3. Materials and Methods
Commercially available starting materials, reagents, catalysts, anhydrous, and degassed solvents were used without further purification. Flash column chromatography was performed with Merck Silica gel 60 (230–400 mesh). The solvents for column chromatography were distilled before the use. Thin layer chromatography was carried out using Merck TLC Silica gel 60 F254 and visualized by short-wavelength ultraviolet light or by treatment with potassium permanganate (KMnO4) stain. 1H, 13C, and 19F-NMR spectra were recorded on a Bruker 250 and 500 MHz at 20 °C. All 1H-NMR spectra are reported in parts per million (ppm) downfield of TMS and were measured relative to the signals for CHCl3 (7.26 ppm) and DMSO (2.50 ppm). All 13C{1H}-NMR spectra were reported in ppm relative to residual CHCl3 (77.00 ppm) or DMSO (39.70 ppm) and were obtained with 1H decoupling. Coupling constants, J, are reported in Hertz (Hz). Gas chromatographic analyses was performed on Gas Chromatograph Mass Spectrometer GCMS-QP2010 Ultra instrument.
The optimal reaction conditions were identified by microscale high-†hroughput experimentation screening. Parallel synthesis was accomplished in an MBraun glovebox operating with a constant Ar-purge (oxygen and water <5 ppm). Screening reactions were carried out in 10 mL vials using suitable heating blocks. Liquid chemicals were dosed using gas tight micro syringes. Isolation of obtained compounds was achieved by column chromatography on Silica gel.
All used boronic acids
2 and some aryl trialkoxysilanes
3 are commercially available and were purchased from appropriate vendors. 2-Bromo-2,2-difluoroacetamides [
49,
50,
51,
52,
53,
54,
55,
56,
57], 1, 2-iodo-2,2-difluoroacetamides [
54], aryl trialkoxysilanes
3 [
58,
59,
60,
61,
62,
63,
64], sulfonium salts
4 [
65,
66,
67], and calixarenes
L1,
L2 [
68,
69] are known compounds in the literature and were prepared according to the known literature, and the spectral data are identical with the corresponding literature. Copies
1H and
13C-NMR spectra are placed in
Supplementary Materials.
General procedure for the synthesis of amides 5 by the reaction of 2-bromo-2,2-difluoroacetamides 1 with aryl boronic acids 2.
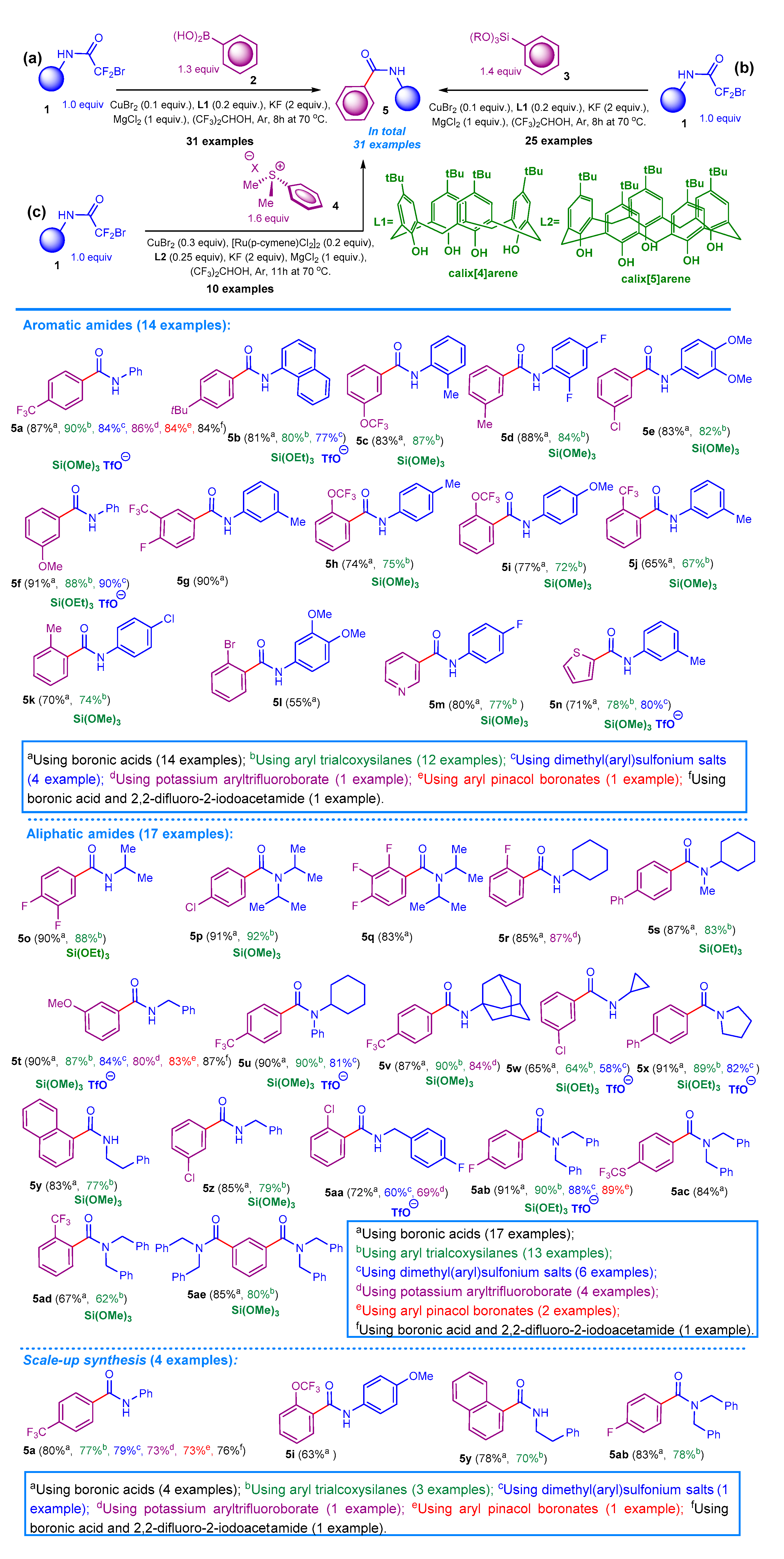
Under inert atmosphere (glovebox operating with a constant Ar-purge), to an 18 mL ACE pressure tube equipped with a stir bar, consequently, an appropriate 2-bromo-2,2-difluoroacetamide (1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid (1.3 mmol, 1.3 equiv.), the L1 (0.2 mmol, 0.2 equiv.), and finally CuBr2 (0.1 mmol, 0.1 equiv.) were placed; then the hexafluoropropanol (0.12 mmol/mL) was added and the reaction vessel was properly capped by Teflon stopper. Finally, the reaction vessel was removed from the glovebox and subjected to heating under vigorous stirring for 8 h. The progress of the reaction was controlled by TLC. After completion, the reaction mixture was evaporated until it reached dryness using a rotary evaporator, the content of the flask was generously treated with distilled water, filtered, and finally properly dried in vacuum. The resulting crude was directly subjected to gradient flash chromatography on silica gel using a mixture of hexane/ethyl acetate as eluent to isolate the desired amide derivative.
The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide.
General procedure for the synthesis of amides 5 by the reaction of 2-bromo-2,2-difluoroacetamides 1 with aryl trialkoxysilanes 3.
Under inert atmosphere (glovebox operating with a constant Ar-purge), to an 18 mL ACE pressure tube equipped with a stir bar, an appropriate 2-bromo-2,2-difluoroacetamide (1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane (1.4 mmol, 1.4 equiv.), the L1 (0.2 mmol, 0.2 equiv.), and finally CuBr2 (0.1 mmol, 0.1 equiv.) was consequently placed; then the hexafluoropropanol (0.12 mmol/mL) was added and the reaction vessel was properly capped by Teflon stopper. Finally, the reaction vessel was removed from the glovebox and subjected to heating under vigorous stirring for 8 h. The progress of the reaction was controlled by TLC. After completion, the reaction mixture was evaporated until it reached dryness using a rotary evaporator, the content of the flask was generously treated with distilled water, filtered, and finally properly dried in vacuum. The resulting crude was directly subjected to gradient flash chromatography on silica gel using a mixture of hexane/ethyl acetate as eluent to isolate the desired amide derivative.
The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide.
General procedure for the synthesis of amides 5 by the reaction of 2-bromo-2,2-difluoroacetamides 1 with (aryl)dimethylsulfonium salts 4.
Under inert atmosphere (glovebox operating with a constant Ar-purge), to an 18 mL ACE pressure equipped with a stir bar, an appropriate 2-bromo-2,2-difluoroacetamide (1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt (1.6 mmol, 1.6 equiv.), the L2 (0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (0.2 mmol, 0.2 equiv.), and finally CuBr2 (0.3 mmol, 0.3 equiv.) was consequently placed; then the hexafluoropropanol (0.12 mmol/mL) was added and the reaction vessel was properly capped by Teflon stopper. Finally, the reaction vessel was removed from the glovebox and subjected to heating under vigorous stirring for 11 h. The progress of the reaction was controlled by TLC. After completion, the reaction mixture was evaporated until it reached dryness using rotary evaporator, the content of the flask was generously treated with distilled water, filtered, and finally properly dried in vacuum. The resulting crude was directly subjected to gradient flash chromatography on silica gel using a mixture of hexane/ethyl acetate as eluent to isolate the desired amide derivative. The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide.
N-phenyl-4-(trifluoromethyl)benzamide 5a. The title compound was prepared, starting with 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl boronic acid 2n (247 mg, 1.3 mmol, 1.3 equiv.), L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5a (231 mg, 0.87 mmol, 87%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1a and the amide 5a was prepared in 80% yield (2.12 g, 8 mmol). Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3k (372 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5a (239 mg, 0.90 mmol, 90%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1a and the amide 5a was prepared in 77% yield (2.04 g, 7.7 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4a (584 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5a (222 mg, 0.84 mmol, 84%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1a and the amide 5a was prepared in 79% yield (2.09 g, 7.9 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate potassium trifluoro(4-(trifluoromethyl)phenyl)borate (328 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5a (228 mg, 0.86 mmol, 86%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1a and the amide 5a was prepared in 73% yield (1.93 g, 7.3 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate 4,4,5,5-tetramethyl-2-(4-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane (354 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5a (222 mg, 0.84 mmol, 84%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1a and the amide 5a was prepared in 73% yield (1.93 g, 7.3 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2,2-difluoro-2-iodo-N-phenylacetamide (297 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2n (247 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5a (222 mg, 0.84 mmol, 84%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1a and the amide 5a was prepared in 76% yield (2.01 g, 7.6 mmol).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 3:1 as an eluent to provide the corresponding amide product.
White solid, mp 184–185 °C. 1H-NMR (500 MHz, DMSO-d6): δ 7.12 (t, 1H, 3J = 7.3 Hz, CHAr), 7.37 (t, 2H, 3J = 8.3 Hz, CHAr), 7.80 (d, 2H, 3J = 7.6 Hz, CHAr), 7.89 (d, 2H, 3J = 8.2 Hz, CHAr), 8.16 (d, 2H, 3J = 8.1 Hz, CHAr), 10.5 (s, 1H, NH).
13C{1H}-NMR (126 MHz, DMSO-d6): δ 120.5, 123.9 (q, 1JCF = 273.8 Hz, CF3), 124.0, 125.4 (d, JCF = 3.1 Hz), 128.6, 128.7, 131.4 (q, 2JCF = 30.3 Hz, CCF3), 138.8 (d, JCF = 11.3 Hz), 164.4.
HRMS (TOF MS ES+): Calcd for C14H11NOF3 (M+H) 266.0809. Found 266.0793.
4-(tert-butyl)-N-(naphthalen-1-yl)benzamide 5b. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1e (300 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2a (231 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5b (245 mg, 0.81 mmol, 81%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1e (300 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3a (414 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5b (242 mg, 0.80 mmol, 80%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1e (300 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4h (312 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5b (233 mg, 0.77 mmol, 77%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 3:1 as an eluent to provide the corresponding amide product.
White solid, mp 146–147 °C. 1H-NMR (500 MHz, CDCl3): δ 1.38 (s, 9H, tBu), 7.46–7.51 (m, 5H, CHAr), 7.72 (d, 1H, 3J = 8.4 Hz, CHAr), 7.87–7.90 (m, 3H, CHAr), 7.92 (s, 1H, CHAr), 7.97 (br. s, 1H, CHAr), 8.33 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 31.2, 35.0, 120.79, 120.80, 121.2, 125.7, 125.9, 126.3, 127.1, 127.5, 127.51, 128.7, 131.9, 132.5, 134.1.
HRMS (TOF MS ES+): Calcd for C21H22NO (M + H) 304.1707. Found 304.1701.
N-(o-tolyl)-3-(trifluoromethoxy)benzamide 5c. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1d (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2r (268 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5c (245 mg, 0.83 mmol, 83%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1d (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3n (395 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.) CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5c (257 mg, 0.87 mmol, 87%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 5:1 as an eluent to provide the corresponding amide product.
White solid, mp 94–95 °C. 1H-NMR (500 MHz, CDCl3): δ 2.30 (s, 3H, Me), 7.12–7.15 (m, 1H, CHAr), 7.21–7.24 (m, 2H, CHAr), 7.40 (d, 1H, 3J = 8.1 Hz, CHAr), 7.50 (t, 1H, 3J = 7.9 Hz, CHAr), 7.74–7.78 (m, 3H, CHAr), 7.81 (br s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 17.8, 120.1, 120.4 (q, 1JCF = 258.6 Hz), 123.6, 124.1, 125.1, 125.9, 126.8, 130.0, 130.2, 130.6, 135.2, 137.0, 149.5, 164.2.
HRMS (TOF MS ES+): Calcd for C15H13NO2F3 (M + H) 296.0901. Found 296.0898.
N-(2,4-difluorophenyl)-3-methylbenzamide 5d. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1j (286 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2b (177 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5d (217 mg, 0.88 mmol, 88%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1j (286 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3b (297 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5d (207 mg, 0.84 mmol, 84%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 4:1 as an eluent to provide the corresponding amide product.
White solid, mp 106–107 °C. 1H-NMR (500 MHz, CDCl3): δ 2.41 (s, 3H, Me), 6.85–6.90 (m, 2H, CHAr), 7.35–7.36 (m, 2H, CHAr), 7.63–7.65 (m, 1H, CHAr), 7.68 (s, 1H, CHAr), 8.02 (br s, 1H, NH), 8.29–8.33 (m, 1H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 21.4, 103.4 (d, JCF = 26.5 Hz), 103.6 (d, JCF = 26.5 Hz), 111.2 (dd, JCF = 21.7 Hz, JCF = 3.6 Hz), 122.6 (dd, JCF = 10.5 Hz, JCF = 3.7 Hz), 123.1 (d, JCF = 9.3 Hz), 124.0, 127.8, 128.6, 132.9, 134.1, 138.7, 152.9 (dd, 1JCF = 246.4 Hz, JCF = 11.9 Hz), 158.6 (dd, 1JCF = 246.5 Hz, JCF = 11.4 Hz), 165.7.
HRMS (TOF MS ES+): Calcd for C14H12NOF2 (M + H) 248.0894. Found 248.0898.
3-chloro-N-(3,4-dimethoxyphenyl)benzamide 5e. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1g (310 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2a (203 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5e (242 mg, 0.83 mmol, 83%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1g (310 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3i (326 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5e (239 mg, 0.82 mmol, 82%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 4:1 as an eluent to provide the corresponding amide product.
Light purple, solid mp 127–128 °C. 1H-NMR (500 MHz, CDCl3): δ 3.79 (s, 3H, OMe), 3.83 (s, 3H, OMe), 6.77 (d, 1H, 3J = 8.4 Hz, CHAr), 7.02 (dd, 1H, 3J = 8.7 Hz, 4J = 2.1 Hz, CHAr), 7.32 (t, 1H, 3J = 7.9 Hz, CHAr), 7.36–7.37 (m, 1H, CHAr), 7.44 (dd, 1H, 3J = 8.0 Hz, 4J = 1.0 Hz, CHAr). 7.70 (d, 1H, 3J = 7.0 Hz, CHAr), 7.81 (s, 1H, CHAr), 8.26 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 55.7, 55.9, 105.2, 111.1, 112.6, 125.1, 127.3, 129.9, 131.1, 131.6, 134.7, 136.6, 146.1, 148.8, 164.5.
HRMS (TOF MS ES+): Calcd for C15H15NO3Cl (M + H) 292.0738. Found 292.0740.
3-methoxy-N-phenylbenzamide 5f. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2w (198 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5f (207 mg, 0.91 mmol, 91%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3j (378 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5f (200 mg, 0.88 mmol, 88%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1a (250 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4b (410 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5f (204 mg, 0.90 mmol, 90%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 4:1 as an eluent to provide the corresponding amide product.
White solid, mp 116–117 °C. 1H-NMR (500 MHz, CDCl3): δ 3.78 (s, 3H, OMe), 7.02 (dd, 1H, 3J = 8.2 Hz, 4J = 2.5 Hz, CHAr), 7.13 (t, 1H, 3J = 7.5 Hz, CHAr), 7.28–7.40 (m, 5H, CHAr), 7.64 (d, 2H, 3J = 7.9 Hz, CHAr), 8.23 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 55.3, 112.3, 117.9, 118.8, 120.3, 124.5, 128.9, 129.6, 136.3, 137.9, 159.8, 165.8.
HRMS (TOF MS ES+): Calcd for C14H14NO2 (M + H) 228.1025. Found 228.1025.
4-fluoro-N-(m-tolyl)-3-(trifluoromethyl)benzamide 5g. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1c (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2p (270 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5g (267 mg, 0.90 mmol, 90%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 5:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 121–122 °C. 1H-NMR (500 MHz, DMSO-d6): δ 2.31 (s, 3H, Me), 6.94 (d, 1H, 3J = 7.4 Hz, CHAr), 7.24 (d, 1H, 3J = 8.2 Hz, CHAr), 7.56 (d, 2H, 3J = 8.3 Hz, CHAr), 7.60 (s, 1H, CHAr), 8.33–8.36 (m, 2H, CHAr), 10.36 (s, 1H, NH),
13C{1H}-NMR (126 MHz, DMSO-d6): δ 21.2, 116.5 (dd, JCF = 33.5 Hz, JCF = 12.1 Hz), 117.5 (d, JCF = 20.8 Hz), 117.7, 121.1, 122.4 (q, 1JCF = 272.3 Hz, CF3), 124.8, 126.9, 128.5, 131.7 (d, JCF = 3.1 Hz), 135.1 (d, JCF = 9.4 Hz), 137.9, 138.7, 160.6 (d, 1JCF = 257.0 Hz), 163.0.
HRMS (TOF MS ES+): Calcd for C15H12NOF4 (M + H) 298.0861. Found 298.0855.
N-(p-tolyl)-2-(trifluoromethoxy)benzamide 5h. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1b (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2q (268 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5h (218 mg, 0.74 mmol, 74%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1b (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3m (395 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5h (221 mg, 0.75 mmol, 75%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 5:1 as an eluent to provide the corresponding amide product.
White solid, mp 108–109 °C. 1H-NMR (500 MHz, CDCl3): δ 2.35 (s, 3H, Me), 7.18 (d, 2H, 3J = 8.0 Hz, CHAr), 7.32 (d, 1H, 3J = 8.0 Hz, CHAr), 7.42 (t, 1H, 3J = 7.5 Hz, CHAr), 7.51–7.55 (m, 3H, CHAr), 8.04 (d, 1H, 3J = 7.7 Hz, CHAr), 8.30 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 20.9, 120.3 (q, 1JCF = 261.0 Hz, OCF3), 120.4, 121.2, 127.6, 128.3, 129.6, 131.9, 132.6, 134.6, 135.0, 145.7, 162.1.
HRMS (TOF MS ES+): Calcd for C15H13NO3F3 (M + H) 312.0847. Found 312.0848.
N-(4-methoxyphenyl)-2-(trifluoromethoxy)benzamide 5i. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1f (280 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2q (268 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5i (239 mg, 0.77 mmol, 77%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1f and the amide 5i was prepared in 63% yield (1.96 g, 0.63 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1f (280 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialckoxysilane 3m (395 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5i (224 mg, 0.72 mmol, 72%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 6:1 as an eluent to provide the corresponding amide product.
Pink solid, mp 117–118 °C. 1H-NMR (500 MHz, CDCl3): δ 3.81 (s, 3H, OMe), 6.88–6.92 (m, 2H, CHAr), 7.32 (d, 1H, 3J = 8.2 Hz, CHAr), 7.42 (dt, 1H, 3J = 7.6 Hz, 4J = 0.7 Hz, CHAr), 7.51–7.54 (m, 3H, CHAr), 8.02 (dd, 1H, 3J = 7.8 Hz, 4J = 1.7 Hz, CHAr), 8.25 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 55.5, 114.2, 120.3 (q, 1JCF = 260.4 Hz, OCF3), 121.2, 122.2, 127.6, 128.3, 130.6, 131.8, 132.5, 145.7, 156.8, 162.1.
HRMS (TOF MS ES+): Calcd for C15H13NO2F3 (M + H) 296.0904. Found 296.0898.
N-(m-tolyl)-2-(trifluoromethyl)benzamide 5j. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1c (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2o (247 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5j (239 mg, 0.65 mmol, 65%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1c (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3m (372 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5j (224 mg, 0.67 mmol, 67%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 5:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 120–121 °C. 1H-NMR (500 MHz, CDCl3): δ 2.35 (s, 3H, Me), 6.98 (d, 1H, 3J = 7.57 Hz, CHAr), 7.22 (t, 1H, 3J = 8.1 Hz, CHAr), 7.33 (d, 1H, 3J = 7.6 Hz, CHAr), 7.44 (s, 1H, CHAr), 7.54–7.57 (m, 3H, CHAr), 7.69–7.71 (m, 3H, NH, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 21.4, 117.3, 120.8, 123.7 (q, 1JCF = 276.1 Hz, CF3), 125.7, 126.4 (q, JCF = 5.2 Hz, CF3), 127.1 (q, 2JCF = 31.5 Hz, CCF3), 128.5, 128.9, 130.0, 132.1, 135.7, 137.4, 139.0, 165.7.
HRMS (TOF MS ES+): Calcd for C15H13NOF3 (M + H) 280.0957. Found 280.0949.
N-(4-chlorophenyl)-2-methylbenzamide 5k. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1h (285 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2c (177 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5k (172 mg, 0.70 mmol, 70%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1h (285 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3c (297 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5k (182 mg, 0.74 mmol, 74%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 3:1 as an eluent to provide the corresponding amide product.
White solid, mp 136–138 °C. 1H-NMR (500 MHz, CDCl3): δ 2.41 (s, 3H, Me), 7.17 (t, 1H, 3J = 7.2 Hz, CHAr), 7.20 (d, 1H, 3J = 7.2 Hz, CHAr), 7.25 (d, 2H, 3J = 8.7 Hz, CHAr), 7.31 (dt, 2H, 3J = 7.6 Hz, 4J = 0.8 Hz, CHAr), 7.36 (d, 1H, 3J = 7.2 Hz, CHAr), 7.50 (d, 1H, 3J = 8.4 Hz, CHAr), 7.87 (br s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 19.7, 121.2, 125.8, 126.6, 129.0, 129.4, 130.3, 131.2, 135.9, 136.3, 136.5, 168.2.
HRMS (TOF MS ES+): Calcd for C14H13NOCl (M + H) 246.0690. Found 246.0686.
2-bromo-N-(3,4-dimethoxyphenyl)benzamide 5l. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1g (310 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2m (261 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5l (185 mg, 0.55 mmol, 55%). Flash column chromatography was performed using a mixture of hexane/ethyl acetate 2:1 as an eluent to provide the corresponding amide product.
Purple solid, mp 140–141 °C. 1H-NMR (500 MHz, CDCl3): δ 3.83 (s, 3H, OMe), 3.84 (s, 3H, OMe), 6.79 (d, 1H, 3J = 8.7 Hz, CHAr), 7.00 (dd, 1H, 3J = 8.4 Hz, 4J = 2.6 Hz, CHAr), 7.22–7.26 (m, 1H, CHAr), 7.29–7.32 (m, 1H, CHAr), 7.41 (d, 1H, 4J = 2.3 Hz, CHAr), 7.51 (dd, 1H, 3J = 7.6 Hz, 4J = 1.6 Hz, CHAr), 7.55 (d, 1H, 3J = 8.3 Hz, CHAr), 7.96 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 55.8, 56.0, 104.8, 111.2, 112.0, 119.2, 127.5, 129.4, 131.2, 131.4, 133.3, 137.7, 146.0, 148.9, 165.5.
HRMS (TOF MS ES+): Calcd for C15H15NO3Br (M + H) 266.0809. Found 266.0793.
N-(4-fluorophenyl)nicotinamide 5m. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1i (268 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2t (160 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5m (173 mg, 0.80 mmol, 80%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1i (268 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3o (277 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5m (166 mg, 0.77 mmol, 77%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 1:1 as an eluent to provide the corresponding amide product.
White solid, mp 130–131 °C. 1H-NMR (500 MHz, CDCl3): δ 7.02 (t, 2H, 3J = 8.7 Hz, CHAr), 7.34–7.37 (m, 1H, CHAr), 7.55–7.58 (m, 2H, CHAr), 8.15 (d, 1H, 3J = 8.3 Hz, CHAr), 8.66 (dd, 1H, 3J = 4.7 Hz, 4J = 1.4 Hz, CHAr), 8.80 (s, 1H, NH), 9.03 (s, 1H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 115.7 (d, JCF = 22.2 Hz), 122.5 (d, JCF = 7.0 Hz), 123.7, 130.6, 133.5, 135.6, 147.9, 152.2, 159.7 (d, 1JCF = 244.1 Hz), 164.1.
HRMS (TOF MS ES+): Calcd for C15H12NOF3Na (M + Na) 302.0769. Found 302.0769.
N-(m-tolyl)thiophene-2-carboxamide 5n. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1c (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2u (166 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5n (154 mg, 0.71 mmol, 71%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1c (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3p (286 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5n (169 mg, 0.78 mmol, 78%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1c (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4c (470 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5n (174 mg, 0.80 mmol, 80%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 6:1 as an eluent to provide the corresponding amide product.
Brownish solid, mp 160–161 °C. 1H-NMR (500 MHz, CDCl3): δ 2.32 (s, 3H, Me), 6.94 (t, 1H, 3J = 7.8 Hz, CHAr), 7.01–7.08 (m, 1H, Thiophene), 7.21 (t, 1H, 3J = 7.8 Hz, CHAr), 7.39 (d, 1H, 3J = 8.1 Hz, CHAr), 7.47 (s, 1H, CHAr), 7.51 (d, 1H, 3J = 4.8 Hz, Thiophene), 7.64 (d, 1H, 3J = 3.6 Hz, Thiophene).
13C{1H}-NMR (126 MHz, CDCl3): δ 21.4, 117.4, 121.0, 125.4, 127.8, 128.4, 128.8, 130.7, 137.5, 138.9, 139.4, 160.1.
HRMS (TOF MS ES+): Calcd for C12H12NOS (M + H) 240.1394. Found 218.0640.
3,4-difluoro-N-isopropylbenzamide 5o. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1k (216 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2g (205 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5o (238 mg, 0.90 mmol, 90%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1k (216 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3g (386 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5o (233 mg, 0.88 mmol, 88%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 8:1 as an eluent to provide the corresponding amide product.
White solid, mp 105–106 °C. 1H-NMR (500 MHz, CDCl3): δ 1.14 (d, 6H, 3J = 6.2 Hz, Me), 4.08 (m, 1H, CH), 7.49–7.54 (m, 1H, CHAr), 7.73–7374 (m, 1H, CHAr), 7.87–7.91 (m, 1H, CHAr), 8.32 (d, 1H, 3J = 6.8 Hz, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 22.2, 41.3, 116.6 (d, JCF = 18.3 Hz), 117.3 (d, JCF = 17.5 Hz), 124.7 (m), 132.2 (m), 149.1 (dd, 1JCF = 247.3 Hz, 1JCF = 12.3 Hz), 151.2 (dd, 1JCF = 250.2 Hz, 1JCF = 12.3 Hz), 163.0.
HRMS (TOF MS ES+): Calcd for C13H19NOCl (M + H) 240.1157. Found 240.1155.
4-chloro-N,N-diisopropylbenzamide 5p. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1l (258 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2j (203 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5p (263 mg, 0.91 mmol, 91%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1l (258 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3h (326 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5p (266 mg, 0.92 mmol, 92%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 8:1 as an eluent to provide the corresponding amide product.
White solid, mp 129–130 °C. 1H-NMR (500 MHz, CDCl3): δ 1.15 (s, 6H, Me), 1.48 (s, 6H, Me), 3.58 (m, 2H, 2xCH), 7.24 (dt, 2H, 3J = 8.5 Hz, 4J = 2.0 Hz, CHAr), 7.34 (dt, 2H, 3J = 8.3 Hz, 4J = 1.7 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 20.7, 127.1, 128.7, 134.6, 137.2, 169.9.
HRMS (TOF MS ES+): Calcd for C13H19NOCl (M + H) 240.1157. Found 240.1155.
2,3,4-trifluoro-N,N-diisopropylbenzamide 5q. Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1l (258 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2h (229 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5q (215 mg, 0.83 mmol, 83%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 11:1 as an eluent to provide the corresponding amide product.
White solid, mp 48–50 °C. 1H-NMR (500 MHz, CDCl3): δ 1.10 (m, 6H, 2xMe), 1.50 (s, 3H, Me), 1.52 (s, 3H, Me), 3.50–3.53 (m, 1H, NCH), 3.65–3.68 53 (m, 1H, NCH), 6.97–7.00 (m, 2H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 20.5 (m), 46.2, 51.3, 112.9 (dd, JCF = 18.0 Hz, JCF = 3.3 Hz), 121.3 (m), 124.3 (dd, JCF = 16.4 Hz, JCF = 2.2 Hz), 139.7 (dt, 1JCF = 253.7 Hz, JCF = 15.2 Hz), 147.2 (ddd, 1JCF = 249.8 Hz, JCF = 10.9 Hz, JCF = 3.1 Hz), 151.2 (ddd, 1JCF = 251.3 Hz, JCF = 9.8 Hz, JCF = 2.4 Hz), 163.5.
HRMS (TOF MS ES+): Calcd for C13H17NOF3 (M + H) 260.1262. Found 260.1262.
N-cyclohexyl-2-fluorobenzamide 5r. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1p (258 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2i (203 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5r (188 mg, 0.85 mmol, 85%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1p (258 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate potassium trifluoro(2-fluorophenyl)borate (263 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5r (192 mg, 0.87 mmol, 87%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 10:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 45–46 °C. 1H-NMR (500 MHz, CDCl3): δ 1.19–1.29 (m, 3H, Cy), 1.36–1.46 (m, 2H, Cy), 1.58–1.62 (m, 1H, Cy), 1.69–1.73 (m, 2H, Cy), 1.98–2.01 (m, 2H, Cy), 3.98–4.00 (m, 1H, Cy), 6.61 (s, 1H, NH), 7.04–7.08 (m, 1H, CHAr), 7.19–7.22 (m, 1H, CHAr), 7.38–7.42 (m, 1H, CHAr), 8.03 (dd, 1H, 3J = 7.9 Hz, 4J = 1.8 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 24.7, 25.5, 32.9, 48.5, 115.8 (d, JCF = 23.6 Hz), 121.5 (d, JCF = 11.0 Hz), 124.6 (d, JCF = 2.7 Hz), 131.9, 133.0 (d, JCF = 9.0 Hz), 160.4 (d, 1JCF = 246.0 Hz, CF), 162.1 (d, JCF = 2.4 Hz).
HRMS (TOF MS ES+): Calcd for C13H17NOF (M + H) 222.1294. Found 222.1294.
N-cyclohexyl-N-methyl-[1,1′-biphenyl]-4-carboxamide 5s. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1o (270 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2e (257 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5s (255 mg, 0.87 mmol, 87%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1o (270 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3e (442 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5s (243 mg, 0.83 mmol, 83%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 8:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 105 - 106 °C. 1H-NMR (500 MHz, CDCl3): δ 1.07–1.09 (m, 2H, Cy), 1.47–1.56 (m, 4H, Cy), 1.72–1.82 (m, 4H, Cy), 2.83, 3.00 (s, 3H, Me cis/trans), 3.54, 4.51 (s, 1H, Cy cis/trans), 7.33–7.36 (m, 1H, CHAr), 7.42–7.45 (m, 4H, CHAr), 7.60–7.61 (m, 4H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 25.0, 25.3, 25.4, 27.4, 29.4, 29.5, 30.7, 31.9, 52.7, 58.1, 126.5 126.7, 126.9, 127.1, 127.2, 127.5, 128.7, 135.8, 140.1, 141.8, 171.4.
HRMS (TOF MS ES+): Calcd for C20H24NO (M + H) 294.1856. Found 294.1858.
N-benzyl-3-methoxybenzamide 5t. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2w (198 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5t (217 mg, 0.90 mmol, 90%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3j (378 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5t (210 mg, 0.87 mmol, 87%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4b (410 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.17 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5t (202 mg, 0.84 mmol, 84%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate potassium trifluoro(3-methoxyphenyl)borate (278 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5t (193 mg, 0.80 mmol, 80%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate 2-(3-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (304 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5t (200 mg, 0.83 mmol, 83%).
Alternatively, the title compound was prepared starting with an appropriate N-benzyl-2,2-difluoro-2-iodoacetamide (311 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2w (198 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5t (210 mg, 0.87 mmol, 87%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 7:1 as an eluent to provide the corresponding amide product.
White solid, mp 77–78 °C. 1H-NMR (500 MHz, CDCl3): δ 2.36 (s, 3H, Me), 4.60 (d, 2H, 3J = 5.6 Hz, CH2), 6.78 (br s, 1H, NH), 7.26–7.33 (m, 7H, CHAr), 7.57 (d, 1H, 3J = 7.2 Hz, CHAr), 7.63 (s, 1H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 21.2, 43.9, 123.9, 127.4, 127.7, 127.8, 128.3, 128.6, 132.1, 134.2, 138.3, 167.6.
HRMS (TOF MS ES+): Calcd for C15H16NO (M + H) 226.1234. Found 226.1232.
N-cyclohexyl-N-phenyl-4-(trifluoromethyl)benzamide 5u. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1n (332 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2n (247 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5u (312 mg, 0.90 mmol, 90%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1n (332 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3k (372 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5u (312 mg, 0.90 mmol, 90%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1n (332 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4a (584 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5u (281 mg, 0.81 mmol, 81%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 8:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 195–196 °C. 1H-NMR (500 MHz, CDCl3): δ 0.97 (tq, 1H, 3J = 13.4 Hz, 4J = 2.9 Hz, Cy), 1.22 (dq, 2H, 3J = 12.9 Hz, 4J = 2.9 Hz, Cy), 1.45 (d, 2H, 3J = 12.4 Hz, Cy), 1.61 (d, 1H, 3J = 13.3 Hz, Cy), 1.78 (d, 2H, 3J = 13.3 Hz, Cy), 1.96 (d, 2H, 3J = 11.1 Hz, Cy), 4.72 (s, 1H, Cy), 7.00 (d, 2H, 3J = 6.9 Hz, CHAr), 7.19–7.20 (m, 3H, CHAr), 7.31 (d, 2H, 3J = 7.3 Hz, CHAr), 7.36 (d, 2H, 3J = 6.9 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 25.3, 25.8, 31.5, 55.3, 123.7 (q, 1JCF = 271.2 Hz, CF3), 124.6, 127.8, 128.3, 128.6, 130.4 (q, 2JCF = 28.0 Hz, CCF3), 130.6, 139.2, 140.8, 169.1.
HRMS (TOF MS ES+): Calcd for C20H21NOF3 (M + H) 348.1581. Found 348.1575.
N-((1s,3s)-adamantan-1-yl)-4-(trifluoromethyl)benzamide 5v. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1q (308 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2n (247 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5v (281 mg, 0.87 mmol, 87%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1q (308 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3k (372 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5v (291 mg, 0.90 mmol, 90%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1q (308 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate potassium trifluoro(4-(trifluoromethyl)phenyl)borate (328 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5v (271 mg, 0.84 mmol, 84%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 12:1 as an eluent to provide the corresponding amide product.
White solid, mp 158–159 °C. 1H-NMR (500 MHz, CDCl3): δ 1.70 (s, 6H, Adam), 2.11 (s, 9H, Adam), 5.91 (s, 1H, NH), 7.61 (d, 2H, 3J = 8.6 Hz, CHAr), 7.78 (d, 2H, 3J = 7.9 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 29.4, 36.2, 41.5, 52.6, 123.7 (q, 1JCF = 273.6 Hz, CF3), 125.4 (m), 127.2, 132.6 (q, 2JCF = 31.1 Hz, CCF3), 139.3.
HRMS (TOF MS ES+): Calcd for C18H21NOF3 (M + H) 324.1583. Found 324.1575.
3-chloro-N-cyclopropylbenzamide 5w. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1r (214 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2k (203 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5w (127 mg, 0.65 mmol, 65%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1r (214 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3i (326 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5w (125 mg, 0.64 mmol, 64%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1r (214 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4d (516 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5w (113 mg, 0.58 mmol, 58%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 7:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 142—143 °C. 1H-NMR (500 MHz, CDCl3): δ 0.83–0.86 (m, 2H, CH2), 1.06–1.09 (m, 2H, CH2), 1.48- 1.53 (m, 1H, CH), 7.04 (d, 1H, 3J = 7.9 Hz, CHAr), 7.20 (t, 1H, 3J = 8.7 Hz, CHAr), 7.32 (d, 1H, 3J = 7.9 Hz, CHAr), 7.63 (br. s, 1H, CHAr), 7.74 (s, 1H, NH).
13C{1H}-NMR (126 MHz, CDCl3): δ 8.2, 15.7, 17.7, 19.9, 124.0, 129.9, 134.6, 139.2, 172.3.
HRMS (TOF MS ES+): Calcd for C10H11NOCl (M + H) 196.0532. Found 196.0529.
[1,1′-biphenyl]-4-yl(pyrrolidin-1-yl)methanone 5x. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1s (228 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2e (257 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5x (326 mg, 0.91 mmol, 91%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1s (228 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3e (442 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5x (223 mg, 0.89 mmol, 89%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1s (228 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4e (582 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5x (206 mg, 0.82 mmol, 82%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 8:1 as an eluent to provide the corresponding amide product.
White solid, mp 139–140 °C. 1H-NMR (500 MHz, CDCl3): δ 1.86 (m, 4H, Pyrr), 3.39 (s, 2H, Pyrr), 3.58 (s, 2H, Pyrr), 7.34 (t, 1H, 3J = 7.7 Hz, CHAr), 7.43 (t, 2H, 3J = 7.7 Hz, CHAr), 7.57 – 7.59 (m, 6H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 24.3, 26.3, 46.1, 49.5, 126.7, 127.0, 127.6, 128.7, 135.8, 140.1, 142.4, 169.3.
HRMS (TOF MS ES+): Calcd for C17H18NO (M + H) 252.1391. Found 252.1388.
N-phenethyl-1-naphthamide 5y. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1t (278 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2d (224 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5y (228 mg, 0.83 mmol, 83%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1t and the amide 5y was prepared in 78% yield (2.15 g, 7.8 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1t (278 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3d (347 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5y (212 mg, 0.77 mmol, 77%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1t and the amide 5y was prepared in 70% yield (1.93 g, 7 mmol).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 5:1 as an eluent to provide the corresponding amide product.
White solid, mp 117–118 °C. 1H-NMR (500 MHz, CDCl3): δ 3.00 (t, 2H, 3J = 6.1 Hz, CH2), 3.80 (q, 2H, 3J = 7.2 Hz, CH2), 6.13 (s, 1H, NH), 7.27–7.28 (m, 3H, CHAr), 7.33–7.37 (m, 2H, CHAr), 7.40–7.43 (m, 1H, CHAr), 7.48–7.54 (m, 3H, CHAr), 7.86–7.88 (m, 1H, CHAr), 7.89 (d, 1H, 3J = 8.0 Hz, CHAr), 8.20–8.22 (m, 1H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 35.6, 41.0, 124.6, 124.85, 125.3, 126.3, 126.5, 127.0, 128.2, 128.7, 128.8, 130.0, 130.4, 133.6, 134.5, 138.7.
HRMS (TOF MS ES+): Calcd for C19H18NO (M + H) 276.1396. Found 276.1388.
N-benzyl-3-chlorobenzamide 5z. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2k (203 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5z (209 mg, 0.85 mmol, 85%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1u (264 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3i (372 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5z (194 mg, 0.79 mmol, 79%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 2:1 as an eluent to provide the corresponding amide product.
Yellowish solid, mp 93–94 °C. 1H-NMR (500 MHz, CDCl3): δ 4.57 (d, 2H, 3J = 5.5 Hz, CH2), 6.77 (s, 1H, NH), 7.27–7.35 (m, 6H, CHAr), 7.44 (dd, 1H, 3J = 8.0 Hz, 4J = 1.0 Hz, CHAr), 7.64 (d, 1H, 3J = 8.0 Hz, CHAr), 7.77 (d, 1H, 4J = 1.8 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 44.1, 125.1, 127.3, 127.6, 127.8, 128.7, 129.8, 131.5, 134.7, 136.1, 137.8, 166.1.
HRMS (TOF MS ES+): Calcd for C14H13NOCl (M + H) 246.0686. Found 246.0686.
N-(4-fluorobenzyl)-2-methylbenzamide 5aa. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1v (282 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2l (203 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5aa (189 mg, 0.72 mmol, 72%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1v (282 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4f (516 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5aa (158 mg, 0.60 mmol, 60%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1v (282 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate potassium (2-chlorophenyl)trifluoroborate (283 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5aa (181 mg, 0.69 mmol, 69%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 4:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 119–120 °C. 1H-NMR (500 MHz, CDCl3): δ 2.36 (s, 3H, Me), 4.46 (d, 2H, 3J = 5.7 Hz, CH2), 6.40 (s, 1H, NH), 6.97 (t, 2H, 3J = 8.7 Hz, CHAr), 7.11–7.17 (m, 2H, CHAr), 7.23–7.28 (m, 4H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 19.7, 42.9, 115.4 (d, JCF = 22.0 Hz), 125.6, 126.6, 129.3 (d, JCF = 8.9 Hz), 129.9, 130.9, 134.1 (d, JCF = 2.3 Hz), 136.0 (d, JCF = 2.7 Hz), 126.1 (d, 1JCF = 243.3 Hz), 169.9.
HRMS (TOF MS ES+): Calcd for C15H15NOF (M + H) 244.1143. Found 244.1138.
N,N-dibenzyl-4-fluorobenzamide 5ab. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2f (174 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ab (290 mg, 0.91 mmol, 91%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1m and the amide 5ab was prepared in 83% yield (2.65 g, 8.3 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3f (361 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ab (287 mg, 0.90 mmol, 90%). The gram scale synthesis was performed on 10 mmol of the starting 2-bromo-2,2-difluoroacetamide 1m and the amide 5ab was prepared in 78% yield (2.45 g, 7.8 mmol).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), aryl sulphonium salt 4g (490 mg, 1.6 mmol, 1.6 equiv.), the L2 (202 mg, 0.25 mmol, 0.25 equiv.), [Ru(p-cymene)Cl2]2 (122 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (67 mg, 0.3 mmol, 0.3 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ab (281 mg, 0.88 mmol, 88%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate 2-(4-fluorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (289 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ab (284 mg, 0.89 mmol, 89%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 3:1 as an eluent to provide the corresponding amide product.
White solid, mp 86–87 °C. 1H-NMR (500 MHz, CDCl3): δ 4.20 (s, 2H, CH2), 4.71 (s, 2H, CH2), 7.07 (t, 2H, 3J = 7.6 Hz, CHAr), 7.15 (br. s, 2H, CHAr), 7.30–7.33 (m, 4H, CHAr), 7.36–7.39 (m, 4H, CHAr), 8.50–7.53 (m, 2H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 47.2, 51.6, 115.6 (d, JCF = 22.2 Hz), 126.8 (m), 127.7 (m), 128.4 (m), 128.7 (m), 128.9, 129.0, 132.0 (m), 136.5 (d, JCF = 67.9 Hz), 163.3 (d, 1JCF = 247.5 Hz), 171.3.
HRMS (TOF MS ES+): Calcd for C21H19NOF (M + H) 320.1455. Found 320.1451.
N,N-dibenzyl-4-((trifluoromethyl)thio)benzamide 5ac. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2s (289 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ac (337 mg, 0.84 mmol, 84%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 3:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 68–70 °C. 1H-NMR (500 MHz, CDCl3): δ 4.39 (s, 2H, CH2), 4.74 (s, 2H, CH2), 7.13 (d, 2H, 3J = 6.9 Hz, CHAr), 7.30–7.34 (m, 4H, CHAr), 7.37–7.40 (m, 4H, CHAr), 7.54 (d, 2H, 3J = 8.1 Hz, CHAr), 7.67 (d, 2H, 3J = 8.1 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 47.1, 51.5, 127.1 (q, 1JCF = 258.2 Hz, SCF3), 126.9, 127.7, 128.2, 128.4, 128.8, 129.0, 135.9, 136.2, 136.5, 138.6, 170.8.
HRMS (TOF MS ES+): Calcd for C22H19NOF3S (M + H) 402.1141. Found 402.1139.
N,N-dibenzyl-2-(trifluoromethyl)benzamide 5ad. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate aryl boronic acid 2o (247 mg, 1.3 mmol, 1.3 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ac (247 mg, 0.67 mmol, 67%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (354 mg, 1.0 mmol, 1.0 equiv.), KF (116 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (95 mg, 1.0 mmol, 1.0 equiv.), appropriate trialkoxysilane 3l (372 mg, 1.4 mmol, 1.4 equiv.), the L1 (130 mg, 0.2 mmol, 0.2 equiv.), CuBr2 (22.3 mg, 0.1 mmol, 0.1 equiv.), and hexafluoropropanol (0.12 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ab (229 mg, 0.62 mmol, 62%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 4:1 as an eluent to provide the corresponding amide product.
Colorless solid, mp 135–136 °C. 1H-NMR (500 MHz, CDCl3): δ 4.11 (t, 2H, 3J = 15.3 Hz, CH2), 4.25 (d, 1H, 3J = 15.8 Hz, CH2), 5.33 (d, 1H, 3J = 14.8 Hz, CH2), 7.11 (m, 2H, 3J = 7.1 Hz, CHAr), 7.29–7.37 (m, 8H, CHAr), 7.47–7.51 (m, 2H, CHAr), 7.56 (t, 1H, 3J = 7.1 Hz, CHAr), 7.71 (d, 1H, 3J = 7.8 Hz, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 46.4, 51.1, 123.7 (q, 1JCF = 274.8 Hz, CF3), 126.6 (m), 127.3, 127.4, 127.7, 127.8, 128.5, 128.8, 129.1, 129.1, 132.1, 135.0, 135.5, 136.1, 169.2.
HRMS (TOF MS ES+): Calcd for C22H19NOF3 (M + H) 370.1422. Found 370.1419.
N1,N1,N3,N3-tetrabenzylisophthalamide 5ae. The title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (885 mg, 2.5 mmol, 2.5 equiv.), KF (232 mg, 2.0 mmol, 2.0 equiv.), MgCl2 (190 mg, 2.0 mmol, 2.0 equiv.), appropriate aryl boronic acid 2v (166 mg, 1.0 mmol, 1.0 equiv.), the L1 (260 mg, 0.4 mmol, 0.4 equiv.), CuBr2 (44.6 mg, 0.2 mmol, 0.2 equiv.), and hexafluoropropanol (0.08 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ae (445 mg, 0.85 mmol, 85%).
Alternatively, the title compound was prepared starting with an appropriate 2-bromo-2,2-difluoroacetamide 1m (885 mg, 2.5 mmol, 2.5 equiv.), KF (232 mg, 4.0 mmol, 4.0 equiv.), MgCl2 (190 mg, 2.0 mmol, 2.0 equiv.), appropriate trialkoxysilane 3q (318 mg, 1.0 mmol, 1.0 equiv.), the L1 (260 mg, 0.4 mmol, 0.4 equiv.), CuBr2 (44.6 mg, 0.2 mmol, 0.2 equiv.), and hexafluoropropanol (0.08 mmol/mL). The purification of the dry crude performed by column chromatography on silica gel provides the amide 5ae (419 mg, 0.80 mmol, 80%).
Flash column chromatography was performed using a mixture of hexane/ethyl acetate 1:2 as an eluent to provide the corresponding amide product.
White solid, mp 172–173 °C. 1H-NMR (500 MHz, CDCl3): δ 4.39 (s, 4H, 2xCH2), 4.71 (s, 4H, 2xCH2), 7.12 (d, 4H, 3J = 6.8 Hz, CHAr), 7.28–7.37 (m, 16H, CHAr), 7.53 (s, 4H, CHAr).
13C{1H}-NMR (126 MHz, CDCl3): δ 46.9, 51.4, 126.8, 126.9, 127.6, 127.7, 128.4, 128.7, 128.9, 136.0, 136.6, 137.4, 171.3.
HRMS (TOF MS ES+): Calcd for C36H33N2O2 (M + H) 525.2540. Found 525.2539.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}