
The Crystal Structure and Intermolecular Interactions in Fenamic Acids–Acridine Complexes


Abstract
1. Introduction
2. Results
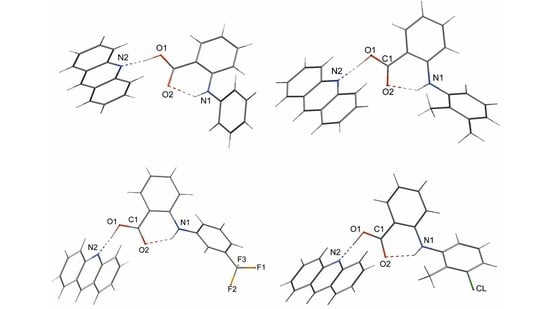
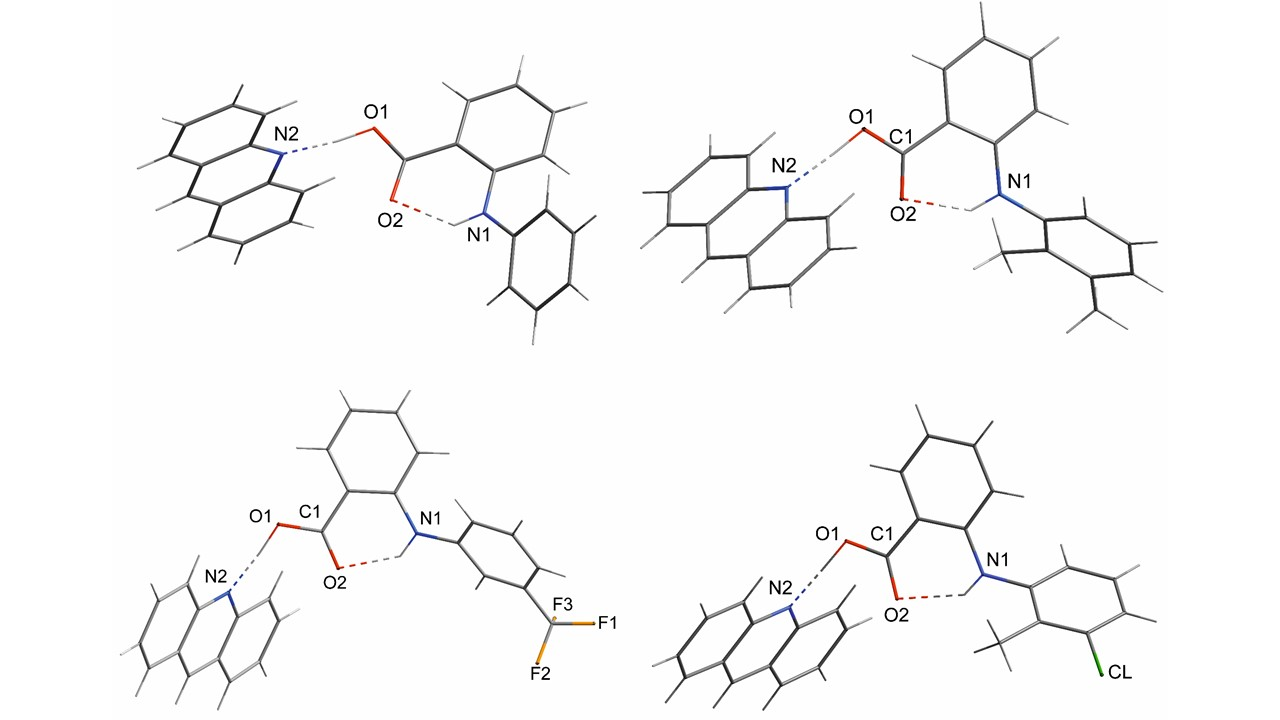
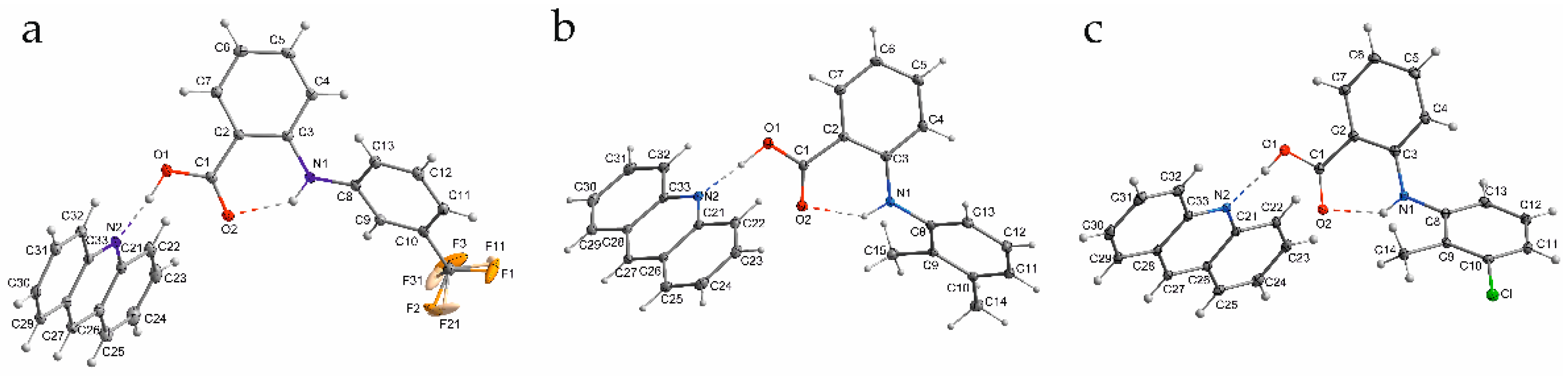
2.1. The Inter- and Intramolecular Hydrogen Bonds of Fenamic Acids–Acridine Complexes
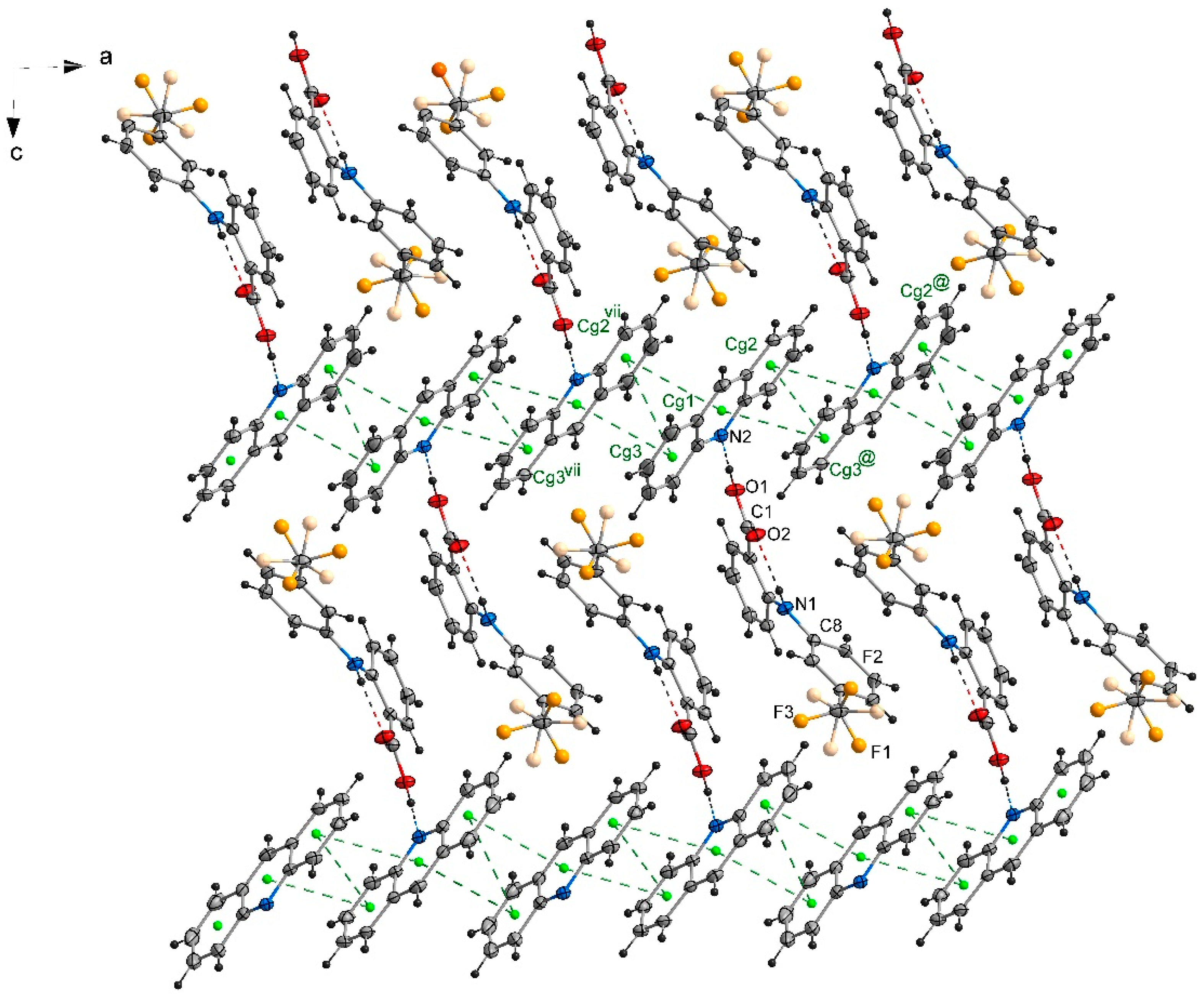
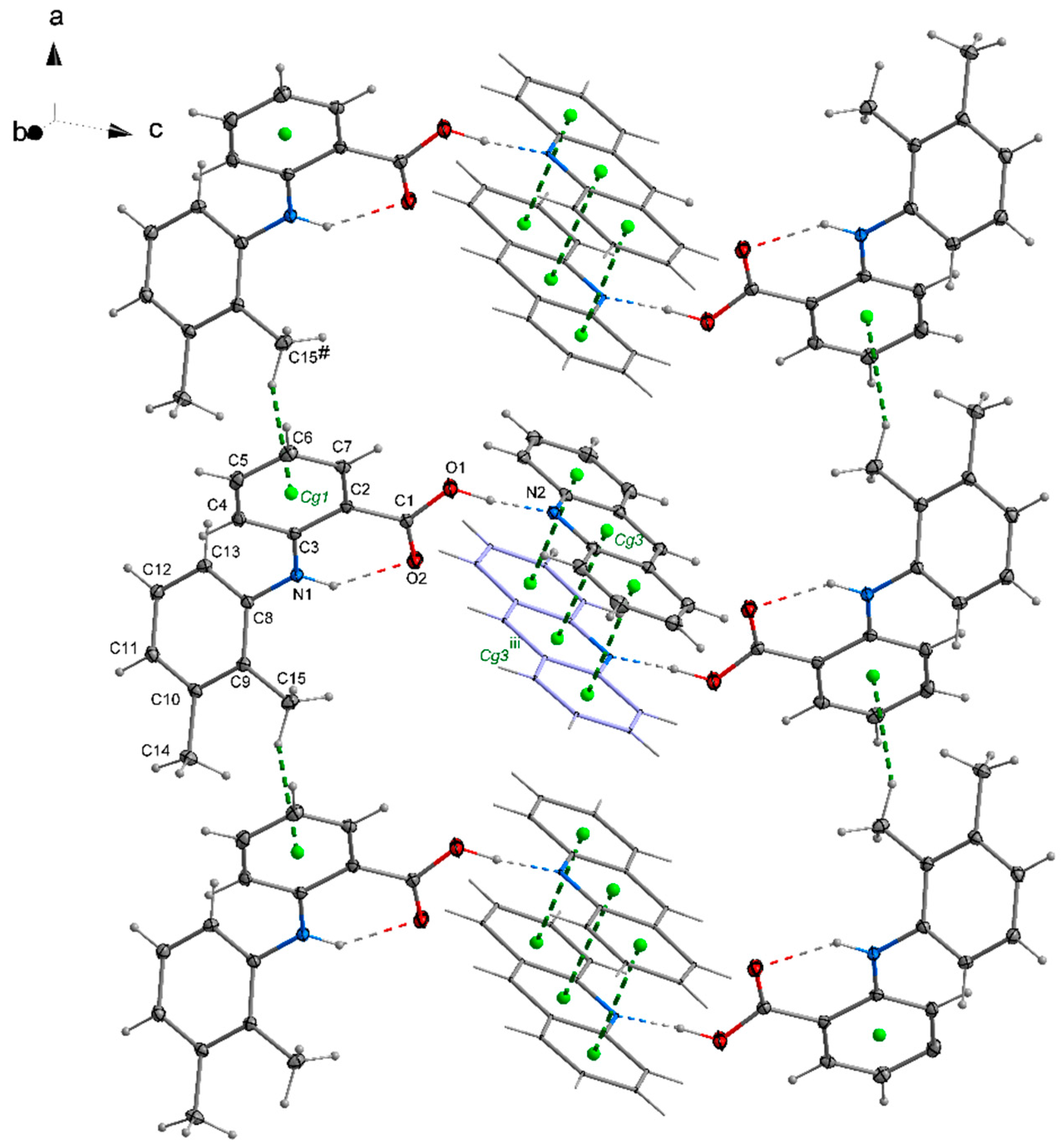
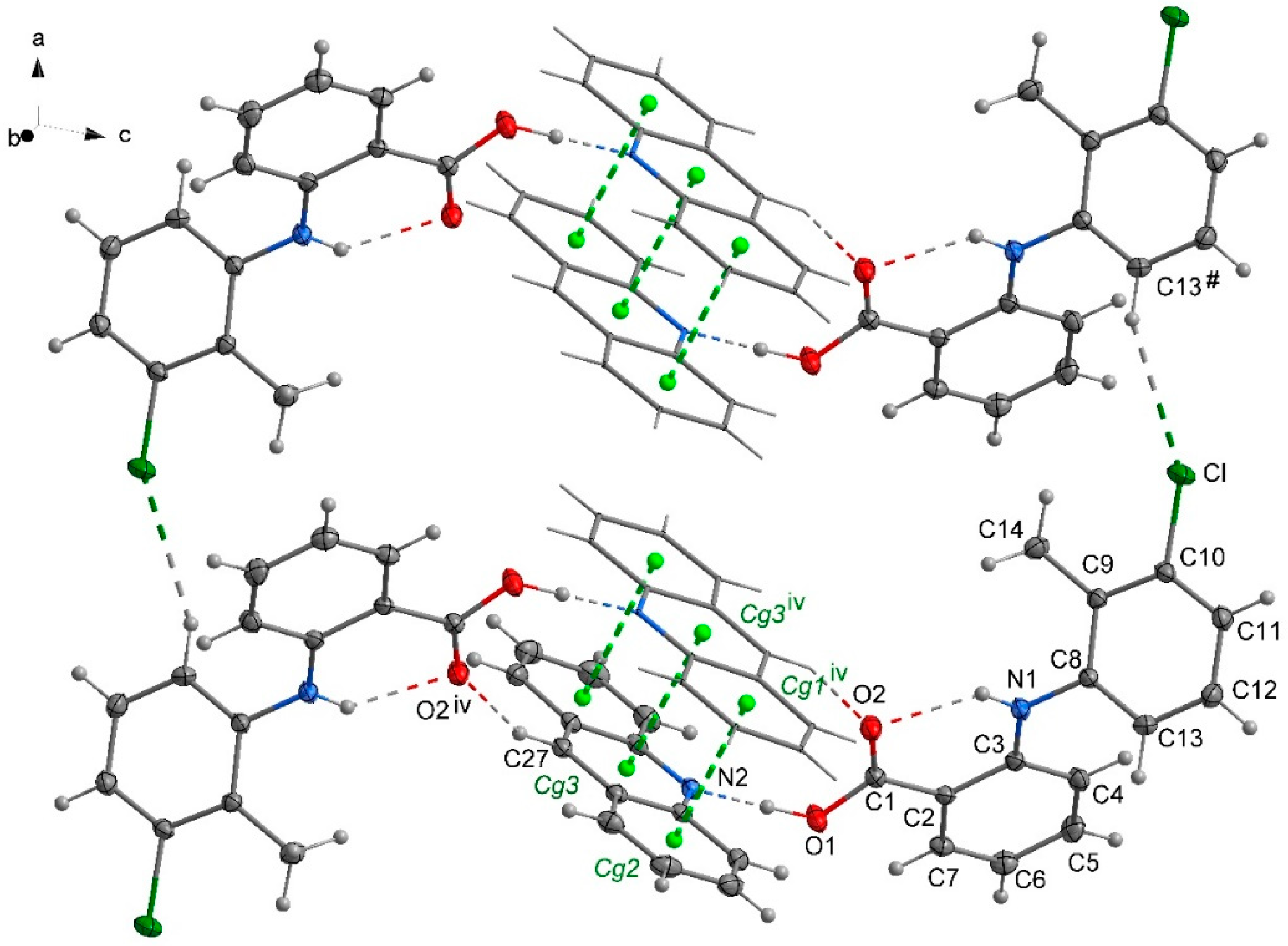
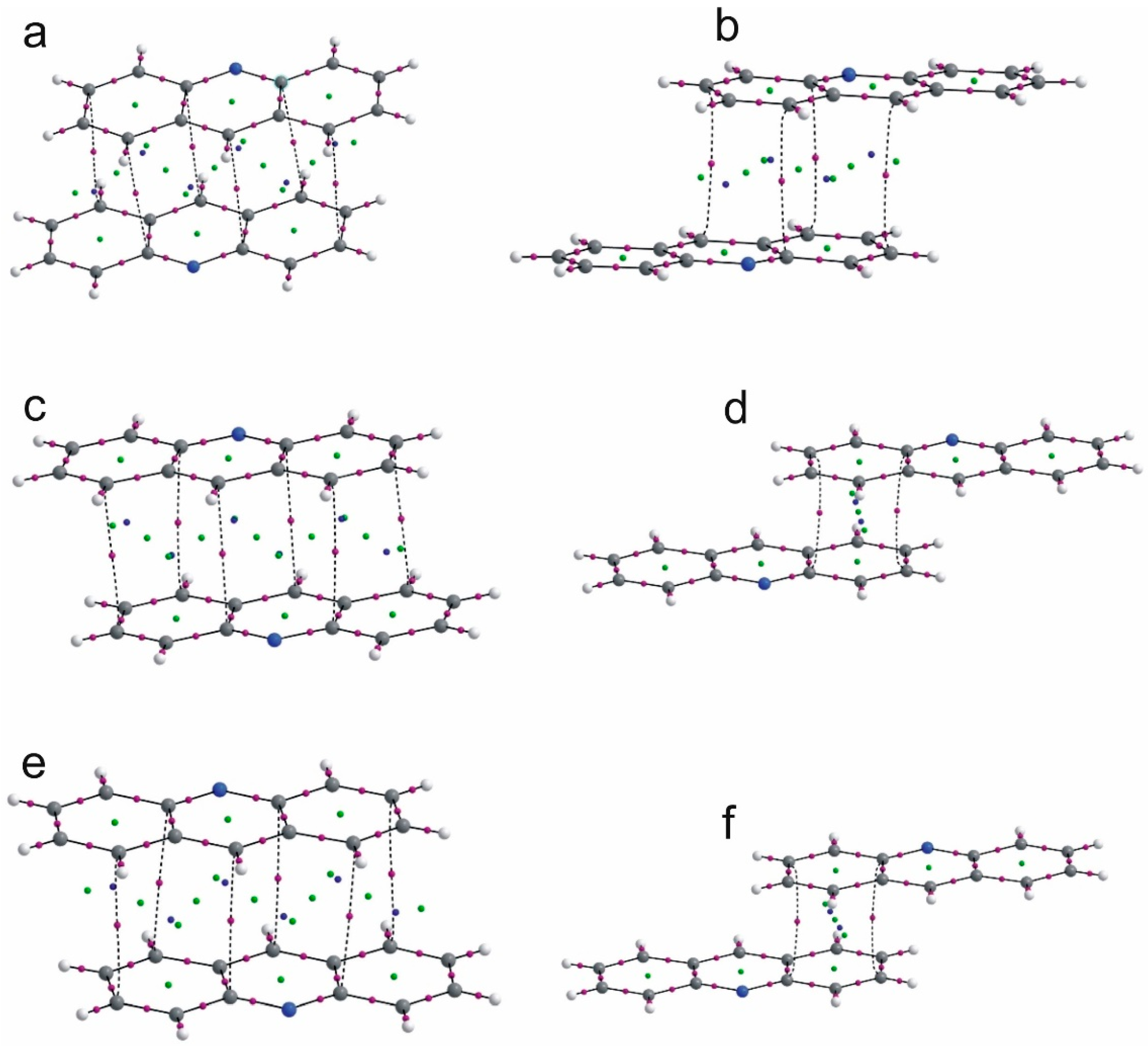
2.2. The Packing of the Molecules in Crystals
2.3. General Motifs in the Crystals of Acridine–Fenamic Acids
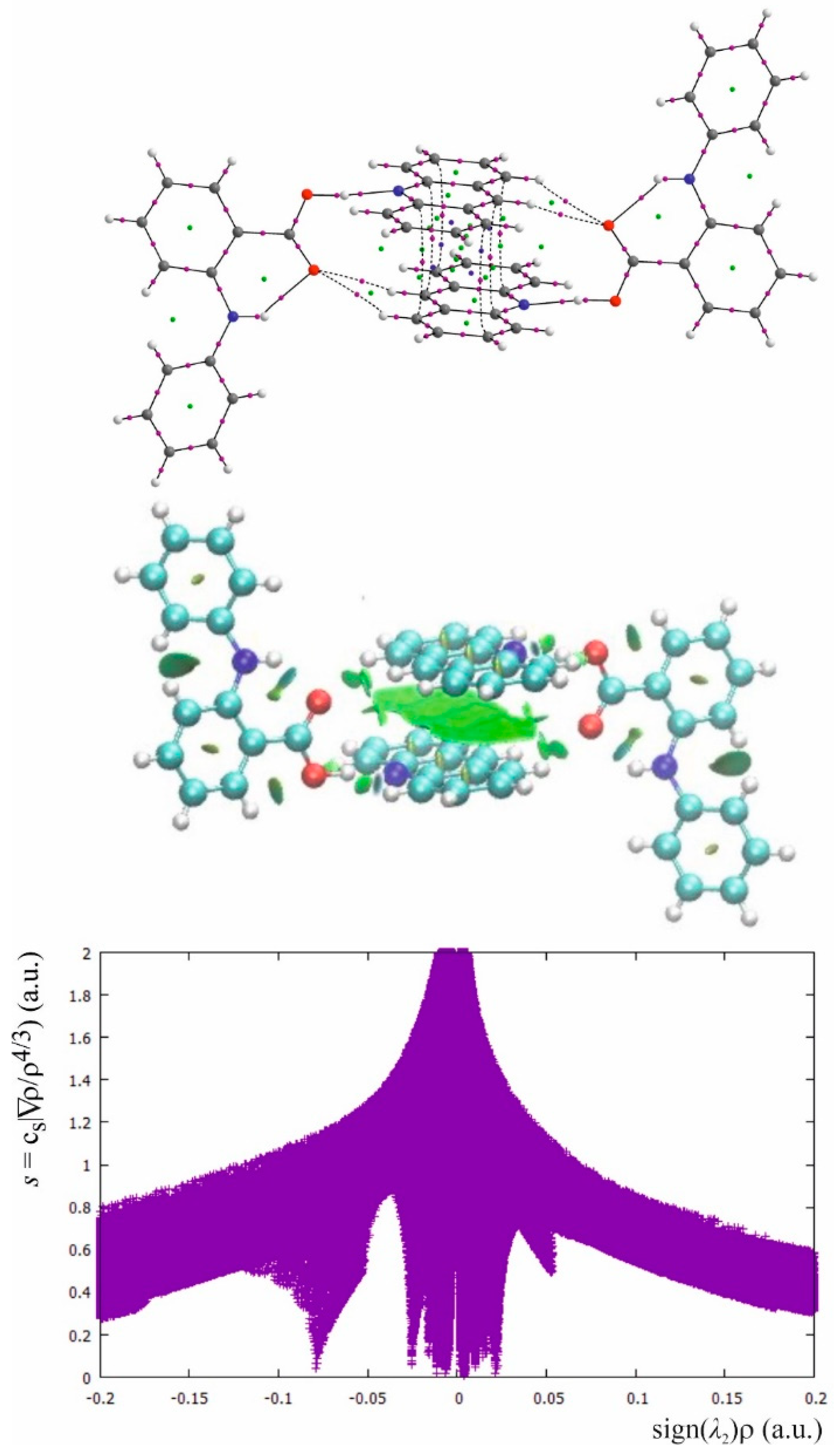
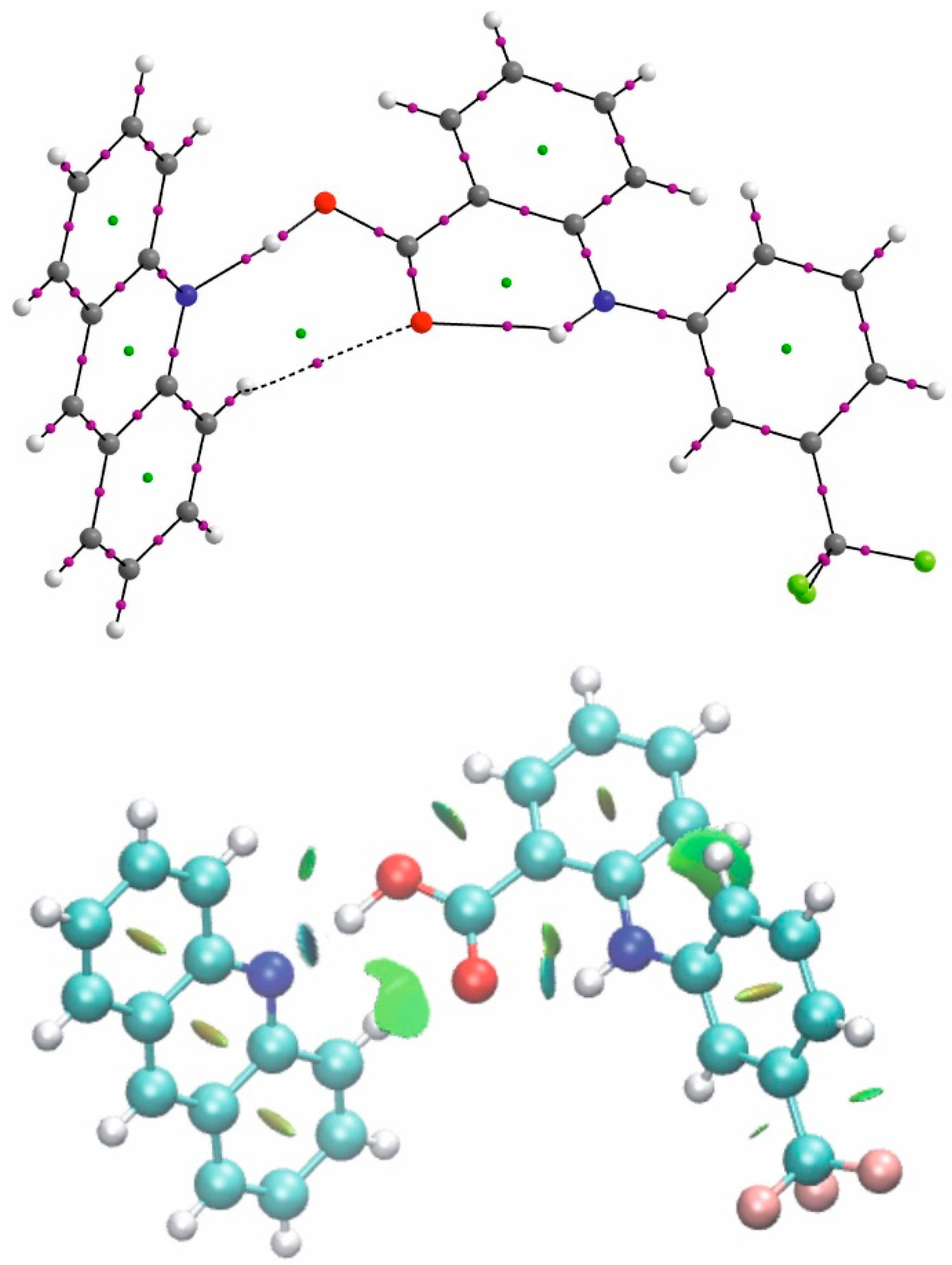
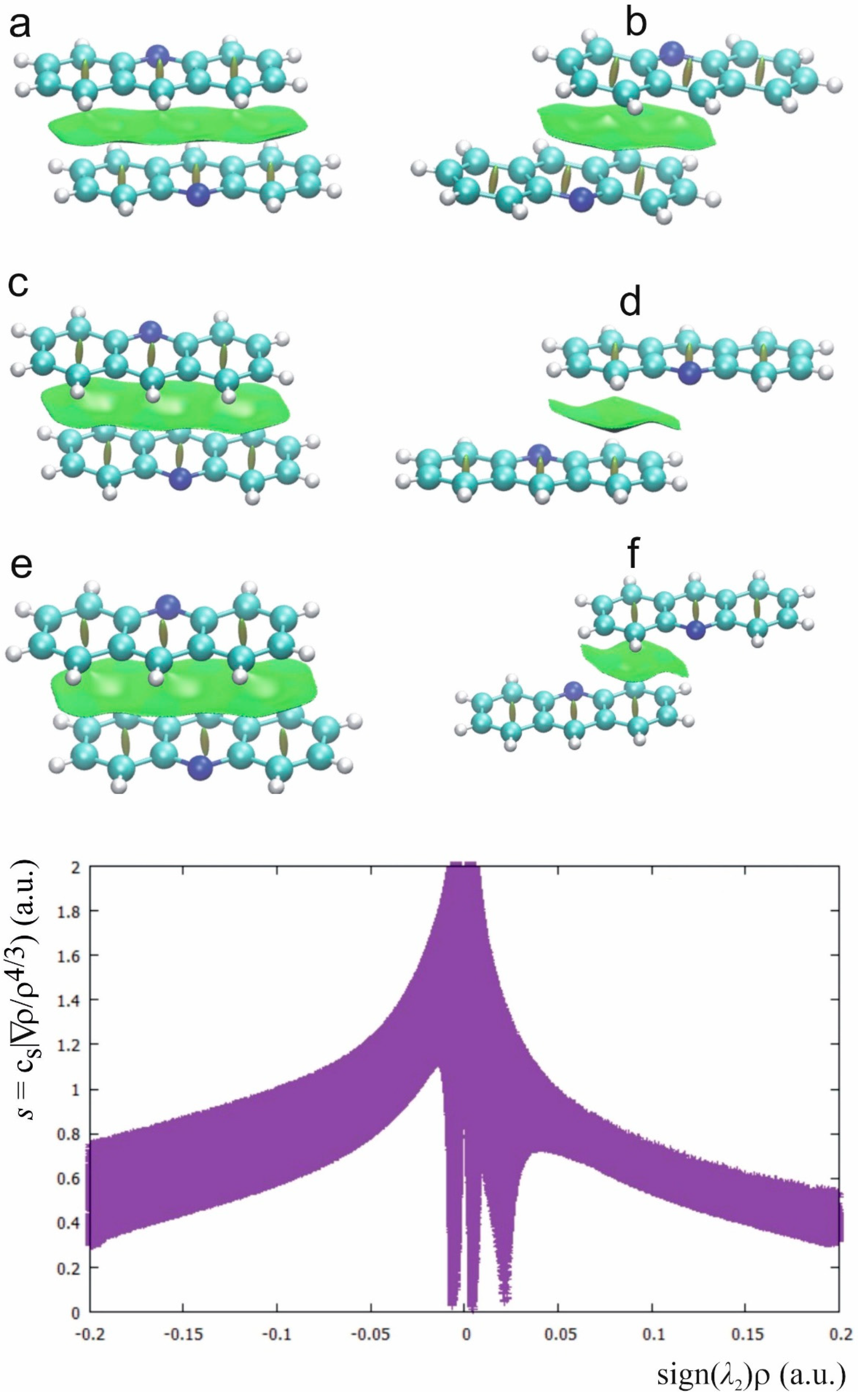
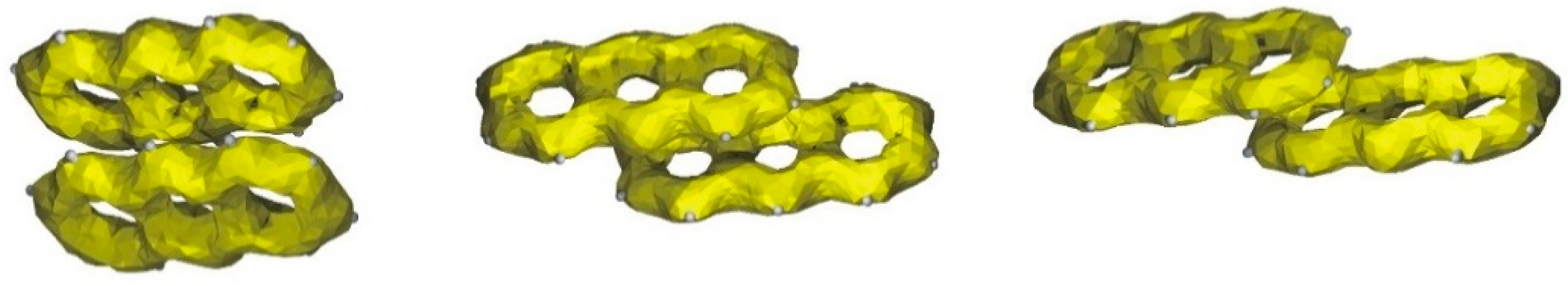
2.4. Stacking Interactions in Acridine–Fenamic Acids Complexes
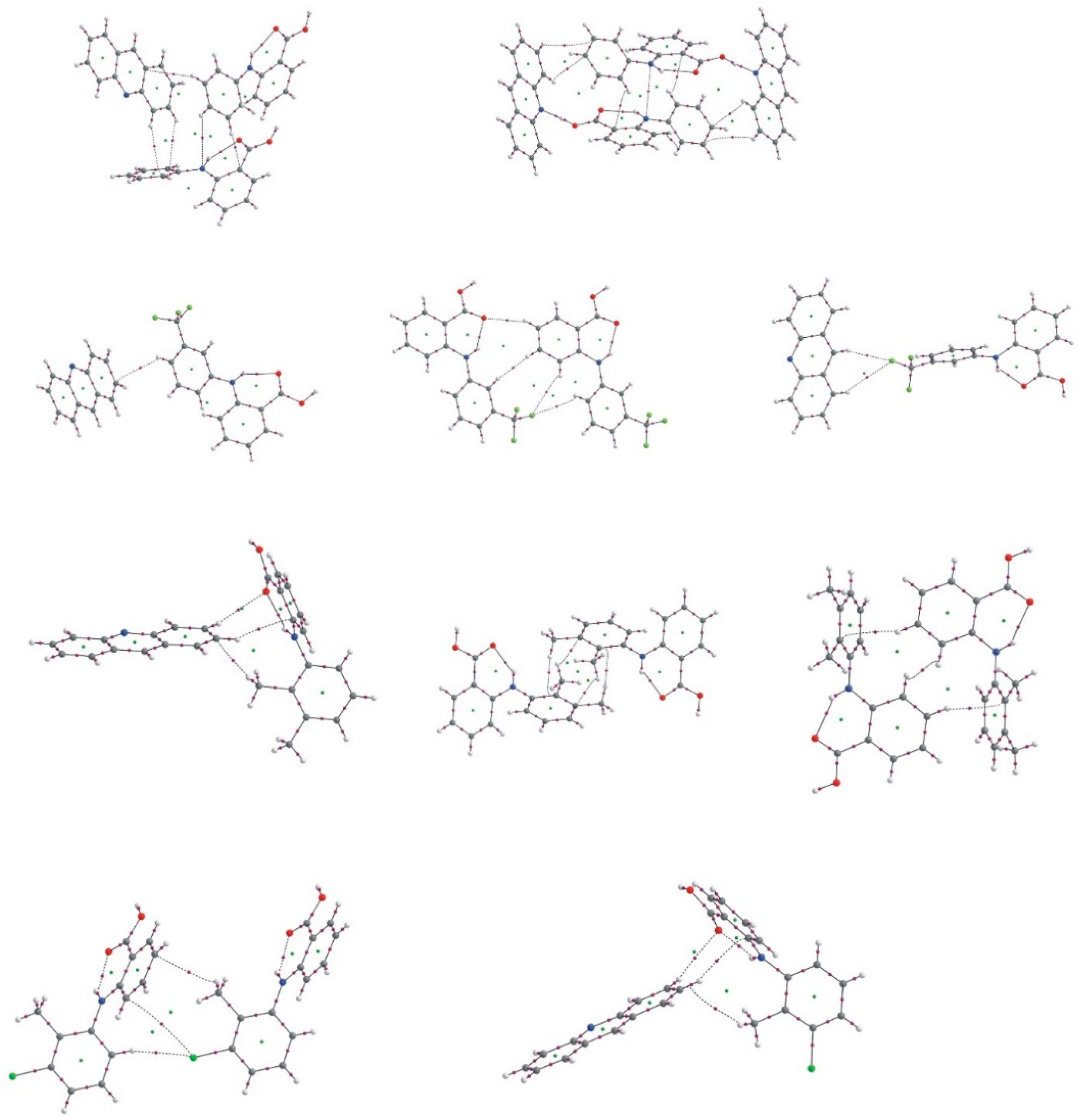
2.5. Additional Weak Interactions in the Crystal Structure of the Investigated Acridine Complexes
3. Materials and Methods
3.1. Crystal Preparation
3.2. X-ray Diffraction
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Khansari, P.S.; Halliwell, R.F. Evidence for neuroprotection by the fenamate NSAID, mefenamic acid. Neurochem. Int. 2009, 55, 683–688. [Google Scholar] [CrossRef]
- Lee, S.H.; Bahn, J.H.; Whitlock, N.C.; Baek, S.J. Actvating transcription factor 2 (ATF2) controlstolfenamic acid–induced ATF3 expression via MAP kinase pathways. Oncogene 2010, 29, 5182–5192. [Google Scholar] [CrossRef] [PubMed]
- Steed, J.W. The role of co-crystals in pharmaceutical design. Trends Pharmacol. Sci. 2013, 34, 185–193. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Poperties. Cryst. Growth Des. 2009, 2950–2967. [Google Scholar] [CrossRef] [PubMed]
- Jerzykiewicz, L.; Sroka, A.; Majerz, I. The Crystal Structure and Behavior of Fenamic Acid-Acridine Complex under High Pressure. J. Pharm Sci. 2016, 105, 3487–3495. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Woller, T.; Martin-Martinez, F.J.; Contreras-García, J.; Geerlings, P.; De Proft, F. Understanding the Fundamental Role of π/π, σ/σ, and σ/π Dispersion Interactions in Shaping Carbon-Based Materials. Chem. Eur. J. 2014, 20, 4931–4941. [Google Scholar] [CrossRef]
- Bone, R.G.A.; Bader, R.F. Identifying and Analyzing Intermolecular Bonding Interactions in van der Waals Molecules. J. Phys. Chem. 1996, 100, 10892–11091. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Altun, A.; Neese, F.; Bistoni, G. The Effect of Electron Correlation on Intermolecular Interactions: A Pair Natual Orbitals Coupled Cluster Based Local Energy Decomposition Study. J. Chem. Theory Comput. 2019, 15, 1761–1776. [Google Scholar] [CrossRef] [PubMed]
- Majerz, I.; Gutmann, M.J. Mechanism of proton transfer in the strong OHN intermolecular hydrogen bond. RSC Adv. 2011, 1, 219–228. [Google Scholar] [CrossRef]
- Zhou, T.; Li, F.; Fan, Y.; Song, W.; Mu, X.; Zhang, H.; Wang, Y. Hydrogen-bonded dimer stacking induced emission of aminobenzoic acid compounds. Chem. Commun. 2009, 3199–3201. [Google Scholar] [CrossRef] [PubMed]
- López-Mejías, V.; Kampf, J.W.; Matzger, A.J. Nonamorphism in Flufenamic Acid and a New Record for a Polymorphic Compound with Solved Structures. J. Am. Chem. Soc. 2012, 134, 9872–9875. [Google Scholar] [CrossRef] [PubMed]
- SeethaLekshmi, S.; Guru Row, T.N. Conformational Polymorphism in a Non-steroidal Anti-inflammatory Drug, Mefenamic Acid. Cryst. Growth Des. 2012, 12, 4283–4289. [Google Scholar] [CrossRef]
- López-Mejías, V.; Kampf, J.W.; Matzger, A.J. Polymer-induced heteronucleation of tolfenamic acid: Structural investigation of a pentamorph. J. Am. Chem. Soc. 2009, 131, 4554–4555. [Google Scholar] [CrossRef] [PubMed]
- Case, D.H.; Srirambhatla, V.K.; Guo, R.; Watson, R.E.; Price, L.S.; Polyzois, H.; Cockcroft, J.K.; Florence, A.J.; Tocher, D.A.; Price, S.L. Successful Computationally Directed Templating of Metastable Pharmaceutical Polymorphs. Cryst. Growth Des. 2018, 18, 5322–5331. [Google Scholar] [CrossRef]
- Zhikol, O.A.; Shishkin, O.V.; Lyssenko, K.A.; Leszczynski, J. Electron density distribution in stacked benzene dimers: A new approach towards the estimation of stacking interaction energies. J. Chem. Phys. 2005, 122, 144104–144108. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Bloom, J.W.G. Toward a More Complete Understanding of Noncovalent Interactions Involving Aromatic Rings. J. Phys. Chem. A 2014, 118, 6133–6147. [Google Scholar] [CrossRef]
- Kim, K.S.; Tarakeshwar, P.; Lee, J.Y. Molecular Clusters of π-Systems: Theoretical Studies of Structures, Spectra, and Origin of Interaction Energies. Chem. Rev. 2000, 100, 4145–4186. [Google Scholar] [CrossRef]
- Główka, M.L.; Martynowski, D.; Kozłowska, K. Stacking of six-membered aromatic rings in crystals. J. Mol. Struct. 1999, 474, 81–89. [Google Scholar] [CrossRef]
- Bora, P.; Saikia, B.; Sarma, B. Regulation of π…π Stacking Interactions in Small Molecule Cocrystals and/or Salts for Physiochemical Property Modulation. Cryst. Growth Des. 2018, 18, 1448–1458. [Google Scholar] [CrossRef]
- Morokuma, K.J. Molecular Orbital Studies of Hydrogen Bonds. III. C=O…H–O Hydrogen Bond in H2CO…H2O and H2CO…2H2O. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1997, 46, 1–10. [Google Scholar] [CrossRef]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2014. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Keith, T.A. AIMALL; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | 1 | 2 | 3 |
|---|---|---|---|
| Chemical formula | C14H10F3NO2 · C13H9N | C15H15NO2 · C13H9N | C14H12ClNO2 · C13H9N |
| Mr | 460.44 | 420.49 | 440.91 |
| Crystal system, space group | Monoclinic, P21/n | Triclinic, P-1 | Triclinic, P-1 |
| Temperature (K) | 100(2) | 100(2) | 100(2) |
| a (Å) | 9.348(3) | 8.052(3) | 8.0560(18) |
| b (Å) | 7.976(2) | 9.788(3) | 9.6596(19) |
| c (Å) | 29.100(9) | 14.695(4) | 14.880(3) |
| α (°) | 103.10(3) | 103.69(2) | |
| β (°) | 92.86(3) | 101.01(3) | 101.73 (2) |
| γ (°) | 98.15(3) | 99.44(2) | |
| V (Å 3) | 2167.0(11) | 1086.3(6) | 1073.8(4) |
| Z | 4 | 2 | 2 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα |
| Crystal size (mm) | 0.37 × 0.30 × 0.28 | 0.70 × 0.67 x 0.60 | 0.18 × 0.18 × 0.15 |
| Data collection | |||
| Diffractometer | Xcalibur, Ruby, Gemini ultra-CCD κ-geometry diffractometer | Xcalibur, Ruby-CCD κ-geometry diffractometer | XtaLAB Synergy R, DW system, HyPix-Arc 150 diffractometer |
| No. of measured, independent and observed [I >2σ(I)] reflections | 24291, 6404, 5076 | 9125, 5884, 4667 | 15700, 5380, 4656 |
| Rint | 0.025 | 0.020 | 0.017 |
| Refinement | |||
| R[F2 > 2σ(F2)], wR(F2), S | 0.047, 0.118, 1.03 | 0.047, 0.126, 1.04 | 0.040, 0.115, 1.04 |
| No. of parameters | 342 | 297 | 296 |
| No. of restraints | 0 | 0 | 0 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.37, −0.27 | 0.38, −0.25 | 1.18, −0.36 |
| O1–H1O…N2 | N1–H1…O2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | C1-O1 [Å] | C1=O2 [Å] | O1–H1O [Å] | H1O…N2 [Å] | O1…N2 [Å] | OHN [°] | N1–H1 [Å] | H1…O2 [Å] | N1… O2 [Å] | NHO [°] |
| acridine–fenamic acid [5] | 1.3162(18) | 1.2336(18) | 1.06(2) | 1.55(2) | 2.609(2) | 173(2) | 0.886(16) | 1.99(2) | 2.691(2) | 135(2) |
| Acridine–flufenamic acid (1) | 1.3221(14) | 1.2292(15) | 0.990(18) | 1.626(18) | 2.6118(14) | 173.2(16) | 0.872(16) | 1.958(15) | 2.6714(15) | 138.0(14) |
| Acridine–mefenamic acid (2) | 1.3172(15) | 1.2317(16) | 1.021(19) | 1.597(19) | 2.6163(16) | 175.0(16) | 0.876(17) | 1.944(16) | 2.6660(16) | 138.8(14) |
| Acridine–tolfenamic acid (3) | 1.3153(16) | 1.2322(16) | 0.89(2) | 1.72(2) | 2.6103(16) | 175(2) | 0.812(19) | 2.011(19) | 2.6692(16) | 137.8(17) |
| Interaction | Bond Length/Interatomic Distance [Å] | ρ(r) | ∇2(r) | V(r) | G(r) | ε(r) | d [Å] |
|---|---|---|---|---|---|---|---|
| Fenamic acid…acridine | |||||||
| O1–H1O | 1.0617 | 0.2637 | −1.4900 | −0.5182 | 0.0728 | 0.0148 | 0.0006 |
| H1O∙∙∙N2 i | 1.5508 | 0.0789 | 0.0795 | −0.0792 | 0.0495 | 0.0291 | 0.0004 |
| O2∙∙∙H27A ii | 2.4505 | 0.0088 | 0.0315 | −0.0051 | 0.0065 | 0.0542 | 0.0127 |
| O2∙∙∙H25A ii | 2.7734 | 0.0048 | 0.0170 | −0.0028 | 0.0035 | 0.0884 | 0.0229 |
| 1 | |||||||
| O1–H1O | 0.990(18) | 0.2659 | −1.5189 | −0.5191 | 0.0697 | 0.0156 | 0.0006 |
| H1O∙∙∙N2 | 1.626(18) | 0.0731 | 0.0857 | −0.0718 | 0.0466 | 0.0337 | 0.0007 |
| O2∙∙∙H22 | 2.860 | 0.0043 | 0.0162 | −0.0025 | 0.0033 | 0.2766 | 0.1311 |
| 2 | |||||||
| O1–H1O | 1.021(19) | 0.2981 | −1.9274 | −0.6299 | 0.0740 | 0.0144 | 0.0004 |
| H1O∙∙∙N2 | 1.597(19) | 0.0699 | 0.0992 | −0.0697 | 0.0472 | 0.0288 | 0.0002 |
| O2∙∙∙H27 iii | 2.4288 | 0.0089 | 0.0328 | −0.0052 | 0.0067 | 0.0531 | 0.0132 |
| H25∙∙∙O2 iii | 3.0584 | 0.0028 | 0.0108 | −0.0015 | 0.0021 | 0.3773 | 0.0381 |
| 3 | |||||||
| O1∙∙∙H1O | 0.89(2) | 0.2705 | −1.5980 | −0.5406 | 0.0706 | 0.0147 | 0.0007 |
| N2∙∙∙H1O | 1.72(2) | 0.0727 | 0.0851 | −0.0712 | 0.0462 | 0.0295 | 0.0001 |
| O2∙∙∙H27 iv | 2.441 | 0.0068 | 0.0243 | −0.0040 | 0.0050 | 0.0529 | 0.0160 |
| O2∙∙∙H25 iv | 3.098 | 0.0024 | 0.0094 | −0.0013 | 0.0018 | 0.3232 | 0.0388 |
| Interaction | r [Å] | CC Distance with Bond Path | ρ(r) | ∇2(r) | ∇(r) | G(r) | ε(r) | d [Å] | |
|---|---|---|---|---|---|---|---|---|---|
| Fenamic acid…acridine | |||||||||
| 3.3157 | 1.4503 | C23–C29 viii | 3.3128 | 0.0066 | 0.0180 | −0.0029 | 0.0037 | 0.8757 | 0.0555 |
| C25–C33 viii | 3.3167 | 0.0063 | 0.0179 | −0.0028 | 0.0037 | 0.2187 | 0.0343 | ||
| C21–C27 viii | 3.3209 | 0.0061 | 0.0177 | −0.0028 | 0.0036 | 0.0386 | 0.0287 | ||
| C27–C21 viii | 3.3209 | 0.0061 | 0.0177 | −0.0028 | 0.0036 | 0.0387 | 0.0287 | ||
| C33–C25 viii | 3.3167 | 0.0063 | 0.0179 | −0.0028 | 0.0037 | 0.2188 | 0.0344 | ||
| C29–C23 viii | 3.3128 | 0.0066 | 0.0180 | −0.0029 | 0.0037 | 0.8760 | 0.0555 | ||
| 1 | |||||||||
| 3.3968 | 1.3990 | C27–C31 vii | 3.4323 | 0.0056 | 0.0144 | −0.0024 | 0.0030 | 0.9382 | 0.0640 |
| C29–C33 vii | 3.3838 | 0.0056 | 0.0162 | −0.0025 | 0.0033 | 0.9147 | 0.0459 | ||
| C33–C29 vii | 3.3838 | 0.0056 | 0.0162 | −0.0025 | 0.0033 | 0.9147 | 0.0459 | ||
| C31–C27 vii | 3.4323 | 0.0056 | 0.0144 | −0.0024 | 0.0030 | 0.9382 | 0.0640 | ||
| 2_stack1 | 0.0089 | 0.0328 | −0.0052 | 0.0067 | 0.0531 | 0.0132 | |||
| 3.3645 | 1.4652 | C23–C29 iii | 3.3719 | 0.0060 | 0.0161 | −0.0026 | 0.0033 | 0.9936 | 0.0579 |
| C25–C33 iii | 3.3931 | 0.0056 | 0.0156 | −0.0025 | 0.0032 | 0.3451 | 0.0396 | ||
| C21–C27 iii | 3.3661 | 0.0057 | 0.0162 | −0.0025 | 0.0033 | 0.1225 | 0.0345 | ||
| C27–C21 iii | 3.3661 | 0.0057 | 0.0162 | −0.0025 | 0.0033 | 0.1225 | 0.0345 | ||
| C33–C25 iii | 3.3931 | 0.0056 | 0.0156 | −0.0025 | 0.0032 | 0.3451 | 0.0396 | ||
| C29–C23 iii | 3.3719 | 0.0060 | 0.0161 | −0.0026 | 0.0033 | 0.9936 | 0.0579 | ||
| 2_stack2 | |||||||||
| 3.2858 | 1.7052 | C21–C23 vi | 3.301 | 0.0066 | 0.0189 | −0.0029 | 0.0038 | 0.6102 | 0.1315 |
| C23–C21 vi | 3.301 | 0.0066 | 0.0189 | −0.0029 | 0.0038 | 0.6102 | 0.1315 | ||
| 3_stack1 | |||||||||
| 3.3645 | 1.4652 | C21–C23 v | 3.3422 | 0.0060 | 0.0173 | −0.0027 | 0.0035 | 0.4528 | 0.1053 |
| C23–C21 v | 3.3422 | 0.0060 | 0.0173 | −0.0027 | 0.0035 | 0.4528 | 0.1053 | ||
| 3_stack2 | |||||||||
| 3.4202 | 1.3813 | C23–C29 iv | 3.4052 | 0.0057 | 0.0149 | −0.0024 | 0.0031 | 0.8305 | 0.0300 |
| C33–C25 iv | 3.4363 | 0.0052 | 0.0144 | −0.0023 | 0.0029 | 0.3814 | 0.0308 | ||
| C21–C27 iv | 3.4015 | 0.0053 | 0.0151 | −0.0023 | 0.0031 | 0.1980 | 0.0235 | ||
| C27–C21 iv | 3.4015 | 0.0053 | 0.0151 | −0.0023 | 0.0031 | 0.1980 | 0.0235 | ||
| C25–C33 iv | 3.4363 | 0.0052 | 0.0144 | −0.0023 | 0.0029 | 0.3814 | 0.0308 | ||
| C29–C23 iv | 3.4052 | 0.0057 | 0.0149 | −0.0024 | 0.0031 | 0.8305 | 0.0300 |
| Eelect | EPauli | Esteric | Eorb | Edysp | Etotal | Eelect | |
|---|---|---|---|---|---|---|---|
| Fenamic acid…acridine | −10.51 | 21.97 | 11.46 | −4.34 | −18.54 | −11.42 | −10.51 |
| 1 | −6.19 | 12.46 | 6.27 | −2.45 | −13.39 | −9.57 | −6.19 |
| 2_stack1 | −9.08 | 18.62 | 9.54 | −3.73 | −17.47 | −11.65 | −9.08 |
| 2_stack2 | −4.63 | 9.64 | 5.01 | −1.81 | −9.67 | −6.47 | −4.63 |
| 3_stack1 | −4.02 | 8.33 | 4.31 | −1.66 | −9.26 | −6.61 | −4.02 |
| 3_stack2 | −7.97 | 16.47 | 8.5 | −3.37 | −16.91 | −11.77 | −7.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawczyk, M.S.; Sroka, A.; Majerz, I. The Crystal Structure and Intermolecular Interactions in Fenamic Acids–Acridine Complexes. Molecules 2021, 26, 2956. https://doi.org/10.3390/molecules26102956
Krawczyk MS, Sroka A, Majerz I. The Crystal Structure and Intermolecular Interactions in Fenamic Acids–Acridine Complexes. Molecules. 2021; 26(10):2956. https://doi.org/10.3390/molecules26102956
Chicago/Turabian StyleKrawczyk, Marta S., Adam Sroka, and Irena Majerz. 2021. "The Crystal Structure and Intermolecular Interactions in Fenamic Acids–Acridine Complexes" Molecules 26, no. 10: 2956. https://doi.org/10.3390/molecules26102956
APA StyleKrawczyk, M. S., Sroka, A., & Majerz, I. (2021). The Crystal Structure and Intermolecular Interactions in Fenamic Acids–Acridine Complexes. Molecules, 26(10), 2956. https://doi.org/10.3390/molecules26102956