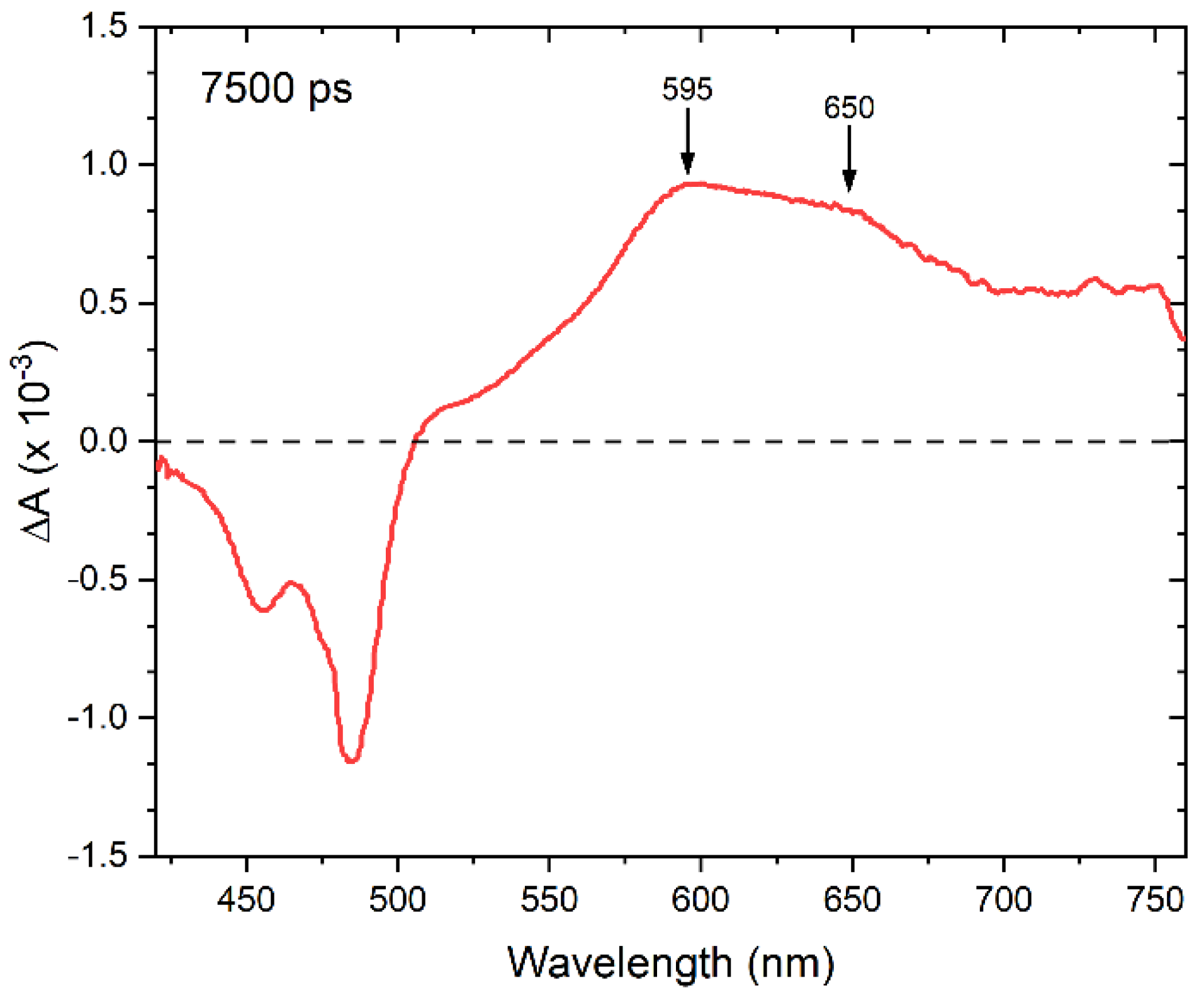
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hanna, M.C.; Nozik, A.J. Solar Conversion Efficiency of Photovoltaic and Photoelectrolysis Cells with Carrier Multiplication Absorbers. J. Appl. Phys. 2006, 100, 074510. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef] [PubMed]
- Burdett, J.J.; Bardeen, C.J. Quantum Beats in Crystalline Tetracene Delayed Fluorescence Due to Triplet Pair Coherences Produced by Direct Singlet Fission. J. Am. Chem. Soc. 2012, 134, 8597–8607. [Google Scholar] [CrossRef]
- Bardeen, C.J. Time Dependent Correlations of Entangled States with Nondegenerate Branches and Possible Experimental Realization Using Singlet Fission. J. Chem. Phys. 2019, 151, 124503. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Recent Advances in Singlet Fission. Annu. Rev. Phys. Chem. 2013, 64, 361–386. [Google Scholar] [CrossRef]
- Piland, G.B.; Burdett, J.J.; Dillon, R.J.; Bardeen, C.J. Singlet Fission: From Coherences to Kinetics. J. Phys. Chem. Lett. 2014, 5, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.W.B.; Rao, A.; Clark, J.; Kumar, R.S.S.; Brida, D.; Cerullo, G.; Friend, R.H. Ultrafast Dynamics of Exciton Fission in Polycrystalline Pentacene. J. Am. Chem. Soc. 2011, 133, 11830–11833. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.W.; Shoer, L.E.; Karlen, S.D.; Dyar, S.M.; Margulies, E.A.; Veldkamp, B.S.; Ramanan, C.; Hartzler, D.A.; Savikhin, S.; Marks, T.J.; et al. Singlet Exciton Fission in Polycrystalline Thin Films of a Slip-Stacked Perylenediimide. J. Am. Chem. Soc. 2013, 135, 14701–14712. [Google Scholar] [CrossRef] [PubMed]
- Busby, E.; Berkelbach, T.C.; Kumar, B.; Chernikov, A.; Zhong, Y.; Hlaing, H.; Zhu, X.-Y.; Heinz, T.F.; Hybertsen, M.S.; Sfeir, M.Y.; et al. Multiphonon Relaxation Slows Singlet Fission in Crystalline Hexacene. J. Am. Chem. Soc. 2014, 136, 10654–10660. [Google Scholar] [CrossRef] [PubMed]
- Pensack, R.D.; Tilley, A.J.; Parkin, S.R.; Lee, T.S.; Payne, M.M.; Gao, D.; Jahnke, A.A.; Oblinsky, D.G.; Li, P.-F.; Anthony, J.E.; et al. Exciton Delocalization Drives Rapid Singlet Fission in Nanoparticles of Acene Derivatives. J. Am. Chem. Soc. 2015, 137, 6790–6803. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.N.; Kumarasamy, E.; Pun, A.B.; Trinh, M.T.; Choi, B.; Xia, J.; Taffet, E.J.; Low, J.Z.; Miller, J.R.; Roy, X.; et al. Quantitative Intramolecular Singlet Fission in Bipentacenes. J. Am. Chem. Soc. 2015, 137, 8965–8972. [Google Scholar] [CrossRef] [PubMed]
- Piland, G.B.; Bardeen, C.J. How Morphology Affects Singlet Fission in Crystalline Tetracene. J. Phys. Chem. Lett. 2015, 6, 1841–1846. [Google Scholar] [CrossRef]
- Korovina, N.V.; Das, S.; Nett, Z.; Feng, X.; Joy, J.; Haiges, R.; Krylov, A.I.; Bradforth, S.E.; Thompson, M.E. Singlet Fission in a Covalently Linked Cofacial Alkynyltetracene Dimer. J. Am. Chem. Soc. 2016, 138, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Margulies, E.A.; Miller, C.E.; Wu, Y.; Ma, L.; Schatz, G.C.; Young, R.M.; Wasielewski, M.R. Enabling Singlet Fission by Controlling Intramolecular Charge Transfer in π-Stacked Covalent Terrylenediimide Dimers. Nat. Chem. 2016, 8, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Lukman, S.; Chen, K.; Hodgkiss, J.M.; Turban, D.H.P.; Hine, N.D.M.; Dong, S.; Wu, J.; Greenham, N.C.; Musser, A.J. Tuning the Role of Charge-Transfer States in Intramolecular Singlet Exciton Fission through Side-Group Engineering. Nat. Commun. 2016, 7, 13622. [Google Scholar] [CrossRef] [PubMed]
- Margulies, E.A.; Logsdon, J.L.; Miller, C.E.; Ma, L.; Simonoff, E.; Young, R.M.; Schatz, G.C.; Wasielewski, M.R. Direct Observation of a Charge-Transfer State Preceding High-Yield Singlet Fission in Terrylenediimide Thin Films. J. Am. Chem. Soc. 2017, 139, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Le, A.K.; Bender, J.A.; Arias, D.H.; Cotton, D.E.; Johnson, J.C.; Roberts, S.T. Singlet Fission Involves an Interplay between Energetic Driving Force and Electronic Coupling in Perylenediimide Films. J. Am. Chem. Soc. 2018, 140, 814–826. [Google Scholar] [CrossRef]
- Frankevich, E.L.; Lesin, V.I.; Pristupa, A.I. Rate Constants of Singlet Exciton Fission in a Tetracene Crystal Determined from the RYDMR Spectral Linewidth. Chem. Phys. Lett. 1978, 58, 127–131. [Google Scholar] [CrossRef]
- Pensack, R.D.; Ostroumov, E.E.; Tilley, A.J.; Mazza, S.; Grieco, C.; Thorley, K.J.; Asbury, J.B.; Seferos, D.S.; Anthony, J.E.; Scholes, G.D. Observation of Two Triplet-Pair Intermediates in Singlet Exciton Fission. J. Phys. Chem. Lett. 2016, 7, 2370–2375. [Google Scholar] [CrossRef]
- Pensack, R.D.; Grieco, C.; Purdum, G.E.; Mazza, S.M.; Tilley, A.J.; Ostroumov, E.E.; Seferos, D.S.; Loo, Y.-L.; Asbury, J.B.; Anthony, J.E.; et al. Solution-Processable, Crystalline Material for Quantitative Singlet Fission. Mater. Horiz. 2017, 4, 915–923. [Google Scholar] [CrossRef]
- Pensack, R.D.; Tilley, A.J.; Grieco, C.; Purdum, G.E.; Ostroumov, E.E.; Granger, D.B.; Oblinsky, D.G.; Dean, J.C.; Doucette, G.S.; Asbury, J.B.; et al. Striking the Right Balance of Intermolecular Coupling for High-Efficiency Singlet Fission. Chem. Sci. 2018, 9, 6240–6259. [Google Scholar] [CrossRef] [PubMed]
- Pensack, R.D.; Purdum, G.E.; Mazza, S.M.; Grieco, C.; Asbury, J.B.; Anthony, J.E.; Loo, Y.-L.; Scholes, G.D. Excited-State Dynamics of 5,14- vs 6,13-Bis(Trialkylsilylethynyl)-Substituted Pentacenes: Implications for Singlet Fission. J. Phys. Chem. C 2022, 126, 9784–9793. [Google Scholar] [CrossRef] [PubMed]
- Carrod, A.J.; Cravcenco, A.; Ye, C.; Börjesson, K. Modulating TTA Efficiency through Control of High Energy Triplet States. J. Mater. Chem. C 2022, 10, 4923–4928. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Sukhanov, A.A.; Zhang, X.; Elmali, A.; Zhao, J.; Ji, S.; Karatay, A.; Voronkova, V.K. Spiro Rhodamine-Perylene Compact Electron Donor–Acceptor Dyads: Conformation Restriction, Charge Separation, and Spin–Orbit Charge Transfer Intersystem Crossing. J. Phys. Chem. B 2021, 125, 4187–4203. [Google Scholar] [CrossRef]
- Kasha, M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55. [Google Scholar] [CrossRef]
- Kasha, M.; Rawls, H.R.; Ashraf El-Bayoumi, M. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef]
- Komfort, M.; Löhmannsröben, H.-G.; Salthammer, T. The Temperature Dependence of Photophysical Processes in Perylene, Tetracene and Some of Their Derivatives. J. Photochem. Photobiol. Chem. 1990, 51, 215–227. [Google Scholar] [CrossRef]
- Zhong, F.; Zhao, J. Phenyleneanthracene Derivatives as Triplet Energy Acceptor/Emitter in Red Light Excitable Triplet-Triplet-Annihilation Upconversion. Dyes Pigments 2017, 136, 909–918. [Google Scholar] [CrossRef]
- Cui, X.; Charaf-Eddin, A.; Wang, J.; Le Guennic, B.; Zhao, J.; Jacquemin, D. Perylene-Derived Triplet Acceptors with Optimized Excited State Energy Levels for Triplet–Triplet Annihilation Assisted Upconversion. J. Org. Chem. 2014, 79, 2038–2048. [Google Scholar] [CrossRef]
- Albrecht, W.G.; Michel-Beyerle, M.E.; Yakhot, V. Exciton Fission in Excimer Forming Crystal. Dynamics of an Excimer Build-up in α-Perylene. Chem. Phys. 1978, 35, 193–200. [Google Scholar] [CrossRef]
- Piland, G.B.; Burdett, J.J.; Hung, T.-Y.; Chen, P.-H.; Lin, C.-F.; Chiu, T.-L.; Lee, J.-H.; Bardeen, C.J. Dynamics of Molecular Excitons near a Semiconductor Surface Studied by Fluorescence Quenching of Polycrystalline Tetracene on Silicon. Chem. Phys. Lett. 2014, 601, 33–38. [Google Scholar] [CrossRef]
- Daiber, B.; van den Hoven, K.; Futscher, M.H.; Ehrler, B. Realistic Efficiency Limits for Singlet-Fission Silicon Solar Cells. ACS Energy Lett. 2021, 6, 2800–2808. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Koh, T.T.; Schwan, J.; Tran, T.T.-T.; Xia, P.; Wang, K.; Mangolini, L.; Tang, M.L.; Roberts, S.T. Bidirectional Triplet Exciton Transfer between Silicon Nanocrystals and Perylene. Chem. Sci. 2021, 12, 6737–6746. [Google Scholar] [CrossRef]
- Jelley, E.E. Spectral Absorption and Fluorescence of Dyes in the Molecular State. Nature 1936, 138, 1009–1010. [Google Scholar] [CrossRef]
- Jelley, E.E. Molecular, Nematic and Crystal States of I: I-Diethyl-Ψ-Cyanine Chloride. Nature 1937, 139, 631–632. [Google Scholar] [CrossRef]
- Scholes, G.D.; Fleming, G.R.; Olaya-Castro, A.; van Grondelle, R. Lessons from Nature about Solar Light Harvesting. Nat. Chem. 2011, 3, 763–774. [Google Scholar] [CrossRef]
- Mirkovic, T.; Ostroumov, E.E.; Anna, J.M.; van Grondelle, R.; Govindjee; Scholes, G.D. Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms. Chem. Rev. 2017, 117, 249–293. [Google Scholar] [CrossRef]
- Seeman, N.C.; Kallenbach, N.R. Design of Immobile Nucleic Acid Junctions. Biophys. J. 1983, 44, 201–209. [Google Scholar] [CrossRef]
- Winfree, E.; Liu, F.; Wenzler, L.A.; Seeman, N.C. Design and Self-Assembly of Two-Dimensional DNA Crystals. Nature 1998, 394, 539–544. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ong, L.L.; Shih, W.M.; Yin, P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.L.; Hanikel, N.; Yaghi, O.K.; Grun, C.; Strauss, M.T.; Bron, P.; Lai-Kee-Him, J.; Schueder, F.; Wang, B.; Wang, P.; et al. Programmable Self-Assembly of Three-Dimensional Nanostructures from 10,000 Unique Components. Nature 2017, 552, 72–77. [Google Scholar] [CrossRef]
- Wang, D.; Yu, L.; Huang, C.-M.; Arya, G.; Chang, S.; Ke, Y. Programmable Transformations of DNA Origami Made of Small Modular Dynamic Units. J. Am. Chem. Soc. 2021, 143, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Seifert, J.L.; Connor, R.E.; Kushon, S.A.; Wang, M.; Armitage, B.A. Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc. 1999, 121, 2987–2995. [Google Scholar] [CrossRef]
- Markova, L.I.; Malinovskii, V.L.; Patsenker, L.D.; Häner, R. Synthesis and Properties of Squaraine-Modified DNA. Org. Biomol. Chem. 2012, 10, 8944. [Google Scholar] [CrossRef]
- Markova, L.I.; Malinovskii, V.L.; Patsenker, L.D.; Häner, R. J- vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a near-IR Chiroptical Reporter. Chem. Commun. 2013, 49, 5298. [Google Scholar] [CrossRef]
- Nicoli, F.; Roos, M.K.; Hemmig, E.A.; Di Antonio, M.; de Vivie-Riedle, R.; Liedl, T. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes. J. Phys. Chem. A 2016, 120, 9941–9947. [Google Scholar] [CrossRef]
- Cannon, B.L.; Kellis, D.L.; Patten, L.K.; Davis, P.H.; Lee, J.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. [Google Scholar] [CrossRef]
- Cannon, B.L.; Patten, L.K.; Kellis, D.L.; Davis, P.H.; Lee, J.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. [Google Scholar] [CrossRef]
- Asanuma, H.; Murayama, K.; Kamiya, Y.; Kashida, H. The DNA Duplex as an Aqueous One-Dimensional Soft Crystal Scaffold for Photochemistry. Bull. Chem. Soc. Jpn. 2018, 91, 1739–1748. [Google Scholar] [CrossRef]
- Cunningham, P.D.; Kim, Y.C.; Díaz, S.A.; Buckhout-White, S.; Mathur, D.; Medintz, I.L.; Melinger, J.S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. [Google Scholar] [CrossRef]
- Kringle, L.; Sawaya, N.P.D.; Widom, J.; Adams, C.; Raymer, M.G.; Aspuru-Guzik, A.; Marcus, A.H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148, 085101. [Google Scholar] [CrossRef] [PubMed]
- Huff, J.S.; Davis, P.H.; Christy, A.; Kellis, D.L.; Kandadai, N.; Toa, Z.S.D.; Scholes, G.D.; Yurke, B.; Knowlton, W.B.; Pensack, R.D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10, 2386–2392. [Google Scholar] [CrossRef]
- Heussman, D.; Kittell, J.; Kringle, L.; Tamimi, A.; von Hippel, P.H.; Marcus, A.H. Measuring Local Conformations and Conformational Disorder of (Cy3)2 Dimer Labeled DNA Fork Junctions Using Absorbance, Circular Dichroism and Two-Dimensional Fluorescence Spectroscopy. Faraday Discuss. 2019, 216, 211–235. [Google Scholar] [CrossRef]
- Cunningham, P.D.; Díaz, S.A.; Yurke, B.; Medintz, I.L.; Melinger, J.S. Delocalized Two-Exciton States in DNA Scaffolded Cyanine Dimers. J. Phys. Chem. B 2020, 124, 8042–8049. [Google Scholar] [CrossRef] [PubMed]
- Mazuski, R.J.; Díaz, S.A.; Wood, R.E.; Lloyd, L.T.; Klein, W.P.; Mathur, D.; Melinger, J.S.; Engel, G.S.; Medintz, I.L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11, 4163–4172. [Google Scholar] [CrossRef] [PubMed]
- Mass, O.A.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Patten, L.K.; Terpetschnig, E.A.; Lee, J.; Pensack, R.D.; Yurke, B.; Knowlton, W.B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. [Google Scholar] [CrossRef]
- Rolczynski, B.S.; Díaz, S.A.; Kim, Y.C.; Medintz, I.L.; Cunningham, P.D.; Melinger, J.S. Understanding Disorder, Vibronic Structure, and Delocalization in Electronically Coupled Dimers on DNA Duplexes. J. Phys. Chem. A 2021, 125, 9632–9644. [Google Scholar] [CrossRef]
- Huff, J.S.; Turner, D.B.; Mass, O.A.; Patten, L.K.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Yurke, B.; Knowlton, W.B.; Davis, P.H.; et al. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. [Google Scholar] [CrossRef]
- Barclay, M.S.; Roy, S.K.; Huff, J.S.; Mass, O.A.; Turner, D.B.; Wilson, C.K.; Kellis, D.L.; Terpetschnig, E.A.; Lee, J.; Davis, P.H.; et al. Rotaxane Rings Promote Oblique Packing and Extended Lifetimes in DNA-Templated Molecular Dye Aggregates. Commun. Chem. 2021, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.M.; Chen, W.J.; Banal, J.L.; Bricker, W.P.; Dodin, A.; Markova, L.; Vyborna, Y.; Willard, A.P.; Häner, R.; Bathe, M.; et al. Engineering Couplings for Exciton Transport Using Synthetic DNA Scaffolds. Chem 2021, 7, 752–773. [Google Scholar] [CrossRef]
- Mass, O.A.; Wilson, C.K.; Barcenas, G.; Terpetschnig, E.A.; Obukhova, O.M.; Kolosova, O.S.; Tatarets, A.L.; Li, L.; Yurke, B.; Knowlton, W.B.; et al. Influence of Hydrophobicity on Excitonic Coupling in DNA-Templated Indolenine Squaraine Dye Aggregates. J. Phys. Chem. C 2022, 126, 3475–3488. [Google Scholar] [CrossRef]
- Barclay, M.S.; Wilson, C.K.; Roy, S.K.; Mass, O.A.; Obukhova, O.M.; Svoiakov, R.P.; Tatarets, A.L.; Chowdhury, A.U.; Huff, J.S.; Turner, D.B.; et al. Oblique Packing and Tunable Excitonic Coupling in DNA-Templated Squaraine Rotaxane Dimer Aggregates. ChemPhotoChem 2022, 6, e202200039. [Google Scholar] [CrossRef]
- Chowdhury, A.U.; Díaz, S.A.; Huff, J.S.; Barclay, M.S.; Chiriboga, M.; Ellis, G.A.; Mathur, D.; Patten, L.K.; Sup, A.; Hallstrom, N.; et al. Tuning between Quenching and Energy Transfer in DNA-Templated Heterodimer Aggregates. J. Phys. Chem. Lett. 2022, 13, 2782–2791. [Google Scholar] [CrossRef]
- Heussman, D.; Kittell, J.; von Hippel, P.H.; Marcus, A.H. Temperature-Dependent Local Conformations and Conformational Distributions of Cyanine Dimer Labeled Single-Stranded–Double-Stranded DNA Junctions by 2D Fluorescence Spectroscopy. J. Chem. Phys. 2022, 156, 045101. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.M.; Wang, X.; Guo, J.; Bathe, M.; Schlau-Cohen, G.S. Tuning Optical Absorption and Emission Using Strongly Coupled Dimers in Programmable DNA Scaffolds. J. Phys. Chem. Lett. 2022, 13, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.S.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. NUPACK: Analysis and Design of Nucleic Acid Systems. J. Comput. Chem. 2011, 32, 170–173. [Google Scholar] [CrossRef]
- Skorobogatyi, M.V.; Malakhov, A.D.; Pchelintseva, A.A.; Turban, A.A.; Bondarev, S.L.; Korshun, V.A. Fluorescent 5-Alkynyl-2′-Deoxyuridines: High Emission Efficiency of a Conjugated Perylene Nucleoside in a DNA Duplex. ChemBioChem 2006, 7, 810–816. [Google Scholar] [CrossRef]
- Oleson, A.; Zhu, T.; Dunn, I.S.; Bialas, D.; Bai, Y.; Zhang, W.; Dai, M.; Reichman, D.R.; Tempelaar, R.; Huang, L.; et al. Perylene Diimide-Based Hj- and hJ-Aggregates: The Prospect of Exciton Band Shape Engineering in Organic Materials. J. Phys. Chem. C 2019, 123, 20567–20578. [Google Scholar] [CrossRef]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C.; Yamagata, H. Vibronic Coupling in J-Aggregates and Beyond: A Direct Means of Determining the Exciton Coherence Length from the Photoluminescence Spectrum. J. Phys. Chem. B 2011, 115, 5133–5143. [Google Scholar] [CrossRef] [PubMed]
- Hestand, N.J.; Spano, F.C. Molecular Aggregate Photophysics beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res. 2017, 50, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Niles, E.T.; Roehling, J.D.; Yamagata, H.; Wise, A.J.; Spano, F.C.; Moulé, A.J.; Grey, J.K. J-Aggregate Behavior in Poly-3-Hexylthiophene Nanofibers. J. Phys. Chem. Lett. 2012, 3, 259–263. [Google Scholar] [CrossRef]
- Furube, A.; Murai, M.; Tamaki, Y.; Watanabe, S.; Katoh, R. Effect of Aggregation on the Excited-State Electronic Structure of Perylene Studied by Transient Absorption Spectroscopy. J. Phys. Chem. A 2006, 110, 6465–6471. [Google Scholar] [CrossRef]
- Van Stokkum, I.H.M.; Larsen, D.S.; van Grondelle, R. Global and Target Analysis of Time-Resolved Spectra. Biochim. Biophys. Acta Bioenerg. 2004, 1657, 82–104. [Google Scholar] [CrossRef]
- Holzwarth, A.R. Data Analysis of Time-Resolved Measurements. In Biophysical Techniques in Photosynthesis; Amesz, J., Hoff, A.J., Eds.; Springer: Dordrecht, The Netherlands, 1996; pp. 75–92. [Google Scholar]
- Tilley, A.J.; Pensack, R.D.; Kynaston, E.L.; Scholes, G.D.; Seferos, D.S. Singlet Fission in Core–Shell Micelles of End-Functionalized Polymers. Chem. Mater. 2018, 30, 4409–4421. [Google Scholar] [CrossRef]
- Markovic, V.; Villamaina, D.; Barabanov, I.; Lawson Daku, L.M.; Vauthey, E. Photoinduced Symmetry-Breaking Charge Separation: The Direction of the Charge Transfer. Angew. Chem. Int. Ed. 2011, 50, 7596–7598. [Google Scholar] [CrossRef]
- Scholes, G.D. Chemical Applications of Lasers: Pump and Proble Studies of Femtoseconed Kinetics. In Encyclopedia of Modern Optics; Guenther, R.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 62–68. [Google Scholar] [CrossRef]
- Kawai, K.; Yamamoto, N.; Tsubomura, H. Simultaneous Formation of Perylene Cation and Anion by Flash Excitation of Perylene in Solutions. Bull. Chem. Soc. Jpn. 1970, 43, 2266–2268. [Google Scholar] [CrossRef]
- Carmichael, I.; Helman, W.P.; Hug, G.L. Extinction Coefficients of Triplet–Triplet Absorption Spectra of Organic Molecules in Condensed Phases: A Least-Squares Analysis. J. Phys. Chem. Ref. Data 1987, 16, 239–260. [Google Scholar] [CrossRef]
- Bensasson, R.; Land, E.J. Triplet-Triplet Extinction Coefficients via Energy Transfer. Trans. Faraday Soc. 1971, 67, 1904. [Google Scholar] [CrossRef]
- Porter, G.; Windsor, M.W. The Triplet State in Fluid Media. Proc. R. Soc. Lond. A 1958, 245, 238–258. [Google Scholar] [CrossRef]
- Cook, R.E.; Phelan, B.T.; Kamire, R.J.; Majewski, M.B.; Young, R.M.; Wasielewski, M.R. Excimer Formation and Symmetry-Breaking Charge Transfer in Cofacial Perylene Dimers. J. Phys. Chem. A 2017, 121, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.B.; Kazzaz, A.A.; King, T.A. ‘Excimer’ Fluorescence—IX. Lifetime Studies of Pyrene Crystals. Proc. R. Soc. Lond. A 1966, 291, 556–569. [Google Scholar] [CrossRef]
- Pensack, R.D.; Ashmore, R.J.; Paoletta, A.L.; Scholes, G.D. The Nature of Excimer Formation in Crystalline Pyrene Nanoparticles. J. Phys. Chem. C 2018, 122, 21004–21017. [Google Scholar] [CrossRef]
- Beckwith, J.S.; Lang, B.; Grilj, J.; Vauthey, E. Ion-Pair Dynamics upon Photoinduced Electron Transfer Monitored by Pump–Pump–Probe Spectroscopy. J. Phys. Chem. Lett. 2019, 10, 3688–3693. [Google Scholar] [CrossRef] [PubMed]
- Korovina, N.V.; Chang, C.H.; Johnson, J.C. Spatial Separation of Triplet Excitons Drives Endothermic Singlet Fission. Nat. Chem. 2020, 12, 391–398. [Google Scholar] [CrossRef]
- Vauthey, E. Photoinduced Symmetry-Breaking Charge Separation. ChemPhysChem 2012, 13, 2001–2011. [Google Scholar] [CrossRef]
- Giaimo, J.M.; Gusev, A.V.; Wasielewski, M.R. Excited-State Symmetry Breaking in Cofacial and Linear Dimers of a Green Perylenediimide Chlorophyll Analogue Leading to Ultrafast Charge Separation. J. Am. Chem. Soc. 2002, 124, 8530–8531. [Google Scholar] [CrossRef]
- Levanon, H.; Möbius, K. Advanced EPR Spectroscopy on Electron Transfer Processes in Photosynthesis and Biomimetic Model Systems. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 495–540. [Google Scholar] [CrossRef]
- Sundström, V. Femtobiology. Annu. Rev. Phys. Chem. 2008, 59, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Kumagai, T.; Shimizu, M.; Itaya, A.; Schweitzer, G.; De Schryver, F.C.; Ashai, T.; Masuhara, H.; Miyasaka, H. Picosecond Dynamics of Excited 9,9′-Bianthryl Adsorbed on Porous Glass: Role of Symmetry Breaking in the Ground State. J Phys Chem A 2002, 106, 2067–2073. [Google Scholar] [CrossRef]
- Wu, Y.; Young, R.M.; Frasconi, M.; Schneebeli, S.T.; Spenst, P.; Gardner, D.M.; Brown, K.E.; Würthner, F.; Stoddart, J.F.; Wasielewski, M.R. Ultrafast Photoinduced Symmetry-Breaking Charge Separation and Electron Sharing in Perylenediimide Molecular Triangles. J. Am. Chem. Soc. 2015, 137, 13236–13239. [Google Scholar] [CrossRef]
- Kellis, D.L.; Sarter, C.; Cannon, B.L.; Davis, P.H.; Graugnard, E.; Lee, J.; Pensack, R.D.; Kolmar, T.; Jäschke, A.; Yurke, B.; et al. An All-Optical Excitonic Switch Operated in the Liquid and Solid Phases. ACS Nano 2019, 13, 2986–2994. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
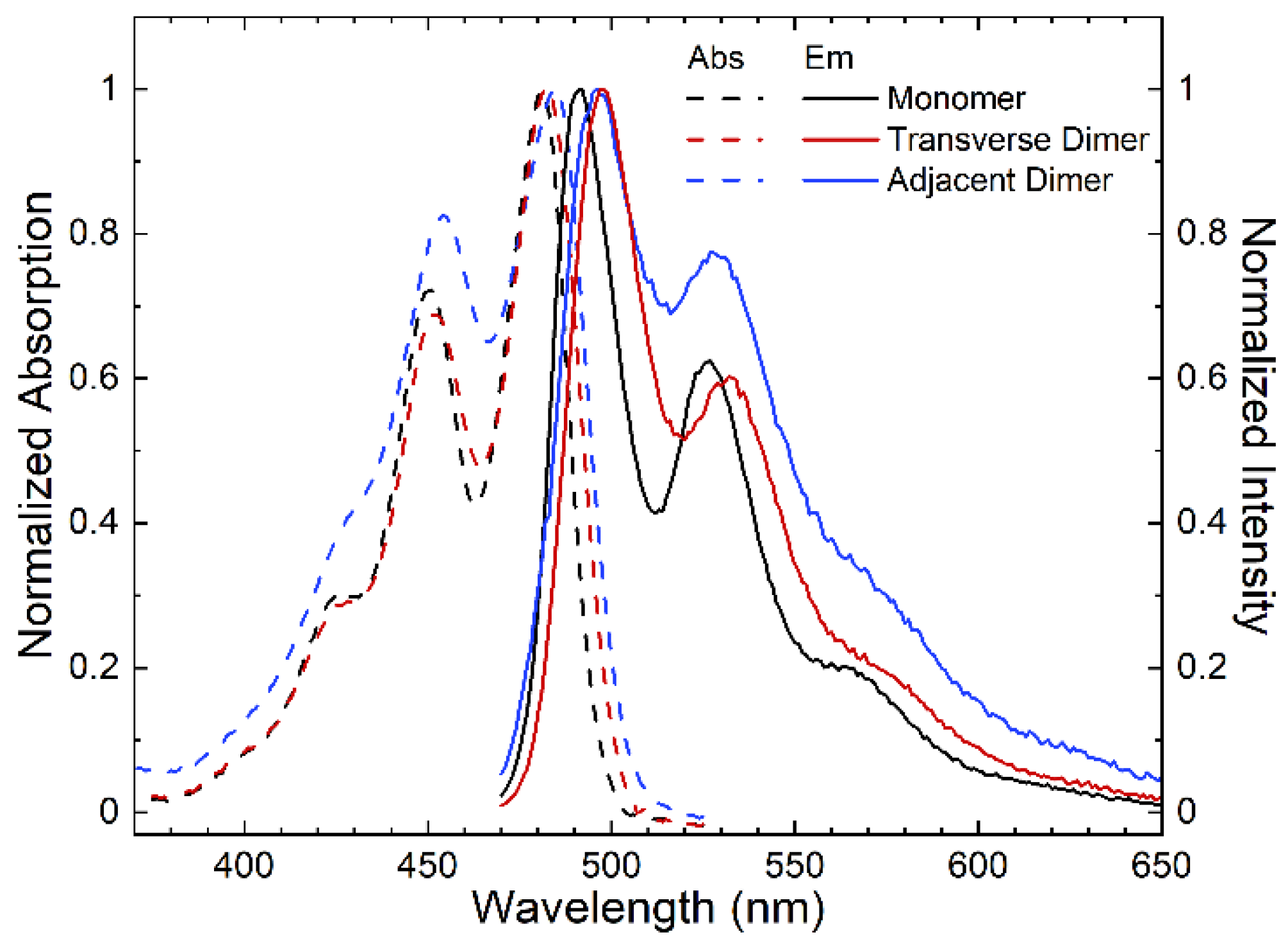
| Structure | λabs, 0–0 (nm) | λem, 0–0 (nm) | Stokes Shift (cm−1) | Stokes Shift (meV) | A0–0/A0–1 | I0–0/I0–1 |
|---|---|---|---|---|---|---|
| Monomer | 481 | 491 | 420 | 50 | 1.38 | 1.60 |
| Transverse Dimer | 482 | 496 | 590 | 70 | 1.45 | 1.67 |
| Adjacent Dimer | 485 | 495 | 420 | 50 | 1.21 | 1.29 |
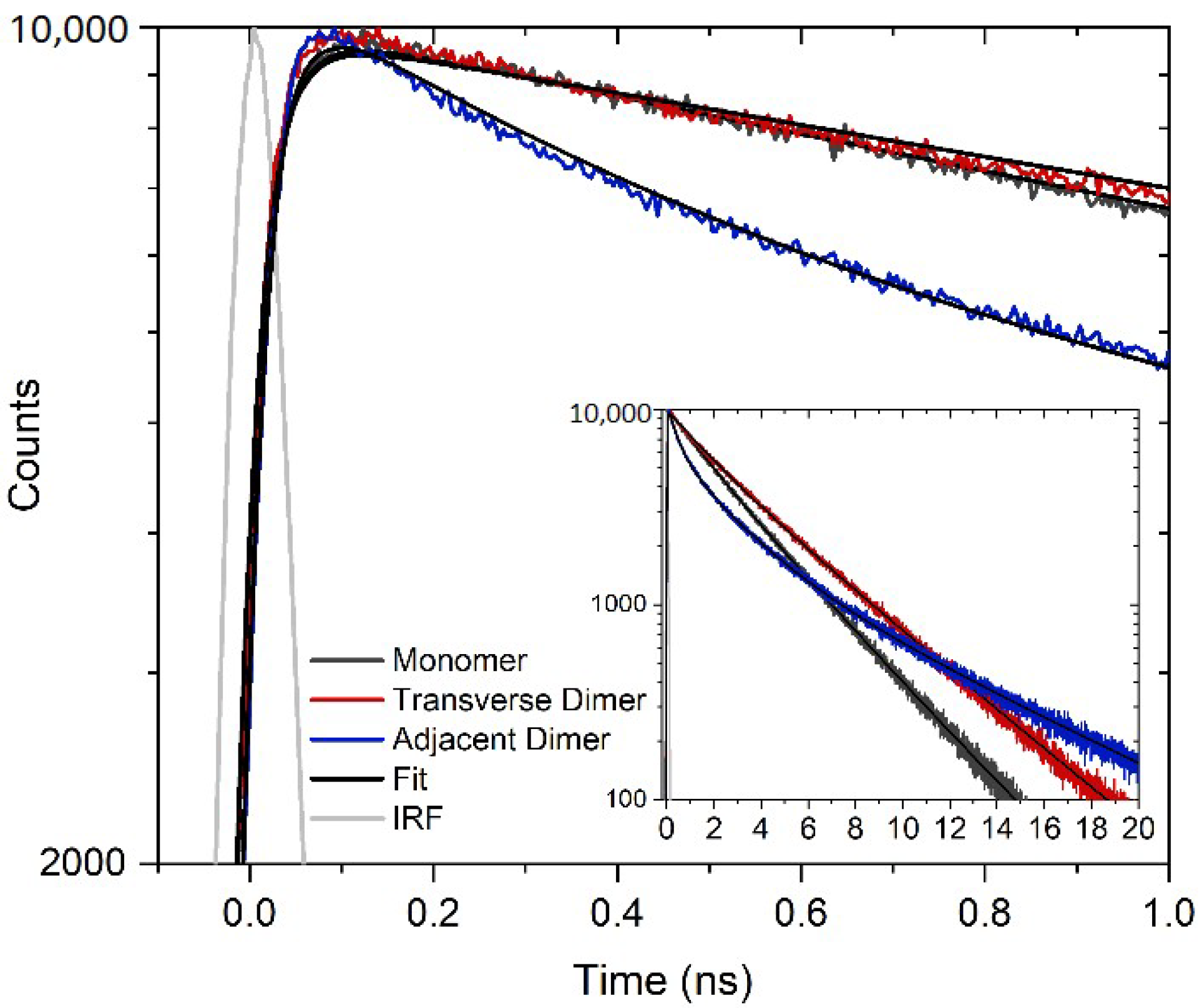
| Structure | τ1 (ns) | A1 (%) | τ2 (ns) | A2 (%) | τ3 (ns) | A3 (%) | τavg 2,3 (ns) |
|---|---|---|---|---|---|---|---|
| Monomer | - | - | 1.69 | 26 | 3.39 | 74 | 2.95 |
| Transverse Dimer | 0.28 | 12 | 2.50 | 31 | 4.49 | 57 | 3.79 |
| Adjacent Dimer | 0.39 | 39 | 2.21 | 40 | 7.25 | 21 | 3.89 |
| Structure | τ1 (ps) | τ2 (ps) |
|---|---|---|
| Monomer | - | >1500 |
| Transverse Dimer | 30 | >1500 |
| Adjacent Dimer | 50 | >1500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duncan, K.M.; Kellis, D.L.; Huff, J.S.; Barclay, M.S.; Lee, J.; Turner, D.B.; Davis, P.H.; Yurke, B.; Knowlton, W.B.; Pensack, R.D. Symmetry Breaking Charge Transfer in DNA-Templated Perylene Dimer Aggregates. Molecules 2022, 27, 6612. https://doi.org/10.3390/molecules27196612
Duncan KM, Kellis DL, Huff JS, Barclay MS, Lee J, Turner DB, Davis PH, Yurke B, Knowlton WB, Pensack RD. Symmetry Breaking Charge Transfer in DNA-Templated Perylene Dimer Aggregates. Molecules. 2022; 27(19):6612. https://doi.org/10.3390/molecules27196612
Chicago/Turabian StyleDuncan, Katelyn M., Donald L. Kellis, Jonathan S. Huff, Matthew S. Barclay, Jeunghoon Lee, Daniel B. Turner, Paul H. Davis, Bernard Yurke, William B. Knowlton, and Ryan D. Pensack. 2022. "Symmetry Breaking Charge Transfer in DNA-Templated Perylene Dimer Aggregates" Molecules 27, no. 19: 6612. https://doi.org/10.3390/molecules27196612
APA StyleDuncan, K. M., Kellis, D. L., Huff, J. S., Barclay, M. S., Lee, J., Turner, D. B., Davis, P. H., Yurke, B., Knowlton, W. B., & Pensack, R. D. (2022). Symmetry Breaking Charge Transfer in DNA-Templated Perylene Dimer Aggregates. Molecules, 27(19), 6612. https://doi.org/10.3390/molecules27196612