
Dianthiamides A–E, Proline-Containing Orbitides from Dianthus chinensis

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. General Experimental Procedures
3.3. Plant Material
3.4. Isolation and Purification of Compounds 1–5
3.5. Characterization of Compounds 1–5
3.6. Absolute Configuration of Amino Acids in 1–5 Using Marfey’s Method
3.7. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Oshima, Y.; Ohsawa, T.; Oikawa, K.; Konno, C.; Hikino, H. Structures of dianosides A and B, analgesic principles of Dianthus superbus var. longicalycinus herbs. Planta Med. 1984, 50, 40–43. [Google Scholar] [CrossRef]
- Oshima, Y.; Ohsawa, T.; Hikino, H. Structure of dianosides C, D, E and F, triterpenoid saponins of Dianthus superbus var. longicalycinus herb. Planta Med. 1984, 50, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Ohsawa, T.; Hikino, H. Structures of dianosides G, H and I, triterpenoid saponins of Dianthus superbus var. longicalycinus herbs. Planta Med. 1984, 50, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Tan, N.H.; Zhou, J.; Wu, H.M. Cyclopeptides from Dianthus superbus. Phytochemistry 1998, 49, 1453–1456. [Google Scholar] [CrossRef]
- Hsieh, P.W.; Chang, F.R.; Wu, C.C.; Wu, K.Y.; Li, C.M.; Chen, S.L.; Wu, Y.C. New cytotoxic cyclic peptides and dianthramide from Dianthus superbus. J. Nat. Prod. 2004, 67, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.W.; Chang, F.R.; Wu, C.C.; Li, C.M.; Wu, K.Y.; Chen, S.L.; Yen, H.F.; Wu, Y.C. Longicalycinin A, a new cytotoxic cyclic peptide from Dianthus superbus var. longicalycinus (MAXIM.) WILL. Chem. Pharm. Bull. 2005, 53, 336–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Zhang, F.M.; Yang, Y.B.; Yang, X.Q.; Cao, Q.E.; Ding, Z.T. A new cyclopeptide from Dianthus caryophyllus. Chin. Chem. Lett. 2008, 19, 193–195. [Google Scholar] [CrossRef]
- Tong, Y.; Luo, J.G.; Wang, R.; Wang, X.B.; Kong, L.Y. New cyclic peptides with osteoblastic proliferative activity from Dianthus superbus. Bioorg. Med. Chem. Lett. 2012, 22, 1908–1911. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Huang, M.; Wang, Z.; Zheng, Y.; Zeng, G.; He, W.; Tan, N. Cyclopentapeptides from Dianthus chinensis. J. Pept. Sci. 2015, 21, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Ponchet, M.; Martin-Tanguy, J.; Marais, A.; Poupet, A. Dianthramides A and B, two N-benzoylanthranilic acid derivatives from elicited tissues of Dianthus caryophyllus. Phytochemistry 1984, 23, 1901–1903. [Google Scholar] [CrossRef]
- Ponchet, M.; Favre-Bonvin, J.; Hauteville, M.; Ricci, P. Dianthramides (N-benzoyl and N-paracoumarylanthranilic acid derivatives) from elicited tissues of Dianthus caryophyllus. Phytochemistry 1988, 27, 725–730. [Google Scholar] [CrossRef]
- Luo, J.G.; Chen, X.; Kong, L.Y. Three new triterpenoid saponins from Dianthus superbus. Chem. Pharm. Bull. 2011, 59, 518–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Sugimoto, S.; Matsunami, K.; Otsuka, H. Dianthosaponins A-F, triterpene saponins, flavonoid glycoside, aromatic amide glucoside and γ-pyrone glucoside from Dianthus japonicus. Chem. Pharm. Bull. 2011, 59, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terahara, N.; Yamaguchi, M.A. 1H NMR spectral analysis of the malylated anthocyanins from Dianthus. Phytochemistry 1986, 25, 2906–2907. [Google Scholar] [CrossRef]
- Shimizu, M.; Hayashi, T.; Shimizu, K.; Morita, N. A pyran-type glycoside from Dianthus superbus var. longicalycinus. Phytochemistry 1982, 21, 245–247. [Google Scholar] [CrossRef]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, S.D.; Pinto, M.E.F.; Ferreira, D.; Bolzani, V.S. Biologically active orbitides from the Euphorbiaceae family. Planta Med. 2018, 84, 558–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houshdar Tehrani, M.H.; Gholibeikian, M.; Bamoniri, A.; Mirjalili, B.B.F. Cancer treatment by Caryophyllaceae-type cyclopeptides. Front. Endocrinol. 2021, 11, 600856. [Google Scholar] [CrossRef]
- Rubin, G.M.; Ding, Y. Recent advances in the biosynthesis of RiPPs from multicore-containing precursor peptides. J. Ind. Microbiol Biotechnol. 2020, 47, 659–674. [Google Scholar] [CrossRef]
- Gholibeikian, M.; Bamoniri, A.; HoushdarTehrani, M.H.; Fatemeh Mirjalili, B.B.; Bijanzadeh, H.R. Structure-activity relationship studies of longicalcynin A analogues, as anticancer cyclopeptides. Chem. Biol. Interact. 2020, 315, 108902. [Google Scholar] [CrossRef]
- Siemion, I.Z.; Wieland, T.; Pook, K.H. Influence of the distance of the proline carbonyl from the β and γ carbon on the 13C chemical shifts. Angew. Chem. Int. Ed. Engl. 1975, 14, 702–703. [Google Scholar] [CrossRef]
- Schmidt, G.; Grube, A.; Kock, M. Stylissamides A-D-New proline-containing cyclic heptapeptides from the marine sponge Stylissa caribica. Eur. J. Org. Chem. 2007, 24, 4103–4110. [Google Scholar] [CrossRef]
- Iwasaki, K.; Iwasaki, A.; Sumimoto, S.; Sano, T.; Hitomi, Y.; Ohno, O.; Suenaga, K. Croissamide, a proline-rich cyclic peptide with an N-prenylated tryptophan from a marine cyanobacterium Symploca sp. Tetrahedron Lett. 2018, 59, 3806–3809. [Google Scholar] [CrossRef]
- Zhan, K.X.; Jiao, W.H.; Yang, F.; Li, J.; Wang, S.P.; Li, Y.S.; Han, B.N.; Lin, H.W. Reniochalistatins A-E, cyclic peptides from the marine sponge Reniochalina stalagmitis. J. Nat. Prod. 2014, 77, 2678–2684. [Google Scholar] [CrossRef] [PubMed]
- Cândido-Bacani, P.D.M.; Figueiredo, P.D.O.; Matos, M.D.F.; Garcez, F.R.; Garcez, W.S. Cytotoxic orbitide from the latex of Croton urucurana. J. Nat. Prod. 2015, 78, 2754–2760. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Sudoh, Y.; Tsuchiya, Y.; Okuda, T.; Matsuura, N.; Motojima, A.; Oikawa, T.; Igarashi, Y. Hikiamides A-C, cyclic pentadepsipeptides from Fusarium sp. TAMA 456. J. Nat. Prod. 2015, 78, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Yamamoto, K.; Fukuda, T.; Shojima, A.; Nakayama, J.; Carro, L.; Trujillo, M.E. Arthroamide, a cyclic depsipeptide with quorum sensing inhibitory activity from Arthrobacter sp. J. Nat. Prod. 2015, 78, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Lee, J.W.; Jang, H.; Lee, H.L.; Kim, J.G.; Wu, W.; Lee, D.; Kim, E.H.; Kim, Y.; Hong, J.T.; et al. Dimeric- and trimeric sesquiterpenes from the flower of Inula japonica. Phytochemistry 2018, 155, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.J.; Lee, Y.J.; Park, C.W.; Chung, Y.B.; Kim, J.S.; Lee, M.K.; Shin, D.H. Evaluation of the physicochemical properties, pharmacokinetics, and in vitro anticancer effects of docetaxel and osthol encapsulated in methoxy poly(ethylene glycol)-b-poly(caprolactone) polymeric micelles. Int. J. Mol. Sci. 2020, 22, 231. [Google Scholar] [CrossRef] [PubMed]
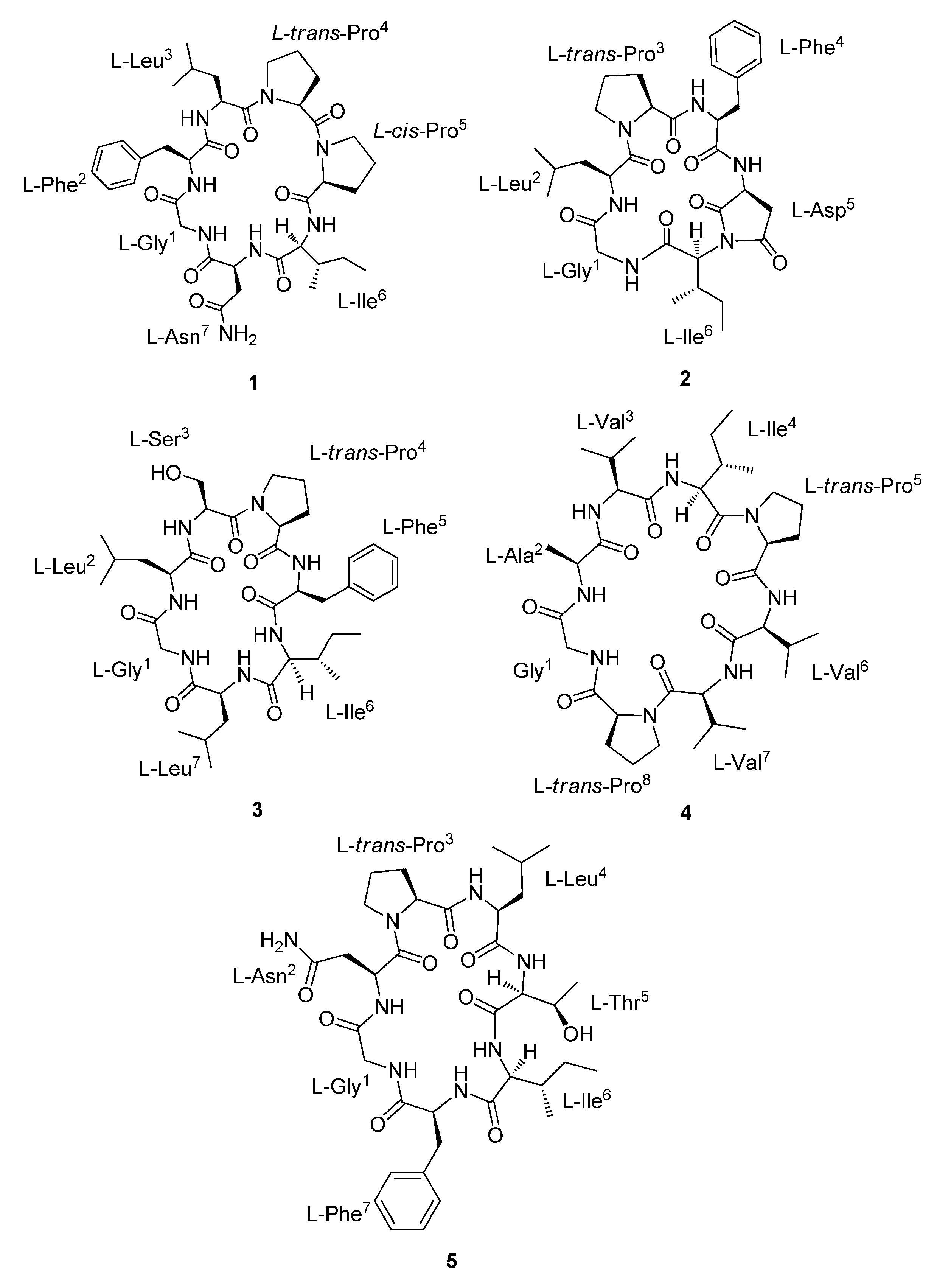
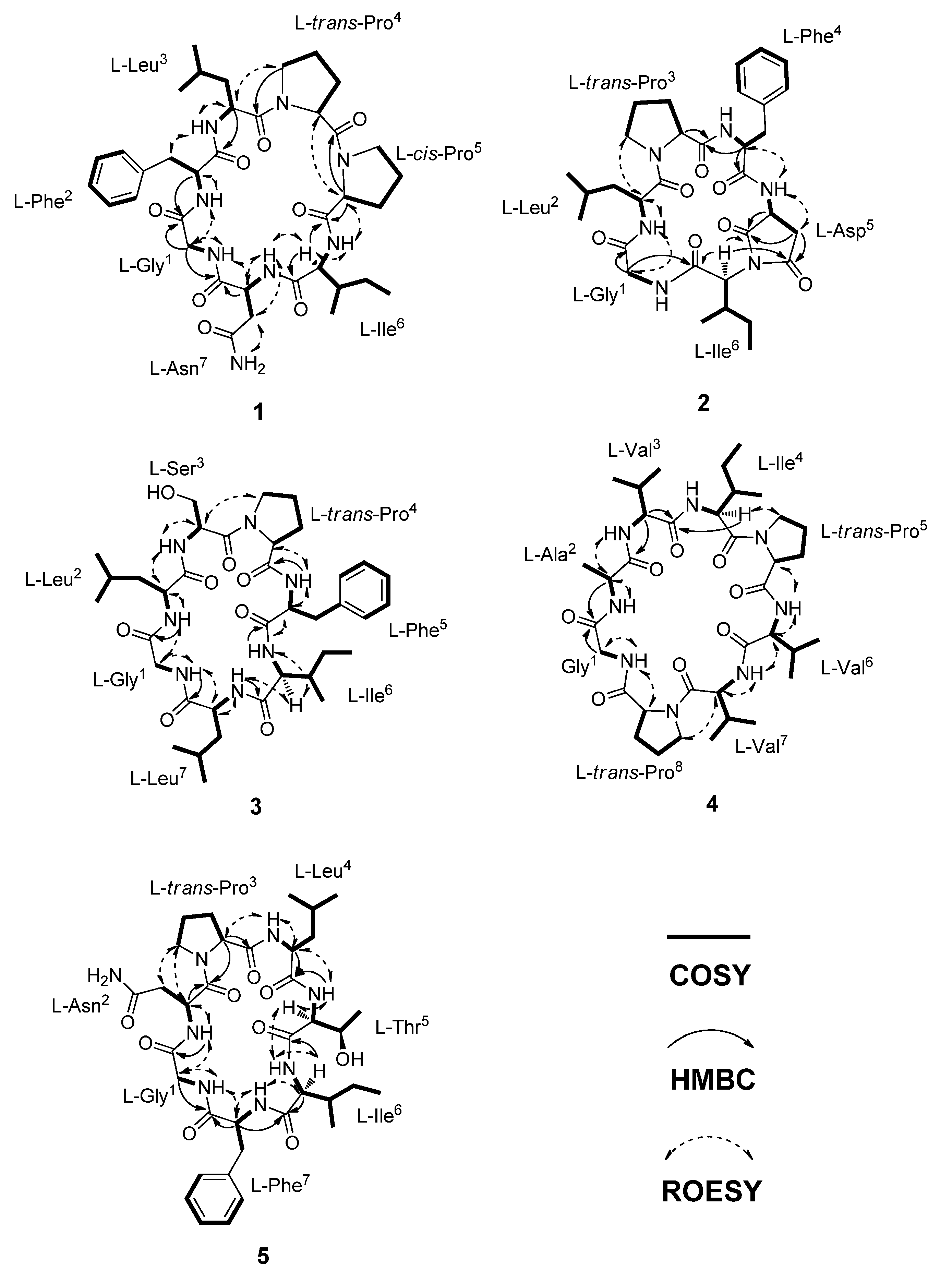
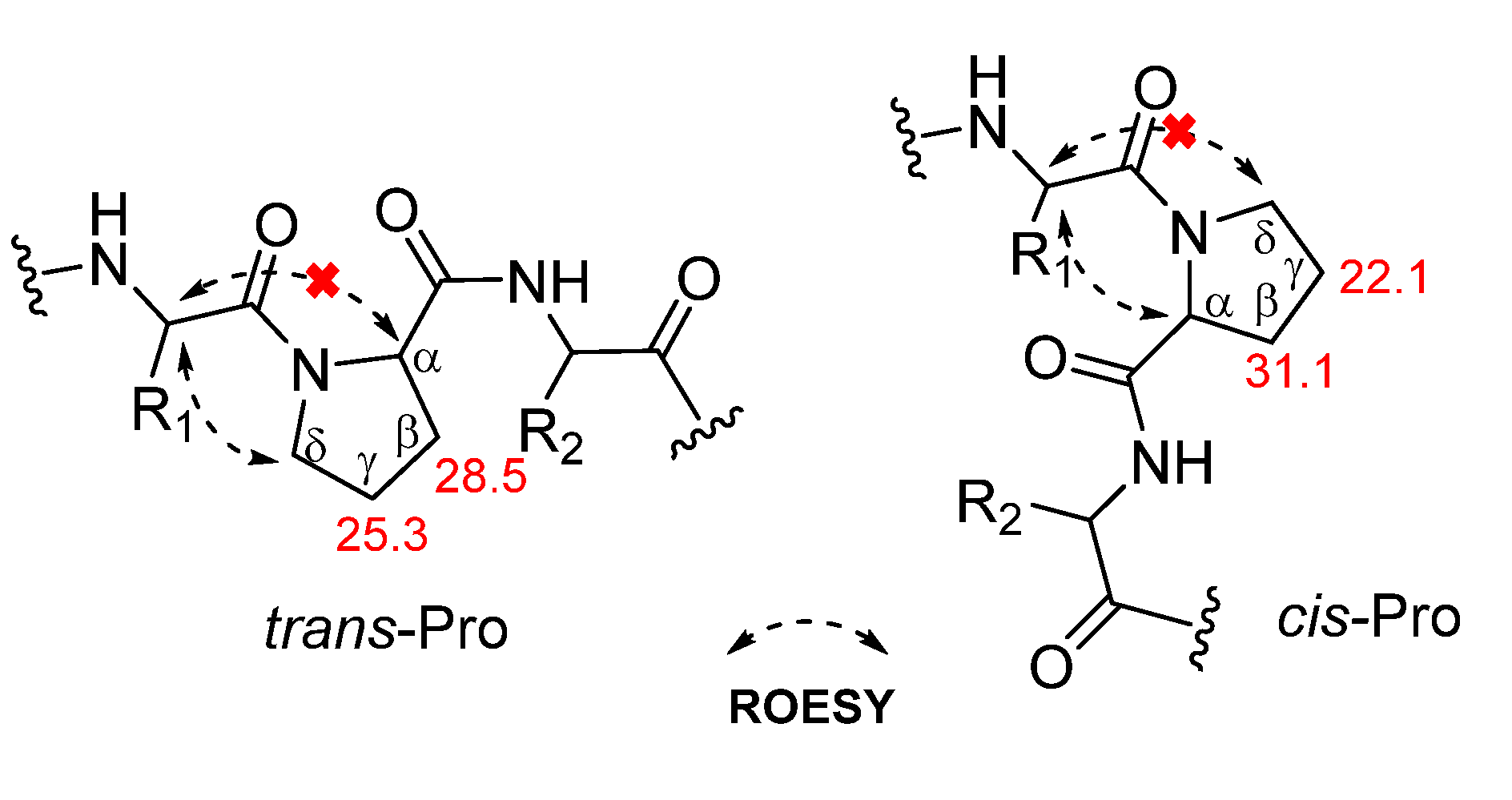

{kind=link}
{kind=link}
{kind=link}
| Position | δC | δH (J in Hz) | Position | δC | δH (J in Hz) |
|---|---|---|---|---|---|
| Gly1 | cis-Prob5 | ||||
| C=O | 169.8, C | C=O | 170.9, C | ||
| NH | 8.70 (t, 4.2) | α | 60.8, CH | 4.55 (d, 7.0) | |
| α | 44.4, CH2 | 3.55 (dd, 16.8, 4.2) | β | 31.1, CH2 | 2.33 (dd, 11.9, 7.0) |
| 3.40 (dd, 16.8, 5.6) | 1.92 (m) | ||||
| Phe2 | γ | 22.1, CH2 | 1.85 (m) | ||
| C=O | 171.1, C | 1.46 (m) | |||
| NH | 7.69 (d, 9.1) | δ | 46.4, CH2 | 3.46 (m) | |
| α | 56.0, CH | 4.38 (m) | 3.25 (t, 9.8) | ||
| β | 37.0, CH2 | 3.01 (m) | Ile6 | ||
| 2.97 (m) | C=O | 171.4, C | |||
| Phe-1′ | 138.4, C | NH | 8.49 (d, 7.7) | ||
| 2′,6′ | 129.1, CH | 7.15–7.27 (m) | α | 61.6, CH | 3.66 (dd, 9.8, 7.7) |
| 3′,5′ | 128.8, CH | 7.15–7.27 (m) | β | 35.3, CH | 2.05 (m) |
| 4′ | 126.8, CH | 7.15–7.27 (m) | γ-CH3 | 16.0, CH3 | 0.90 (t, 7.7) |
| Leu3 | |||||
| C=O | 168.8, C | γ-CH2 | 25.9, CH2 | 1.50 (m) | |
| NH | 7.28 (m) | 1.15 (m) | |||
| α | 48.6, CH | 4.48 (m) | δ-CH3 | 10.9, CH3 | 0.82 (d, 7.7) |
| β | 41.0, CH2 | 1.68 (m) | Asn7 | ||
| 1.19 (m) | C=O | 172.3, C | |||
| γ-CH | 24.6, CH | 1.51 (m) | NH | 7.26 (m) | |
| δ-CH3 | 23.6, CH3 | 0.85 (d, 6.3) | α | 49.6, CH | 4.18 (m) |
| δ-CH3 | 22.6, CH3 | 0.88 (d, 6.3) | β | 35.8, CH2 | 3.15 (m) |
| trans-Proa4 | 3.05 (m) | ||||
| C=O | 170.8, C | C=O | 173.4, C | ||
| α | 59.6, CH | 4.45 (t, 7.0) | NH2 | 7.97 (s) | |
| β | 28.5, CH2 | 2.27 (m) | 7.54 (s) | ||
| 1.64 (m) | |||||
| γ | 25.3, CH2 | 1.96 (m) | |||
| 1.83 (m) | |||||
| δ | 47.1, CH2 | 3.44 (m) |
| Position | δC | δH (J in Hz) | Position | δC | δH (J in Hz) |
|---|---|---|---|---|---|
| Gly1 | Phe4 | ||||
| C=O | 168.7, C | C=O | 170.7, C | ||
| NH | 8.68 (m) | NH | 8.68 (m) | ||
| α | 43.3, CH2 | 3.65 (t, 7.0) | α | 57.2, CH | 3.80 (m) |
| Leu2 | β | 33.8, CH2 | 3.33 (m) | ||
| C=O | 170.7, C | 3.30 (m) | |||
| NH | 6.50 (m) | Phe-1′ | 139.6, C | ||
| α | 48.6, CH | 4.51 (m) | 2′,6′ | 129.6, CH | 7.10–7.30 (m) |
| β | 40.5, CH2 | 1.40 (m) | 3′,5′ | 128.6, CH | 7.10–7.30 (m) |
| 1.20 (m) | 4′ | 126.6, CH | 7.10–7.30 (m) | ||
| γ-CH | 24.6, CH | 1.62 (m) | Asp5 | ||
| δ-CH3 | 23.8, CH3 | 0.91 (d, 7.0) | C=O | 176.2, C | |
| δ-CH3 | 21.3, CH3 | 0.93 (d, 7.0) | NH | 7.95 (d, 8.4) | |
| trans-Pro3 | α | 47.3, CH | 5.23 (m) | ||
| C=O | 171.7, C | β | 36.5, CH2 | 3.22 (m) | |
| α | 61.0, CH | 3.92 (t, 7.0) | 2.05 (d, 3.5) | ||
| β | 29.3, CH2 | 1.85 (m) | C=O | 177.2, C | |
| 1.67 (m) | Ile6 | ||||
| γ | 25.4, CH2 | 2.01 (m) | C=O | 167.0, C | |
| 1.83 (m) | α | 61.5, CH | 4.19 (d, 11.9) | ||
| δ | 47.5, CH2 | 3.68 (m) | β | 30.4, CH | 2.68 (m) |
| 3.42 (m) | γ-CH3 | 16.1, CH3 | 0.87 (t, 7.7) | ||
| γ-CH2 | 24.3, CH2 | 1.38 (m) | |||
| 0.95 (m) | |||||
| δ-CH3 | 10.5, CH3 | 0.79 (d, 7.7) |
| Position | δC | δH (J in Hz) | Position | δC | δH (J in Hz) |
|---|---|---|---|---|---|
| Gly1 | Phe5 | ||||
| C=O | 169.1, C | C=O | 170.7, C | ||
| NH | 8.91 (t, 4.9) | NH | 7.43 (d, 9.8) | ||
| α | 43.2, CH | 4.00 (m) | α | 54.6, CH | 4.38 (m) |
| 3.33 (m) | β | 38.4, CH2 | 3.21 (dd, 13.3, 3.5) | ||
| Leua2 | 2.62 (t, 13.3) | ||||
| C=O | 171.2, C | Phe-1′ | 138.4, C | ||
| NH | 8.10 (d, 10.5) | 2′,6′ | 129.4, CH | 7.18–7.31 (m) | |
| α | 53.0, CH | 4.52 (m) | 3′,5′ | 128.6, CH | 7.18–7.31 (m) |
| β | 43.8, CH2 | 1.42 (m) | 4′ | 126.8, CH | 7.18–7.31 (m) |
| 1.34 (m) | Ile6 | ||||
| γ-CH | 24.5, CH | 1.55 (m) | C=O | 171.6, C | |
| δ-CH3 | 22.9, CH3 | 0.87 (d, 6.3) | NH | 7.01 (d, 9.1) | |
| δ-CH3 | 22.2, CH3 | 0.84 (d, 6.3) | α | 56.4, CH | 4.35 (m) |
| Ser3 | β | 37.9, CH | 1.71 (m) | ||
| C=O | 168.9, C | γ-CH3 | 15.1, CH3 | 0.82 (t, 7.7) | |
| NH | 8.73 (d, 7.0) | γ-CH2 | 24.7, CH2 | 1.39 (m) | |
| α | 54.2, CH | 4.89 (br s) | 1.00 (m) | ||
| β | 61.9, CH2 | 4.24 (m) | δ-CH3 | 11.0, CH3 | 0.81 (d, 7.7) |
| 3.71 (d, 10.5) | Leub7 | ||||
| trans-Pro4 | C=O | 172.3, C | |||
| C=O | 170.5, C | NH | 8.71 (br s) | ||
| α | 62.2, CH | 3.98 (m) | α | 54.2, CH | 3.87 (m) |
| β | 29.0, CH2 | 1.97 (m) | β | 40.1, CH2 | 1.47 (m) |
| 1.24 (m) | γ-CH | 24.4, CH | 1.54 (m) | ||
| γ | 25.8, CH2 | 1.79 (m) | δ-CH3 | 23.4, CH3 | 0.86 (m) |
| 1.68 (m) | δ-CH3 | 22.6, CH3 | 0.93 (m) | ||
| δ | 47.8, CH2 | 3.91 (m) | |||
| 3.42 (m) |
| Position | δC | δH (J in Hz) | Position | δC | δH (J in Hz) |
|---|---|---|---|---|---|
| Gly1 | trans-Pro5 | ||||
| C=O | 168.4, C | C=O | 170.7, C | ||
| NH | 8.97 (t, 4.9) | α | 62.0, CH | 4.29 (m) | |
| α | 43.2, CH | 3.84 (dd, 14.7, 4.9) | β | 27.6, CH2 | 2.11 (m) |
| 3.33 (dd, 14.7, 4.2) | 1.91 (m) | ||||
| Ala2 | γ | 25.0, CH2 | 1.89 (m) | ||
| C=O | 173.1, C | 1.79 (m) | |||
| NH | 7.30 (d, 7.0) | δ | 48.2, CH2 | 3.79 (m) | |
| α | 47.2, CH | 4.62 (t, 7.0) | 3.62 (m) | ||
| β | 19.6, CH3 | 1.42 (d, 7.0) | Val6 | ||
| Val3 | C=O | 171.6, C | |||
| C=O | 171.4, C | NH | 8.24 (d, 7.0) | ||
| NH | 7.99 (d, 4.9) | α | 60.0, CH | 3.89 (dd, 7.0, 4.2) | |
| α | 60.8, CH | 3.71 (t, 4.9) | β | 29.3, CH | 2.23 (m) |
| β | 28.7, CH | 2.14 (m) | γ-CH3 | 20.1, CH3 | 0.90 (m) |
| γ-CH3 | 19.9, CH3 | 0.92 (d, 6.3) | γ-CH3 | 18.4, CH3 | 0.85 (d, 5.6) |
| γ-CH3 | 19.0, CH3 | 0.93 (d, 6.3) | Val7 | ||
| Ile4 | C=O | 169.8, C | |||
| C=O | 173.0, C | NH | 7.16 (d, 7.0) | ||
| NH | 6.70 (d, 7.7) | α | 55.6, CH | 4.43 (t, 7.0) | |
| α | 54.7, CH | 4.48 (d, 7.7) | β | 31.1, CH | 2.02 (m) |
| β | 35.9, CH | 1.81 (m) | γ-CH3 | 19.9, CH3 | 0.86 (m) |
| γ-CH3 | 15.6, CH3 | 0.88 (t, 5.6) | γ-CH3 | 18.4, CH3 | 0.75 (d, 5.6) |
| γ-CH2 | 24.3, CH2 | 1.49 (m) | trans-Pro8 | ||
| 1.08 (m) | C=O | 173.0, C | |||
| δ-CH3 | 11.0, CH3 | 0.83 (d, 7.0) | α | 60.8, CH | 4.18 (m) |
| β | 29.4, CH2 | 2.08 (m) | |||
| 1.74 (m) | |||||
| γ | 25.4, CH2 | 1.99 (m) | |||
| 1.84 (m) | |||||
| δ | 47.9, CH2 | 3.76 (m) | |||
| 3.53 (m) |
| Position | δC | δH (J in Hz) | Position | δC | δH (J in Hz) |
|---|---|---|---|---|---|
| Gly1 | Thr5 | ||||
| C=O | 169.2, C | C=O | 170.7, C | ||
| NH | 8.01 (m) | NH | 7.57 (d, 9.1) | ||
| α | 43.6, CH2 | 4.02 (m) | α | 57.0, CH | 4.67 (m) |
| 3.31 (m) | β | 68.7, CH | 4.25 (m) | ||
| Asn2 | γ-CH3 | 19.4, CH3 | 1.03 (d, 6.3) | ||
| C=O | 171.0, C | OH | 5.26 (d, 6.3) | ||
| NH | 7.15 | Ile6 | |||
| α | 48.8, CH | 4.79 (dd, 14.0, 7.0) | C=O | 171.1, C | |
| β | 37.2, CH2 | 2.75 (m) | NH | 8.00 (m) | |
| 2.57 (m) | α | 59.2, CH | 3.95 (t, 5.6) | ||
| C=O | 172.2, C | β | 35.7, CH | 1.88 (m) | |
| NH2 | 7.75 (br s) | γ-CH3 | 15.9, CH3 | 0.70 (d, 7.0) | |
| 7.20 (m) | γ-CH2 | 23.9, CH2 | 0.97 (m) | ||
| trans-Pro3 | δ-CH3 | 11.8, CH3 | 0.68 (d, 7.0) | ||
| C=O | 171.5, C | Phe7 | |||
| α | 62.1, CH | 4.11 (t, 7.7) | C=O | 171.6, C | |
| β | 29.7, CH2 | 2.20 (m) | NH | 7.78 (d, 2.1) | |
| 1.71 (m) | α | 54.9, CH | 4.41 (m) | ||
| γ | 25.3, CH2 | 1.92 (m) | β | 37.3, CH2 | 3.13 (dd, 14.0, 5.6) |
| δ | 47.5, CH2 | 3.73 (m) | 2.84 (dd, 14.0, 9.1) | ||
| 3.56 (m) | Phe-1′ | 138.0, C | |||
| Leu4 | 2′,6′ | 129.4, CH | 7.17–7.29 (m) | ||
| C=O | 172.2, C | 3′,5′ | 128.7, CH | 7.17–7.29 (m) | |
| NH | 8.39 (br s) | 4′ | 126.8, CH | 7.17–7.29 (m) | |
| α | 52.4, CH | 4.00 (m) | |||
| β | 39.2, CH2 | 1.79 (m) | |||
| 1.63 (m) | |||||
| γ | 25.1, CH | 1.54 (m) | |||
| δ-CH3 | 23.7, CH3 | 0.88 (d, 7.0) | |||
| δ-CH3 | 21.3, CH3 | 0.82 (d, 7.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.W.; Kim, J.G.; Han, J.S.; Cho, Y.B.; Lee, Y.J.; Lee, D.; Shin, D.H.; Hong, J.T.; Lee, M.K.; Hwang, B.Y. Dianthiamides A–E, Proline-Containing Orbitides from Dianthus chinensis. Molecules 2021, 26, 7275. https://doi.org/10.3390/molecules26237275
Lee JW, Kim JG, Han JS, Cho YB, Lee YJ, Lee D, Shin DH, Hong JT, Lee MK, Hwang BY. Dianthiamides A–E, Proline-Containing Orbitides from Dianthus chinensis. Molecules. 2021; 26(23):7275. https://doi.org/10.3390/molecules26237275
Chicago/Turabian StyleLee, Jin Woo, Jun Gu Kim, Jae Sang Han, Yong Beom Cho, Yu Jin Lee, Dongho Lee, Dae Hwan Shin, Jin Tae Hong, Mi Kyeong Lee, and Bang Yeon Hwang. 2021. "Dianthiamides A–E, Proline-Containing Orbitides from Dianthus chinensis" Molecules 26, no. 23: 7275. https://doi.org/10.3390/molecules26237275
APA StyleLee, J. W., Kim, J. G., Han, J. S., Cho, Y. B., Lee, Y. J., Lee, D., Shin, D. H., Hong, J. T., Lee, M. K., & Hwang, B. Y. (2021). Dianthiamides A–E, Proline-Containing Orbitides from Dianthus chinensis. Molecules, 26(23), 7275. https://doi.org/10.3390/molecules26237275