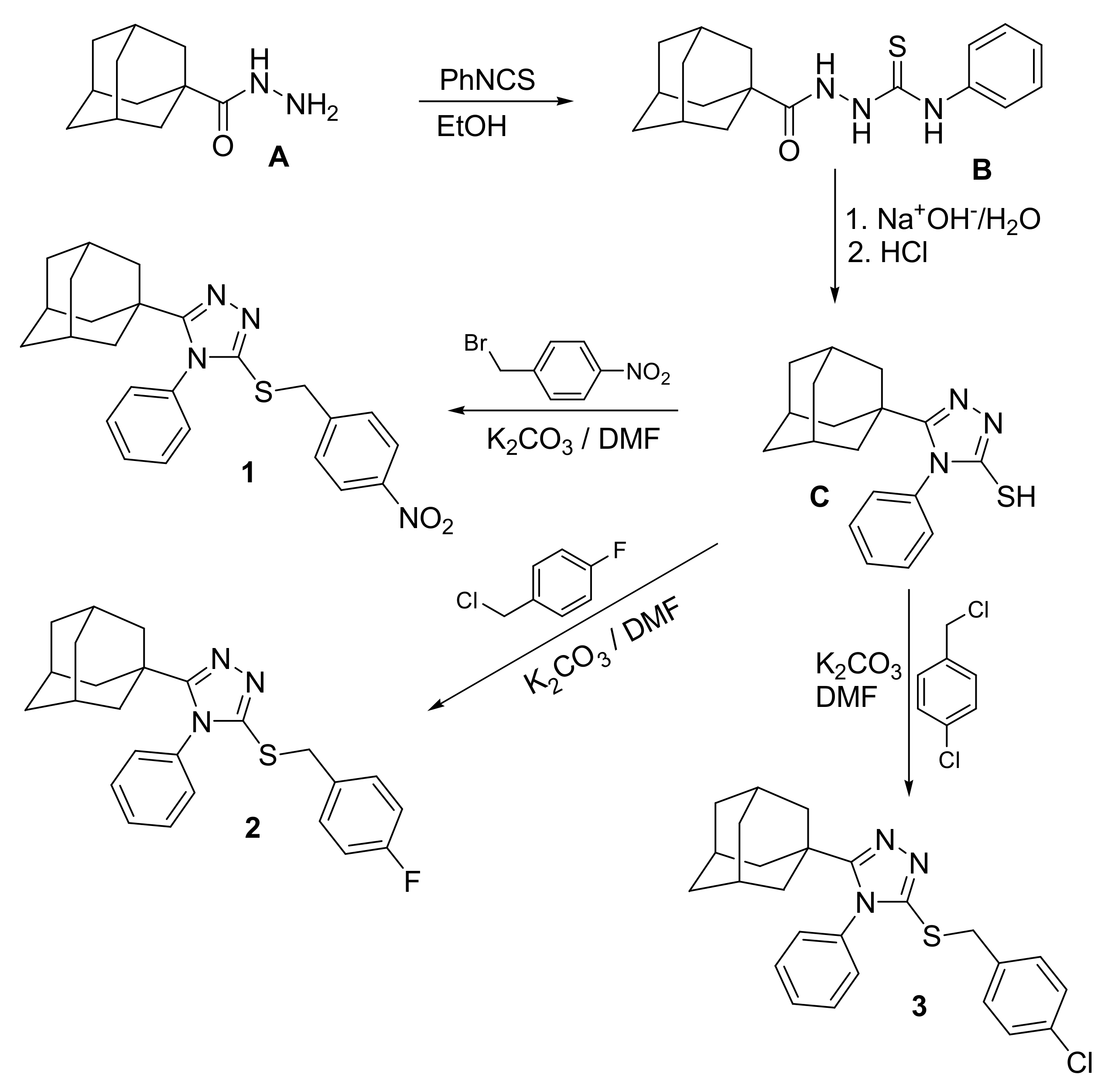
2.2. Crystal Structures
Crystal data, data collection, and structure refinement details of compounds
1–
3 are summarized in
Table 1. The structures of the three compounds consist of three basic fragments; the adamantane cage, the triazole ring and the arylmethylsulfanyl moiety (
Figure 3). A search in the CSD database version 5.41 (1 November 2019 with three updates; accessed on 1 November 2020), through the ConQuest software version 2020.2.0, for molecules with a similar core gave four related structures [
29,
30,
31,
32], but the crystal structures of compounds
1,
2 and
3 have not been reported.
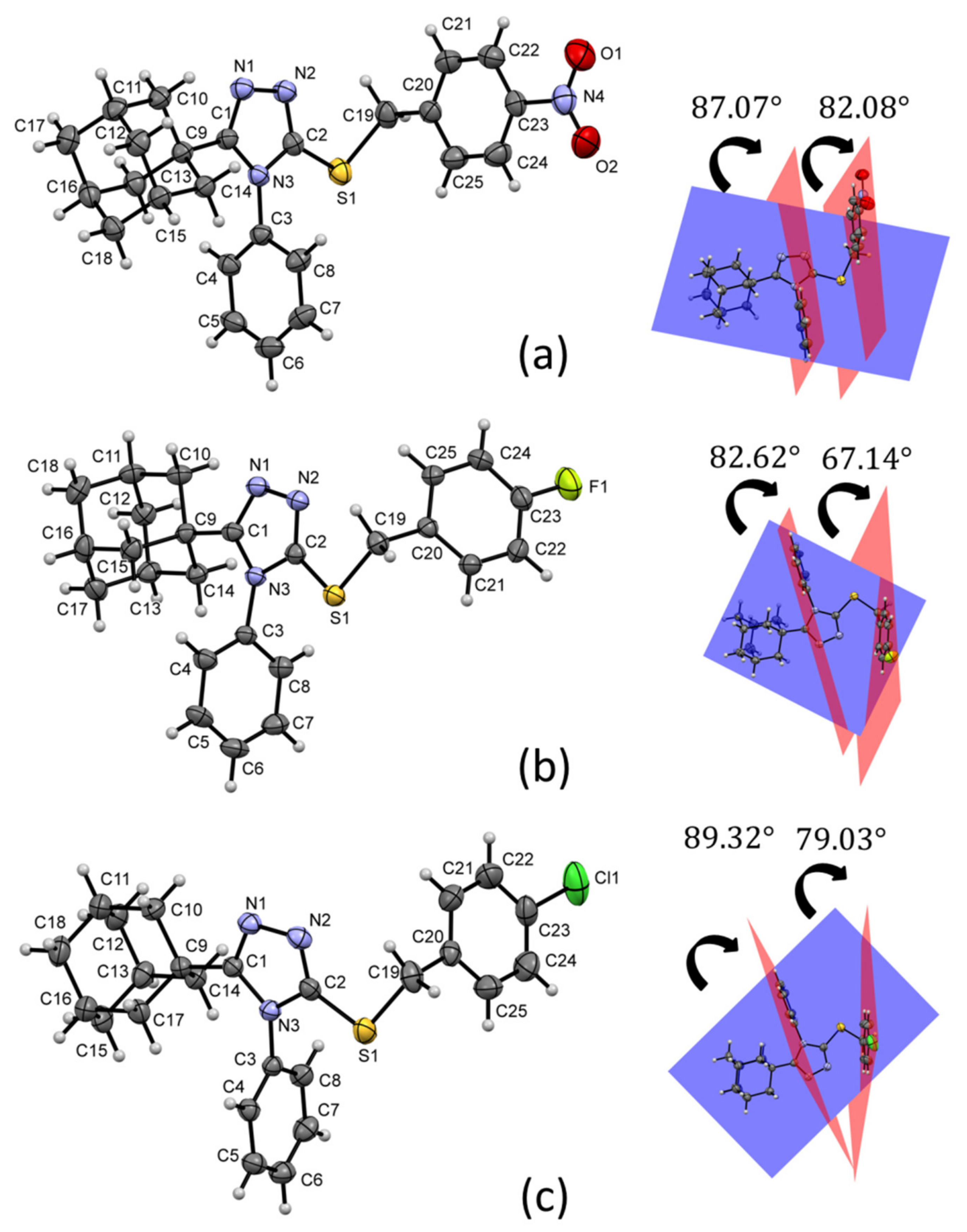
Figure 3 shows the weighted least-squares mean planes that contain the planar fragments of the molecules. The dihedral angles show a change in the molecular conformations of the aryl rings with respect to the triazole-sulfanyl fragment. In the nitrobenzyl analogue
1, there is a tendency to the orthogonality between these groups. Meanwhile, in the halobenzyl derivatives
2 and
3, a clear deviation was observed. The orthogonality between aryl ring is influenced by steric effects between C3/C8 and C20/C25 aryl rings and the adamantane ring.
In the two related molecules 5-(adamantan-1-yl)-3-(benzylsulfanyl)- 4-methyl-4
H-1,2,4-triazole [
29], and 5-(adamantan-1-yl)-3-[(4-chlorobenzyl)- sulfanyl]-4-methyl-4
H-1,2,4-triazole [
30], the equivalent to the C20/C25 ring is parallel to the triazole-sulfanyl fragment due to the absence of an aromatic ring in the N3 position of the triazole group. However, observing the conformational behavior in 3-(adamantan-1-yl)-5-[(2-methoxyethyl)sulfanyl]-4-phenyl-4
H-1,2,4-triazole [
31], a phenyl ring in N3 would constantly be orthogonal due to its steric effect with the adamantane cage.
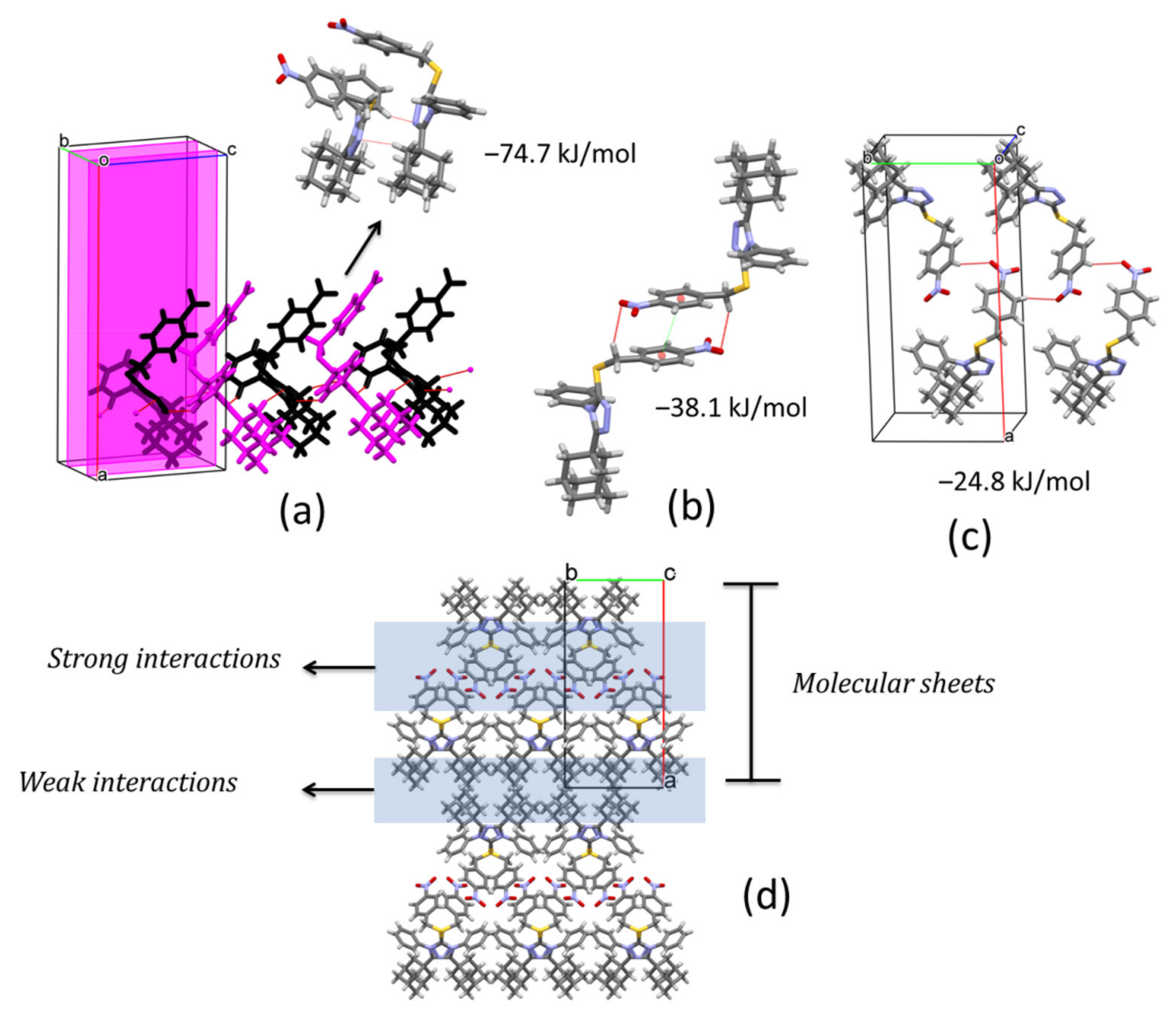
In the crystal structure of compound
1, pairs of C4‒H4···N1
i and C14‒H14B···N2
ii (symmetry codes: (i) x, 1/2 − y, −1/2 + z, (ii) x, 1/2 − y, 1/2 + z) hydrogen bonds connect molecules along [001] direction, related by a glide plane perpendicular to [010], and with distances between their molecular centroids (mean atomic position) of 5.09 Å (
Figure 4a,
Table 2). This interaction is the strongest contact in the crystal with a total pairwise interaction energy of −74.7 kJ/mol, being the dispersion force the principal contributor (E
elec = −25.9 kJ/mol; E
pol = −11.6 kJ/mol; E
dis = −93.9 kJ/mol; E
rep = 69.7 kJ/mol) (
Table 3). The neighboring chains are connected along
a axis through a combination of C19‒H19A···O1
iii and π···π
iii (symmetry code: (iii) 1 − x, −y, 1 − z) interactions linking inversion related molecules (
Figure 4b,
Table 2) with total pairwise interaction energy of −38.1 kJ/mol (E
elec = −13.9 kJ/mol; E
pol = −2.3 kJ/mol; E
dis = −40.8 kJ/mol; E
rep = 22.6 kJ/mol) (
Table 3) and distances between centroids of 12.53 Å. Along [010] direction, C22‒H22···O2
iv (symmetry code: (iv) 1 − x, −1/2 + y, 3/2 − z) hydrogen bonds complement the formation of (100) sheets stacked along
a axis (
Figure 4c,d). The total pairwise interaction energy in this contact involving the nitro group is −24.8 kJ/mol (E
elec = −14.3 kJ/mol; E
pol = −3.7 kJ/mol; E
dis = −14.2 kJ/mol; E
rep = 8.8 kJ/mol) (
Table 3). The packing showed that between molecular sheets, van de Waals forces act to maintain the three-dimensional architecture through dispersion interactions between layers of adamantane rings (
Figure 4d). These contacts are rather weak with total pairwise interaction energies of −9.0 kJ/mol and −14.1 kJ/mol, maintaining larger distances (13–15 Å).
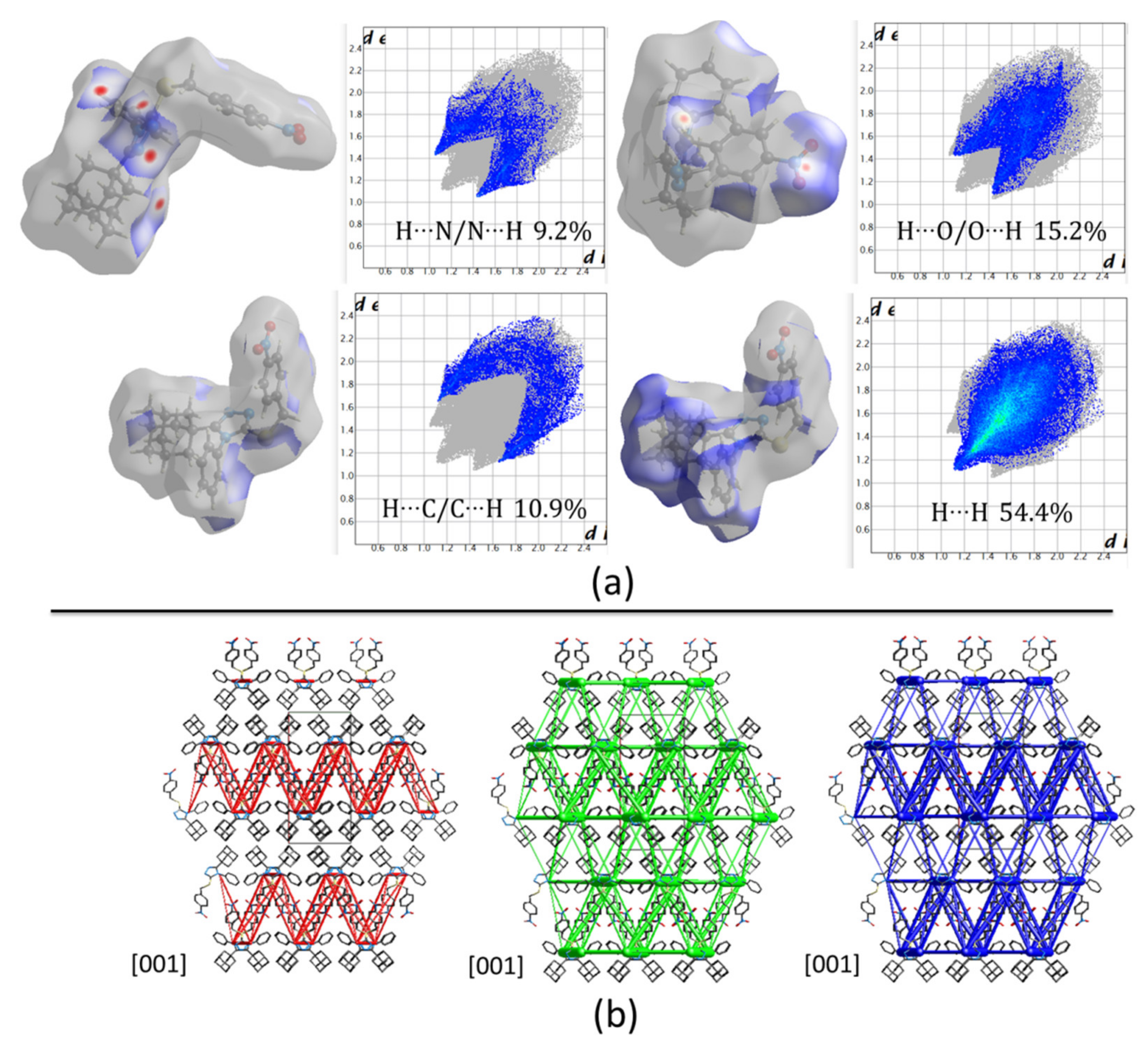
Hirshfeld surface (HFs) maps calculated at the B3LYP/6-31G(d,p) level of theory (
Figure 5a) show that the shortest contacts correspond to the C‒H···(N, O) hydrogen interactions with H···N/N···H and H···O/O···H comprising 9.2% and 15.2% of the total HFs maps. The non-covalent H···H interactions occupy 54.4% of the total HFs maps showing high participation of adamantane rings in the crystal structure (
Figure 4d and
Figure 5a).
Computed energies between molecular pairs are represented using cylinders joining the centroids (molecular center of mass) of the molecules, with a radius proportional to the magnitude of the interaction managing a minimal cut-off of 5 kJ/mol.
Figure 5b shows the energy framework diagrams for pairs of molecules for separate electrostatic (red) and dispersion (green) contributions to the total nearest-neighbor pairwise interaction energies (blue). As it is observed, the electrostatic forces act mainly along the center of the defined unit cell and define molecular sheets (
Figure 4d and
Figure 5b). However, dispersion forces act, not only between adamantane rings, but also in the complete structure. Despite the three-dimensional tendency of the dispersion forces, the total energy framework (blue in
Figure 5b) shows a laminar energetic topology due to the strong contribution of electrostatic forces.
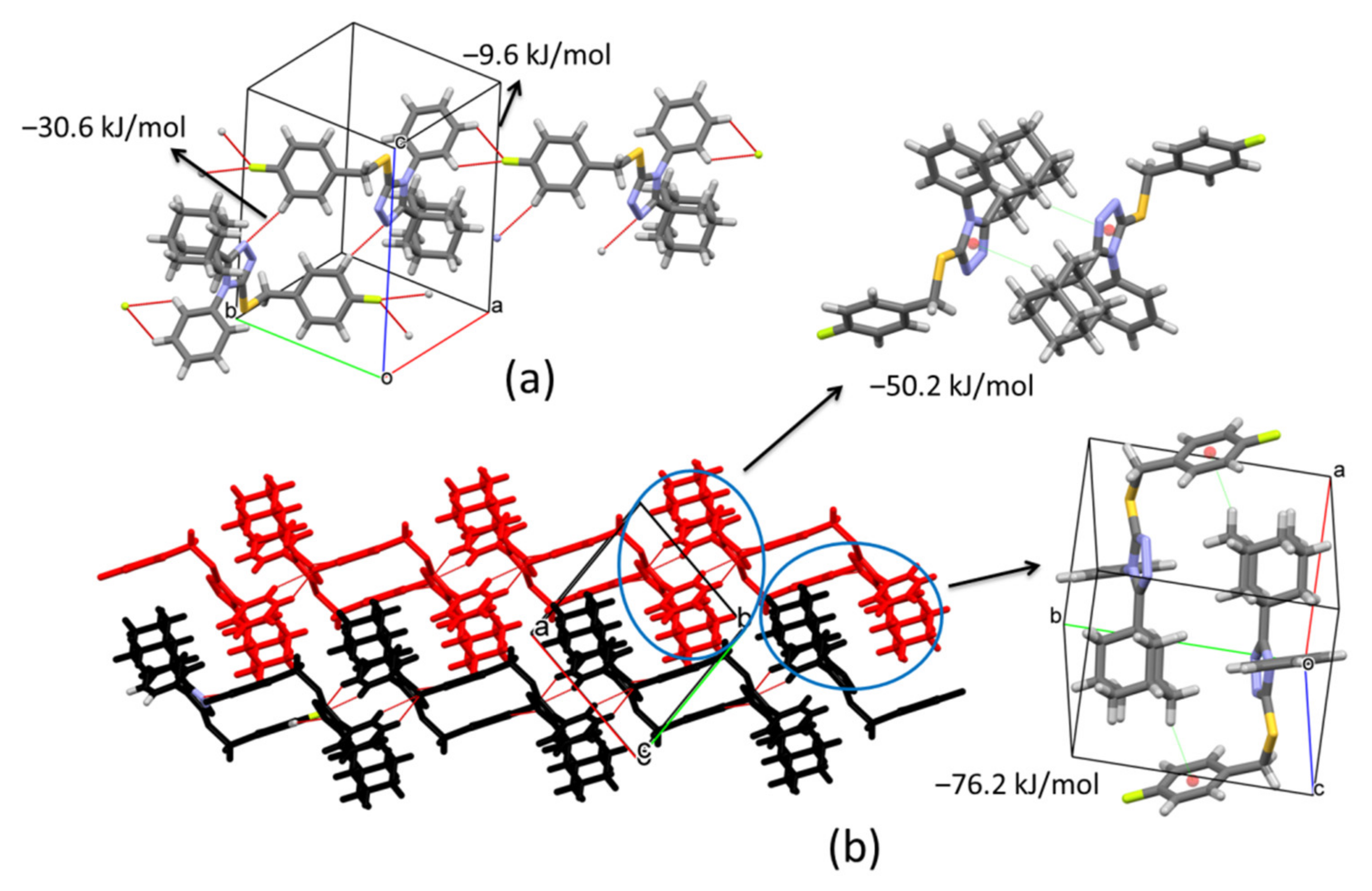
The supramolecular structure in compound
2 changes compared to compound
1. In this case, the presence of the fluorine atom (F) on the aryl ring varies the connection between molecules. Inversion related molecules are linked by pairs of C24‒H24···N1
v (symmetry code: (v) −x, 1 − y, 1 − z) hydrogen bonds forming dimers with distances between their molecular centroids (mean atomic position) of 8.99 Å (
Figure 6a,
Table 2). The total pairwise interaction energy that keeps the dimers connected has a value of −30.6 kJ/mol (E
elec = −15.9 kJ/mol; E
pol = −7.5 kJ/mol; E
dis = −27.7 kJ/mol; E
rep = 25.6 kJ/mol) (
Table 3). These pairs of inversion related molecules are further connected by bifurcated C4‒H4···F1
vi and C5‒H5···F1
vi (symmetry code: (vi) 1 + x, −1 + y, z) hydrogen interactions to build chains along [1–10] direction (
Figure 6a,
Table 2). The connection between dimers through F atoms shows weaker total interaction energy, −9.6 kJ/mol, manifested in the distance between centroids (12.53 Å). Inside chains, the molecular disposition allows the formation of C15‒H15B···Cg2
vii (symmetry code: (vii) 1 − x, −y, 1 − z) contacts with a total interaction energy of −50.2 kJ/mol (E
elec = −14.2 kJ/mol; E
pol = −4.2 kJ/mol; E
dis = −59.8 kJ/mol; E
rep = 32.4 kJ/mol) (
Figure 6b,
Table 2 and
Table 3). The high value of the dispersion term suggests a strong participation of the adamantane rings. Neighboring chains are connected by C12‒H12A···Cg1
viii (symmetry code: (viii) 1 − x, 1 − y, 1 − z) interactions along
b axis forming (001) sheets which correspond to the strongest contacts in the crystal with a total energy of −76.2 kJ/mol (E
elec = −15.3 kJ/mol; E
pol = −5.8 kJ/mol; E
dis = −101.8 kJ/mol; E
rep = 53.3 kJ/mol) (
Figure 6b,
Table 2 and
Table 3). These interactions involve the highest dispersion energy in the solid as a result of the short distance between molecules (5.63 Å) that allows an important closeness between adamantane rings (6.80 Å between their centroids) (
Figure 6b).
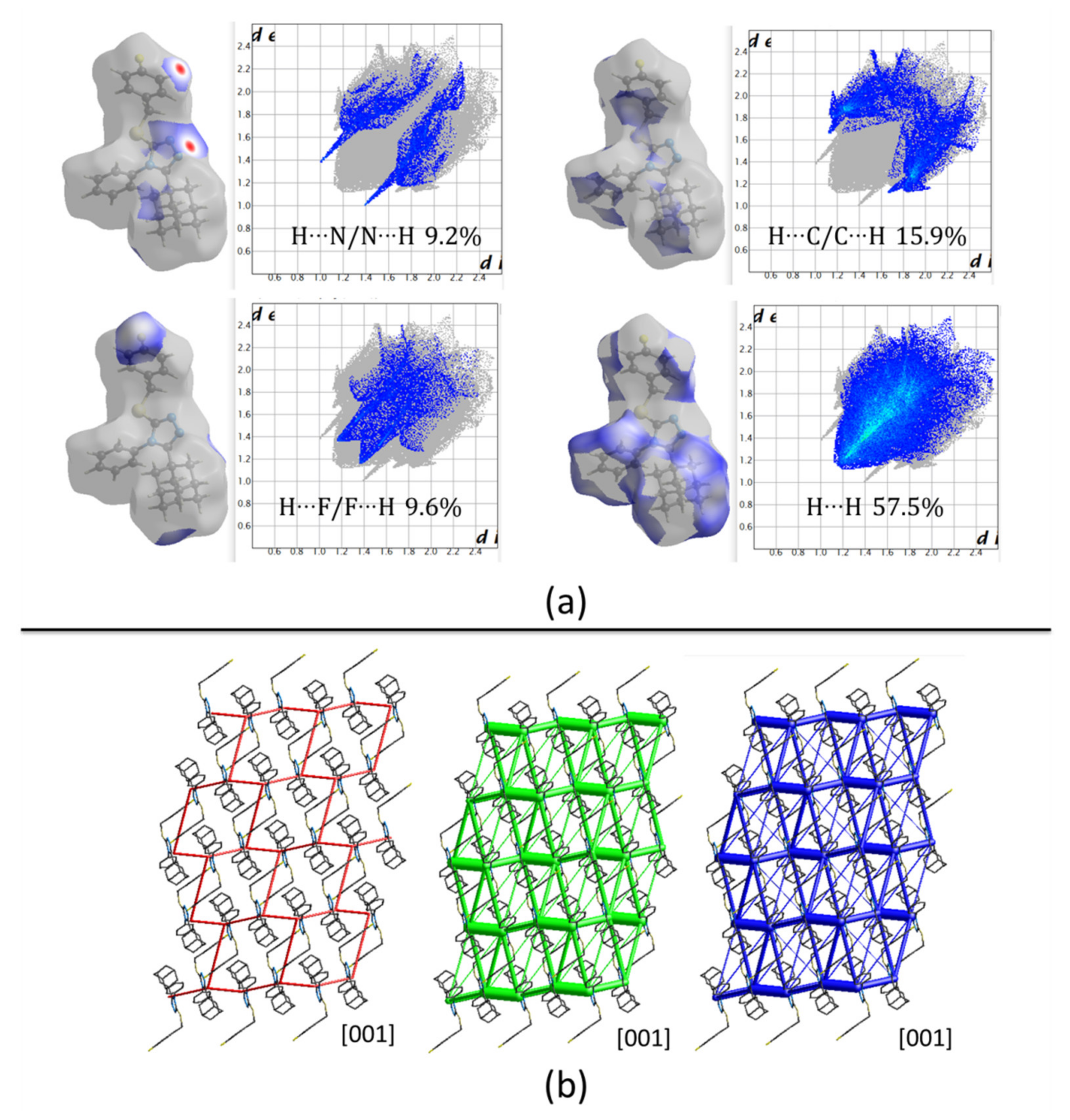
Hirshfeld surfaces (HFs) mapped over d
norm only present two red spots correlated to C24‒H24···N1
v interactions with H···N distances of 2.56 Å. The rest of the surface shows blue/white colors, representing long contacts with non-covalent H···H interactions covering 57.5% of the total surface (
Figure 7a). The energy framework diagrams show that dispersion forces (green) act with a three-dimensional topology. However, the cylinders joining adamantane rings have higher radius as a consequence of the high dispersion forces acting between them (
Figure 7b) showing the importance of these rings in the formation of the crystal.
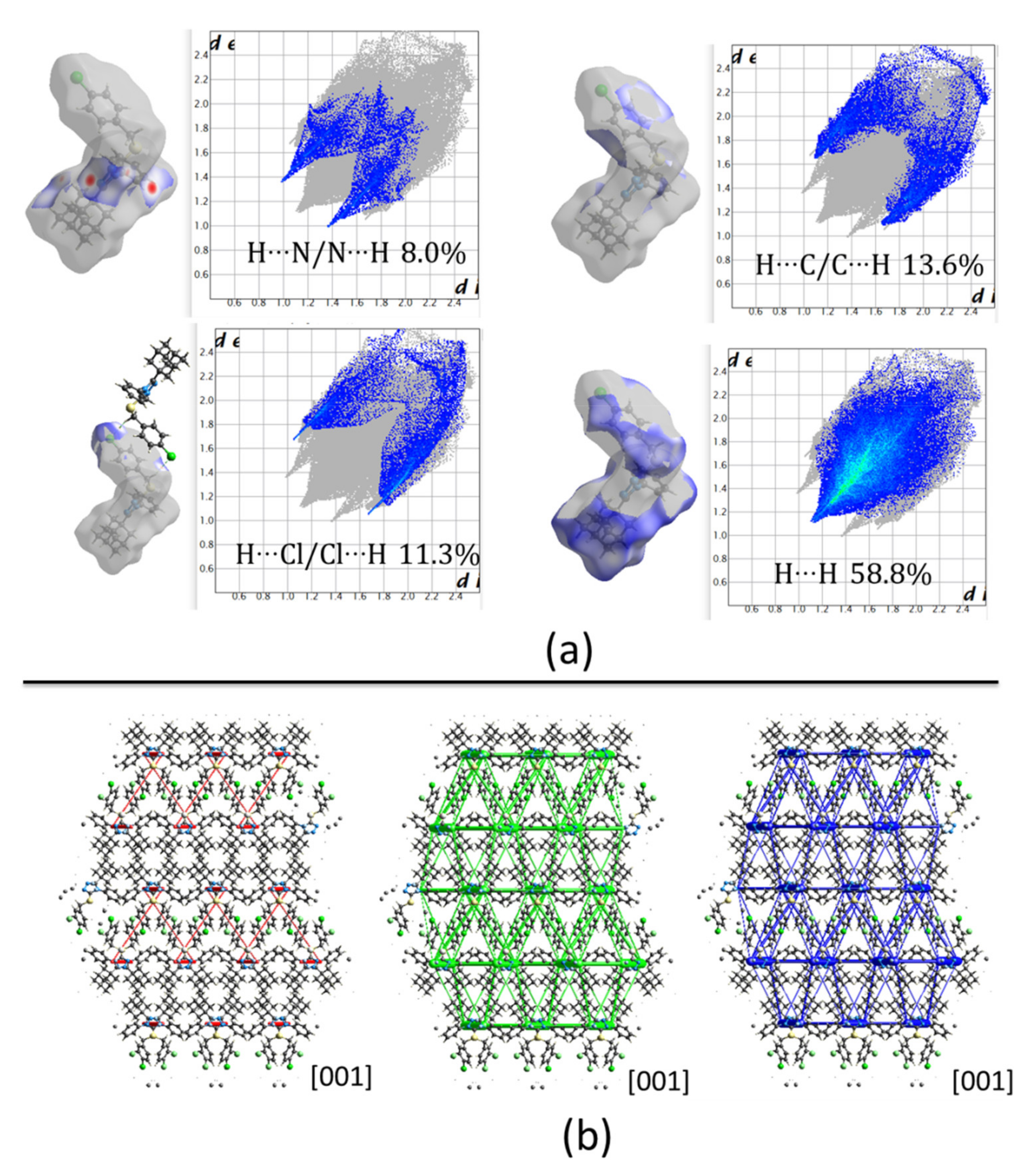
Compound
3 crystallizes in a very similar form as
1, sharing the same monoclinic
P2
1/
c space group and cell parameters with comparable dimensions. As mentioned above for compound
1, in this case, also pairs of C4‒H4···N1
i and C14‒H14A···N2
ii (symmetry codes: (i) x, 1/2 − y, −1/2 + z, (ii) x, 1/2 − y, 1/2 + z) hydrogen bonds link molecules to form chains along [001] direction (See
Figure 4 for reference). The centroids are separated by 5.24 Å and the total pairwise interaction energy is −72.4 kJ/mol (E
elec = −29.8 kJ/mol; E
pol = −11.5 kJ/mol; E
dis = −84.8 kJ/mol; E
rep = 67.1 kJ/mol) (
Table 2 and
Table 3). The high structural similarity is also observed in the C19‒H19B···Cl1
iii and π···π
iii (symmetry code: (iii) 1 − x, −y, 1 − z) stacking interactions joining molecules separated by 12.67 Å and with total pairwise interaction energy −25.2 kJ/mol (E
elec = −11.0 kJ/mol; E
pol = −1.5 kJ/mol; E
dis = −32.3 kJ/mol; E
rep = 25.5 kJ/mol) (
Table 2 and
Table 3). This value is lower compared with the same pair of molecules in compound
1 which is an indication of weaker attractions due to the change of a nitro group by a halogen. In general, the packing in compound
3 is similar to
1. Nevertheless, C‒H···O hydrogen bonds involving the nitro group in compound
1 induce a different energy framework compared to compound
3 (
Figure 8b), being the contribution of electrostatic forces lower in the last case. Also, the 2D fingerprints show differences between both structures. In the present case, the spikes from H···N interactions are most prominent and sharped in compound
3 which is consequent with shortest distances in C4‒H4···N1
i (
Figure 8a).
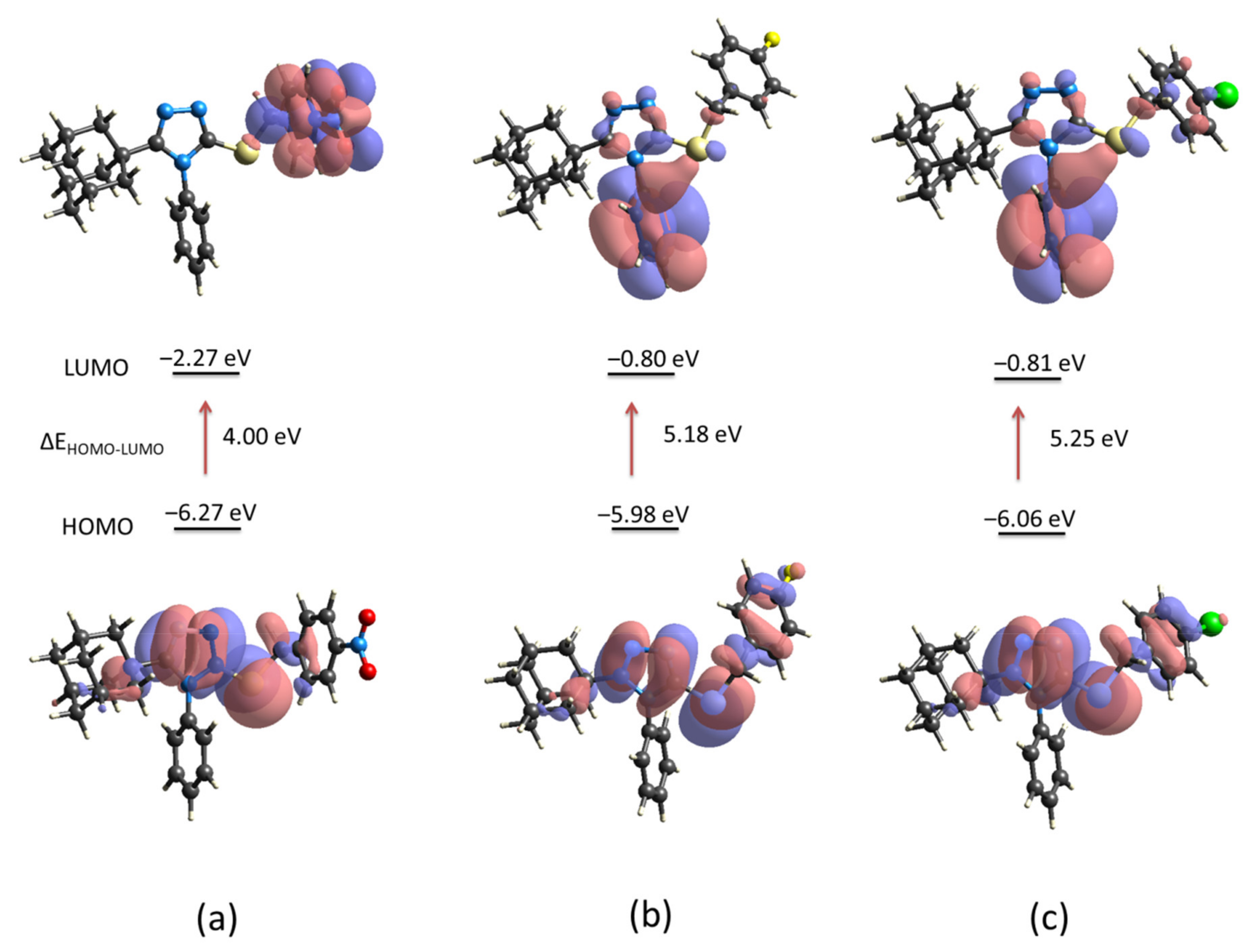
The energy levels of the electron-donor HOMO (highest occupied molecular orbital) and electron-acceptor LUMO (lowest unoccupied molecular orbital) for compounds
1,
2 and
3 were computed using the crystallographic information (
Figure 9). The energy gap between molecules is similar for compounds
2 and
3 but different from compound
1. The presence of the nitro group (NO
2) in the
para position of the aryl group in compound
1 induces the charge-transfer interaction involves mainly the triazole-sulfanyl and aryl (Ar-NO
2) moieties (
Figure 9a). As it is well known, the NO
2 group is an electron withdrawing group which under the description of their resonant structures, induces a concentration of positive charge at the respective
ortho-,
para- positions, being
meta-directors to electrophilic substitutions. On the other hand, halides are
ortho-,
para- directing groups, however, with a mildly deactivation behavior due to their dual properties as inductive withdrawal and resonance donation. This last description is the case of compounds
2 and
3. In these molecules, the charge-transfer interaction involves the triazole-sulfanyl and Ar-halogen moieties in the HOMO level, and the triazole-sulfanyl and aryl moieties in the LUMO level (
Figure 9b,c). The strong electron withdrawing property of NO
2 group is observed in the decrease of the LUMO energy (−2.27 eV). Even, the lower energy gap in compound
1 characterizes this molecule as soft, a property associated with low kinetic stability, high chemical reactivity, and greater polarizability due to the lower energy needed for excitation [
33]. Considering that HOMO and LUMO energies represent the ability to donate and gain an electron, respectively, it is clear that the presence of the NO
2, and F/Cl groups, modify the reactivity of these molecules. In all cases, the triazole-sulfanyl fragment has an active disposition to donate electrons but the molecular disposition towards potential nucleophilic addition reactions is different in compound
1 compared with compounds
2 and
3. Based on the calculated molecular orbitals, some derived parameters were highlighted such as chemical hardness, chemical potential and electrophilic index among others (
Table 4).
The chemical potential and chemical hardness were calculated using Koopmans’ theorem as:
μ = (E
LUMO + E
HOMO)/2, and
η = (E
LUMO − E
HOMO)/2 [
34,
35]. The electrophilicity index was calculated according to Parr et al. as
ω = μ2/2
η [
36]. The propensity to donate charge or electrodonating power,
ω− = [(3IP + EA)
2 / 16(IP − EA)], the propensity to accept charge or electroaccepting power,
ω+ = [(IP + 3EA)
2 / 16(IP − EA)], and net electrophilicity Δ
ω± =
ω+ +
ω−, were calculated according to Gázquez et al. [
37]. The magnitude of hardness
η parameter allows concluding that, effectively, molecules of
1 could be more reactive than
2 and
3. This softness is also observed in the lower value of the LUMO energy (−2.27 eV) which signifies that it is the best electron acceptor. This property suggests that
1 is the strongest electrophile, which is in good agreement with the higher net electrophilicity index (Δ
ω± = 9.61 eV).
2.3. Prediction of Activity Spectra and Molecular Docking Studies
Prediction of Activity Spectra (PASS) is an online structure-activity relationship tool that predicts pharmacological properties of over 4000 types of biological activities and targets based on the structure of the studied compound [
38]. PASS analysis was therefore used to predict the pharmacological properties of compounds
1,
2 and
3. The results indicated that the predicted highest probability of biological activity (Pa) was for anti-obesity and anti-diabetic (type 2) activities (
Table 5). The ability of compounds
1,
2 and
3 to inhibit the 11β-HSD1 enzyme, which is a target that has been identified to potentially treat obesity and type-2 diabetes [
19,
20,
21,
22], was predicted at Pa values of 0.619 to 0.678. These predicted 11β-HSD1 inhibitory abilities fell well within the top predicted activities of each compound (
Table 5).
Molecular docking has become widely used in the development of novel therapeutic agents. These techniques greatly improve our knowledge of the structural activity relationships between ligands and active site residues as well as conformational changes of the active site caused by the complex formed with a ligand [
39]. Therefore, the quality of the three-dimensional (3D) protein X-ray crystal structure is crucial for reproducible and accurate ligand docking. There are several methods in determining the best protein crystal structure with the most common method considering both the resolution (Å) as well as the R-factor values [
40]. Conversely, it has been reported that these metrics are not absolute and that these metrics alone cannot appropriately predict the performance of molecular docking within a specified protein crystal [
41,
42]. Holo X-ray crystal structures with different co-crystallized ligands affect the conformation of the receptors active site residues. Therefore, the native ligand or non-native ligands with similar molecular structures of a ligand-receptor complex will more likely exhibit tighter binding affinities compared to structurally different non-native ligands [
43]. Therefore, it can be proposed that if non-native ligands are able to dock into X-ray protein structure with a similar binding pose to their crystallized binding pose within their native enzyme, then it will probably be more proficient at accurately docking experimental ligands. In this study, the holo crystal structures were selected using the resolution and R-factor scores obtained from the protein data bank (PDB) (
https://www.rcsb.org, accessed on 26 July 2021) [
44]. Thereafter, the native ligands were re-docked into the respective enzymes using the self-docking approach. The self-docking approach assists in validating the docking protocol as well as determining the suitability of the protein structures. Finally, the cross-docking approach was employed to dock non-native 11β-HSD1 ligands into multiple 11β-HSD1 enzymes The cross-dock approach is used to establish the most suitable X-ray crystal structure to be used for experimental ligand docking [
43]. Previous studies have shown that non-native ligands with similar chemical structures to the native ligand, in the cross-docking approach, exhibited similar binding poses in reference to their crystallized binding pose [
45,
46]. Therefore, we extracted holo 11β-HSD1 protein structures that contained co-crystallized ligands that were either similar or diverse in chemical structure.
Sixteen holo 11β-HSD1 X-ray structures and their native ligands were prepared, docked and analyzed using the self-docking protocol and docking evaluation criteria (
Table 6). Ten of the sixteen holo X-ray structures successfully re-docked the native ligands met the criteria requirements. These structures were considered for cross-docking. Protein
4C7J, was selected as the most suitable template for superposition of the other crystal proteins active sites as it exhibited the best overall criteria results.
The successfully self-docked holo protein structures were prepared and superposed onto the X-ray crystal structure of
4C7J. The ligands were prepared and saved in a merged database. The docking protocol was altered to 100 returned poses for re-scoring to improve docking accuracy. To ensure the validity of the docking protocol, the selfdocking criteria was used on the re-docked native ligands. Three of the ten holo X-ray structures (
3HFG,
4C7K and
4K1L) were unable to reproduce the native ligand’s crystallographic binding pose and therefore, were removed from further analysis. The cross-docking results were averaged over all the ligands for comparison (
Table 7 and
Supplementary Materials, Tables S1–S3).
The importance of cross-docking was observed as multiple protein structures were unable to successfully dock non-native ligands that met the docking analysis criteria. Only
4HX5 was able to reproduce an average top pose below 2 Å, 4
C7J and
4HX5 were able to reproduce an average lowest pose RMSD of below 2 Å, 4
C7J,
4HX5,
4IJV,
4IJW and
5QII were able to reproduce 3 or more crystallographic poses of non-native ligands below 2 Å and no protein structures were able to reproduce a RMSD average across the top 5 poses below 3 Å. Most of the protein structures were able reproduce the binding affinity scores of the non-native ligands to within 1.0 kcal/mol when compared to the native binding affinity scores (
Supplementary Materials, Table S4). The majority of the ligands exhibited non-native receptor binding affinity scores higher than the native receptor binding affinity scores. Thus, these scores confirm that ligands exhibit tighter predicted binding affinity when docked within their native receptor when compared to non-native receptors. The results also confirmed that proteins containing co-crystallized ligands with similar chemical structures exhibited RMSD results within the self-docking criteria. Protein structures 4
C7J and
4C7K as well as
4IJW and
5QII exhibited excellent RMSD cross-docking results. Even though
4C7K was unable to reproduce self-docking RMSD values within the docking criteria it was however able to do so for 4
C7J. Protein structures 4
C7J and
4HX5 exhibited the best overall cross-docking results. 4
C7J was chosen as the most appropriate protein structure for the docking of compounds
1,
2 and
3 over
4HX5. Compounds
1–
3 have a number of structural features, e.g., the adamantane- and triazole moieties, that are similar to the potent 11β-HSD1 co-crystallized inhibitor 4-cyclopropyl-
N-(trans-5-hydroxy-2-adamantyl)-2-(2-hydroxyethoxy)-thiazole-5-carboxamide (
4YQ, 11β-HSD1 IC
50 = 9.9 nM) [
47].
4HX5 was also unable to significantly reproduce the crystallized poses for both 4
C7J and
4C7K native ligands. Compounds
1,
2 and
3 were prepared and docked into X-ray protein structure 4
C7J as described in the methods section. The docking results were analyzed using web-based protein-ligand complex analysis server Protein-Ligand Interaction Profiler (PLIP,
https://plip-tool.biotec.tu- dresden.de, accessed on 26 July 2021) [
48]. Compounds
1,
2 and
3 docked conformations were able to bind to the active site with binding affinity scores of −8.30 kcal/mol, −7.70 kcal/mol and −7.83 kcal/mol, respectively. The binding affinity scores are similar to
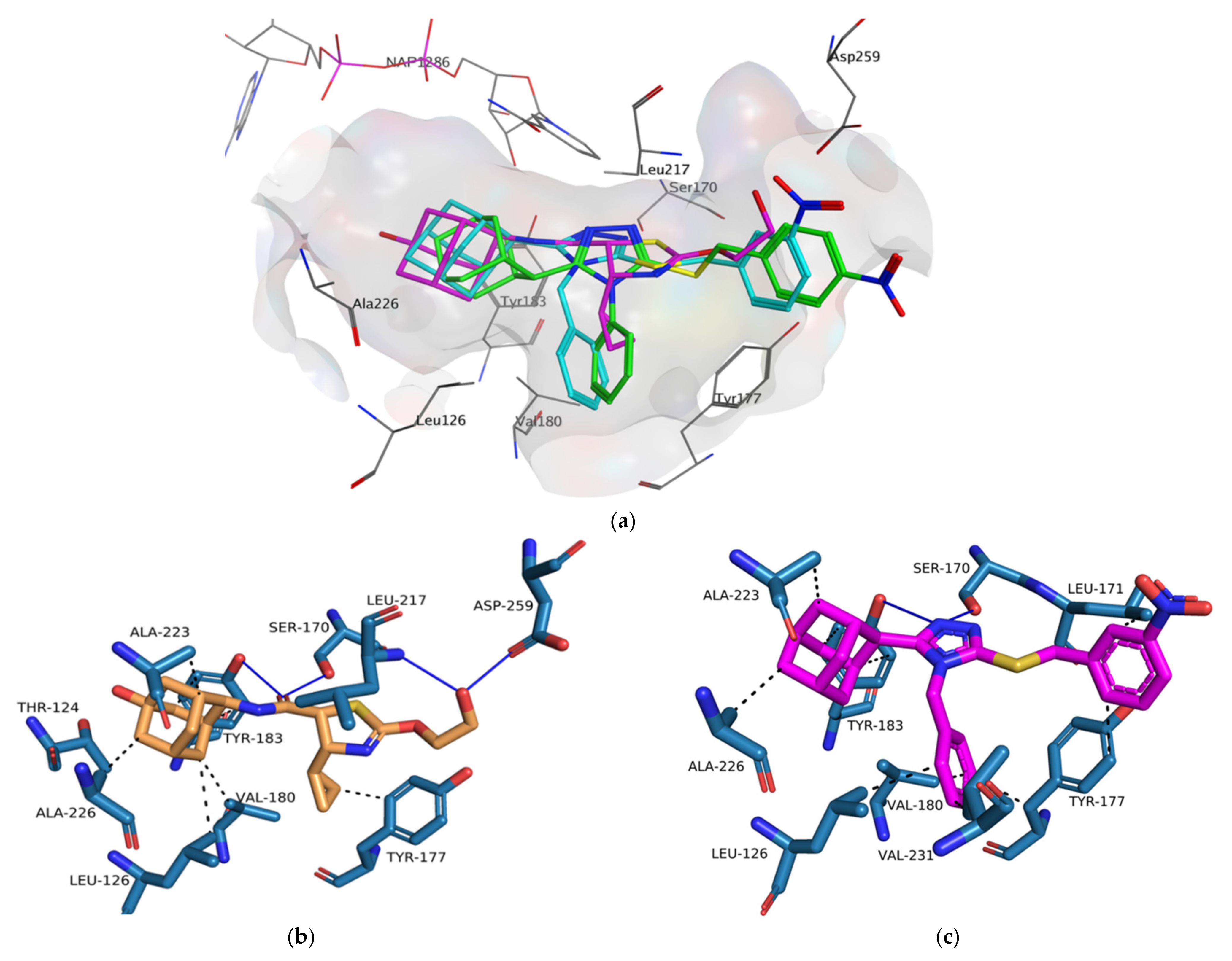
4YQ (−8.20 kcal/mol).
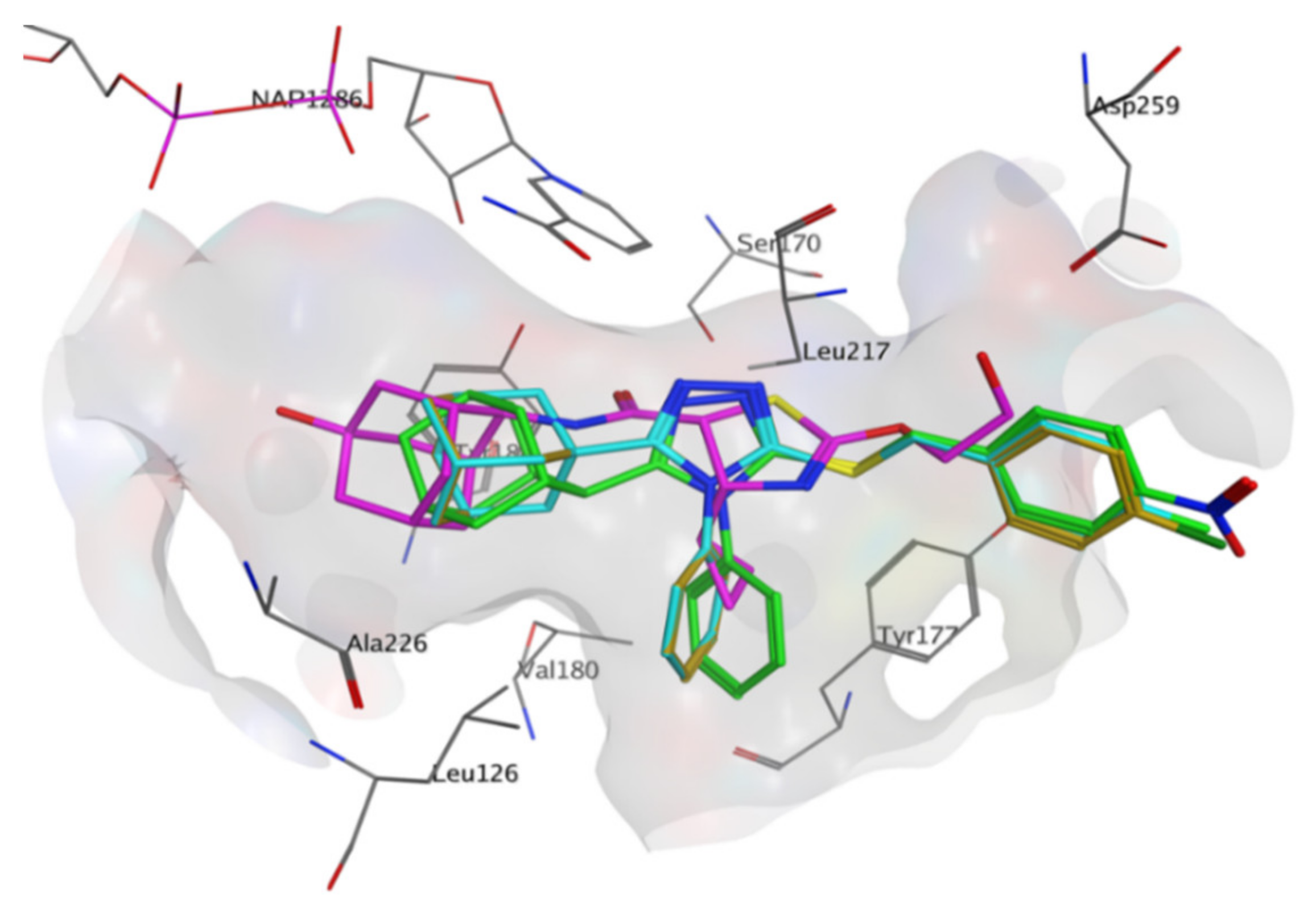
4YQ exhibited hydrogen bond (HB) interactions with important active site residues Ser 170 (HB), Tyr 183 (HB), Asp 259 (HB), Leu 217 (HB) (
Figure 10 and
Figure 11a). The adamantane moiety of
4YQ was buried deep within the hydrophobic pocket exhibiting hydrophobic interactions (HI) with residues Ala 223, Ile 121, Val 180, Tyr 183 and Leu126. The 4-cyclopropylthiazole moiety substituent interacted with Tyr 177 with a hydrophobic interaction within the secondary entrance, positioned center bottom of the active site in
Figure 10. Interactions with these residues have previously shown to be important for 11β-HSD1 inhibition [
49,
50,
51,
52].
Compounds
1–
3 were orientated in a similar manner to
4YQ with the adamantane moiety positioned within the hydrophobic pocket between Tyr183, Ala 226, Ala 223 and Val180, the benzylsulfanyl moiety spanning towards the primary entrance of the active site and the 4-phenyl-1,2,4-triazole moiety occupying a similar position within the secondary entrance as the 4-cyclopropylthiazole moiety of
4YQ (
Figure 10 and
Figure 11). In general, compounds
1–
3 exhibited weak hydrophobic interactions with 11β-HSD1 active site residues with only compound
1 interacting with Tyr 177 through a π-π interaction. The phenyl substitution conjugated to the triazole moiety is positioned within the secondary entrance of the active site and resulted in pulling the compound towards the primary entrance of the active site. The positioning of the phenyl substitution also caused the triazole moiety to rotate out of plane from key residues responsible for active site catalysis Ser 170 and Tyr 183 (
Figure 10 and
Figure 11). The lipophilic adamantane moiety is then removed from the deep pocket within the active site reducing the number of hydrophobic interactions within the hydrophobic pocket. The nitro group and halogen substitutions are exposed to the outside of the active site and therefore lacks the ability to form potential binding interactions with the active site pocket (
Figure 10).
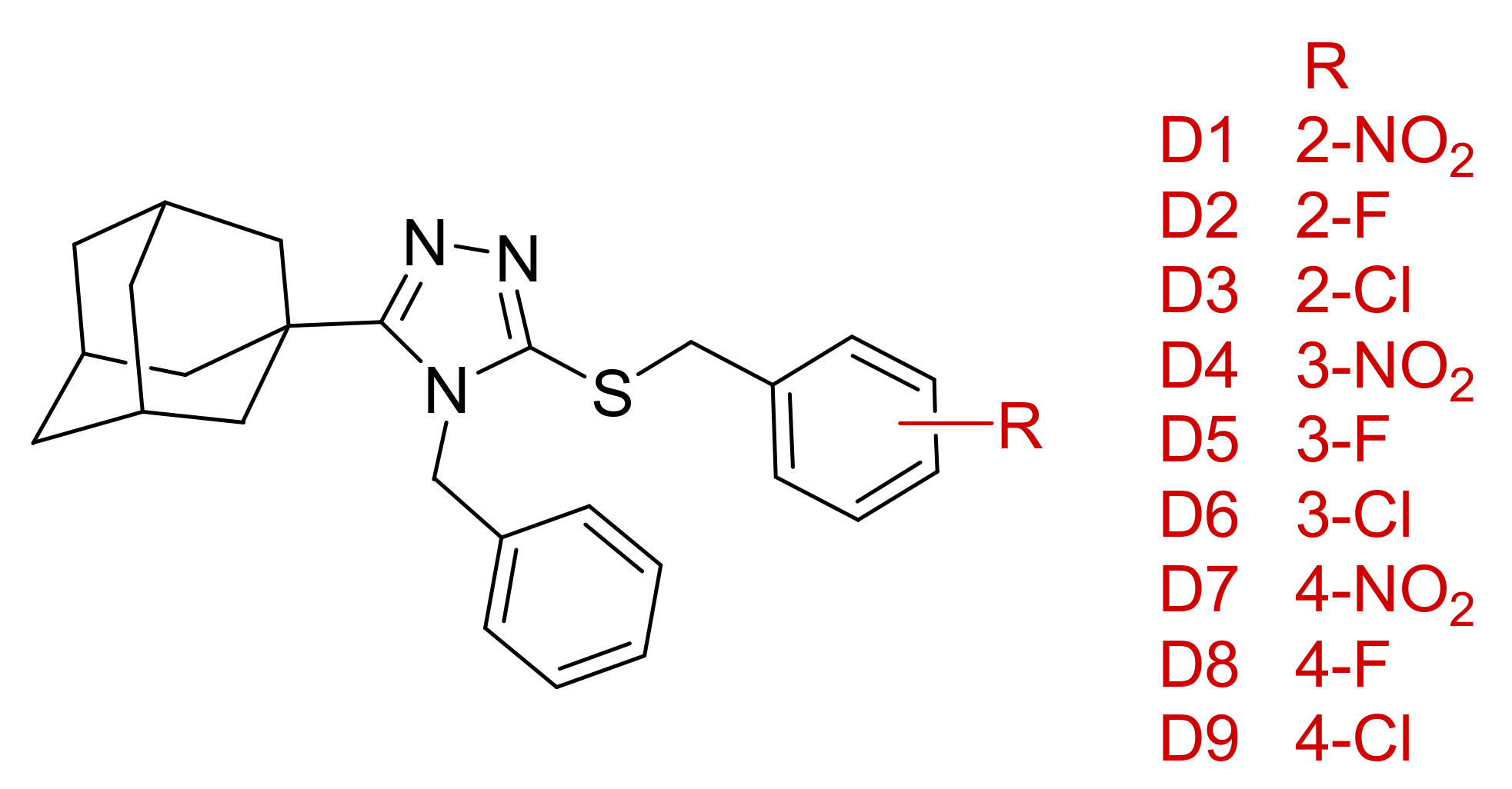
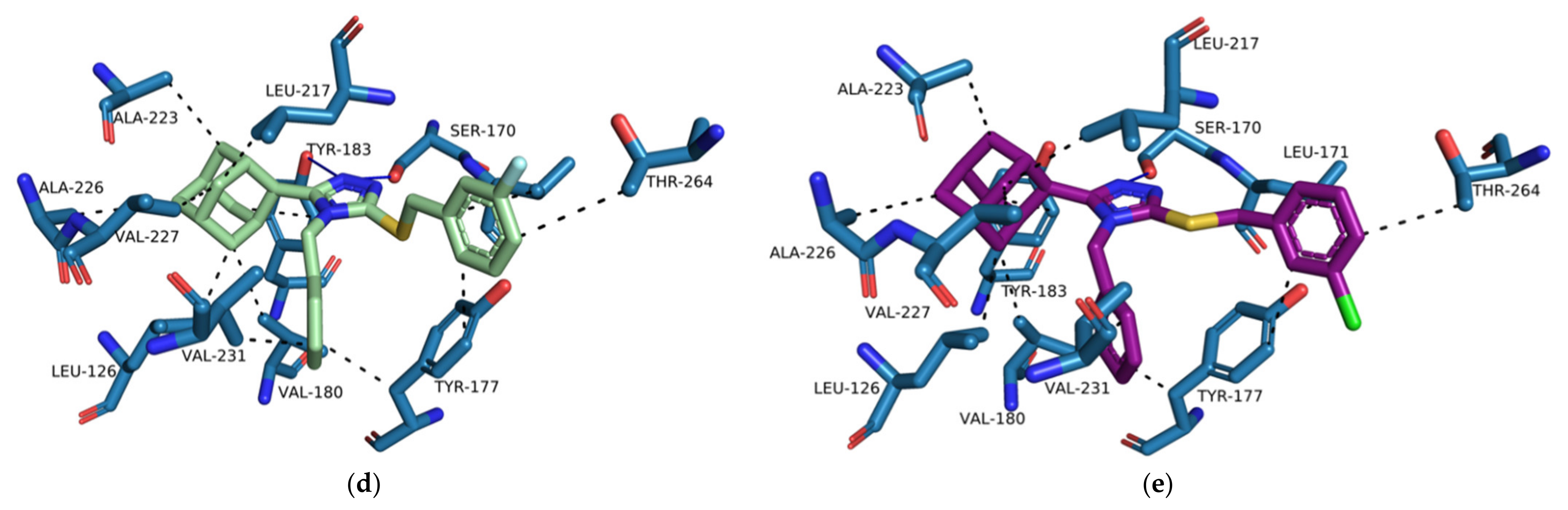
Based on the docking results, we propose a series of compounds (
D1–
D9,
Figure 12) based on the structures of compounds
1–
3 as potential 11β-HSD1 inhibitors with improved binding interactions and binding affinity scores. The proposed compounds were designed through the employment of rational as well as computer-aided drug design (CADD) strategies. The designed compounds were structurally altered by the addition of a single carbon linker between the triazole moiety and the phenyl substitution. The additional carbon linker is designed to improve the compound flexibility and allow the triazole moiety to interact with key residues Ser 170 and Tyr 183 through hydrogen bond interactions. We also varied the positions of the nitro and halogen substitutions on the benzylsulfanyl moiety. The CADD designed compounds’ binding conformations exhibited binding affinity scores between −7.98 to −8.48 kcal/mol (
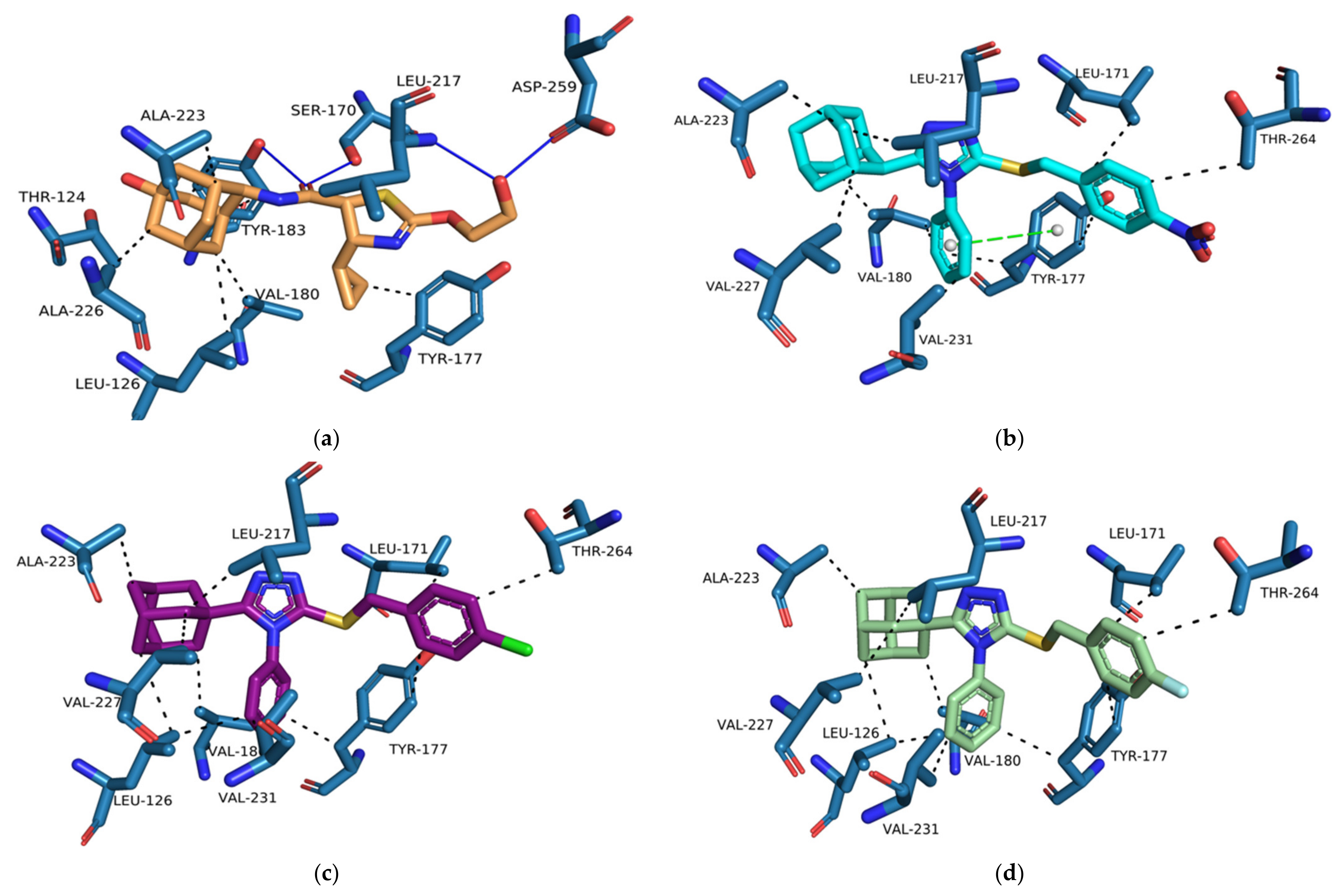
Supplementary Materials, Table S5). The majority of the designed compounds interacted with both Ser 170 and Tyr 183 through hydrogen bond interactions as well as Tyr 177 and Tyr 183 through hydrophobic interactions, similar to
4YQ (
Figure 13b–e and
Supplementary Materials,
Tables S6–S18).
The increased flexibility of the phenyl substitution by the addition of the single carbon linker resulted in the rotation of the triazole moiety into a planar position in reference to the Ser 170 and Tyr 183 residues and pushed the lipophilic adamantane moiety deeper into the hydrophobic pocket (
Figure 13a). The
para-substituted compounds,
D7–
D9, were still exposed to the external environment due to the length of the structures of the compounds. The
ortho- and
meta-substituted compounds,
D1–
D6, reduced the exposure of the substitutions to the external environment. However, the halogen substitutions did not interact with the Asp 259. When halogens are bound to aromatic carbons the electron density of the halogen is redistributed resulting in the formation of an electrophilic region at the distal end of the halogen atom. The electrophilic region known as the sigma-hole contributes to the halogen bond donating capabilities of halogens [
55]. However, since linearity of halogen bond donor angles is crucial for halogen bonding, analysis tool like PLIP have strict linear halogen bond donor angle cut-offs of ≥ 165° [
48]. The angles between the halogen of the halogen substituted series
D compounds and Asp 259 was consistently below 100° and therefore no halogen donor bond interactions were formed. No binding interactions were formed between the nitro substitution and Asp 259, even though they are observed to be in proximity to one another (
Figure 13a). Both the nitro substitution and Asp 259 are anionized within the 11β-HSD1 active site and consequently are unable to form any type of binding interaction. Therefore, proton donors containing substitutions on the benzylsulfanyl moiety should be considered for future studies.


 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}