Structural and Functional Characterization of NadR from Lactococcus lactis


Abstract
1. Introduction
2. Results
2.1. Multiple Sequence Alignment of NadR
2.2. Substrate Specificity of NadR
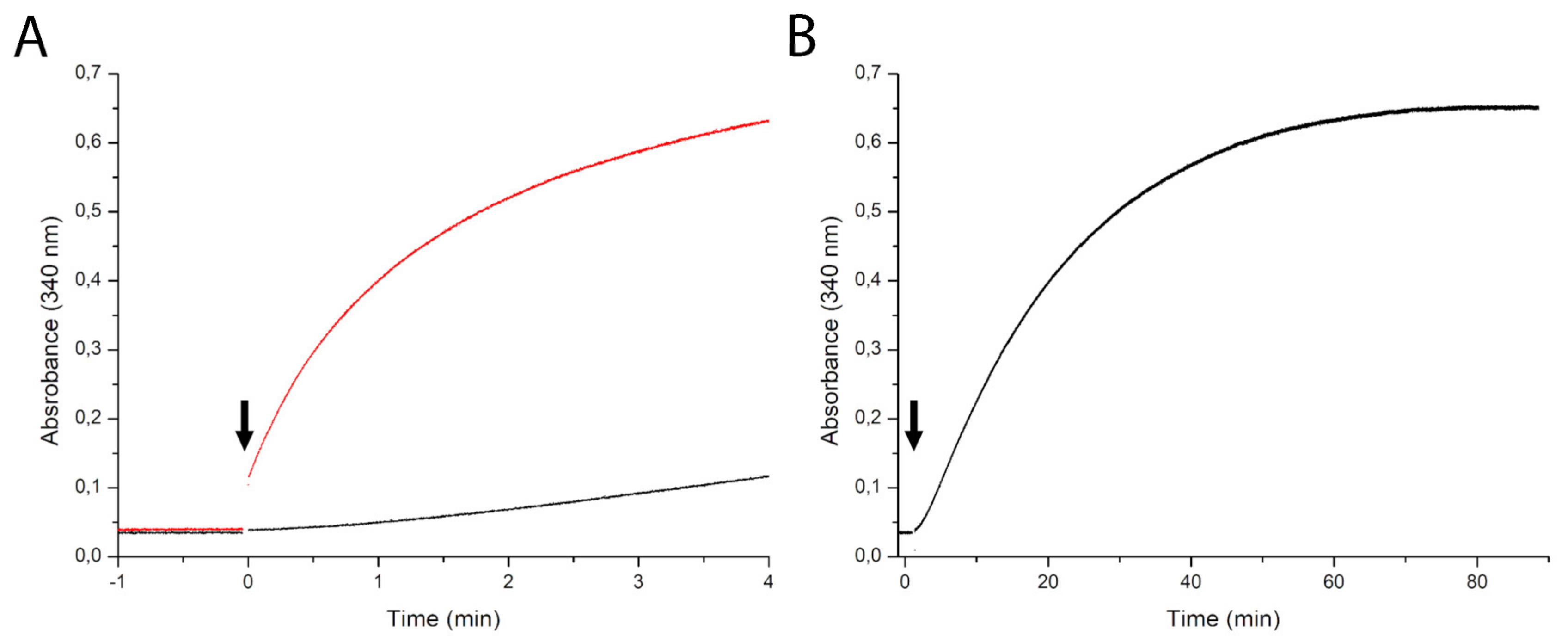
2.3. Steady State Kinetics
2.4. Dependence of the Kinetics of NAD Production on the NadRll Concentration
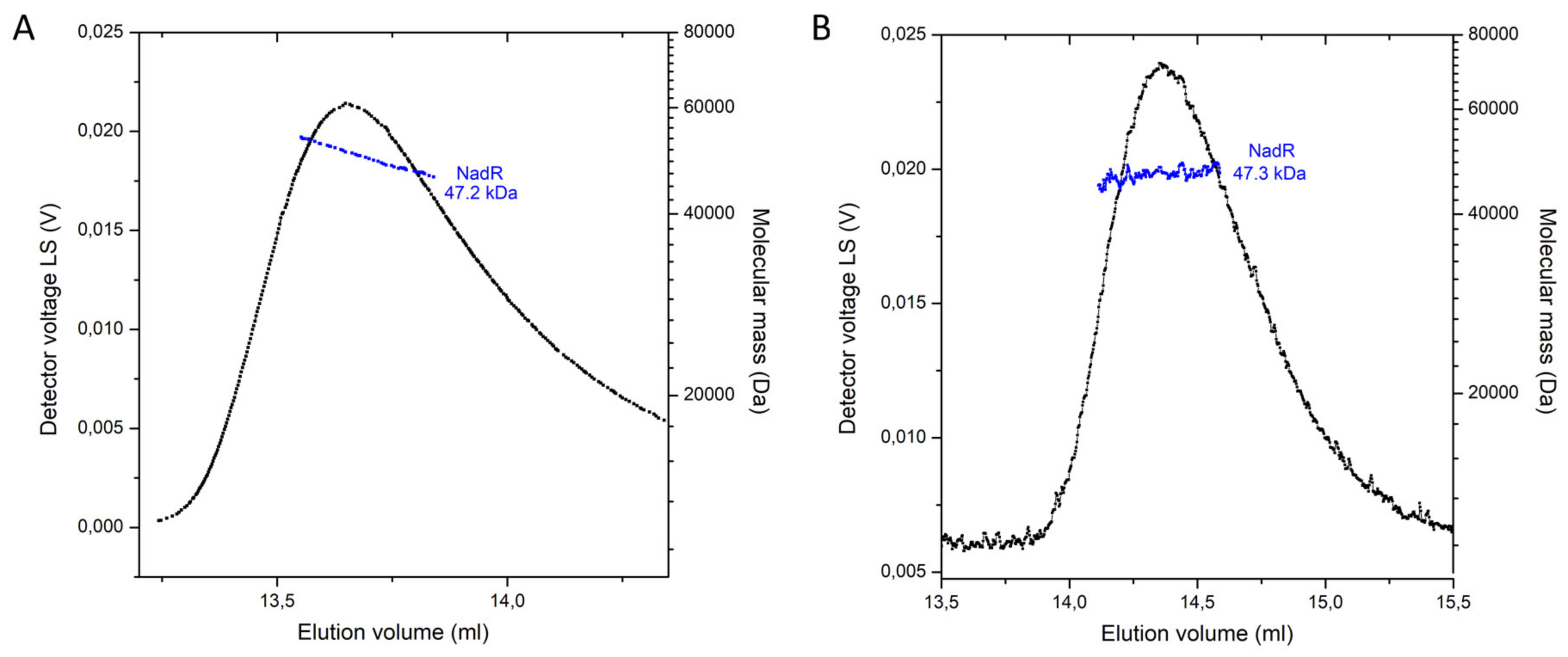
2.5. Oligomeric State of NadR
2.6. Crystal Structures of NadR
3. Discussion
4. Materials and Methods
4.1. Cloning
4.2. Protein Expression and Purification
4.3. Isothermal Calorimetric Titration (ITC) Measurements for Substrate Specificity
4.4. Lactate Dehydrogenase (LDH) Coupled NADH Measurements Assay
4.5. Crystallization
4.6. Static Light Scattering and Refractive Index Measurements
4.7. Data Collection, Structure Determination, and Refinement
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jaehme, M.; Slotboom, D.J. Structure, function, evolution, and application of bacterial Pnu-type vitamin transporters. Biol. Chem. 2015, 396, 955–966. [Google Scholar] [CrossRef]
- Jaehme, M.; Slotboom, D.J. Diversity of membrane transport proteins for vitamins in bacteria and archaea. Biochim. Biophys. Acta (BBA)-Gene. Subj. 2015, 1850, 565–576. [Google Scholar] [CrossRef]
- Bacher, A.; Eberhardt, S.; Fischer, M.; Kis, K.; Richter, G. Biosynthesis of vitamin b2 (riboflavin). Annu. Rev. Nutr. 2000, 20, 153–167. [Google Scholar] [CrossRef]
- Marsili, E.; Baron, D.B.; Shikhare, I.D.; Coursolle, D.; Gralnick, J.A.; Bond, D.R. Shewanella secretes flavins that mediate extracellular electron transfer. Proc. Natl. Acad. Sci. USA 2008, 105, 3968–3973. [Google Scholar] [CrossRef]
- Kemmer, G.; Reilly, T.J.; Schmidt-Brauns, J.; Zlotnik, G.W.; Green, B.A.; Fiske, M.J.; Herbert, M.; Kraiss, A.; Schlor, S.; Smith, A.; et al. NadN and e (P4) are essential for utilization of NAD and nicotinamide mononucleotide but not nicotinamide riboside in Haemophilus influenzae. J. Bacteriol. 2001, 183, 3974–3981. [Google Scholar] [CrossRef]
- Grose, J.H.; Bergthorsson, U.; Xu, Y.; Sterneckert, J.; Khodaverdian, B.; Roth, J.R. Assimilation of nicotinamide mononucleotide requires periplasmic AphA phosphatase in Salmonella enterica. J. Bacteriol. 2005, 187, 4521–4530. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Li, X.; Rodionova, I.A.; Yang, C.; Sorci, L.; Dervyn, E.; Martynowski, D.; Zhang, H.; Gelfand, M.S.; Osterman, A.L. Transcriptional regulation of NAD metabolism in bacteria: Genomic reconstruction of NiaR (YrxA) regulon. Nucleic Acids Res. 2008, 36, 2032–2046. [Google Scholar] [CrossRef]
- Jaehme, M.; Singh, R.; Garaeva, A.A.; Duurkens, R.H.; Slotboom, D.-J. PnuT uses a facilitated diffusion mechanism for thiamine uptake. J. Gene. Physiol. 2018, 150, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Jaehme, M.; Guskov, A.; Slotboom, D.J. Crystal structure of the vitamin B3 transporter PnuC, a full-length SWEET homolog. Nat. Struct. Mol. Biol. 2014, 21, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Slotboom, D.J. Structural and mechanistic insights into prokaryotic energy-coupling factor transporters. Nat. Rev. Microbiol. 2014, 12, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Sauer, E.; Merdanovic, M.; Mortimer, A.P.; Bringmann, G.; Reidl, J. PnuC and the utilization of the nicotinamide riboside analog 3-aminopyridine in Haemophilus influenzae. Antimicrob. Agents Chemother 2004, 48, 4532–4541. [Google Scholar] [CrossRef] [PubMed]
- Cynamon, M.H.; Sorg, T.B.; Patapow, A. Utilization and Metabolism of NAD by Haemophilus parainfluenzae. Microbiology 1988, 134, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Merdanovic, M.; Sauer, E.; Reidl, J. Coupling of NAD+ biosynthesis and nicotinamide ribosyl transport: Characterization of NadR ribonucleotide kinase mutants of Haemophilus influenzae. J. Bacteriol. 2005, 187, 4410–4420. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Bergthorsson, U.; Roth, J.R. Regulation of NAD synthesis by the trifunctional NadR protein of Salmonella enterica. J. Bacteriol. 2005, 187, 2774–2782. [Google Scholar] [CrossRef]
- Gerasimova, A.V.; Gelfand, M.S. Evolution of the NadR regulon in Enterobacteriaceae. J. Bioinform. Comput. Biol. 2005, 3, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Spector, M.P.; Hill, J.M.; Holley, E.A.; Foster, J.W. Genetic characterization of pyridine nucleotide uptake mutants of Salmonella typhimurium. J. Gen. Microbiol. 1985, 131, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Tirgari, S.; Spector, M.P.; Foster, J.W. Genetics of NAD metabolism in Salmonella typhimurium and cloning of the nadA and pnuC loci. J. Bacteriol. 1986, 167, 1086–1088. [Google Scholar] [CrossRef]
- Zhu, N.; Roth, J.R. The nadI region of Salmonella typhimurium encodes a bifunctional regulatory protein. J. Bacteriol. 1991, 173, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Vogl, C.; Grill, S.; Schilling, O.; Stulke, J.; Mack, M.; Stolz, J. Characterization of riboflavin (vitamin B2) transport proteins from Bacillus subtilis and Corynebacterium glutamicum. J. Bacteriol. 2007, 189, 7367–7375. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Moyle, J. Group-translocation: A consequence of enzyme-catalysed group-transfer. Nature 1958, 182, 372–373. [Google Scholar] [CrossRef]
- Kurnasov, O.V.; Polanuyer, B.M.; Ananta, S.; Sloutsky, R.; Tam, A.; Gerdes, S.Y.; Osterman, A.L. Ribosylnicotinamide kinase domain of NadR protein: Identification and implications in NAD biosynthesis. J. Bacteriol. 2002, 184, 6906–6917. [Google Scholar] [CrossRef]
- Singh, S.K.; Kurnasov, O.V.; Chen, B.; Robinson, H.; Grishin, N.V.; Osterman, A.L.; Zhang, H. Crystal structure of Haemophilus influenzae NadR protein. A bifunctional enzyme endowed with NMN adenyltransferase and ribosylnicotinimide kinase activities. J. Biol. Chem. 2002, 277, 33291–33299. [Google Scholar] [CrossRef]
- Raffaelli, N.; Lorenzi, T.; Mariani, P.L.; Emanuelli, M.; Amici, A.; Ruggieri, S.; Magni, G. The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J. Bacteriol. 1999, 181, 5509–5511. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, T.; Kurnasov, O.; Cheek, S.; Grishin, N.V.; Osterman, A. Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure (Lond. Engl. 1993) 2002, 10, 69–79. [Google Scholar] [CrossRef]
- Penfound, T.; Foster, J.W. NAD-dependent DNA-binding activity of the bifunctional NadR regulator of Salmonella typhimurium. J. Bacteriol. 1999, 181, 648–655. [Google Scholar] [CrossRef]
- Foster, J.W.; Penfound, T. The bifunctional NadR regulator of Salmonella typhimurium: Location of regions involved with DNA binding, nucleotide transport and intramolecular communication. FEMS Microbiol. Lett. 1993, 112, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Olivera, B.M.; Roth, J.R. Genetic characterization and regulation of the nadB locus of Salmonella typhimurium. J. Bacteriol. 1987, 169, 4285–4293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Olivera, B.M.; Roth, J.R. Genetic characterization of the pnuC gene, which encodes a component of the nicotinamide mononucleotide transport system in Salmonella typhimurium. J. Bacteriol. 1989, 171, 4402–4409. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.W.; Holley-Guthrie, E.A.; Warren, F. Regulation of NAD metabolism in Salmonella typhimurium: Genetic analysis and cloning of the nadR repressor locus. Mol. Gen. Genet. (MGG) 1987, 208, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Olivera, B.M.; Roth, J.R. Activity of the nicotinamide mononucleotide transport system is regulated in Salmonella typhimurium. J. Bacteriol. 1991, 173, 1311–1320. [Google Scholar] [CrossRef][Green Version]
- Robert, X.; Gouet, O. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acid Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.V. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol (London) 1910, 40, 4–7. [Google Scholar]
- Slotboom, D.J.; Duurkens, R.H.; Olieman, K.; Erkens, G.B. Static light scattering to characterize membrane proteins in detergent solution. Methods 2008, 46, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Beek, J.ter; Duurkens, R.H.; Erkens, G.B.; Slotboom, D.J. Quaternary structure and functional unit of energy coupling factor (ECF)-type transporters. J. Biol. Chem. 2011, 286, 5471–5475. [Google Scholar] [CrossRef]
- Birkner, J.P.; Poolman, B.; Koçer, A. Hydrophobic gating of mechanosensitive channel of large conductance evidenced by single-subunit resolution. Proc. Natl. Acad. Sci. USA 2012, 109, 12944–12949. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Dissociation Constant–KD (µM) |
|---|---|
| ATP | 63 ± 1 |
| AMP-PNP | 13 ± 1 |
| NR | No Binding |
| NMN | No Binding |
| NAD | No Binding |
| Nicotinamide | No Binding |
| Nicotinic acid | No Binding |
| Km, mM | Vmax, µmol·min−1·mg−1 | Ki, mM | |
|---|---|---|---|
| NR | 0.6 ± 0.1 | 0.4 ± 0.1 | 31.4 ± 8.1 |
| NMN | 0.5 ± 0.1 | 1.5 ± 0.1 | 25.4 ± 10.8 |
| Km, mM | Vmax, µmol·min−1·mg−1 | The Hill Coefficient | |
|---|---|---|---|
| NR | 5.2 ± 0.9 | 0.4 ± 0.1 | 2.3 ± 0.5 |
| NMN | 5.2 ± 0.6 | 1.2 ± 0.1 | 2.2 ± 0.3 |
| NadR NR | NadR NMN | NadR NAD AMP-PNP | |
|---|---|---|---|
| PDB code Data collection | 6GYE | 6GYF | 6GZO |
| Space group | P 64 2 2 | P 64 2 2 | P 64 2 2 |
| Unit cell dimensions | |||
| a, b, c (Å) | 165.14, 165.14, 196.55 | 165.09, 165.09, 193.05 | 167.32, 167.32, 192.81 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| Resolution range (Å) | 47.4–2.30 (2.33–2.30) * | 47.83–2.70 (2.76–2.70) * | 45.74–3.00 (3.09–3.00) * |
| Rmeas (%) | 16.6 (>100) * | 13.1 (>100) * | 9.4 (>100) * |
| cc1/2 | 99.8 (29.4) * | 99.8 (33.0) * | 99.8 (72.5) * |
| I/σI | 12.5 (0.9) * | 11 (0.6) * | 5.6 (1.7) * |
| Completeness (%) | 100 (100) * | 100 (99.9) * | 100 (99.4) * |
| Multipilicity | 13.5 (13.4) * | 13.4 (13.1) * | 6.5 (6.4) * |
| Refinement | |||
| Resolution (Å) | 47.4–2.30 | 47.83–2.70 | 45.74–3.00 |
| No. of reflections | 70441 | 41577 | 32439 |
| Rwork/Rfree | 0.18/0.21 | 0.20/0.24 | 0.19/0.24 |
| Number non-hydrogen atoms | 6446 | 6109 | 5993 |
| Protein | 6084 | 6042 | 5838 |
| Ligands/Ions | 36/60 | 44/23 | 88/5 |
| Water | 266 | 0 | 0 |
| B-factors (Å2) | |||
| Protein | 70.9 | 86.5 | 118.8 |
| Ligand/Ions | 109.7/115.1 | 98.6/101.1 | 135.9/178.3 |
| Water | 67.1 | – | – |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.008 | 0.009 | 0.009 |
| Bond angles (°) | 0.958 | 1.054 | 1.275 |
| Molprobity score | 1.83 | 2.08 | 2.56 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stetsenko, A.; Singh, R.; Jaehme, M.; Guskov, A.; Slotboom, D.J. Structural and Functional Characterization of NadR from Lactococcus lactis. Molecules 2020, 25, 1940. https://doi.org/10.3390/molecules25081940
Stetsenko A, Singh R, Jaehme M, Guskov A, Slotboom DJ. Structural and Functional Characterization of NadR from Lactococcus lactis. Molecules. 2020; 25(8):1940. https://doi.org/10.3390/molecules25081940
Chicago/Turabian StyleStetsenko, Artem, Rajkumar Singh, Michael Jaehme, Albert Guskov, and Dirk Jan Slotboom. 2020. "Structural and Functional Characterization of NadR from Lactococcus lactis" Molecules 25, no. 8: 1940. https://doi.org/10.3390/molecules25081940
APA StyleStetsenko, A., Singh, R., Jaehme, M., Guskov, A., & Slotboom, D. J. (2020). Structural and Functional Characterization of NadR from Lactococcus lactis. Molecules, 25(8), 1940. https://doi.org/10.3390/molecules25081940