
Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
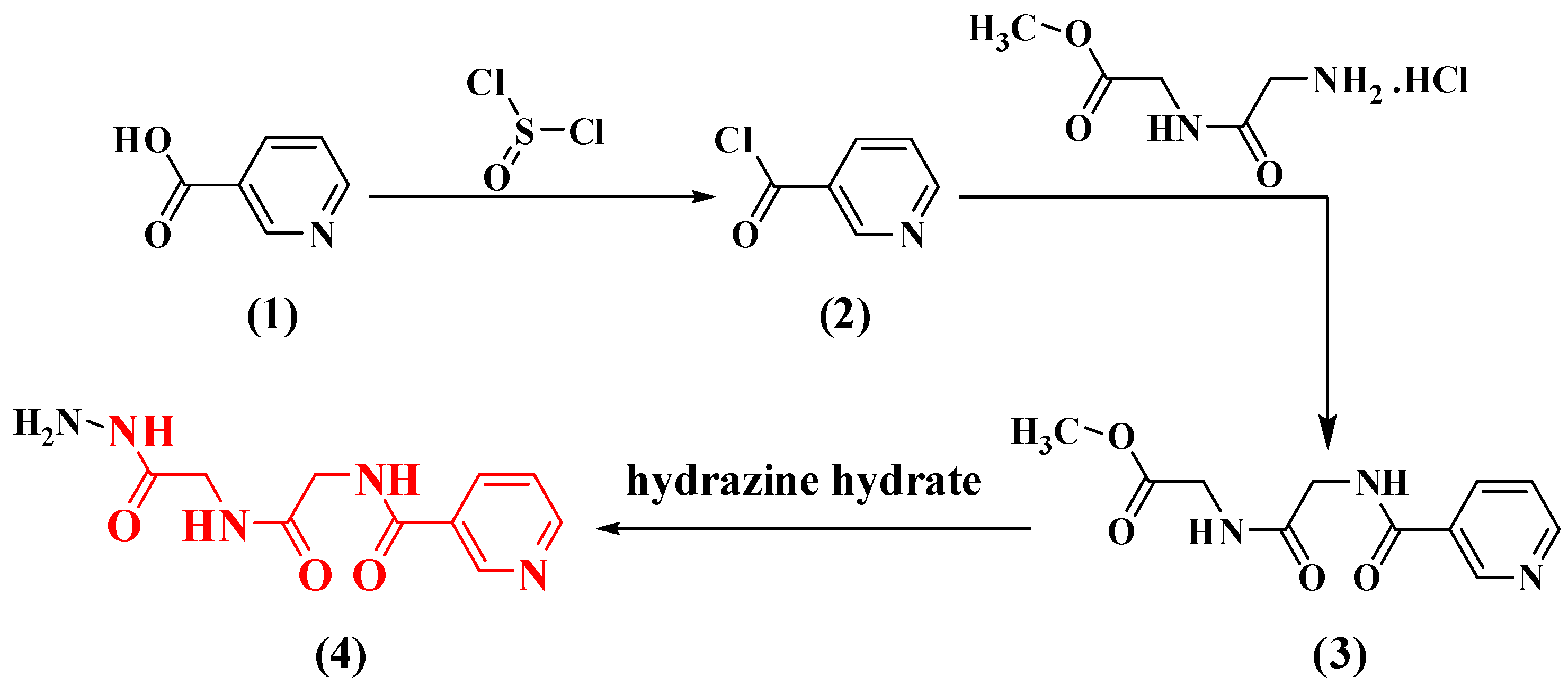
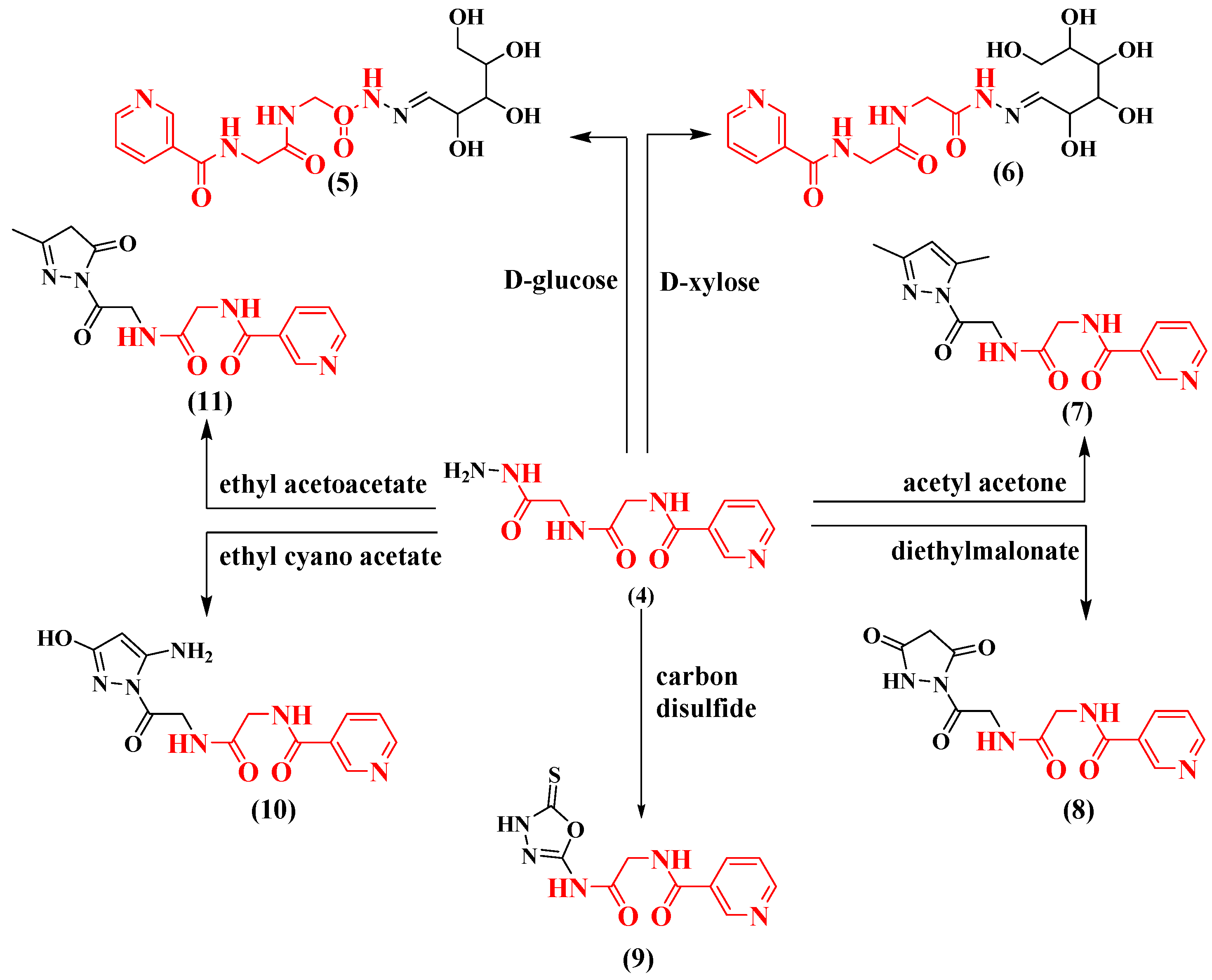
2.1. Chemistry
2.2. Evaluation of Biological Aspects
2.3. Computational Studies
2.3.1. Active Site
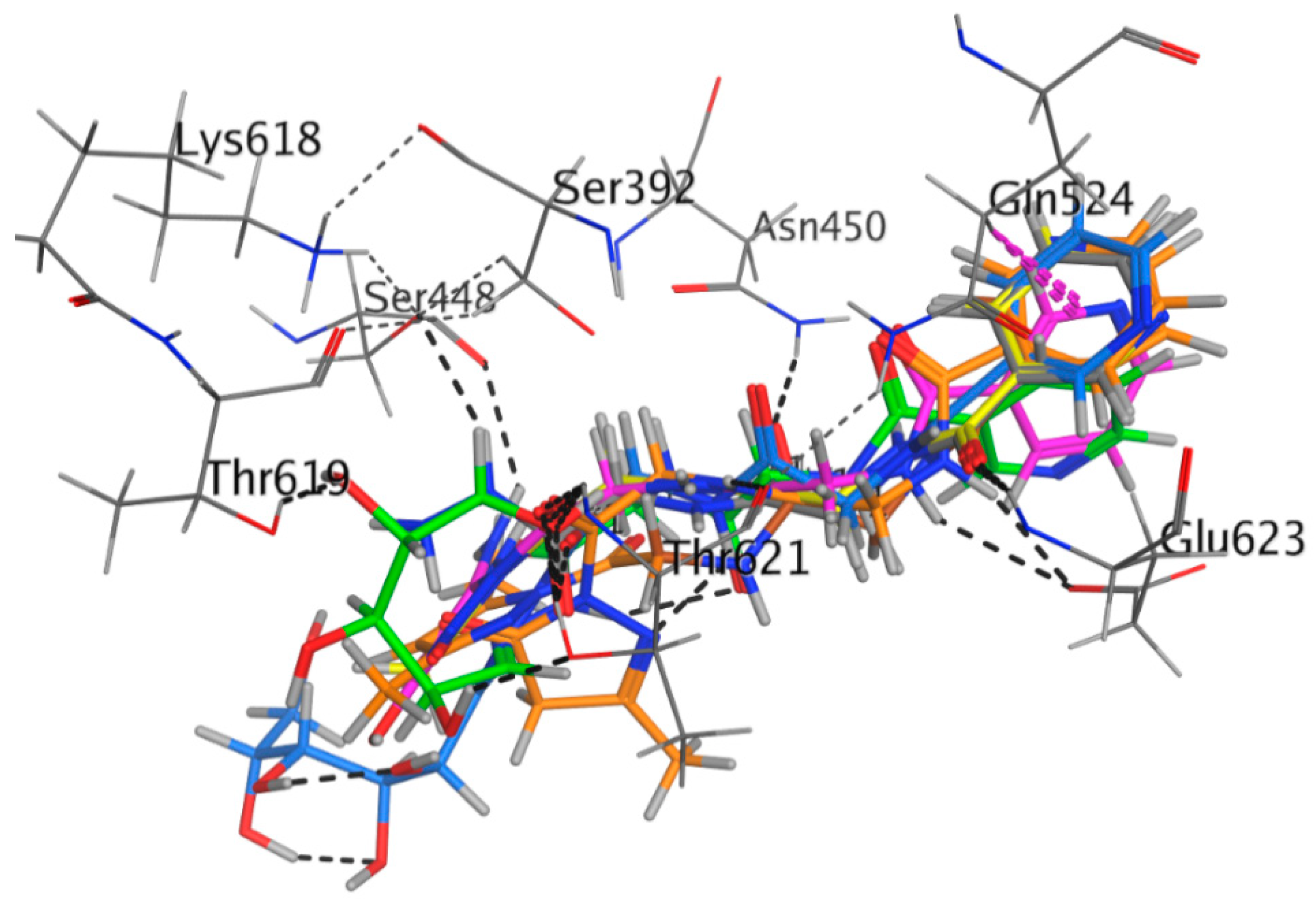
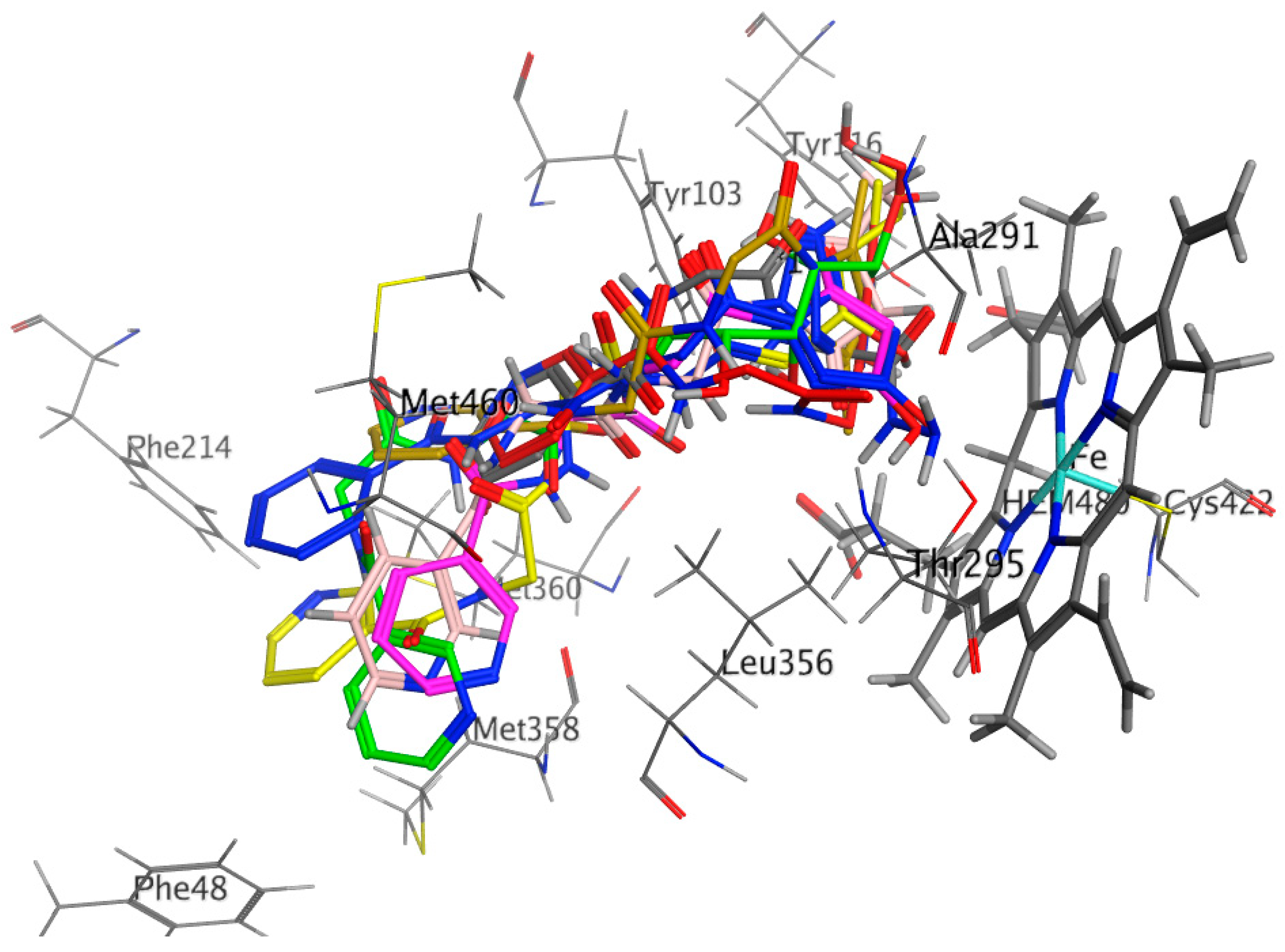
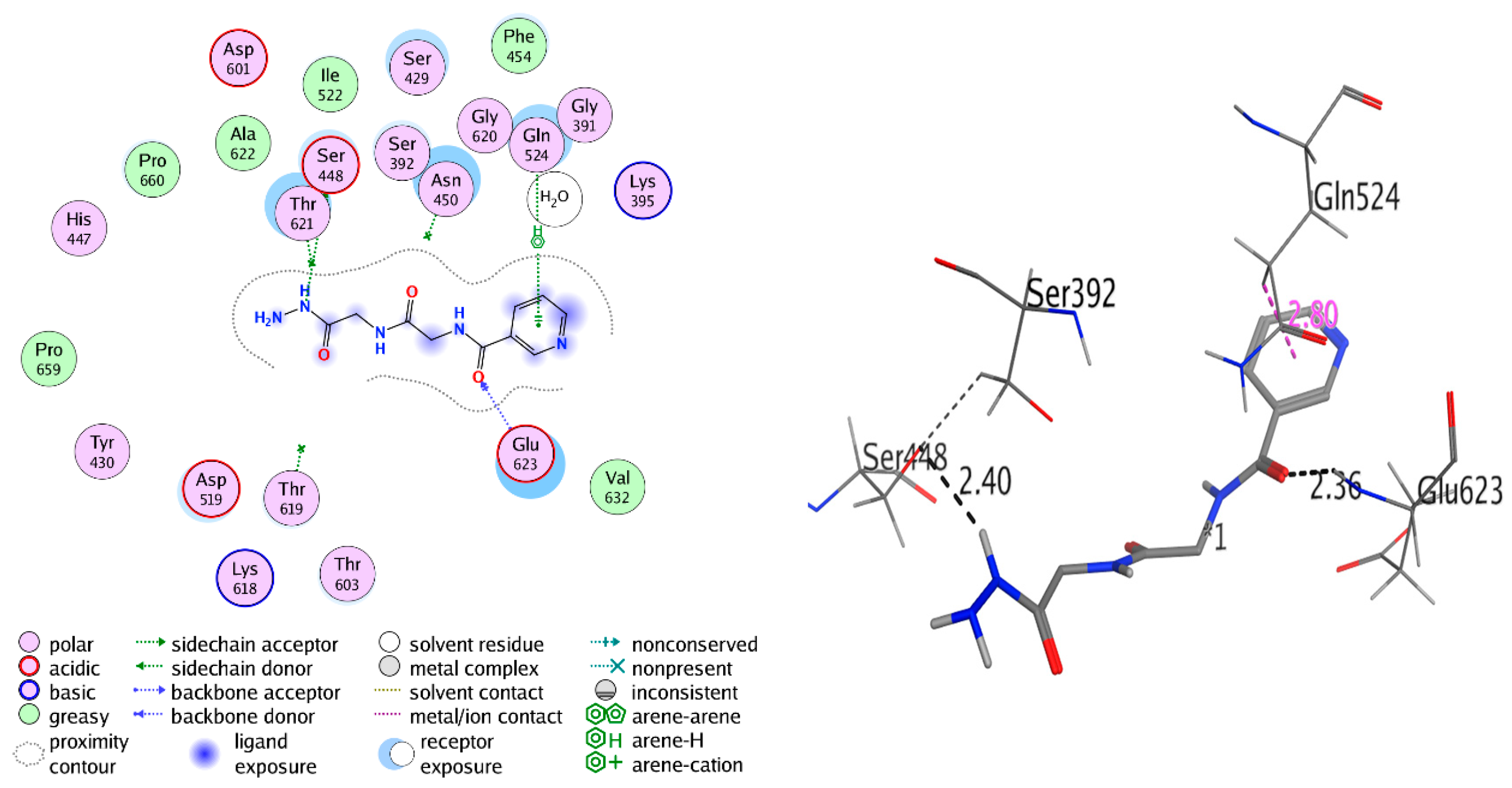
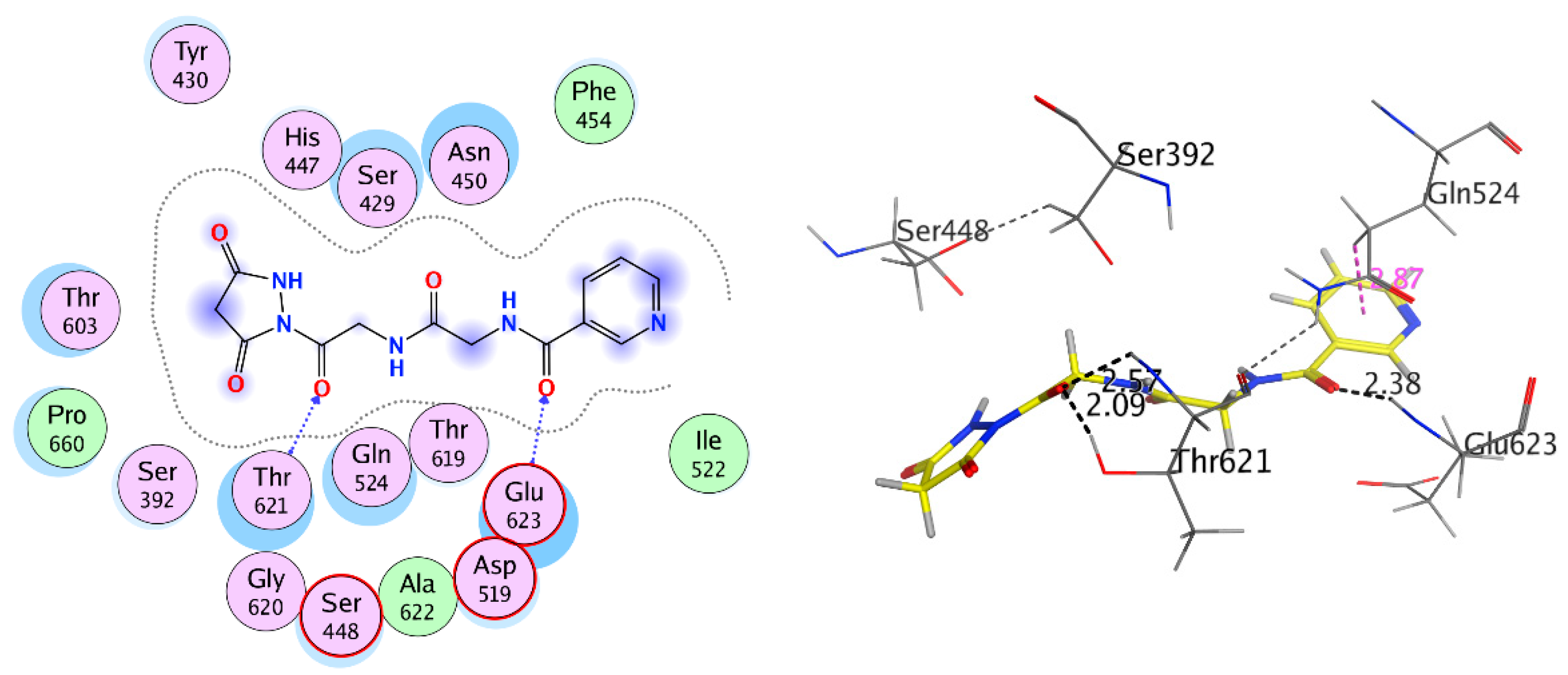
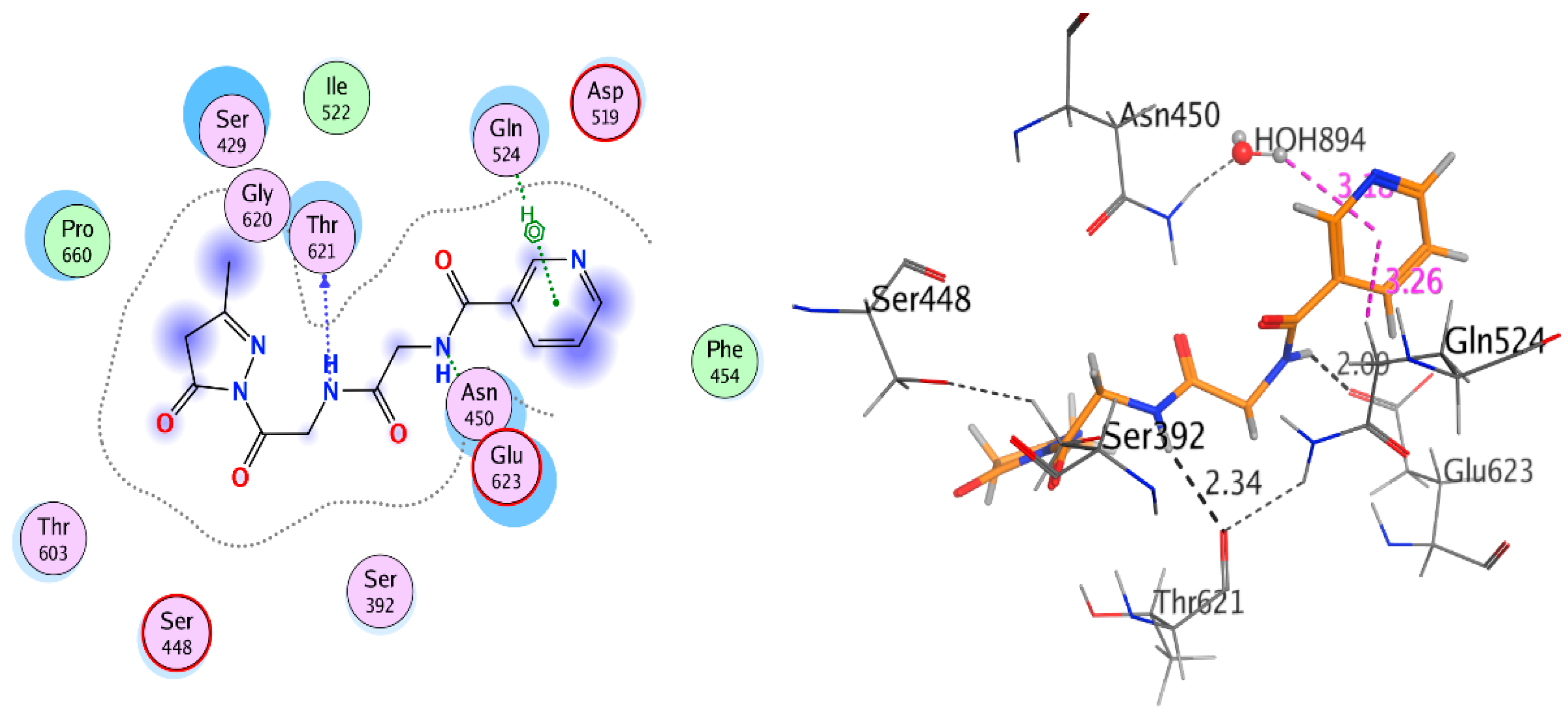
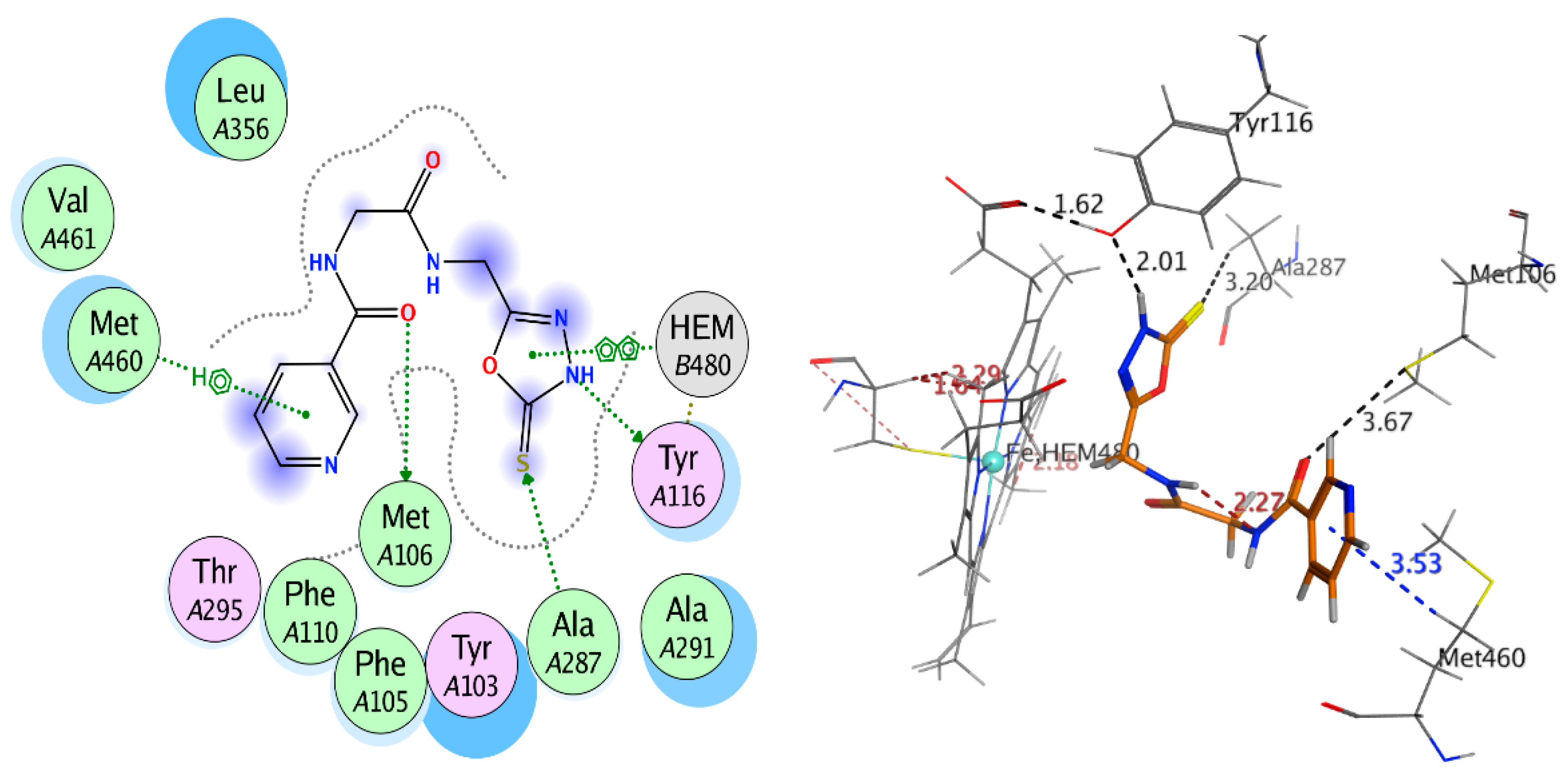
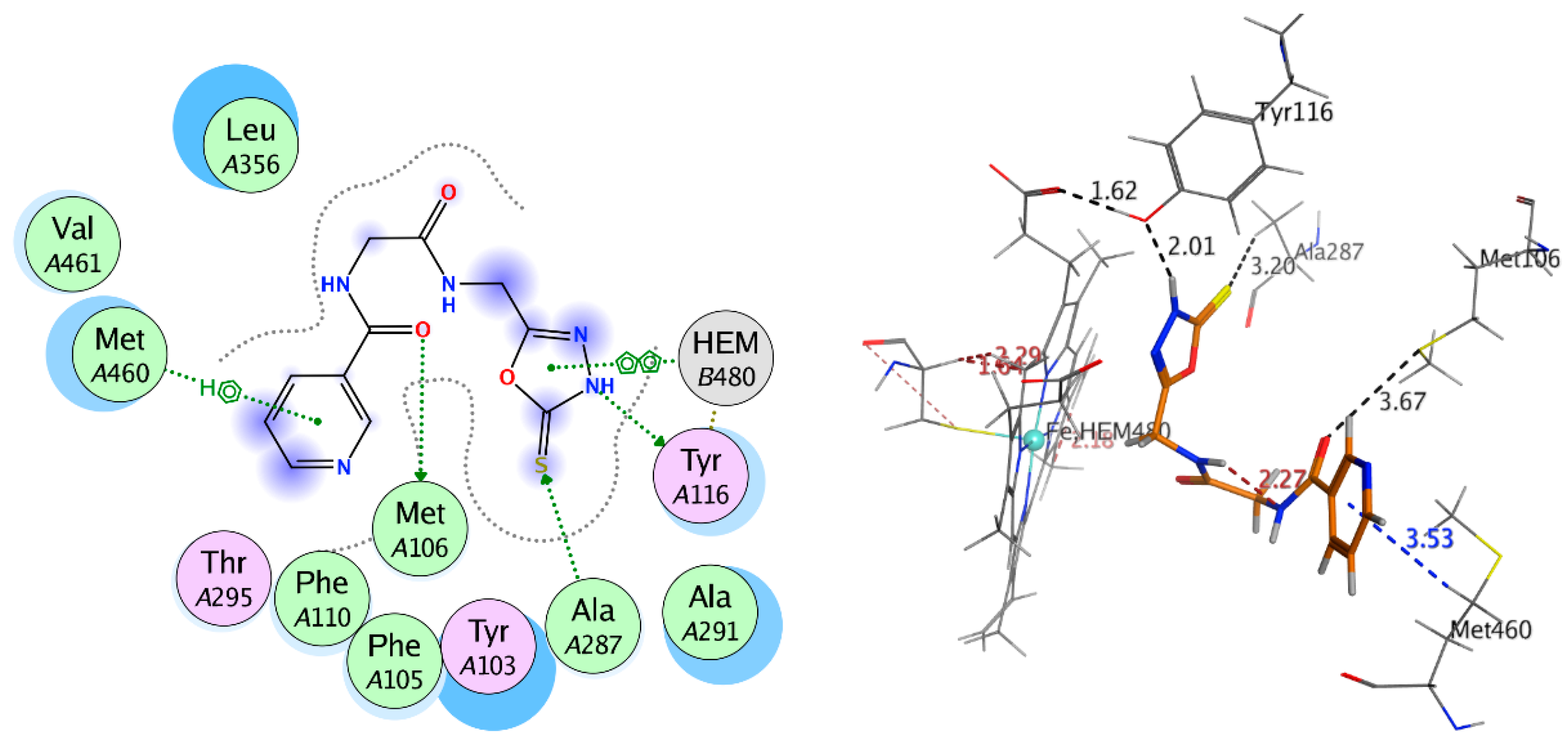
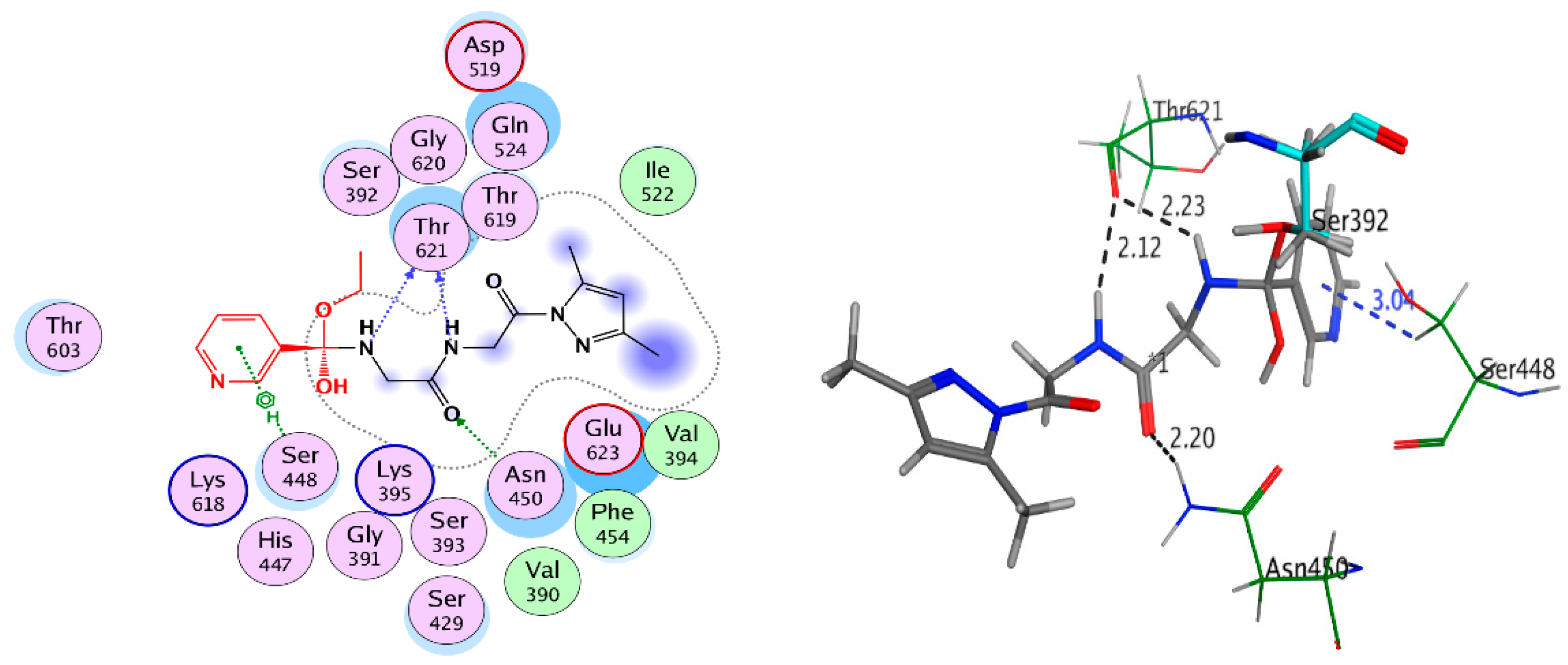
2.3.2. Docking Study
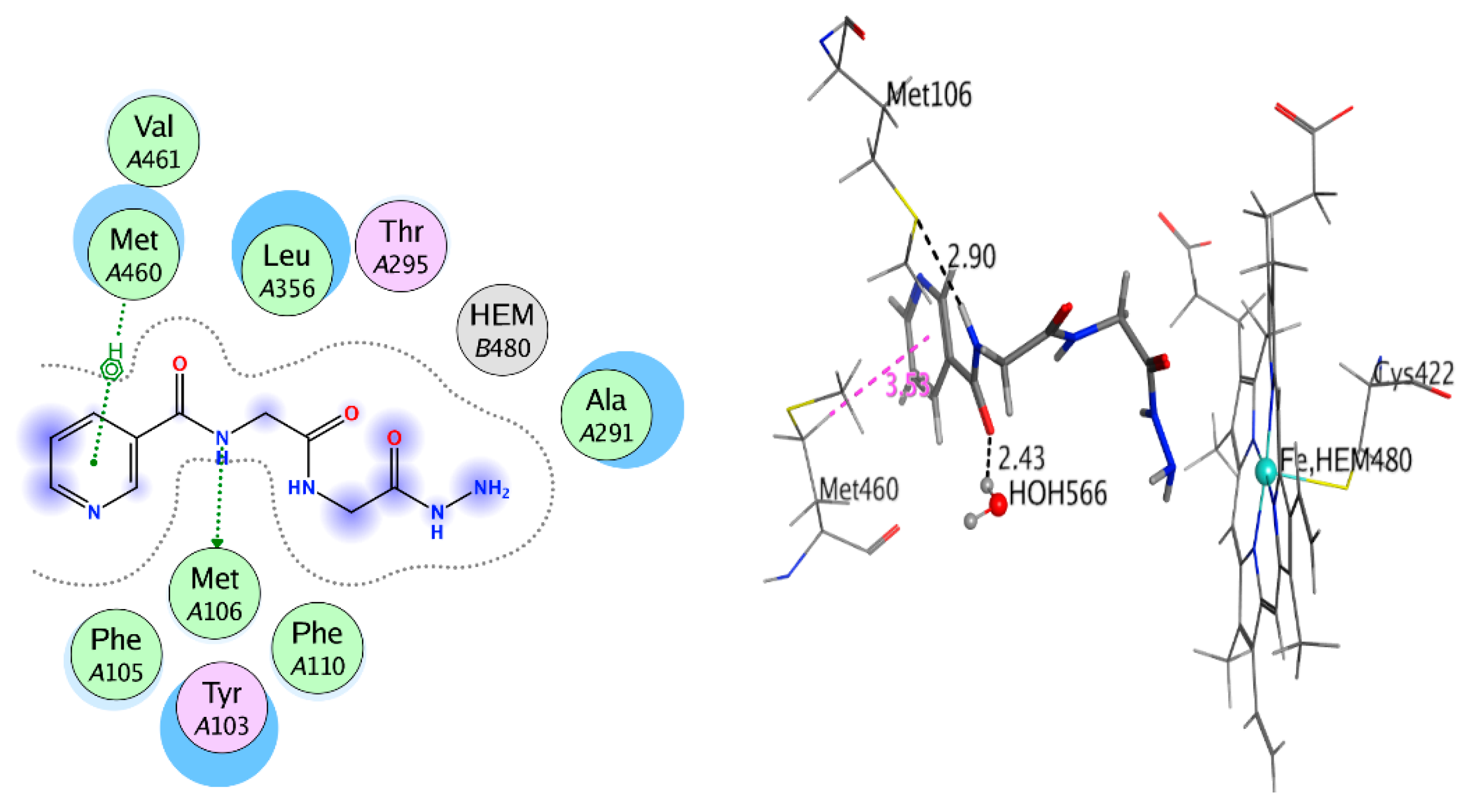
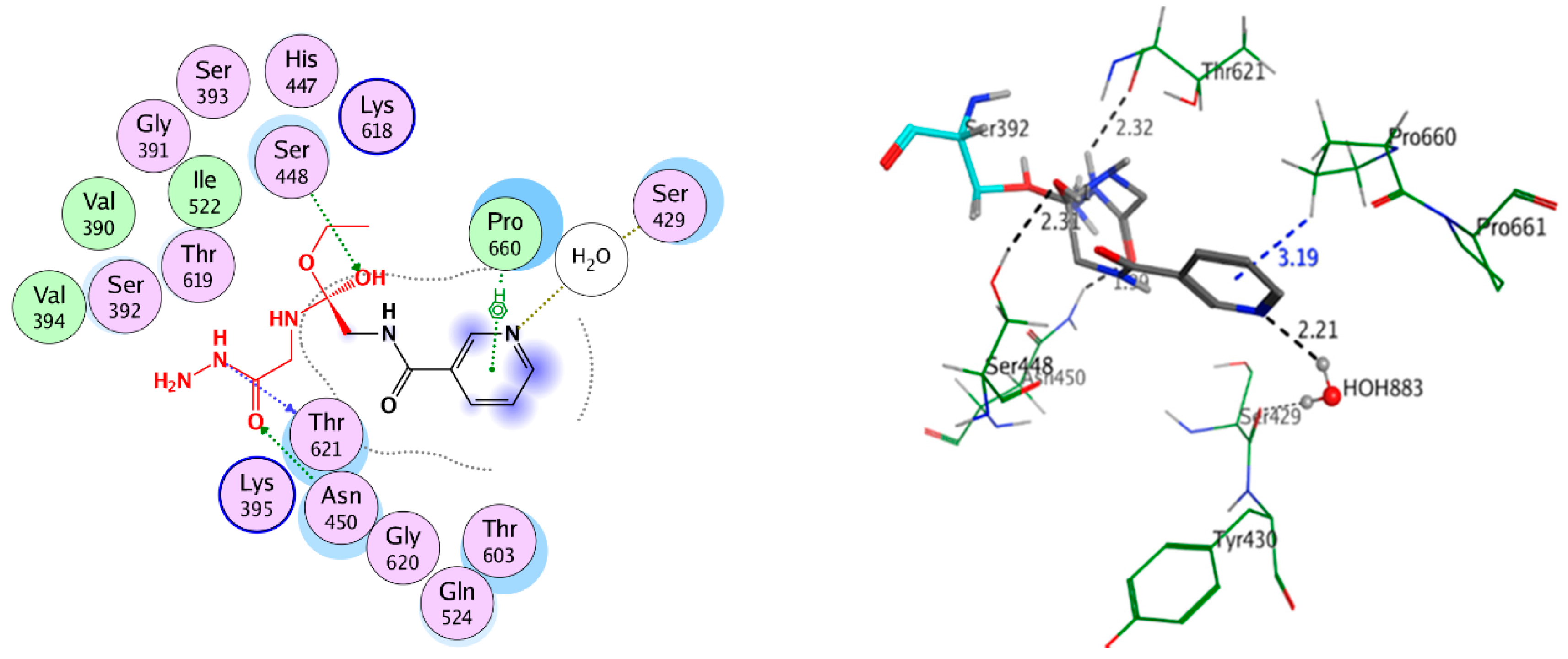
2.3.3. Covalent Docking
Properties of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET)
In Silico Toxicity
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials
3.1.2. Synthesis of Nicotinoyl Chloride (2), Nicotinoyl Glycyl-glycine-methyl Ester Hydrochloride (3), and N-(2-((2-hydrazinyl-2-oxoethyl) amino)-2-oxoethyl) nicotinamide (4)
3.1.3. The General Procedure for the Synthesis of Compounds 5 and 6
3.1.4. The General Procedure for the Synthesis of Compound 7
3.1.5. The General Procedure for the Synthesis of Compound 8
3.1.6. The General Procedure for Compound 9 Synthesis
3.1.7. The General Procedure for Compounds 10 and 11 Synthesis
3.2. Biological Evaluations
Anti-Microbial Activity (Agar Diffusion Assay)
3.3. Computational Studies
3.3.1. Molecular Modeling
Protein and Ligand Preparation for Docking Studies
Non-Covalent Docking Studies
Covalent Docking Studies
ADMET Properties
In Silico Toxicity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of nicotinic acid on serum cholesterol in man. Arch. Biochem. 1955, 54, 558–559. [Google Scholar] [CrossRef]
- Brinton, E.A.; Triscari, J.; Brudi, P.; Chen, E.; Johnson-Levonas, A.O.; Sisk, C.M.; Ruck, R.A.; MacLean, A.A.; Maccubbin, D.; Mitchel, Y.B. Effects of extended-release niacin/laropiprant on correlations between apolipoprotein B, LDL-cholesterol and non-HDL-cholesterol in patients with type 2 diabetes. Lipid Health Dis. 2016, 15, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Blankenhorn, D.H.; Azen, S.P.; Crawford, D.W.; Nessim, S.A.; Sanmarco, M.E.; Selzer, R.H.; Shircore, A.M.; Wickham, E.C. Effects of colestipol–niacin therapy on human femoral atherosclerosis. Circulation 1991, 83, 438–447. [Google Scholar] [CrossRef]
- Blankenhorn, D.H.; Selzer, R.H.; Crawford, D.W.; Barth, J.D.; Liu, C.; Liu, C.; Mack, W.J.; Alaupovic, P. Beneficial effects of colestipol–niacin therapy on the common carotid artery: Twoand four-year reduction of intima-media thickness measured by ultrasound. Circulation 1993, 88, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.G.; Zhao, X.; Chait, A.; Fisher, L.D.; Cheung, M.C.; Morse, J.S.; Dowdy, A.A.; Marino, E.K.; Bolson, E.L.; Alaupovic, P.; et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Engl. J. Med. 2001, 345, 1583–1592. [Google Scholar] [CrossRef]
- Lourenco, M.C.S.; de Souza, M.V.N.; Pinheiro, A.C.; Pinheiro, A.C.; Ferreira, M.D.L.; Gonçalves, R.S.B.; Nogueira, T.C.M.; Peralta, M.A. Evaluation of anti-tubercular activity of nicotinic and isoniazid analogues. Arkivoc 2007, 2007, 181–191. [Google Scholar]
- Hageman, G.J.; Stierum, R.H. Niacin, poly (ADP-ribose) polymerase-1 and genomic stability. Mut. Res.-Fundament. Molec. Mech. Mutagen. 2001, 475, 45–56. [Google Scholar] [CrossRef]
- Carlson, L.A. Nicotinic acid: The broad-spectrumlipid drug.A 50th anniversary review. J. Intern. Med. 2005, 258, 94–114. [Google Scholar] [CrossRef]
- Reinhardt, A.; Neundorf, I. Design and Application of Antimicrobial Peptide Conjugates. Int. J. Mol. Sci. 2016, 17, 701. [Google Scholar] [CrossRef]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. Biophys. 2011, 40, 387–397. [Google Scholar] [CrossRef]
- Reddy, K.V.R.; Yedery, R.D.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Ag. 2004, 24, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trend Biotech. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, N.H.; Labovitiadi, O.; Cushnie, T.P.; Matthews, K.H.; Mercer, D.K.; Lamb, A.J. Production and evaluation of an antimicrobial peptide-containing wafer formulation for topical application. Curr. Microbiol. 2013, 66, 271–278. [Google Scholar]
- Hein, K.Z.; Takahashi, H.; Tsumori, T.; Yasui, Y.; Nanjoh, Y.; Toga, T.; Wu, Z.; Grötzinger, J.; Jung, S. Disulphide-reduced psoriasin is a human apoptosis-inducing broad-spectrum fungicide. Proc. Natl. Acad. Sci. USA 2015, 112, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.G. Mechanistic basis of enzyme-targeted drugs. Biochemistry 2005, 44, 5561–5571. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef]
- Hansen, R.; Peters, U.; Babbar, A.; Chen, Y.; Feng, J.; Janes, M.R.; Li, L.S.; Ren, P.; Liu, Y.; Zarrinkar, P.P. The reactivity-driven biochemical mechanism of covalent KRAS(G12C) inhibitors. Nat. Struct. Mol. Biol. 2018. [Google Scholar]
- Goedken, E.R.; Argiriadi, M.A.; Banach, D.L.; Fiamengo, B.A.; Foley, S.E.; Frank, K.E.; George, J.S.; Harris, C.M.; Hobson, A.D.; Ihle, D.C.; et al. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J. Biol. Chem. 2015, 290, 4573–4589. [Google Scholar] [CrossRef]
- Prats-Gomez, R.M.; Pla, J.; Blasco, B.; Ayala, J. A new beta-lactam-binding protein derived from penicillin-binding protein 3 of Escherichia coli. J. Bacteriol. 1989, 171, 5194–5198. [Google Scholar] [CrossRef]
- Lynch, J.P.; Zhanel, G.G. Escalation of antimicrobial resistance among Streptococcus pneumoniae: Implications for therapy. Semin. Respir. Crit. Care Med. 2005, 26, 575–616. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Kleshchenko, Y.; Nes, W.D.; Villalta, F.; Waterman, M.R. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids 2008, 43, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; York, J.D.; Guengerich, F.P.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef] [PubMed]
- Naglah, A.M.; Moustafa, G.O.; Al-Omar, M.A.; Al-Salem, H.S.A.; Hozzein, W.N. Synthesis, Characterization and In Vitro Antimicrobial Investigation of Novel Amino Acids and Dipeptides Based on Dibenzofuran-2-Sulfonyl-Chloride. J. Comput. Theor. Nanosci. 2017, 14, 3183–3190. [Google Scholar] [CrossRef]
- Al-Salem, H.S.A.; Naglah, A.M.; Moustafa, G.O.; Mahmoud, A.Z.; Al-Omar, M.A. Synthesis of Novel Tripeptides Based on Dibenzofuran-2-Sulfonyl [Aromatic and Hydroxy Aromatic Residues]: Towards Antimicrobial and Antifungal Agents. J. Comput. Theor. Nanosci. 2017, 14, 3958–3966. [Google Scholar] [CrossRef]
- Hassan, A.S.; Askar, A.A.; Nossier, E.S.; Naglah, A.M.; Moustafa, G.O.; Al-Omar, M.A. Antibacterial Evaluation, In Silico Characters and Molecular Docking of Schiff Bases Derived from 5-aminopyrazoles. Molecules 2019, 24, 3130. [Google Scholar] [CrossRef]
- Hasanin, M.S.; Moustafa, G.O. New potential green, bioactive and antimicrobial nanocomposites based on cellulose and amino acid. Int. J. Biol. Macromolec. 2019, 144, 441–448. [Google Scholar] [CrossRef]
- Elsherif, M.A.; Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Morsy, N.M. Antimicrobial Evaluation and Molecular Properties Prediction of Pyrazolines Incorporating Benzofuran and Pyrazole Moieties. Bull. Chem. Soc. Ethiopia. 2020, 10, 037–043. [Google Scholar] [CrossRef]
- Hassan, A.S.; Moustafa, G.O.; Morsy, N.M.; Abdou, A.M.; Hafez, T.S. Design, synthesis and antibacterial activity of N-aryl-3-(arylamino)-5-(((5-substituted furan-2-yl) methylene) amino)-1H-pyrazole-4-carboxamide as Nitrofurantoin® analogues. Egypt. J. Chem. 2020, 63. in press. [Google Scholar] [CrossRef]
- Moustafa, G.; Khalaf, H.; Naglah, A.; Al-Wasidi, A.; Al-Jafshar, N.; Awad, H. The Synthesis of Molecular Docking Studies, In Vitro Antimicrobialand Antifungal Activities of Novel Dipeptide Derivatives Based on N-(2-(2-hydrazinyl-2-oxoethylamino)-2-oxoethyl)-Nicotinamide. Molecules 2018, 23, 761. [Google Scholar] [CrossRef]
- Hassan, A.S.; Moustafa, G.O.; Askar, A.A.; Naglah, A.M.; Al-Omar, M.A. Synthesis and antibacterial evaluation of fused pyrazoles and Schiff bases. Synth. Commun. 2018, 48, 2761–2772. [Google Scholar] [CrossRef]
- Abo-Ghalia, M.H.; Moustafa, G.O.; Alwasidi, A.S.; Naglah, A.M. Cytotoxic Investigation of Isophthaloyl Cyclopentapeptides. Lat. Am. J. Pharm. 2017, 36, 1957–1962. [Google Scholar]
- Moustafa, G.O.; El-Sawy, A.A.; Abo-Ghalia, M.H. Synthesis of novel cyclopeptide candidates: I-cyclo- [N_-isophthaloyl-bis-(Glycine-amino acid)-L-lysine] derivatives with expected anticancer activity. Egypt. J. Chem. 2013, 5, 473–494. [Google Scholar]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M. Synthesis and in vitro anticancer activity of pyrazolo [1,5-a] pyrimidines and pyrazolo [3,4-d] [1, 2, 3] triazines. Synth. Commun. 2017, 47, 1963–1972. [Google Scholar] [CrossRef]
- Amr, A.E.; Abo-Ghalia, M.H.; Moustafa, G.O.; Al-Omar, M.A.; Nossier, E.S.; Elsayed, E.A. Design, synthesis and docking studies of novel macrocyclic pentapeptides as anticancer multi-targeted kinase inhibitors. Molecules 2018, 23, 2416. [Google Scholar] [CrossRef]
- Moustafa, G.O.; Younis, A.; Al-Yousef, S.A.; Mahmoud, S.Y. Design, Synthesis of Novel Cyclic Pentapeptide Derivatives Based on 1, 2-benzenedicarbonyl chloride with Expected Anticancer Activity. J. Comput. Theor. Nanosci. 2019, 16, 1733–1739. [Google Scholar] [CrossRef]
- Elhenawy, A.A.; AL-Harbi, L.M.; Moustafa, G.O.; El-Gazzar, M.A.; Abdel-Rahman, R.F.; Salim, A. Synthesis, comparative dockingand pharmacological activity of naproxenyl amino acid derivatives as possible Anti-Inflammatory and analgesic agents. Drug Des. Dev. Ther. 2019, 13, 1773. [Google Scholar] [CrossRef]
- Kassem, A.F.; Moustafa, G.O.; Nossier, E.S.; Khalaf, H.S.; Mounier, M.M.; Al-Yousef, S.A.; Mahmoud, S.Y. In vitro anticancer potentiality and molecular modelling study of novel amino acid derivatives based on N 1, N 3-bis-(1-hydrazinyl-1-oxopropan-2-yl) isophthalamide. J. Enzym. Inhib. Med. Chem. 2019, 34, 1247–1258. [Google Scholar] [CrossRef]
- Mohamed, F.H.; Shalaby, A.M.; Soliman, H.A.; Abdelazem, A.Z.; Mounier, M.M.; Nossier, E.S.; Moustafa, G.O. Design, Synthesis and Molecular Docking Studies of Novel Cyclic Pentapeptides Based on Phthaloyl Chloride with Expected Anticancer Activity. Egypt. J. Chem. 2020, 63. in press. [Google Scholar] [CrossRef]
- Abo-Ghalia, M.H.; Moustafa, G.O.; Amr, A.E.; Naglah, A.M.; Elsayed, E.A.; Bakheit, A.H. Anticancer activities and 3D-QSAR studies of some new synthesized macrocyclic heptapeptide derivatives. Molecules 2020, 25, 1253. [Google Scholar] [CrossRef]
- Kalmouch, A.; Radwan, M.A.A.; Omran, M.M.; Sharaky, M.; Moustafa, G.O. Synthesis of novel 2, 3’-bipyrrole derivatives from chalcone and amino acids as antitumor agents. Egypt. J. Chem. 2020, 63. in press. [Google Scholar]
- Lepesheva, I.G.; Waterman, M.R. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Cur. Top. Med. Chem. 2011, 11, 2060–2071. [Google Scholar] [CrossRef]
- Hecht, D.; Fogel, G.B. Computational intelligence methods for docking scores. Cur. Comp.-Aided Drug Design 2009, 5, 56–68. [Google Scholar] [CrossRef]
- Boobis, A.; Gundert-Remy, U.; Kremers, P.; Macheras, P.; Pelkonen, O. In silico prediction of ADME and pharmacokinetics: Report of an expert meeting organised by COST B15. Eur. J. Pharmac. Sci. 2002, 17, 183–193. [Google Scholar] [CrossRef]
- Hosea, N.A.; Jones, H.M. Predicting pharmacokinetic profiles using in silico derived parameters. Mol. Pharmac. 2013, 10, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Dearden, J. In silico prediction of drug toxicity. J. Comput. Aided Mol. Des. 2003, 17, 119–127. [Google Scholar] [CrossRef]
- Enslein, K.; Gombar, V.K.; Blake, B.W. Use of SAR in computer-assited prediction of carcinogenicity and mutagenicity of chemicals by the TOPKAT program. Mutat. Res./Fundament. Molec. Mech. Mutagen. 1994, 305, 47–61. [Google Scholar] [CrossRef]
- Ridings, J.E.; Barratt, M.D.; Cary, R.; Earnshaw, C.G.; Eggington, C.E.; Ellis, M.K.; Judson, P.N.; Langowski, J.J.; Marchant, C.A.; Payne, M.P. Computer prediction of possible toxic action from chemical structure: An update on the DEREK system. Toxicology 1996, 106, 267–279. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, L.; Cui, S.; Liang, Y.; Zhang, X. Synthesis and Anti-hypertensive Effects of the Twin Drug of Nicotinic Acid and Quercetin Tetramethyl Ether. Molecules 2014, 19, 4791–4801. [Google Scholar] [CrossRef]
- Dalton, N.; Gordon, C.P.; Boyle, T.P.; Vandegraaf, N.; Deadman, J.; Rhodes, D.I.; Coates, J.A.; Pyne, S.G.; Keller, P.A.; Bremner, J.B. The discovery of allyltyrosine based tripeptides as selective inhibitors of the HIV-1 integrase strandtransfer reaction. Org. Biomol. Chem. 2016, 14, 6010–6023. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arthington-Skaggs, B.A.; Motley, M.; Warnock, D.W.; Morrison, C.J. Comparative Evaluation of PASCO and National Committee for Clinical Laboratory Standards M27-A Broth Microdilution Methods for Antifungal Drug Susceptibility Testing of Yeasts. J. Clin. Microbiol. 2000, 38, 2254–2260. [Google Scholar] [CrossRef] [PubMed]
- Barchiesi, F.; Colombo, A.L.; Mcgough, D.A.; Rinaldi, M.G. Comparative Study of Broth Macrodilution and Microdilution Techniques for In Vitro Antifungal Susceptibility Testing of Yeasts by Using the National Committee for Clinical Laboratory Standards Proposed Standard. J. Clin. Microbiol. 1994, 32, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group, Inc. MOE (the Molecular Operating Environment) Version 2014; Chemical Computing Group, Inc.: Montreal, QC, Canada, 2014. [Google Scholar]
- Dewar, M.J.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J. Development and use of quantum mechanical molecular models. 76. AM1: A new general-purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Yoshida, H.; Kawai, F.; Obayashi, E.; Akashi, S.; Roper, D.I.; Tame, J.R.; Park, S.Y. Crystal Structures of Penicillin-Binding Protein 3 (PBP3) from Methicillin-Resistant Staphylococcus aureus in the Apo and Cefotaxime-Bound Forms. J. Mol. Biol. 2012, 423, 351–364. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Park, H.W.; Hargrove, T.Y.; Vanhollebeke, B.; Wawrzak, Z.; Harp, J.M.; Sundaramoorthy, M.; Nes, W.D.; Pays, E.; Chaudhuri, M. Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 2010, 285, 1773–1780. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second-generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Oda, A.; Yamaotsu, N.; Hirono, S. New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models. J. Comp. Chem. 2005, 26, 818–826. [Google Scholar] [CrossRef]
- Clark, A.M.; Labute, P. 2D depiction of protein− ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comp. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef]
- Young, D.C. Computational Drug Design: A Guide for Computational and Medicinal Chemists; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Zhu, K.B.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Materials Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2015. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all the compounds are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Test Organism | |||
|---|---|---|---|---|
| Bacteria | Fungi | |||
| Gram-Positive | Gram-Negative | Unicellular | Filamentous | |
| B. subtilits | E. coli | C. albicans | A. niger | |
| Inhibition Zone (mm) | ||||
| 4 | 29 | 30 | 28 | 16 |
| 5 | 15 | 00 | 17 | 00 |
| 6 | 15 | 13 | 16 | 00 |
| 7 | 18 | 15 | 20 | 00 |
| 8 | 15 | 14 | 20 | 16 |
| 9 | 19 | 15 | 20 | 00 |
| 10 | 11 | 15 | 15 | 00 |
| 11 | 11 | 12 | 15 | 00 |
| Standard Antibiotics | ||||
| NA = 30 µg | 20 | 16 | 00 | 00 |
| S = 10 µg | 14 | 00 | 12 | 00 |
| N = 30 µg | 00 | 00 | 00 | 16 |
| Ny = 100 µg | 00 | 00 | 00 | 00 |
| NV = 30 µg | 29 | 30 | 00 | 00 |
| T = 30 µg | 30 | 27 | 00 | 00 |
| SDZ = 30 µg | 00 | 20 | 00 | 00 |
| VA = 30 µg | 21 | 23 | 00 | 00 |
| Receptor | Compound | Amino Acid Residues | Type of Interaction | Distance (Å) | Binding Affinity kcal·mol−1 | Docking Score (S) | RMSD Refine |
|---|---|---|---|---|---|---|---|
| Antibacterial Penicillin-Binding Protein 3 (PBP3) (3VSL) | Compound 4 | Ser448 | H-donor | 2.40 | −5.315 | −11.868 | 2.258 |
| Glu623 | H-acceptor | 2.36 | |||||
| Gln524 | pi-H | 2.80 | |||||
| Compound 5 | Thr621 | H-donor | 1.87 | −6.177 | −10.463 | 2.559 | |
| Thr619 | H-acceptor | 1.82 | |||||
| Thr621 | H-acceptor | 2.47 | |||||
| Thr621 | H-acceptor | 193 | |||||
| Compound 6 | Thr621 | H-acceptor | 2.83 | −6.228 | −10.831 | 2.610 | |
| Gln524 | Pi-H | 3.27 | |||||
| Compound 7 | Glu623 | H-acceptor | 2.70 | −5.799 | −10.280 | 1.658 | |
| Asn450 | H-acceptor | 2.10 | |||||
| Gln524 | Pi-H | 2.86 | |||||
| Compound 8 | Glu623 | H-acceptor | 2.37 | −5.822 | −10.287 | 2.194 | |
| Thr621 | H-acceptor | 2.09 | |||||
| Thr621 | H-acceptor | 2.57 | |||||
| Gln524 | Pi-H | 2.87 | |||||
| Compound 9 | Glu623 | H-donor | 1.99 | −5.508 | −10.891 | 2.741 | |
| Ser448 | H-donor | 2.54 | |||||
| Thr621 | H-acceptor | 3.16 | |||||
| Thr621 | H-acceptor | 2.85 | |||||
| HOH894 | Pi-H | 2.75 | |||||
| Compound 10 | Glu623 | H-donor | 2.59 | −5.790 | −9.695 | 1.625 | |
| Thr621 | H-acceptor | 2.00 | |||||
| Thr621 | H-acceptor | 2.24 | |||||
| Compound 11 | Glu623 | H-donor | 3.06 3.22 | −5.47 | −10.372 | 1.5346 | |
| Thr621 | H-donor | ||||||
| Gln524 | pi-H | ||||||
| Standard drug S | Val658 | H-donor | 3.12 | −7.152 | −17.891 | 1.982 | |
| Glu623 | H-donor | 2.9 | |||||
| Thr621 | H-donor | 3.48 | |||||
| Thr621 | H-acceptor | 3.06 | |||||
| Asn450 | H-acceptor | 3.1 | |||||
| Asn450 | H-acceptor | 2.9 | |||||
| Glu623 | ionic | 2.9 | |||||
| Glu623 | ionic | 3.77 | |||||
| Ser448 | ionic | 4.03 | |||||
| Standard drug NA | Ser429 | H-acceptor | 2.84 | −4.781 | −9.597 | 0.551 | |
| Asn450 | H-acceptor | 3.11 | |||||
| Standard drug N | Asp601 | H-donor | 3.17 | −6.739 | −10.590 | 2.123 | |
| His447 | H-donor | 3.21 | |||||
| Thr621 | H-donor | 3.44 | |||||
| Asn450 | H-donor | 3.12 | |||||
| Glu623 | H-donor | 2.86 | |||||
| Glu623 | H-donor | 3.43 | |||||
| Glu623 | ionic | 2.86 | |||||
| Glu623 | ionic | 3.43 |
| Receptor | Compound | Amino Acid Residues | Type of Interaction | Distance (Å) | Docking Score (S) | Binding Affinity kcal·mol−1 | RMSD Refine |
|---|---|---|---|---|---|---|---|
| Antifungal Sterol 14-Alpha Demethylase (CYP51) (3GW9) | Compound 4 | Met106 Met460 HOH566 | H-donor Pi-H H-acceptor | 2.90 3.53 | −9.333 | −5.886 | 2.145 |
| Compound 5 | Met 358 | Pi-H | 3.34 | −9.690 | −5.674 | 1.693 | |
| HOH546 | H-acceptor | 1.79 | |||||
| Compound 6 | Met106 | H-donor | 4.82 | −9.531 | −6.132 | 3.165 | |
| Leu357 | H-acceptor | 2.63 | |||||
| Compound 7 | Met106 | H-donor | 4.47 | −9.043 | −5.940 | 2.008 | |
| Met358 | pi-H | 3.95 | |||||
| Compound 8 | Met106 | H-donor | 3.44 | −10.359 | −6.686 | 2.01 | |
| Leu357 | H-acceptor | 1.87 | |||||
| HEM480 | ionic | 2.85 | |||||
| Compound 9 | Met106 | H-donor | 3.67 | −9.401 | −6.157 | 1.042 | |
| Tyr116 | H-donor | 2.01 | |||||
| Ala287 | H-acceptor | 3.20 | |||||
| Met460 | Pi-H | 3.53 | |||||
| HEM480 | Pi-H | 2.89 | |||||
| Compound 10 | Met106 | H-donor | 3.43 | −9.884 | −5.783 | 2.341 | |
| Leu357 | H-acceptor | 1.97 | |||||
| HEM480 | H-pi | 2.95 | |||||
| Compound 11 | Met106 | H-donor | 3.62 | −9.503 | −5.407 | 1.176 | |
| Standard drug S | Met360 | H-donor | 2.89 | −12.996 | −5.573 | 2.291 | |
| Met358 | H-acceptor | 3.16 | |||||
| Standard drug NA | Ala291 | pi-H | 4.18 | −10.169 | −6.329 | 1.145 | |
| Standard drug N | Met106 | H-donor | 3.7 | −11.891 | −6.792 | 2.75 | |
| Tyr116 | H-donor | 2.92 |
| Receptor | Compound | Amino Acid Residues | Type of Interaction | Distance (Å) | Binding Affinity Kcal/moL | Docking Score (S) | RMSD Refine |
|---|---|---|---|---|---|---|---|
| Antibacterial Penicillin-Binding Protein 3 (PBP3) (3VSL) | Compound 4 | Ser392 | Covalent bond | −4.315 | −10.254 | 2.258 | |
| Asn450 | H-acceptor | 1.99 | |||||
| Ser448 | H-acceptor | 2.31 | |||||
| Thr621 | H-donor | 2.32 | |||||
| Pro660 | Pi-H | 3.19 | |||||
| HOH883 | H-acceptor | 2.21 | |||||
| Compound 5 | Ser392 | Covalent bond | −5.031 | −9.73 | 1.322 | ||
| Thr621 | H-acceptor | 2.56 | |||||
| Glu623 | H-donor | 1.96 | |||||
| Compound 6 | Ser392 | Covalent bond | −5.966 | −9.94 | 0.6999 | ||
| Thr603 | H-donor | 2.08 | |||||
| Compound 7 | Ser392 | Covalent bond | −4.782 | −9.466 | 1.472 | ||
| Asn450 | H-acceptor | 2.13 | |||||
| Thr621 | H-donor | 2.13 | |||||
| Compound 8 | Ser392 | Covalent bond | −5.73 | −9.161 | 0.7531 | ||
| Asn450 | H-acceptor | 2.12 | |||||
| Thr621 | H-acceptor | 2.14 | |||||
| Thr621 | H-acceptor | 2.38 | |||||
| Ser448 | pi-H | 3.89 | |||||
| Compound 9 | Ser392 | Covalent bond | 1.60 | −4.97 | −9.234 | 1.012 | |
| Thr621 | H-acceptor | 1.83 | |||||
| Thr621 | H-acceptor | 2.04 | |||||
| Asn450 | H-donor | 3.10 | |||||
| Compound 10 | Ser392 | Covalent bond | −5.602 | −11.84 | 0.8385 | ||
| Thr621 | H-acceptor | 2.05 | |||||
| Thr621 | H-acceptor | 2.27 | |||||
| Asn450 | H-acceptor | 2.22 | |||||
| Ser448 | Pi-H | 2.91 | |||||
| Compound 11 | Ser392 | Covalent bond | −5.777 | −9.822 | 1.155 | ||
| Thr621 | H-acceptor | 2.06 | |||||
| Thr621 | H-acceptor | 2.26 | |||||
| Asn450 | H-acceptor | 2.26 | |||||
| Ser448 | Pi-H | 2.83 | |||||
| NA | Ser392 | Covalent bond | −4.528 | −7.344 | 1.003 | ||
| Thr621 | H-donor | 2.02 |
| Name | Comp. 4 | Comp. 5 | Comp. 6 | Comp. 7 | Comp. 8 | Comp. 9 | Comp. 10 | Comp. 11 |
|---|---|---|---|---|---|---|---|---|
| Formula | C10H13N5O3 | C15H21N5O7 | C16H23N5O8 | C15H17N5O3 | C13H13N5O5 | C11H11N5O3S | C13H14N6O4 | C14H15N5O4 |
| MWt (≤450 g/mol) | 251.24 | 383.36 | 413.38 | 315.33 | 319.27 | 293.3 | 318.29 | 317.3 |
| Heavy Atoms | 18 | 27 | 29 | 23 | 23 | 20 | 23 | 23 |
| Aromatic Heavy Atoms | 6 | 6 | 6 | 11 | 6 | 11 | 11 | 6 |
| Rotatable Bonds (Nrot < 10) | 8 | 13 | 14 | 8 | 8 | 7 | 8 | 8 |
| H-bond Acceptors (HBA ≤ 8) | 5 | 9 | 10 | 5 | 6 | 5 | 6 | 6 |
| H-Bond Donors (HBD ≤ 5) | 4 | 7 | 8 | 2 | 3 | 3 | 4 | 2 |
| The distribution coefficient (LogD 3.5) | 2.588 | 4.217 | 4.728 | 0.675 | 2.332 | 0.488 | 1.428 | 2.037 |
| the topological polar surface area (TPSA ≤ 140) | 126.2 | 193.47 | 213.7 | 105.98 | 137.57 | 145 | 152.23 | 120.83 |
| LogPo/w | 0.41 | 0.06 | 0.91 | 2.09 | 0.14 | 1.36 | 1.25 | 1.55 |
| log Kp (cm/s) | −9.2 | −11.29 | −11.92 | −8.09 | −9.53 | −8.34 | −8.54 | −9.11 |
| Lipinski Violations | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| Ghose Violations | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 |
| Bioavailability Score | 0.55 | 0.17 | 0.17 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| PAINS Alerts | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Brenk Alerts | 2 | 1 | 1 | 0 | 1 | 1 | 0 | 0 |
| Solubility (logS ≥ 0.010 mg/mL) | 3.16 | 1.26 | 5.73 | 1.42 | 1.21 | 1.34 | 6.37 | |
| Solubility Level | 5 | 5 | 5 | 4 | 4 | 4 | 4 | 4 |
| Unknown, AlogP98 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 1 |
| EXT_PPB | −8.478 | −13.247 | −15.348 | −5.4949 | −8.288 | −8.3825 | −9.958 | −7.986 |
| AlogP98 | −2.076 | −4.217 | −4.728 | −0.675 | −1.819 | −0.611 | −1.428 | −1.525 |
| BBB Level | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| undefined | undefined | undefined | undefined | undefined | undefined | undefined | undefined | |
| EXT Hepatotoxic | 0.427 | -1.967 | -2.013 | 2.865 | -0.2698 | 2.3875 | 3.387 | 0.8041 |
| Absorption Level | 2 | 3 | 3 | 0 | 2 | 0 | 3 | 1 |
| Low | Very low | Very low | very good | Low | very good | Very low | moderate | |
| CYP2D6 | false | false | false | false | false | false | false | False |
| Hepatotoxic | true | true | true | true | true | true | true | true |
| PPB | false | false | false | false | false | false | false | false |
| Name | Comp. 4 | Comp. 5 | Comp. 6 | Comp. 7 | Comp. 8 | Comp. 9 | Comp. 10 | Comp. 11 |
|---|---|---|---|---|---|---|---|---|
| Mouse Female NTP ** | 0.575 | 0.527 | 0.502 | 0.571 | 0.57 | 0.599 | 0.516 | 0.501 |
| Mouse Male NTP | 0.576 | 0.247 | 0.189 | 0.539 | 0.578 | 0.469 | 0.546 | 0.403 |
| Rat Female NTP | 0.478 | 0.381 | 0.391 | 0.462 | 0.446 | 0.482 | 0.417 | 0.318 |
| Rat Male NTP | 0.353 | 0.2 | 0.222 | 0.348 | 0.384 | 0.385 | 0.319 | 0.237 |
| Mouse Female FDA * None vs. Carcinogen | 0.209 | 0.206 | 0.205 | 0.205 | 0.21 | 0.218 | 0.205 | 0.207 |
| Mouse Male FDA None vs. Carcinogen | 0.294 | 0.198 | 0.187 | 0.334 | 0.259 | 0.281 | 0.275 | 0.296 |
| Rat Female FDA None vs. Carcinogen | 0.273 | 0.226 | 0.225 | 0.292 | 0.283 | 0.307 | 0.3 | 0.291 |
| Rat Male FDA None vs. Carcinogen | 0.294 | 0.299 | 0.324 | 0.366 | 0.313 | 0.328 | 0.367 | 0.324 |
| Mouse Male FDA Single vs. Multiple | 0.162 | - | - | 0.148 | - | 0.185 | 0.149 | 0.161 |
| Rat Female FDA Single vs. Multiple | 0.529 | - | - | 0.55 | 0.53 | 0.583 | 0.566 | 0.533 |
| WOE | 0.469 | 0.384 | 0.368 | 0.475 | 0.471 | 0.461 | 0.477 | 0.434 |
| Ames | 0.621 | 0.495 | 0.454 | 0.533 | 0.521 | 0.684 | 0.581 | 0.544 |
| DTP | 0.567 | 0.573 | 0.607 | 0.485 | 0.503 | 0.507 | 0.502 | 0.501 |
| Skin Irritancy None vs. Irritant | 0.972 | 0.973 | 0.973 | 0.973 | 0.972 | 0.974 | 0.962 | 0.974 |
| Skin Sensitization None vs. Sensitizer | 0.669 | 0.545 | 0.572 | 0.612 | 0.659 | 0.744 | 0.629 | 0.658 |
| Ocular Irritancy None vs. Irritant | 0.999975 | 0.999942 | 0.999942 | 0.999976 | 0.999967 | 0.999988 | 0.999975 | 0.999977 |
| Ocular Irritancy Mild vs. Moderate Severe | 0.836 | 0.891 | 0.891 | 0.838 | 0.866 | 0.834 | 0.834 | 0.83 |
| Ocular Irritancy Moderate vs. Severe | 0.663 | 0.662 | 0.665 | 0.69 | 0.645 | 0.668 | 0.613 | 0.636 |
| Aerobic Biodegradability | 0.472 | 0.648 | 0.653 | 0.548 | 0.55 | 0.439 | 0.318 | 0.563 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalaf, H.S.; Naglah, A.M.; Al-Omar, M.A.; Moustafa, G.O.; Awad, H.M.; Bakheit, A.H. Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide. Molecules 2020, 25, 3589. https://doi.org/10.3390/molecules25163589
Khalaf HS, Naglah AM, Al-Omar MA, Moustafa GO, Awad HM, Bakheit AH. Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide. Molecules. 2020; 25(16):3589. https://doi.org/10.3390/molecules25163589
Chicago/Turabian StyleKhalaf, Hemat S., Ahmed M. Naglah, Mohamed A. Al-Omar, Gaber O. Moustafa, Hassan M. Awad, and Ahmed H. Bakheit. 2020. "Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide" Molecules 25, no. 16: 3589. https://doi.org/10.3390/molecules25163589
APA StyleKhalaf, H. S., Naglah, A. M., Al-Omar, M. A., Moustafa, G. O., Awad, H. M., & Bakheit, A. H. (2020). Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide. Molecules, 25(16), 3589. https://doi.org/10.3390/molecules25163589