Abstract
Rearrangements of o-tolyl aryl ethers, amines, and sulfides with the Grubbs–Stoltz reagent (Et3SiH + KOtBu) were recently announced, in which the ethers were converted to o-hydroxydiarylmethanes, while the (o-tol)(Ar)NH amines were transformed into dihydroacridines. Radical mechanisms were proposed, based on prior evidence for triethylsilyl radicals in this reagent system. A detailed computational investigation of the rearrangements of the aryl tolyl ethers now instead supports an anionic Truce–Smiles rearrangement, where the initial benzyl anion can be formed by either of two pathways: (i) direct deprotonation of the tolyl methyl group under basic conditions or (ii) electron transfer to an initially formed benzyl radical. By contrast, the rearrangements of o-tolyl aryl amines depend on the nature of the amine. Secondary amines undergo deprotonation of the N-H followed by a radical rearrangement, to form dihydroacridines, while tertiary amines form both dihydroacridines and diarylmethanes through radical and/or anionic pathways. Overall, this study highlights the competition between the reactive intermediates formed by the Et3SiH/KOtBu system.
1. Introduction
The first aryl migration reaction was published by Wieland in 1911 [1]. Since then, many studies have graced the literature, presenting synthetically useful transformations [2,3]. The Smiles rearrangement, discovered in 1930 [4,5], an intramolecular SNAr reaction taking place at the ipso position of a substituted aromatic system, is an example of an aryl migration under basic conditions (Scheme 1) [6,7]. This SNAr reaction, like the vicarious nucleophilic substitution, investigated by the team of Mąkosza [8], often features activation using electron-withdrawing groups, usually a nitro group [6,7,9,10,11,12]. However, the first identification of this rearrangement by Smiles was on naphthalene derivatives which lacked activating groups [5].
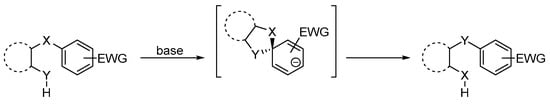
Scheme 1.
General representation of a Smiles rearrangement [9].
The Truce–Smiles rearrangement, discovered in 1958, is a derivative of the Smiles rearrangement in which the attacking atom is a carbanion [7]. The rearrangement of unactivated substrates in the Smiles rearrangements requires the use of strong bases and forcing conditions [13,14,15,16]. The Truce–Smiles rearrangement does not require the use of activating groups in the migrating aryl group, however, forcing conditions are still necessary for the generation of a carbanion [7,9,17]. The Smiles rearrangement, originally a two-electron process, has since then also been developed as a radical rearrangement [3,9,17,18,19,20,21,22,23,24,25,26,27]. Very recently, a series of radical cation Smiles rearrangements was reported [19] and a DFT study of the radical Smiles rearrangement has also been published [28].
In our recent publication [29], o-tolylaryl ethers 1 and sulfides 3 underwent rearrangement to diarylmethanes 2 and 4, respectively, while o-tolylaryl amine 5 yielded oxidatively cyclised products 6 and 7 (Scheme 2A). The reactions were mediated by triethylsilane and potassium tert-butoxide. This novel reagent pair was first reported by Grubbs and Stoltz in 2013 [30]. The original discovery presented a new method for the cleavage of strong C–O bonds in aryl ethers (8→9, Scheme 2B). Since then, the reagent pair has proven to be remarkably versatile by facilitating the wide range of transformations shown in Scheme 2B. Three reaction intermediates 24a–26a (Scheme 2C) are proposed to be responsible for the diverse chemistry observed [30,31,32,33,34,35,36,37,38]. Triethylsilyl radicals 24a were previously identified by detection of a TEMPO-SiEt3 adduct [32]. In addition, a ReactIR study on the combination of triethylsilane and potassium tert-butoxide had revealed the formation of a new species in situ, suggested to be pentavalent silicate 25a. [33]. This intermediate can be a source of a hydrogen atom or a hydride ion [33,34]. Smith et al. proposed radical anion 26a as an intermediate in the debenzylation of N-benzylindoles [35]. Accordingly, substrates treated with the triethylsilane/potassium tert-butoxide system are subjected to radical, base, hydrogen atom transfer, hydride ion, and electron transfer chemistry simultaneously, allowing for diverse reaction outcomes and mechanisms. Following our publication [29], we decided to launch a computational and experimental study to understand the difference in reactivity between the ether and amine substrate classes. The results of this investigation are presented within this paper.
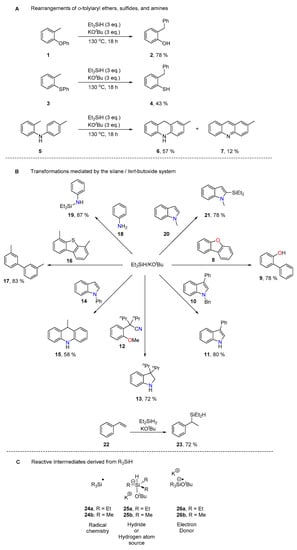
Scheme 2.
Chemistry of the Et3SiH/KOtBu system. (A) Rearrangements of o-tolylaryl ethers, sulfides, and amines, (B) Transformations mediated by the silane/tert-butoxide system, (C) Reactive Intermediates derived from R3SiH.
Theoretical details: DFT calculations were carried out using the M06-2X functional [39,40] with the 6-311++G(d,p) [41,42,43,44] basis set on all atoms. All calculations were performed using the C-PCM [44] implicit solvent model with parameters for triethylamine as solvent. No silane (Me3SiH or Et3SiH) solvents are parametrised in Gaussian 16, so triethylamine was chosen as the closest model to actual silane solvent since it has a similar dielectric constant (ε = 2.3832) compared to triethylsilane (ε = 2.323) [45]. All calculations were performed in Gaussian 16 [46] at 403.15 K. While experimental reactions used triethylsilane, yielding intermediates 24a–26a, theoretical studies made use of the corresponding trimethylsilane-derived intermediates, 24b–26b which were used for computational economy.
2. Results and Discussion
The o-tolylaryl ethers, represented by 1, were considered first (Scheme 3). Thus, after hydrogen atom abstraction by triethylsilyl radicals 24a to form benzyl radical 27, two possibilities for cyclisation were considered. 5-Exo-trig cyclisation gives the spiro intermediate 28, which then fragments to yield phenoxyl radical 29. This species is transformed to the isolated product 2 either by electron transfer followed by protonation, or by hydrogen atom transfer. In the alternative route, benzyl radical 27 undergoes a 6-aryl cyclisation (we prefer to refer to such cyclisations as ‘6-aryl’, since they could potentially be regarded as 6-exo or 6-endo depending on the initial Kekulé representation of the Ph group in 27) to give cyclohexadienyl radical 30, which likely suffers rapid deprotonation by either KOtBu or pentavalent silicate 25b to form radical anion 31 [47]. To proceed to product 2, this would be followed by C–O fragmentation to give distal radical anion 32. Hydrogen atom abstraction and protonation would then yield product 2. Scheme 3 reports the energy changes for the two competing cyclisation steps. The 6-aryl cyclisation is favoured here, with a lower transition state for 27→30 (22.5 kcal mol−1) than for 27→28 (25.2 kcal mol−1). In addition, the formation of 30 is less endergonic (1.8 kcal mol−1) compared to 28 (7.3 kcal mol−1). Based on these results, the 6-aryl cyclisation is kinetically and thermodynamically favoured; however, the steps following either cyclisation mode towards the product are exergonic (28→29, 30→31). Given these figures, and the accuracy of computational predictions (accuracy to within 2.0 kcal mol−1 [39]) one might expect that the 6-aryl cyclisation is favoured or that both cyclisation routes are in contention.
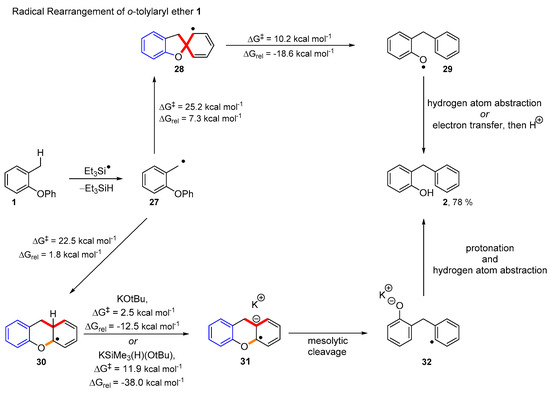
Scheme 3.
Energy barriers and relative energy changes for rearrangement of o-tolylaryl ether 1.
However, our recent paper showed that the rearranged ethers must arise only by the 5-exo cyclisation route using substrates 33 (Scheme 4). In these cases, a different product 34 or 39, would arise, depending on the cyclisation mode. Thus, 5-exo cyclisation of 35 would give spiro intermediate 36, resulting in product 34, where the R group is para- to the benzylic CH2. Alternatively, 6-aryl cyclisation of 35 would lead to product 39, where the relationship is meta. The outcome of these experiments was that products 34 were isolated and no 39 was ever detected. Therefore, the laboratory reaction must proceed through a 5-exo cyclisation. The discrepancy between this result and the energy-based predictions of Scheme 3 suggested that further mechanistic possibilities should be considered.
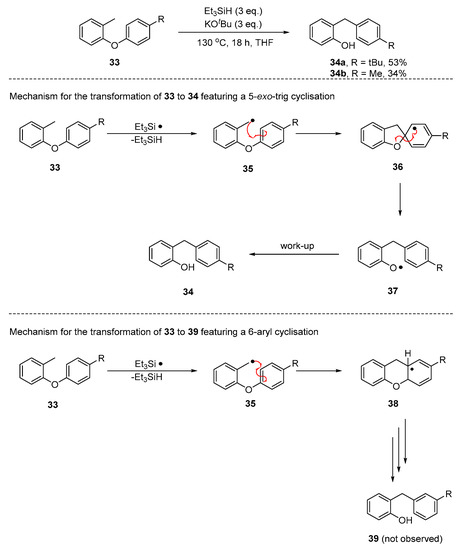
Scheme 4.
Reaction of o-tolylaryl ether 33 with the Et3SiH/KOtBu [25].
The simplest change to consider was how the competition between the two modes of cyclisation would be affected by complexation to a potassium cation. Experiments and computational evidence testify to the important role that π-cation complexes involving K+ ions can have on organic reactions of aryl substrates [37,48,49,50,51,52].
Scheme 5 indicates that the presence of a potassium cation does affect the energy profile of the reactions, but with the numbers still favouring the 6-aryl case (42→43) slightly over 5-exo (40→41). Thus, the computational results did not reflect the experimental results.

Scheme 5.
Probing potential cation-π interactions.
Accordingly, the investigation of the mechanism was extended to anionic intermediates, and pathways to access benzyl anions were considered. As mentioned above, radical anion 26a has been proposed as an intermediate that is formed from heating triethylsilane and KOtBu. According to our computational studies, 26b (without a potassium counterion) is an extremely strong electron donor [31,53], with Eox = −3.74 V vs. SCE (MeCN). Therefore, it would be capable of reducing an intermediate benzylic radical [Ered = −1.43 V vs. SCE (MeCN)] [54] to an anion. This single electron transfer (SET) was probed using the Nelsen Four-Point method [55] (Scheme 6).

Scheme 6.
SET reduction of benzylic radical 27 to benzylic anion 44.
The reduction of benzyl radical 27 was almost barrierless with an activation energy of 0.3 kcal mol−1. The reduction was also exergonic and, so, it is likely to happen in situ prior to the cyclisation. Therefore, it was appropriate to explore the energy profiles for cyclisations of benzyl anions.
Anion 44 is likely to complex with a potassium cation in situ to form a salt, 46. This salt was used to investigate the two cyclisation modes available to it (Scheme 7).
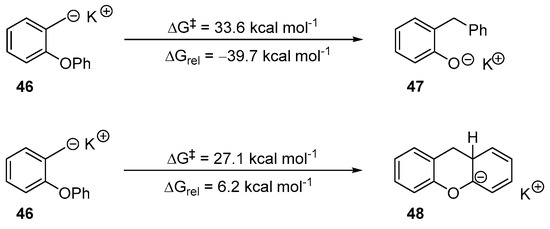
Scheme 7.
Examination of the two cyclisation modes available to salt 46.
Thermodynamics predict that the 5-exo-trig cyclisation [ΔGrel = −39.7 kcal mol−1] leading to 47 is favoured over the 6-aryl cyclisation [ΔGrel = +6.2 kcal mol−1] that would lead to 48. The activation energy for the 5-exo-trig pathway (ΔG‡ = 33.6 kcal mol−1) is achievable under the conditions of the reaction. The 6-aryl cyclisation does possess a lower activation energy favouring it as the kinetic cyclisation. However, it is also endergonic by 6.2 kcal mol−1. The experimental results have already shown that this cyclisation mode is not featured in the products (Scheme 3). Therefore, if it occurs, intermediate 48 reverts to intermediate 46 which undergoes an irreversible 5-exo-trig cyclisation.
Our computations also show that this Truce–Smiles rearrangement from 46 to 47 is concerted. Generally, Smiles and Truce–Smiles rearrangements proceed through the formation of a spiro intermediate, which subsequently undergoes a ring-opening to yield the product [6,17]. Mechanisms featuring a spiro transition state rather than a spiro intermediate have been proposed [7], with several examples being identified by Clayden et al. for the Smiles and Truce–Smiles rearrangements [56,57,58,59,60,61,62]. Concerted reaction pathways have also been identified for other Smiles-type rearrangements [63,64,65]. In our case, the conversion of salt 46 to 47 presents a new example of a concerted Truce–Smiles rearrangement. This was confirmed through an intrinsic reaction coordinate (IRC) calculation which displayed the simultaneous contraction of the C–C bond being formed and the elongation C–O bond being broken. Attempts to optimise a spiro intermediate all resulted in the ring-opening of the dihydrofuran ring yielding salt 47.
The formation of anion 46 through a radical-polar crossover from radical 27 however, is not the only possible route for formation of 46, and so we considered the direct deprotonation of the methyl group of aryl ether 1 by either pentavalent silicate 25 or KOtBu (Scheme 8). The initiation route featuring pentavalent silicate 25b was found to be exergonic (ΔGrel = −10.7 kcal mol−1) with an attainable activation energy (ΔG‡ = 23.6 kcal mol−1), making it a competitive initiation route with the radical-polar crossover pathway. KOtBu was also identified as a base potentially capable of initiating the reaction; the deprotonation has a surmountable barrier, but it is accompanied by an endergonic change in energy disfavouring the formation of salt 46. The following concerted Truce–Smiles rearrangement is sufficiently exergonic to push the reaction forward. Hence, a control experiment using only KOtBu and aryl ether 1 was carried out to probe this possibility. However, the reaction did not yield the product and only starting material was recovered. This result rules out KOtBu as a base capable of initiating the reaction via direct deprotonation of the methyl group of the o-tolyl ring. Complete reaction coordinate diagrams utilising both initiation routes are shown in the SI (Figures S3 and S4).
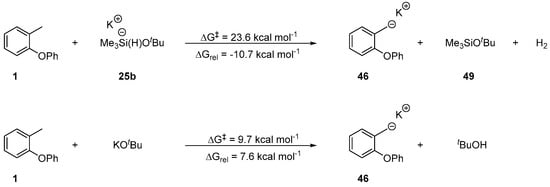
Scheme 8.
Anionic rearrangement route for 1 utilising pentavalent silicate 25b or KOtBu as the active base.
In summary, o-tolyl aryl ethers were identified to yield o-hydroxydiarylmethanes through a concerted Truce–Smiles rearrangement. The o-tolyl anion responsible for the rearrangement is generated through two competitive pathways: (i) a radical polar crossover route featuring a SET reduction of the initial benzylic radical formed via hydrogen atom abstraction by a triethylsilyl radical 24a (Scheme 6), (ii) the direct deprotonation of the o-tolyl methyl group by pentavalent silicate 25a (Scheme 8). The carbanion generated by these initiation pathways has the option to undergo either a 5-exo-trig cyclisation or a 6-aryl cyclisation. The former cyclisation mode is supported by experimental evidence from the treatment of strategically substituted aryl ether 33 (Scheme 4). Computationally, the 6-aryl cyclisation was established to be kinetically favoured over the 5-exo-trig cyclisation. However, the 6-aryl cyclisation was ruled out based on experimental evidence (Scheme 4). This is compatible with an endergonic and reversible 6-aryl cyclisation, which ultimately results in the carbanion undergoing the thermodynamically favoured irreversible 5-exo trig cyclisation (Scheme 7).
Nitrogen series: Our recent experimental findings showed that the structure of the o-tolyl amine substrate governs which type(s) of product are formed. When the starting amine is secondary (50a) or when it has a group bonded to the nitrogen, as in 51a, which is cleavable under the reaction conditions, only acridine-type products are formed (Scheme 9). To test what happens when the starting amine is tertiary, the N-methyl amine 52 was prepared and subjected to the reaction conditions. This yielded both dihydroacridine 55 (10%) and diarylmethane 56 (14%).
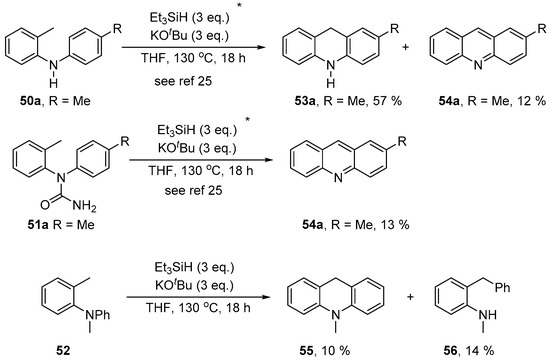
Scheme 9.
Treatment of o-tolyl aryl amines with the Et3SiH/KOtBu system.
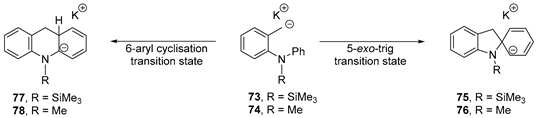
Substrates 50a and 51a will be considered first. Under strongly basic conditions, they are converted to potassium salt 57a. A radical mechanism for the transformation of salt 57 was initially probed (Scheme 10). For computational economy, the simpler case 57b, derived from 50b and 51b was explored. Benzyl radical 58b, formed via hydrogen atom abstraction by a trimethylsilyl radical (ΔG‡ = 19.4 kcal mol−1; ΔGrel = 0.9 kcal mol−1) could undergo either a 5-exo-trig cyclisation to 59b or a 6-aryl cyclisation to 61b. The latter is preferred, having a lower activation (ΔG‡ = 22.8 kcal mol−1) and a favourable change in Gibbs free energy (ΔGrel = −2.5 kcal mol−1) versus the 5-exo-trig cyclisation mode (ΔG‡ = 31.5 kcal mol−1, (ΔGrel = 11.7 kcal mol−1). The 6-aryl cyclisation intermediate 61b is subsequently deprotonated by either pentavalent silicate 25a or KOtBu, yielding the corresponding radical anion 62b. Oxidation and protonation of 62 on workup yields dihydroacridine 53b which can be further oxidised by air during purification to yield acridine 54b.
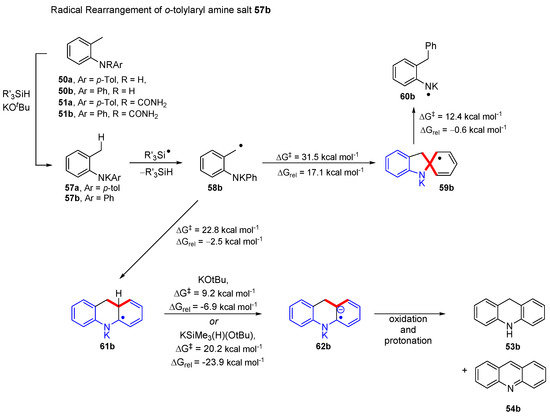
Scheme 10.
Energy barriers and relative changes in energy for rearrangement of o-tolylaryl amine salts 57.
Having studied the behaviour of the benzyl radicals, the next stage was to study the corresponding benzyl anions. These might be formed by: (i) reduction of the initially formed benzyl radical to a benzyl anion by single electron transfer and (ii) formation of the benzyl anion by direct deprotonation of the methyl group of the o-tolyl ring. Both of these routes were also now investigated for salt 57b (Scheme 11).
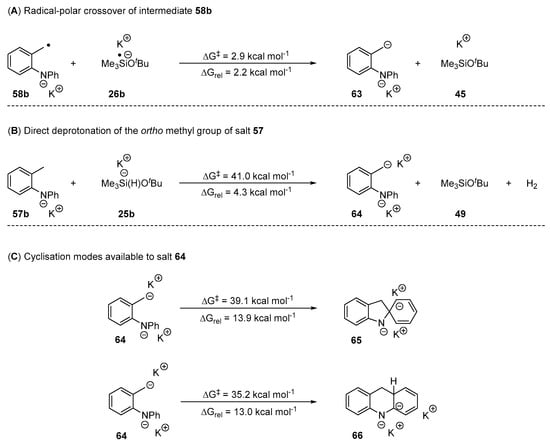
Scheme 11.
(A) Investigation of a radical-polar crossover of benzylic radical 58b to anion 63 via SET from silyl radical anion 26b. (B) Investigation into the direct deprotonation of the ortho methyl group of salt 57b by pentavalent silicate 25b. (C) Investigation of the two cyclisation modes available to salt 64.
The conversion of 58b to 63 by electron transfer (Nelsen Four-Point method) [55] was endergonic (Scheme 11A). In solution, the product anion might be rapidly stabilised by complexation with a potassium cation to form 64 (for an analogous stabilisation, see Figure S14). The alternative route to the benzyl anion 64 utilising pentavalent silicate 25b as base, was also found to be unproductive, as the activation energy (ΔG‡ = 41.0 kcal mol−1) exceeded the attainable limit at 130 °C. Assuming 64 was formed by the electron transfer route, its cyclisation by 5-exo-trig or 6-aryl cyclisation was not feasible due to the high activation barriers in both cases (Scheme 11C); this rules out an anionic cyclisation mechanism for o-tolylaryl amines that are converted to the analogous potassium salt 57 under the reaction conditions. Therefore, o-tolyl aryl amines which yield the corresponding amide salt in situ prior to the rearrangement proceed through a radical mechanism by 6-aryl cyclisation to yield the observed acridine-type products (Scheme 10).
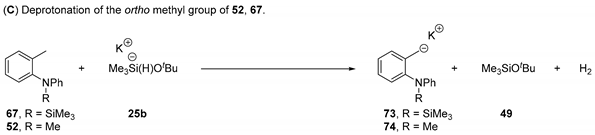
The above discussion assumes that salt 57 is the reactive species in solution. However, it has recently been shown by Palumbo et al. [36] that amide anions can be silylated by Et3SiH/KOtBu. Therefore, a substrate containing a SiMe3 group bonded to the nitrogen atom, 67, was explored (Figure 1). Effectively, substrate 67 features a tertiary amine, as does substrate 52, so the reactivity of substrate 52 is considered below, after that of 67. Subsequently, our studies on an additional substrate, 68, will be reported below. Its relevance lies in the fact that, although all of our substrates to date have been ortho-tolyl amines and ethers, our experimental interests lie in extending studies to more complex substrates, where the tolyl methyl group is replaced by an extended chain, for which substrate 68 would be the simplest computational model.
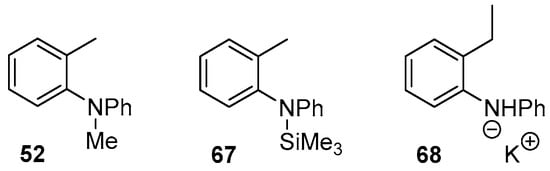
Figure 1.
Additional o-tolyl aryl amine substrates investigated.
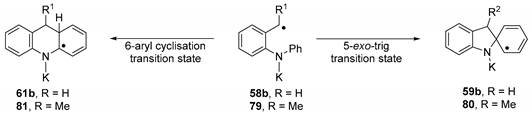
Cyclisation of substrate 67 was studied via both benzyl radical and benzyl anion intermediates and the energy profiles for formation of these intermediates are shown in Table 1. The initial hydrogen atom abstraction of the ortho methyl by a trimethylsilyl radical 24b has attainable activation energy (17.8 kcal mol−1) and is almost thermoneutral (entry 1, Table 1A).

Table 1.
Investigation into (A) the hydrogen atom abstraction facilitated by 24b, (B) the radical-polar crossover mediated by 26b, and (C) direct deprotonation of the ortho methyl group via pentavalent silicate 25b for substrates 65 and 58.
The energy profiles for the cyclisation of benzyl radical 69 were closely balanced: for 6-aryl cyclisation, ΔG‡ = 24.6 kcal mol−1 and ΔGrel = 2.3 kcal mol−1, while for 5-exo cyclisation, ΔG‡ = 25.1 kcal mol−1 and ΔGrel = 7.9 kcal mol−1, (see Figures S7 and S8) and so the expectation would be that a mixture of diarylmethane and dihydroacridine products would be produced by this pathway.
Addressing possible anionic pathways, reduction of radical 69 to anion 71 was found to be feasible (entry 1, Table 1B), making the benzyl anion a likely participant in the reaction. Direct deprotonation of the methyl group of the o-tolyl ring of 67 by pentavalent silicate 25b was also energetically viable (entry 1, Table 1C). Anionic cyclisations of the benzyl anion intermediates formed from these initiation routes were investigated next (Table 2). The anionic 5-exo-trig cyclisation of 73 has a very achievable activation energy (ΔG‡ = 26.5 kcal mol−1) [66,67] similar to the alternative 6-aryl cyclisation (ΔG‡ = 27.7 kcal mol−1), and so the expectation would be that a mixture of diarylmethane and dihydroacridine products would also be produced by this pathway.
Table 2.
Summary of activation energies and relative changes in Gibbs free energy for the cyclisation step of the anionic mechanisms for anions 73 and 74.
As both the radical and the anionic routes predict that a diarylmethane product should be formed from the silylated substrate 67, and as no such product is observed experimentally, we conclude that silylation of secondary amine substrates plays no role in their conversion to products.
Turning now to examine substrate 52, conversion to the benzyl radical 70 is easily achieved (Table 1A, entry 2). Radical cyclisations of substrate 52 showed a preference for 6-aryl cyclisation (ΔG‡= 19.8 kcal mol−1, ΔGrel = −2.7 kcal mol−1) versus 5-exo cyclisation (ΔG‡= 23.1 kcal mol−1, ΔGrel = 4.8 kcal mol−1) (see Figures S11 and S12). Accordingly, the radical cyclisation pathway favours formation of dihydroacridine product, 55 (Scheme 9).
Reduction of radical 70 to anion 72 witnesses a low barrier (3.8 kcal mol−1) and is exergonic. Anion 72 would be further stabilised by complexing with a potassium ion to form 74 (see Figures S15 and S16). Salt 74 could alternatively arise by direct deprotonation of substrate 52 by strong base 25b. Table 1C (entry 2) shows that this is also an energetically accessible route.
Table 2 (entry 2) reports the energy profile for 5-exo and 6-aryl cyclisations of salt 74. 5-Exo-trig cyclisation is almost thermoneutral and has an accessible barrier, while 6-aryl cyclisation is not at all favoured, with its very high barrier (ΔG‡ = 31.0 kcal mol−1); furthermore, it is quite endergonic (ΔGrel = 8.5 kcal mol−1). Therefore, the anionic pathway favours the pathway that leads towards a diarylmethane product 56
Scheme 9 shows that both dihydroacridine 55 and diarylmethane 56 are isolated from the reaction of substrate 52, suggesting that both radical and anionic pathways contribute to product formation. Computation clearly shows that both cyclisation routes are accessible, but it is challenging to define the relative contribution of each pathway.
The final substrate to be examined was substrate 68 (Figure 1). It is a close analogue of substrate 50b. In that case, we have reported above that the pathway through benzyl radical was operative, while that through a benzyl anion was not. Accordingly, here we investigated solely the radical pathway. Table 3 presents the energy changes for the cyclisation step of the radical pathway for radical 79 derived from substrate 68 and compares them with radical 58b derived from substrate 50b. As for 50b, the 6-aryl cyclisation is the preferred pathway, with the cyclisation being somewhat facilitated for the more highly substituted 68.
Table 3.
Summary of activation energies and relative change in Gibbs free energy for the cyclisation steps for radical 79 compared to 58b.
The computations report that the cyclisation of substrates like 68 proceed through a benzylic radical intermediate rather than through an anionic counterpart. In terms of future plans, this informs us that more complex sidechains for analogues of 68 should be designed with substituents that do not intercept the benzyl radical more rapidly than it attacks the target aryl ring.
In this paper, we have relied on computational assessment of the mechanism. This is appropriate, as the computation can compare the favourability of cyclisation of radical and anionic intermediates that could arise from identical substrates. There are of course, experimental approaches to determining whether a reaction is radical [68] or anionic [69], and indeed we have used such methods in previous studies on different chemistry [70,71]. A benzyl radical intermediate might be probed by means of an adjacent radical clock or by trapping with a commercial persistent radical, e.g., TEMPO. However, TEMPO would likely intercept the first radical intermediates in our pathway (silyl radicals) and this would inhibit that pathway before the benzyl radicals were formed, while radical clocks would necessarily intercept the relevant benzyl radical intermediate prior to cyclisation, and thus the test substrates would not actually cyclise (detecting a benzyl radical would not mean that it was normally responsible for the cyclisation). On the other hand, a benzyl anion intermediate might be reported by a leaving group (e.g., OMe) on the adjacent carbon, elimination of which would give rise to a styrene. However, this substrate would then not cyclise, and so it would not be clear which intermediate was responsible for the cyclisation.
3. Materials and Methods
3.1. Experimental Details
All reagents and solvents were obtained from commercial suppliers and were used without further purification unless mentioned otherwise. Anhydrous diethyl ether, THF and dichloromethane (DCM) were dried using a Pure-Solv 400 solvent purification system (Innovative Technology Inc., USA). DMF was dried over 3 Å pre-activated molecular sieves. Molecular sieves were activated by three heating cycles in the microwave, followed by evacuation under vacuum.
The glovebox was supplied by Innovative Technology Inc., Herndon, VA, USA, which is operated with a nitrogen atmosphere.
Thin Layer Chromatography was performed on silica gel pre-coated aluminium plates (60 Å, F254 UV indicator) purchased from Merck. The thin layer chromatograms were analysed by UV (254 nm, UVP mineralight UVG-11 lamp) and staining either with basic KMnO4 [KMnO4 (6 g), K2CO3 (40 g), NaOH (5 mL, 10% w/w) in water (600 mL)] or an ethanolic solution of phosphomolybdic acid [phosphomolybdic acid hydrate (10 g) in ethanol (100 mL)].
Flash Column Chromatography purification was performed with 35-70 μm particle size silica gel 60 Å (200–400 mesh) purchased from Prolabo.
NMR spectra were measured on a Bruker AV400 instrument. 1H and 13C NMR spectra were obtained at 400 and 101 MHz, respectively. Spectra were recorded in chloroform-d1. The frequency was locked against the deuterated solvent signal and the final spectra were referenced against the residual non-deuterated solvent signal (for 1H spectra) or the deuterated solvent signal (for 13C spectra). Chemical shifts are reported as δ (ppm) with respect to tetramethylsilane. The following multiplet abbreviations are used: s = singlet, d = doublet, t = triplet, td = triplet of doublets, m = multiplet, b = broad.
Infrared spectra were recorded on a Shimadzu IRAffinity-1 instrument. GC-(EI)MS analysis was performed on an Agilent Technologies 7890A GC system connected to an Agilent Technologies 5975C inert XL EI/CI MSD triple axis-mass detector. The GC was equipped with a Rxi-5Sil MS column (30 m × 0.25 mm × 0.25 μm). Helium was used as the carrier gas (1.0 mL/min flow rate). The injector temperature was 320 °C and was operated in splitless mode.
High resolution mass spectrometry (HRMS) was conducted at the University of Glasgow using a Bruker microTOFq High Resolution Mass Spectrometer. This instrument has an Electrospray (ESI) ion source coupled to a time-of-flight (ToF) analyser.
3.2. Substrate Synthesis
3.2.1. Preparation of 1-methyl-2-phenoxybenzene (1)
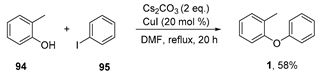
This substrate was prepared according to a literature procedure [29].
1 1H NMR (400MHz, CDCl3) δ 7.32–7.26 (m, 2H), 7.25–7.23 (m, 1 H), 7.19–7.13 (m, 1 H), 7.11–6.99 (m, 2H), 6.93–6.87 (m, 3H), 2.24 (s, 3H); 13C NMR (101MHz, CDCl3) δ 158.0, 154.6, 131.6, 130.2, 129.8, 127.2, 124.1, 122.4, 119.9, 117.4, 16.3; ATR-IR νmax (neat)/cm−1 1582, 1485, 1233, 1111, 874, 748, 691; GC-MS [m/z (%)] (11.33 min): 184.3 (99, [M]+), 165.2 (73), 155.2 (60), 141.2 (100), 128.2 (51), 115.2 (76), 106.2 (80), 91.2 (88), 78.2 (89), 65.2 (96), 50.2 (73). Analytical data in agreement with those reported in the literature [29].
3.2.2. Preparation of N,2-dimethyl-N-phenylaniline (52)
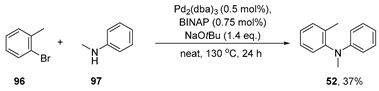
This substrate was prepared according to a literature procedure [72]. To an oven-dried pressure tube equipped with a stirrer bar was added NaOtBu (2.11 g, 22 mmol, 1.4 equiv.), Pd2(dba)3 (73 mg, 0.5 mol %), BINAP (74 mg, 0.75 mol %), N-methylaniline 97 (2 mL, 19 mmol, 1.2 equiv.), and 2-bromotoluene 96 (1.93 mL, 16 mmol, 1 equiv.). The liquid substrates were added last. The vial was flushed with a stream of argon and tightly capped. The mixture was refluxed in a pre-heated oil bath at 130 ℃ for 24 h. The mixture was allowed to cool to room temperature, taken up in ether (50 mL), filtered, and concentrated. The crude product was then purified by column chromatography (100% hexane 3% EtOAc in hexane) to afford N,2-dimethyl-N-phenylaniline 52 as a colourless oil (1.16 g, 37%).
52 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 1H), 7.25–7.12 (m, 5H), 6.74–6.68 (tt, J = 7.3 Hz, 1.0 Hz, 1H), 6.56–6.50 (dd, J = 8.8, 1.0 Hz, 2H), 3.22 (s, 3H), 2.15 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 148.7, 146.3, 136.3, 130.9, 128.5, 127.8, 127.0, 125.9, 116.3, 112.3, 38.5, 17.3; ATR-IR νmax (neat)/cm−1 3022, 2877, 1593, 1490, 1338, 1251, 1112, 746, 727, 691; GC-MS [m/z (%)] (12.56) min: 198.3 (100, [M]+), 183.2 (89), 165.2 (62), 155.2 (66), 107.2 (63), 91.2 (97), 77.2 (93), 65.2 (96), 51.2 (82). Analytical data are in agreement with those reported in the literature [73].
3.3. Reactions of Substrates
3.3.1. Reaction of 1-methyl-2-phenoxybenzene 1 with KOtBu—Neat
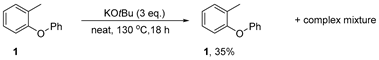
Substrate 1 (92 mg, 1.0 equiv., 0.5 mmol) and KOtBu (3.0 equiv., 1.5 mmol, 168 mg) were sealed in a pressure tube in a nitrogen-filled glovebox. This experiment was carried out neat and the contents subjected to the reaction conditions as is. The mixture was stirred at 130 °C for 18 h before the pressure tube was cooled to room temperature, opened to air and diluted with water (50 mL). The organic products were extracted into Et2O (3 × 50 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. Analysis of the crude mixture afforded starting material 1 (34 mg, 35%) alongside traces of an unidentifiable complex mixture.
3.3.2. Reaction of 1-methyl-2-phenoxybenzene 1 with KOtBu—in THF
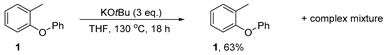
Substrate 1 (92 mg, 1.0 equiv., 0.5 mmol) and KOtBu (3.0 equiv., 1.5 mmol, 168 mg) were added to a pressure tube, followed by THF (5 mL) in a nitrogen-filled glovebox. The tube was sealed, removed from the glovebox, and the mixture stirred at 130 °C for 18 h. After reaction, the pressure tube was cooled to room temperature, opened to air and diluted with water (50 mL). The organic products were extracted into Et2O (3 × 50 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. Analysis of the crude mixture afforded starting material 1 (58.4 mg, 63%) alongside traces of an unidentifiable complex mixture.
3.3.3. Reaction of N,2-dimethyl-N-phenylaniline 52 with KOtBu/Et3SiH—in THF
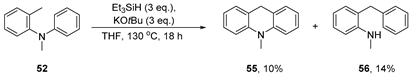
Substrate 52 (99 mg, 1.0 equiv., 0.5 mmol), KOtBu (3.0 equiv., 1.5 mmol, 168 mg), and Et3SiH (3.0 equiv., 1.5 mmol, 240 μL) were dissolved in THF (5 mL) and sealed in a pressure tube in a nitrogen-filled glovebox. The contents of the pressure tube were stirred at 130 °C for 18 h before the pressure tube was cooled to room temperature, opened to air and diluted with water (50 mL). The organic products were extracted into Et2O (3 × 50 mL). The combined organic phases were dried over Na2SO4 and concentrated in vacuo. The crude material was purified using column chromatography (50% hexane in toluene→100% toluene) affording 10-methyl-9,10-dihydroacridine 55 as a yellow oil (10.8 mg, 10%) and 2-benzyl-N-methylaniline 56 as a yellow oil (15.4 mg, 14%).
55 1H NMR (400 MHz, CDCl3) δ 7.22–7.14 (m, 4H), 6.96–6.85 (m, 4H), 3.89 (s, 2H), 3.38 (s, 3H); 13C NMR (101 MHz, CDCl3) 143.8, 127.7, 127.0, 124.5, 120.7, 112.0, 33.4, 33.3 ATR-IR νmax (neat)/cm−1 2922, 1635, 1595, 1494, 1460, 1367, 1178, 752; GC-MS [m/z (%)] (14.37 min): 194 (100, [M-H]+), 176 (42), 152 (14), 126 (4), 97 (9), 63 (10). Analytical data are in agreement with those reported in the literature [74].
56 1H NMR (400 MHz, CDCl3) δ 7.32–7.26 (m, 2H), 7.24–7.18 (m, 2H), 7.16 (d, J = 7.3 Hz, 2H), 7.05–7.00 (d, J = 7.1 Hz, 1H), 6.77 (t, J = 7.4 Hz, 1H), 6.65 (d, J = 8.1 Hz, 1H), 3.87 (s, 2H), 3.53 (bs, 1H), 2.77 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 147.3, 139.4, 130.6, 128.8, 128.6, 128.0, 126.5, 124.7, 117.1, 110.1, 38.0, 30.9; ATR-IR νmax (neat)/cm−1: 3431, 2893, 1604, 1512, 1307, 1161, 729. HRMS (ESI): calculated for C14H16N ([M+H]+): 198.1277 found: 198.1277. NMR data are in agreement with those reported in the literature [75].
3.3.4. Reaction of N,2-dimethyl-N-phenylaniline 52 with KOtBu/Et3SiH—Neat
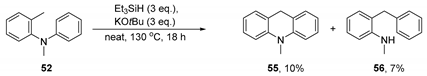
Substrate 52 (99 mg, 1.0 equiv., 0.5 mmol), KOtBu (3.0 equiv., 1.5 mmol, 168 mg), and Et3SiH (3.0 equiv., 1.5 mmol, 240 μL) were sealed in a pressure tube in a nitrogen-filled glovebox. The contents of the pressure tube were stirred at 130 °C for 18 h before the pressure tube was cooled to room temperature, opened to air and diluted with water (50 mL). The organic products were extracted into Et2O (3 × 50 mL). The combined organic phases were dried over Na2SO4 and concentrated in vacuo. The crude material was purified using column chromatography (50% hexane in toluene→100% toluene), affording 10-methyl-9,10-dihydroacridine 55 as a yellow oil (10.6 mg, 10%) and 2-benzyl-N-methylaniline 56 as a yellow oil (6.8 mg, 7%).
Analytical data for 61 and 62 are in agreement with the corresponding data reported above.
4. Conclusions
In summary, subjecting o-tolylaryl ethers and amines to the Et3SiH/KOtBu system yields rearranged products. o-Tolylaryl ethers undergo a concerted Truce–Smiles rearrangement to yield diarylmethane products that is initiated by benzyl anions formed by two competitive routes: a radical-polar crossover consisting of a hydrogen atom abstraction by a trialkylsilyl radical 24a followed by a SET reduction via silyl radical anion 26a, and/or the direct deprotonation of the ortho methyl group by the pentavalent silicate base that is formed in situ.
o-Tolyl arylamines that are secondary, or that contain a labile group bonded to the nitrogen atom, result in the formation of dihydroacridine products through a radical pathway when treated with the Et3SiH/KOtBu system. Tertiary amines form both dihydroacridines and diarylmethanes through radical and anionic pathways respectively. Overall, this study showcases how the reactive intermediates of the Et3SiH/KOtBu compete with one another during the reaction mechanisms allowing for a broad range of chemical outcomes and possibilities.
This research provides mechanistic detail on another of the expanding family of transformations that can be achieved by KOtBu + Et3SiH. This reagent pair unusually produces at least three silicon-based reactive intermediates, making determination of mechanism both challenging and important; the knowledge from our study can contribute to future understanding of the Grubbs–Stoltz system. In terms of the development of this specific project, the results reported here allow us to plan the synthesis of more complex substrates, e.g., based on 68. Knowing that a benzyl radical is the intermediate that cyclises allows us to plan extended side-chains that will not intercept the benzyl radical before it cyclises onto the target arene ring.
Supplementary Materials
The following are available online, xyz coordinates of all computed structures, and NMR spectra.
Author Contributions
Conceptualization, J.A.M.; Data curation, J.A.M., K.K. and A.J.S.; Funding acquisition, J.A.M. and T.T.; Investigation, K.K. and A.J.S.; Resources, T.T.; Supervision, J.A.M. and T.T.; Writing—original draft, K.K.; Writing—review & editing, J.A.M., K.K., A.J.S. and T.T. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the University of Strathclyde for funding and the EPSRC-funded ARCHIE-WeSt High Performance Computer (www.archie-west.ac.uk, accessed on 13 October 2021) for computational resource via EPSRC grant no. EP/K000586/1.
Data Availability Statement
Data are contained within the article or Supplementary Material.
Acknowledgments
We thank John Parkinson, Craig Irving and Patricia Keating for assistance with spectroscopic provision.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Wieland, H. Über Triphenylmethyl-peroxyd, Ein Beitrag zur Chemie der freien Radikale. Chem. Ber. 1911, 44, 2550. [Google Scholar] [CrossRef] [Green Version]
- Studer, A.; Bossart, M. Radical aryl migration reactions. Tetrahedron 2001, 57, 9649–9667. [Google Scholar] [CrossRef]
- Chen, Z.-M.; Zhang, X.-M.; Tu, Y.Q. Radical aryl migration reactions and synthetic applications. Chem. Soc. Rev. 2015, 44, 5220–5245. [Google Scholar] [CrossRef]
- Warren, L.A.; Samuel, S. CLXXI.—Dehydro-2-naphtholsulphone. J. Chem. Soc. 1930, 1327–1331. [Google Scholar] [CrossRef]
- Warren, W.A.; Smiles, S. CXXII.—The Conversion of iso-β-Naphthol Sulphide into 2-Naphthol 1-Sulphide. J. Chem. Soc. 1931, 914–922. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Zahler, R.E. Aromatic nucleophilic substitution reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Truce, E.W.; Kreider, M.E.; Brand, W.W. The Smiles and Related Rearrangements of Aromatic Systems. Org. React. 1970, 18, 99–215. [Google Scholar]
- Makosza, M. Nucleophilic substitution in nitroarenes: A general corrected mechanism. ChemTexts 2019, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Holden, C.M.; Greaney, M.F. Modern Aspects of the Smiles Rearrangement. Chem. Eur. J. 2017, 23, 8992–9008. [Google Scholar] [CrossRef] [PubMed]
- Whalley, D.M.; Seayad, J.; Greaney, M.F. Truce–Smiles Rearrangements by Strain Release: Harnessing Primary Alkyl Radicals for Metal-Free Arylation. Angew. Chem. Int. Ed. 2021, 60, 22219–22223. [Google Scholar] [CrossRef] [PubMed]
- Whalley, D.M.; Duong, H.A.; Greaney, M.F. A visible light-mediated, decarboxylative, desulfonylative Smiles rearrangement for general arylethylamine syntheses. Chem. Commun. 2020, 56, 11493–11496. [Google Scholar] [CrossRef] [PubMed]
- Whalley, D.M.; Duong, H.A.; Greaney, M.F. Alkene Carboarylation through Catalyst-Free, Visible Light-Mediated Smiles Rearrangement. Chem. Eur. J. 2019, 25, 1927–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayles, R.; Johnson, M.C.; Maisey, R.F.; Turner, R.W. A Smiles Rearrangement Involving Non-Activated Aromatic Systems; the Facile Conversion of Phenols to Anilines. Synthesis 1977, 33–34. Available online: https://pascal-francis.inist.fr/vibad/index.php?action=getRecordDetail&idt=PASCAL7760286965 (accessed on 13 October 2021). [CrossRef]
- Coutts, I.G.C.; Southcott, M.R. The conversion of phenols to primary and secondary aromatic amines via a Smiles rearrangement. J. Chem. Soc. Perkin Trans. 1 1990, 767–771. [Google Scholar] [CrossRef]
- Ten Hoeve, W.; Kruse, C.G.; Luteyn, J.M.; Thiecke, J.R.G.; Wynberg, H. Direct Substitution of Aromatic Ethers by Lithium Amides. A New Aromatic Amination Reaction. J. Org. Chem. 1993, 58, 5101–5106. [Google Scholar] [CrossRef]
- Bonini, C.; Cristiani, G.; Funicello, M.; Viggiani, L. Facile entry to 4- and 5-hydroxybenzofuran and to their amino derivatives. Synth. Commun. 2006, 36, 1983–1990. [Google Scholar] [CrossRef]
- Snape, T.J. A truce on the Smiles rearrangement: Revisiting an old reaction–the Truce–Smiles rearrangement. Chem. Soc. Rev. 2008, 37, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Monos, T.M.; McAtee, R.C.; Stephenson, C.R.J. Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation. Science 2018, 361, 1369–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.A.; Dominey, A.P.; Williams, G.D.; Murphy, J.A. Visible light-mediated Smiles rearrangements and annulations of non-activated aromatics. Chem. Commun. 2020, 56, 11445–11448. [Google Scholar] [CrossRef] [PubMed]
- Loven, R.; Speckamp, W.N. A Novel 1,4 arylradical rearrangement. Tetrahedron Lett. 1972, 1567–1570. [Google Scholar] [CrossRef]
- Köhler, J.J.; Speckamp, W.N. Intramolecular Radical Reactions in α-halomethyl substituted piperidine sulfonamides. Tetrahedron Lett. 1977, 631–634. [Google Scholar] [CrossRef]
- Motherwell, W.B.; Pennell, A.M.K. A novel route to biaryls via intramolecular free radical ipso substitution reactions. J. Chem. Soc. Chem. Commun. 1991, 877–879. [Google Scholar] [CrossRef]
- Da Mata, M.L.E.N.; Motherwell, W.B.; Ujjainwalla, F. Steric and electronic effects in the synthesis of biaryls and their heterocyclic congeners using intramolecular free radical [1,5] ipso substitution reactions. Tetrahedron Lett. 1997, 38, 137–140. [Google Scholar] [CrossRef]
- Kong, W.; Casimiro, M.; Merino, E.; Nevado, C. Copper-catalyzed one-Pot trifluoromethylation/aryl migration/desulfonylation and C(sp2)-N bond formation of conjugated tosyl amides. J. Am. Chem. Soc. 2013, 135, 14480–14483. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Merino, E.; Nevado, C. Arylphosphonylation and arylazidation of activated alkenes. Angew. Chem. Int. Ed. 2014, 53, 5078–5082. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Kong, W.; Fernández-Sánchez, L.; Merino, E.; Nevado, C. Cyclization cascades via N-amidyl radicals toward highly functionalized heterocyclic scaffolds. J. Am. Chem. Soc. 2015, 137, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.J.; Albright, H.; Sevrin, M.J.; Cole, K.P.; Stephenson, C.R.J. A visible-light-mediated radical Smiles rearrangement and its application to the synthesis of a difluoro-substituted spirocyclic ORL-1 antagonist. Angew. Chem. Int. Ed. 2015, 54, 14898–14902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khartabil, H.; Doudet, L.; Allart-Simon, I.; Ponce-Vargas, M.; Gérard, S.; Hénon, E. Mechanistic insights into Smiles rearrangement. Focus on π–π stacking interactions along the radical cascade. Org. Biomol. Chem. 2020, 18, 6840–6848. [Google Scholar] [CrossRef]
- Arokianathar, J.N.; Kolodziejczak, K.; Bugden, F.E.; Clark, K.F.; Tuttle, T.; Murphy, J.A. Benzylic C−H Functionalisation by [Et3SiH+KOtBu] leads to Radical Rearrangements in o-tolyl Aryl Ethers, Amines and Sulfides. Adv. Synth. Catal. 2020, 362, 2260–2267. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, A.; Toutov, A.A.; Swisher, N.A.; Grubbs, R.H. Lewis-base silane activation: From reductive cleavage of aryl ethers to selective ortho-silylation. Chem. Sci. 2013, 4, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Toutov, A.A.; Salata, M.; Fedorov, A.; Yang, Y.F.; Liang, Y.; Cariou, R.; Betz, K.N.; Couzijn, E.P.A.; Shabaker, J.W.; Houk, K.N.; et al. A potassium tert-butoxide and hydrosilane system for ultra-deep desulfurization of fuels. Nat. Energy 2017, 2, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.B.; Schuman, D.P.; Yang, Y.F.; Toutov, A.A.; Liang, Y.; Klare, H.F.T.; Nesnas, N.; Oestreich, M.; Blackmond, D.G.; Virgil, S.C.; et al. Potassium tert-Butoxide-Catalyzed Dehydrogenative C-H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study. J. Am. Chem. Soc. 2017, 139, 6867–6879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Yang, Y.F.; Jenkins, I.D.; Liang, Y.; Toutov, A.A.; Liu, W.B.; Schuman, D.P.; Grubbs, R.H.; Stoltz, B.M.; Krenske, E.H.; et al. Ionic and Neutral Mechanisms for C-H Bond Silylation of Aromatic Heterocycles Catalyzed by Potassium tert-Butoxide. J. Am. Chem. Soc. 2017, 139, 6880–6887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgari, P.; Hua, Y.; Bokka, A.; Thiamsiri, C.; Prasitwatcharakorn, W.; Karedath, A.; Chen, X.; Sardar, S.; Yum, K.; Leem, G.; et al. Catalytic hydrogen atom transfer from hydrosilanes to vinylarenes for hydrosilylation and polymerization. Nat. Catal. 2019, 2, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Young, A.; Rohrbach, S.; O’Connor, E.F.; Allison, M.; Wang, H.S.; Poole, D.L.; Tuttle, T.; Murphy, J.A. Electron-Transfer and Hydride-Transfer Pathways in the Stoltz–Grubbs Reducing System (KOtBu/Et3SiH). Angew. Chem. Int. Ed. 2017, 56, 13747–13751. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, F.; Rohrbach, S.; Tuttle, T.; Murphy, J.A. N-silylation of amines mediated by Et3SiH/KOtBu. Helv. Chim. Acta. 2019, 102, e1900235. [Google Scholar] [CrossRef]
- Smith, A.J.; Dimitrova, D.; Arokianathar, J.N.; Clark, K.F.; Poole, D.L.; Leach, S.G.; Murphy, J.A. Et3SiH + KOtBu provide multiple reactive intermediates that compete in the reactions and rearrangements of benzylnitriles and indolenines. Chem. Sci. 2020, 11, 12364–12370. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Dimitrova, D.; Arokianathar, J.N.; Kolodziejczak, K.; Young, A.; Allison, M.; Poole, D.L.; Leach, S.G.; Parkinson, J.A.; Tuttle, T.; et al. New reductive rearrangement of: N-arylindoles triggered by the Grubbs–Stoltz reagent Et3SiH/KOtBu. Chem. Sci. 2020, 11, 3719–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, K.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital stud-ies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Andersson, M.P.; Uvdal, P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis Set 6-311+G.(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Altshuller, A.P.; Rosenblum, L. Dielectric Properties of Some Alkylsilanes. J. Am. Chem. Soc. 1955, 77, 272–274. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: www.gaussian.com (accessed on 13 October 2021).
- Studer, A.; Curran, D.P. Organocatalysis and C-H activation meet radical- and electron-transfer reactions. Angew. Chem. Int. Ed. 2011, 50, 5018–5022. [Google Scholar] [CrossRef]
- Dub, P.A.; Henson, N.J.; Martin, R.L.; Gordon, J.C. Unravelling the mechanism of the asymmetric hydrogenation of acetophenone by [RuX2(diphosphine)(1,2-diamine)] catalysts. J. Am. Chem. Soc. 2014, 136, 3505–3521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sha, S.-C.; Bellomo, A.; Trongsiriwat, N.; Gao, F.; Tomson, N.C.; Walsh, P.J. Positional Selectivity in C-H Functionalizations of 2-Benzylfurans with Bimetallic Catalysts. J. Am. Chem. Soc. 2016, 138, 4260–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaga, A.; Peng, X.; Hirao, H.; Chiba, S. Diastereo-Divergent Synthesis of Saturated Azaheterocycles Enabled by tBuOK-Mediated Hydroamination of Alkenyl Hydrazones. Chem. Eur. J. 2015, 21, 19112–19118. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Tong, B.M.K.; Hirao, H.; Chiba, S. Inorganic-base-mediated hydroamination of alkenyl oximes for the synthesis of cyclic nitrones. Angew. Chem. Int. Ed. 2014, 53, 1959–1962. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S. Cation-π Interactions in Organic Synthesis. Chem. Rev. 2018, 118, 11353–11432. [Google Scholar] [CrossRef] [PubMed]
- Roth, H.G.; Roth, H.G.; Romero, N.A.; Nicewicz, D.A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Sim, B.A.; Griller, D.; Wayner, D.D.M. Reduction Potential for Substituted Benzyl Radicals: PKa values for the Corresponding Toluenes. J. Am. Chem. Soc. 1989, 111, 754–755. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of Inner Shell Marcus Terms for Amino Nitrogen Compounds by Molecular Orbital Calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Costil, R.; Dale, H.J.A.; Fey, N.; Whitcombe, G.; Matlock, J.V.; Clayden, J. Heavily Substituted Atropisomeric Diarylamines by Unactivated Smiles Rearrangement of N-Aryl Anthranilamides. Angew. Chem. Int. Ed. 2017, 56, 12533–12537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costil, R.; Lefebvre, Q.; Clayden, J. Medium-Sized-Ring Analogues of Dibenzodiazepines by a Conformationally Induced Smiles Ring Expansion. Angew. Chem. Int. Ed. 2017, 56, 14602–14606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, D.J.; Ward, J.W.; Clayden, J. Asymmetric α-arylation of amino acids. Nature 2018, 562, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Mas-Roselló, J.; Okoh, M.; Clayden, J. Enantioselectively functionalised phenytoin derivatives by auxiliary-directed N to C aryl migration in lithiated α-amino nitriles. Chem. Commun. 2018, 54, 10985–10988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, R.; Clayden, J. Photocatalytic Difunctionalization of Vinyl Ureas by Radical Addition Polar Truce–Smiles Rearrangement Cascades. Angew. Chem. Int. Ed. 2020, 59, 11600–11606. [Google Scholar] [CrossRef]
- Millward, M.J.; Ellis, E.; Ward, J.W.; Clayden, J. Hydantoin-bridged medium ring scaffolds by migratory insertion of urea-tethered nitrile anions into aromatic C–N bonds. Chem. Sci. 2021, 12, 2091–2096. [Google Scholar] [CrossRef]
- Abrams, R.; Jesani, M.H.; Browning, A.; Clayden, J. Triarylmethanes and their Medium-Ring Analogues by Unactivated Truce–Smiles Rearrangement of Benzanilides. Angew. Chem. Int. Ed. 2021, 60, 11272–11277. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, S.; Smith, A.J.; Pang, J.H.; Poole, D.L.; Tuttle, T.; Chiba, S.; Murphy, J.A. Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem. Int. Ed. 2019, 58, 16368–16388. [Google Scholar] [CrossRef] [Green Version]
- Legnani, L.; Porta, A.; Caramella, P.; Toma, L.; Zanoni, G.; Vidari, G. Computational mechanistic study of the Julia-Kocieński reaction. J. Org. Chem. 2015, 80, 3092–3100. [Google Scholar] [CrossRef]
- Nawaz, F.; Mohanan, K.; Charles, L.; Rajzmann, M.; Bonne, D.; Chuzel, O.; Rodriguez, J.; Coquerel, Y. Temporary intramolecular generation of pyridine carbenes in metal-free three-component C–H bond functionalisation/aryl-transfer reactions. Chem. Eur. J. 2013, 19, 17578–17583. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, A.; García, J.J. Bond Activation with Low-Valent Nickel in Homogeneous Systems. Eur. J. Inorg. Chem. 2010, 2010, 4063–4074. [Google Scholar] [CrossRef]
- Estimation of Approximate Figures for Rate Constants for Unimolecular Reactions with Particular Energy Barriers. Using This Tool, We Calculate a Ball-Park Figure of 3 × 10−2 s−1 for a Unimolecular Reaction with a Barrier of 26 kcal mol−1 Carried Out at 130 °C. Available online: https://www.unige.ch/sciences/chiorg/lacour/correl (accessed on 13 October 2021).
- Newcomb, M. Radical Kinetics and Clocks. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 1, Chapter 5; ISBN 9781119953678. [Google Scholar] [CrossRef]
- Crombie, L.; Wyvill, R.D. β-Halogeno ether synthesis of olefinic alcohols: Stereochemistry and conformation of 2-substituted 3-halogenotetrahydro-pyran and -furan precursors. J. Chem. Soc. Perkin Trans. 1985, 1, 1971–1981. [Google Scholar] [CrossRef]
- Murphy, J.A.; Khan, T.A.; Zhou, S.; Thomson, D.W.; Mahesh, M. Highly Efficient Reduction of Unactivated Aryl and Alkyl Iodides by a Ground-State Neutral Organic Electron Donor. Angew. Chem. Int. Ed. 2005, 44, 1356–1360. [Google Scholar] [CrossRef]
- Murphy, J.A.; Zhou, S.; Thomson, D.W.; Schoenebeck, F.; Mahesh, M.; Park, S.R.; Tuttle, T.; Berlouis, L.E.A. The Generation of Aryl Anions by Double Electron Transfer to Aryl Iodides from a Neutral Ground-State Organic Super-Electron Donor. Angew. Chem. Int. Ed. 2007, 46, 5178–5183. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. An Improved Catalyst System for Aromatic Carbon−Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates. J Am Chem Soc. 1996, 118, 7215–7216. [Google Scholar] [CrossRef]
- Weber, P.; Scherpf, T.; Rodstein, I.; Lichte, D.; Scharf, L.T.; Gooßen, L.J.; Gessner, V.H. A Highly Active Ylide-Functionalized Phosphine for Palladium-Catalyzed Aminations of Aryl Chlorides. Angew. Chem. Int. Ed. 2019, 58, 3203–3207. [Google Scholar] [CrossRef]
- Stopka, T.; Marzo, L.; Zurro, M.; Janich, S.; Würthwein, E.U.; Daniliuc, C.G.; Alemán, J.; Mancheño, O.G. Oxidative C-H Bond Functionalization and Ring Expansion with TMSCHN2: A Copper(I)-Catalyzed Approach to Dibenzoxepines and Dibenzoazepines. Angew. Chem. Int. Ed. 2015, 54, 5049–5053. [Google Scholar] [CrossRef] [PubMed]
- Creencia, E.C.; Taguchi, K.; Horaguchi, T. Thermal reactions of N-alkyl-2-benzylaniline and N-alkyl-N′-phenyl-o-phenylenediamine: An unusual route to 2-phenylindole and 2-phenylbenzimidazole. J. Heterocycl. Chem. 2008, 45, 837–843. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).