
Antibody-Drug Conjugates and Targeted Treatment Strategies for Hepatocellular Carcinoma: A Drug-Delivery Perspective


Abstract
1. Introduction
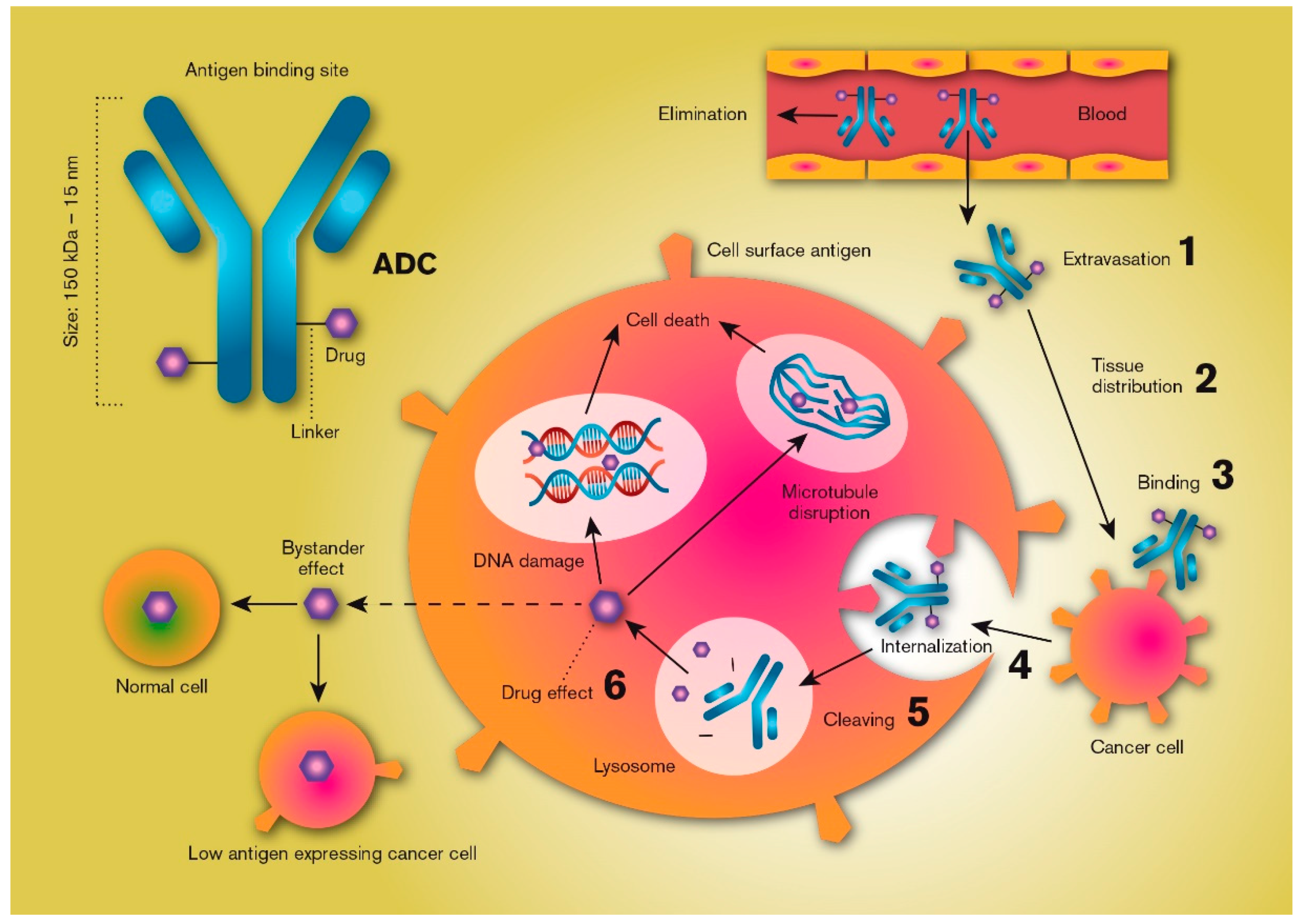
2. Theory
3. Vascular Endothelial Transport and Target Tissue Distribution
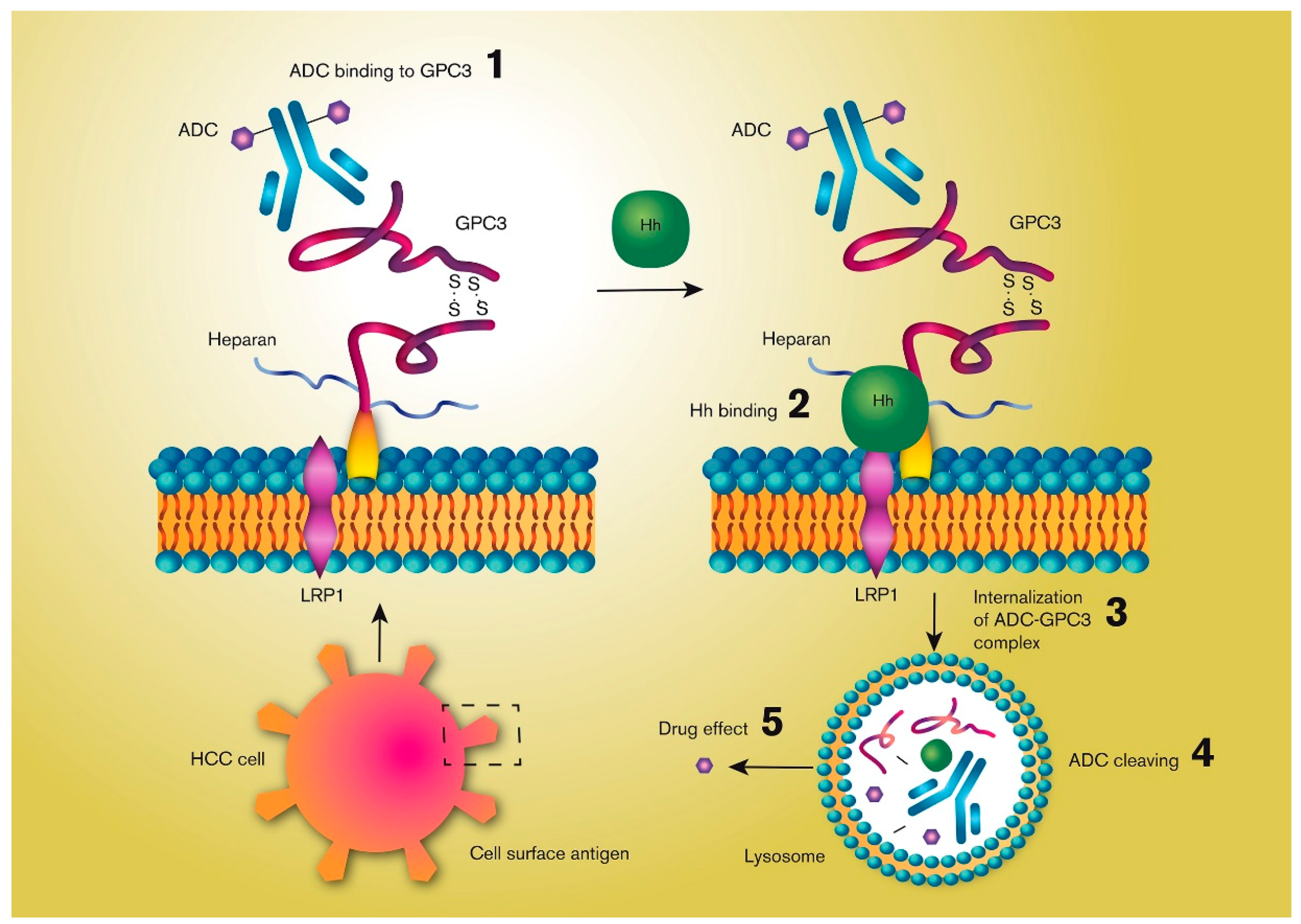
4. The Antigen
5. Internalization
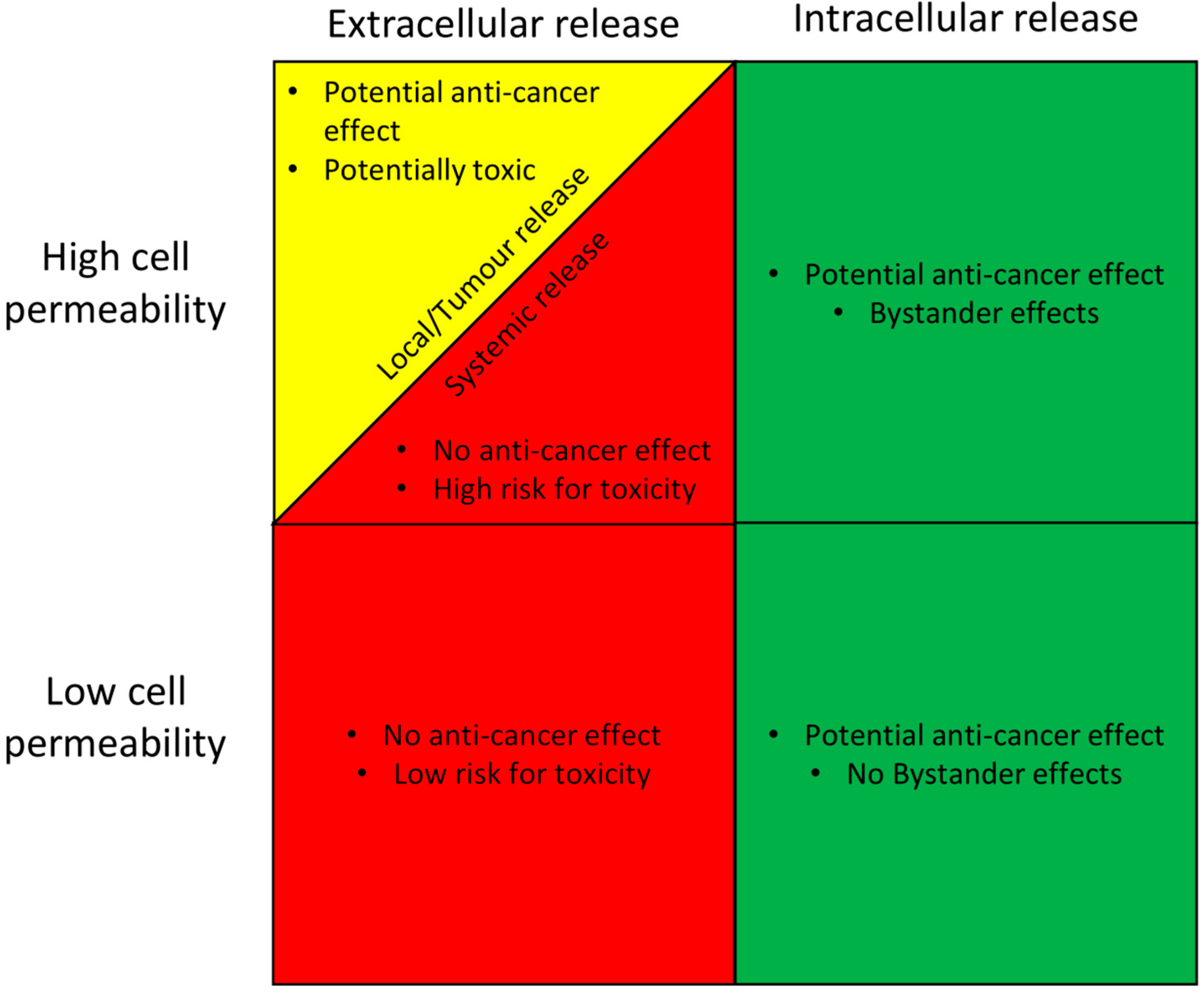
6. Drug Release
7. The Active Drug
8. Computational Molecular Docking
9. Hepatocellular Carcinoma
Approved and Experimental Pharmacological Treatment Strategies
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sau, S.; Alsaab, H.O.; Kashaw, S.K.; Tatiparti, K.; Iyer, A.K. Advances in antibody–drug conjugates: A new era of targeted cancer therapy. Drug Discov. Today 2017, 22, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Monoclonal antibodies in the clinic. Nat. Biotechnol. 2001, 19, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. New Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. New Engl. J. Med. 2019, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Yamashita-Kashima, Y.; Fujimura, T.; Shu, S.; Yanagisawa, M.; Mochizuki, M.; Yorozu, K.; Kouno, M.; Furugaki, K.; Higuchi, R.; Shoda, J.; et al. Preclinical study of antitumor activity of trastuzumab emtansine in HER2-positive biliary tract cancer. J. Clin. Oncol. 2018, 36, 256. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Krop, I.E.; Lorusso, P.; Miller, K.D.; Modi, S.; Yardley, D.; Rodriguez, G.; Guardino, E.; Lu, M.; Zheng, M.; Girish, S.; et al. A Phase II Study of Trastuzumab Emtansine in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer Who Were Previously Treated With Trastuzumab, Lapatinib, an Anthracycline, a Taxane, and Capecitabine. J. Clin. Oncol. 2012, 30, 3234–3241. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Govindan, S.V.; Cardillo, T.M.; Rossi, E.A.; Trisal, P.; McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M. Improving the Therapeutic Index in Cancer Therapy by Using Antibody–Drug Conjugates Designed with a Moderately Cytotoxic Drug. Mol. Pharm. 2014, 12, 1836–1847. [Google Scholar] [CrossRef]
- Boni, V.; Sharma, M.R.; Patnaik, A. The Resurgence of Antibody Drug Conjugates in Cancer Therapeutics: Novel Targets and Payloads. Am. Soc. Clin. Oncol. Educ. Book 2020. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Lamb, Y.N. Inotuzumab ozogamicin: First global approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef]
- Hanna, K. Enfortumab vedotin to treat urothelial carcinoma. Drugs Today 2020, 56, 329–335. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Sacituzumab govitecan, a novel, third-generation, antibody-drug conjugate (ADC) for cancer therapy. Expert Opin. Biol. Ther. 2020. [Google Scholar] [CrossRef]
- Sassoon, I.; Blanc, V. Antibody–Drug Conjugate (ADC) Clinical Pipeline: A Review; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of Linker Stability to the Activities of Anticancer Immunoconjugates. Bioconjugate Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef]
- Klein, J.S.; Gnanapragasam, P.N.P.; Galimidi, R.P.; Foglesong, C.P.; West, A.P.; Bjorkman, P.J. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc. Natl. Acad. Sci. USA 2009, 106, 7385–7390. [Google Scholar]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors. Microvasc. Res. 1991, 41, 5–23. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; A Berk, D.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.E.; Wu, A.M.; Yazaki, P.J.; Liu, A.; Raubitschek, A.A.; Shively, J.E.; Wong, J.Y.C. Numerical Selection of Optimal Tumor Imaging Agents with Application to Engineered Antibodies. Cancer Biotherapy Radiopharm. 2001, 16, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Maeda, H.; Bharate, G.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef]
- Debie, P.; Lafont, C.; Defrise, M.; Hansen, I.; Van Willigen, D.M.; Van Leeuwen, F.W.; Gijsbers, R.; D’Huyvetter, M.; Devoogdt, N.; Lahoutte, T.; et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J. Control. Release 2020, 317, 34–42. [Google Scholar] [CrossRef]
- Chauhan, V.; Boucher, Y.; Ferrone, C.R.; Roberge, S.; Martin, J.D.; Stylianopoulos, T.; Bardeesy, N.; Depinho, R.A.; Padera, T.P.; Munn, L.L.; et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 2014, 26, 14–15. [Google Scholar] [CrossRef]
- Chauhan, V.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Lu, Y.; Mahato, R.I. Pharmaceutical Perspectives of Cancer Therapeutics, 1st ed.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Gabizon, A. Pegylated liposomal doxorubicin: Metamorphosis of an old drug into a new form of chemotherapy. Cancer Investig. 2001, 19, 424–436. [Google Scholar] [CrossRef]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin. Cancer Res. 2001, 7, 243–254. [Google Scholar]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016. [Google Scholar] [CrossRef]
- Forster, J.; Harriss-Phillips, W.M.; Douglass, M.J.J.; Bezak, E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia 2017, 5, 21–32. [Google Scholar] [CrossRef]
- Stapleton, S.; Milosevic, M.; Tannock, I.F.; Allen, C.; A Jaffray, D. The intra-tumoral relationship between microcirculation, interstitial fluid pressure and liposome accumulation. J. Control. Release 2015, 211, 163–170. [Google Scholar] [CrossRef]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; Macmillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Oh, P.; E Testa, J.; Borgström, P.; Witkiewicz, H.; Li, Y.; E Schnitzer, J. In vivo proteomic imaging analysis of caveolae reveals pumping system to penetrate solid tumors. Nat. Med. 2014, 20, 1062–1068. [Google Scholar] [CrossRef]
- Kim, S.M.; Faix, P.H.; E Schnitzer, J. Overcoming key biological barriers to cancer drug delivery and efficacy. J. Control. Release 2017, 267, 15–30. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2010. mAbs 2010, 2, 84–100. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Lapusan, S.; Vidriales, M.-B.; Thomas, X.; De Botton, S.; Vekhoff, A.; Tang, R.; Dumontet, C.; Morariu-Zamfir, R.; Lambert, J.M.; Ozoux, M.-L.; et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Investig. New Drugs 2011, 30, 1121–1131. [Google Scholar] [CrossRef]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef]
- Slamon, D.J.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martín, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in HER2-positive breast cancer. New Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef]
- Lambert, J.M.; Berkenblit, A. Antibody–Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; Van Osdol, W.; Weinstein, J.N. Micropharmacology of monoclonal antibodies in solid tumors: Direct experimental evidence for a binding site barrier. Cancer Res. 1992, 52, 5144–5153. [Google Scholar]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015. [Google Scholar] [CrossRef]
- Austin, C.D.; De Mazière, A.M.; Pisacane, P.I.; Van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and Sorting of ErbB2 and the Site of Action of Cancer Therapeutics Trastuzumab and Geldanamycin. Mol. Boil. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef]
- Kyriakos, R.J.; Shih, L.B.; Ong, G.L.; Patel, K.; Goldenberg, D.M.; Mattes, M.J. The fate of antibodies bound to the surface of tumor cells in vitro. Cancer Res. 1992, 52, 835–842. [Google Scholar]
- Xu, S. Internalization, Trafficking, Intracellular Processing and Actions of Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3577–3583. [Google Scholar] [CrossRef]
- Zuckier, L.S.; Berkowitz, E.Z.; Sattenberg, R.J.; Zhao, Q.H.; Deng, H.F.; Scharff, M.D. Influence of affinity and antigen density on antibody localization in a modifiable tumor targeting model. Cancer Res. 2000, 60, 7008–7013. [Google Scholar] [PubMed]
- Ackerman, M.E.; Pawlowski, D.; Wittrup, K.D. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Mol. Cancer Ther. 2008, 7, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of Affinity and Antigen Internalization on the Uptake and Penetration of Anti-HER2 Antibodies in Solid Tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef]
- Wang, W.; Singh, M. Biological drug products: Development and strategies, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Maass, K.; Kulkarni, C.; Betts, A.M.; Wittrup, K.D. Determination of Cellular Processing Rates for a Trastuzumab-Maytansinoid Antibody-Drug Conjugate (ADC) Highlights Key Parameters for ADC Design. AAPS J. 2016, 18, 635–646. [Google Scholar] [CrossRef]
- Walter, R.B.; Raden, B.W.; Kamikura, D.M.; A Cooper, J.; Bernstein, I.D. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin–induced cytotoxicity. Blood 2005, 105, 1295–1302. [Google Scholar] [CrossRef]
- Capurro, M.I.; Shi, W.; Filmus, J. LRP1 mediates Hedgehog-induced endocytosis of the GPC3–Hedgehog complex. J. Cell Sci. 2012, 125, 3380–3389. [Google Scholar] [CrossRef]
- Berger, C.; Madshus, I.H.; Stang, E. Cetuximab in combination with anti-human IgG antibodies efficiently down-regulates the EGF receptor by macropinocytosis. Exp. Cell Res. 2012, 318, 2578–2591. [Google Scholar] [CrossRef]
- Van Bueren, J.J.L.; Bleeker, W.K.; Bøgh, H.O.; Houtkamp, M.; Schuurman, J.; Van De Winkel, J.; Parren, P.W. Effect of Target Dynamics on Pharmacokinetics of a Novel Therapeutic Antibody against the Epidermal Growth Factor Receptor: Implications for the Mechanisms of Action. Cancer Res. 2006, 66, 7630–7638. [Google Scholar] [CrossRef]
- Thurber, G.M.; Zajic, S.C.; Wittrup, K.D. Theoretic Criteria for Antibody Penetration into Solid Tumors and Micrometastases. J. Nucl. Med. 2007, 48, 995–999. [Google Scholar] [CrossRef]
- Liu, G.; He, J.; Dou, S.; Gupta, S.; Rusckowski, M.; Hnatowich, D.J. Further investigations of morpholino pretargeting in mice—establishing quantitative relations in tumor. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 1115–1123. [Google Scholar] [CrossRef][Green Version]
- Tian, F.; Jackson, D.; Bai, Y. Site-Specific Antibody-Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Springer: Berlin/Heidelberg, Germany; pp. 241–265. [CrossRef]
- Feld, J.; Barta, S.; Schinke, C.; Braunschweig, I.; Zhou, Y.; Verma, A. Linked-In: Design and Efficacy of Antibody Drug Conjugates in Oncology. Oncotarget 2013, 4, 397–412. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; Wong, W.L.T.; et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody-Cytotoxic Drug Conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blättler, W.A.; Goldmacher, V.S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; De Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-Drug Conjugates for the Treatment of Non-Hodgkin’s Lymphoma: Target and Linker-Drug Selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Noninternalizing Targeted Cytotoxics for Cancer Therapy. Mol. Pharm. 2015, 12, 1880–1884. [Google Scholar] [CrossRef]
- Corso, A.D.; Gébleux, R.; Murer, P.; Soltermann, A.; Neri, D. A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo. J. Control. Release 2017, 264, 211–218. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Ziffels, B.; Widmayer, F.; Murer, P.; Pellegrini, G.; Pretto, F.; Wulhfard, S.; Neri, D. Enhanced Therapeutic Activity of Non-Internalizing Small-Molecule-Drug Conjugates Targeting Carbonic Anhydrase IX in Combination with Targeted Interleukin-2. Clin. Cancer Res. 2018, 24, 3656–3667. [Google Scholar] [CrossRef]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J.L.; Supuran, C.T.; Neri, D. A Small-Molecule Drug Conjugate for the Treatment of Carbonic Anhydrase IX Expressing Tumors. Angew. Chem. Int. Ed. 2014, 53, 4231–4235. [Google Scholar] [CrossRef]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody-Drug Conjugates for the Treatment of Cancer. Chem. Boil. Drug Des. 2012, 81, 113–121. [Google Scholar] [CrossRef]
- Varbanov, H.P.; Kuttler, F.; Banfi, D.; Turcatti, G.; Dyson, P.J. Repositioning approved drugs for the treatment of problematic cancers using a screening approach. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Dubbelboer, I.R.; Pavlovic, N.; Heindryckx, F.; Sjögren, E.; Lennernäs, H. Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile. Cancers 2019, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Uddin, Z.; Li, X.; Jasti, B. In Vitro Cytotoxicity and In Vivo Anti-Tumor Efficacy of CD13 Targeted Peptide-Monomethyl Auristatin E (MMAE) Conjugates. Curr. Trends Biotechnol. Pharm. 2019, 13, 146–155. [Google Scholar]
- Wang, Y.; Benz, F.W.; Wu, Y.; Wang, Q.; Chen, Y.; Chen, X.; Li, H.; Zhang, Y.; Zhang, R.; Yang, J. Structural Insights into the Pharmacophore of Vinca Domain Inhibitors of Microtubules. Mol. Pharmacol. 2015, 89, 233–242. [Google Scholar] [CrossRef]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor Delivery and In Vivo Processing of Disulfide-Linked and Thioether-Linked Antibody−Maytansinoid Conjugates. Bioconjugate Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Benet, L.Z.; Amidon, G.L.; Barends, D.M.; Lennernäs, H.; Polli, J.E.; Shah, V.P.; Stavchansky, S.A.; Yu, L.X. The Use of BDDCS in Classifying the Permeability of Marketed Drugs. Pharm. Res. 2008, 25, 483–488. [Google Scholar] [CrossRef]
- Melo, R.; Lemos, A.; Preto, A.; Almeida, J.G.; Correia, J.D.; Sensoy, O.; Moreira, I.S. Computational Approaches in Antibody-drug Conjugate Optimization for Targeted Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 1091–1109. [Google Scholar] [CrossRef]
- A Norman, R.; Ambrosetti, F.; Bonvin, A.M.J.J.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational approaches to therapeutic antibody design: Established methods and emerging trends. Briefings Bioinform. 2019. [Google Scholar] [CrossRef]
- Carr, B.I. Hepatocellular Carcinoma: Diagnosis and Treatment, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- McGlynn, K.A.; Tsao, L.; Hsing, A.W.; Devesa, S.S.; Fraumeni, J.F. International trends and patterns of primary liver cancer. Int. J. Cancer 2001, 94, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, N.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzmán, N.R.; Amoako, Y.A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; European Organisation for Research and Treatment of Cancer EASL–EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [CrossRef]
- Lencioni, R.; De Baere, T.; Soulen, M.; Rilling, W.S.; Geschwind, J.H. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: A systematic review of efficacy and safety data. Hepatol. 2016, 64, 106–116. [Google Scholar] [CrossRef]
- Marisi, G.; Cucchetti, A.; Ulivi, P.; Canale, M.; Cabibbo, G.; Solaini, L.; Foschi, F.G.; De Matteis, S.; Ercolani, G.; Valgiusti, M.; et al. Ten years of sorafenib in hepatocellular carcinoma: Are there any predictive and/or prognostic markers? World J. Gastroenterol. 2018, 24, 4152–4163. [Google Scholar] [CrossRef]
- Xiong, Y.-Q.; Sun, H.-C.; Zhang, W.; Zhu, X.-D.; Zhuang, P.-Y.; Zhang, J.-B.; Wang, L.; Wu, W.-Z.; Qin, L.; Tang, Z.-Y. Human Hepatocellular Carcinoma Tumor-derived Endothelial Cells Manifest Increased Angiogenesis Capability and Drug Resistance Compared with Normal Endothelial Cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. European Association for the Study of the Liver EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Capurro, M.; Wanless, I.R.; Sherman, M.; DeBoer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Gao, W.; Tang, Z.; Zhang, Y.-F.; Feng, M.; Qian, M.; Dimitrov, D.S.; Ho, M. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat. Commun. 2015. [Google Scholar] [CrossRef]
- Feng, M.; Gao, W.; Wang, R.; Chen, W.; Man, Y.-G.; Figg, W.D.; Wang, X.W.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, E1083–E1091. [Google Scholar]
- Guo, M.; Zhang, H.; Zheng, J.; Liu, Y. Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer 2020, 11, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Ho, M. Humanization of high-affinity antibodies targeting glypican-3 in hepatocellular carcinoma. Sci. Rep. 2016, 6, 33878. [Google Scholar] [CrossRef] [PubMed]
- Phung, Y.; Gao, W.; Man, Y.-G.; Nagata, S.; Ho, M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs 2012, 4, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Urban, D.J.; Nani, R.R.; Zhang, Y.-F.; Li, N.; Fu, H.; Shah, H.; Gorka, A.P.; Guha, R.; Chen, L.; et al. Glypican-3-Specific Antibody Drug Conjugates Targeting Hepatocellular Carcinoma. Hepatol. 2019, 70, 563–576. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific In Vitro Anticancer Activity. Bioconjugate Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Ohsawa, T.; Higashi, T.; Tsuji, T. The secretion of high molecular weight cathepsin B from cultured human liver cancers. Acta Med. Okayama 1989, 43, 9–15. [Google Scholar]
- Huang, L.R.; Hsu, H.C. Cloning and expression of CD24 gene in human hepatocellular carcinoma: A potential early tumor marker gene correlates with p53 mutation and tumor differentiation. Cancer Res. 1995, 55, 4717–4721. [Google Scholar]
- Ma, Z.; He, H.; Sun, F.; Xu, Y.; Huang, X.; Ma, Y.; Zhao, H.; Wang, Y.; Wang, M.; Zhang, J. Selective targeted delivery of doxorubicin via conjugating to anti-CD24 antibody results in enhanced antitumor potency for hepatocellular carcinoma both in vitro and in vivo. J. Cancer Res. Clin. Oncol. 2017, 143, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, T.; Jiang, J.; Wang, Y.; Ma, Z.; Li, Z.; Han, Y.; Pan, M.; Cai, J.; Wang, M.; et al. Engineering a high-affinity humanized anti-CD24 antibody to target hepatocellular carcinoma by a novel CDR grafting design. Oncotarget 2017, 8, 51238–51252. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ADC | Antigen Target | Drug | Drug Mechanism | Indication |
|---|---|---|---|---|
| Brentuximab vedotin [11] | CD30 | Auristatin | Microtubule disruptor | Hodgkin lymphoma |
| Gemtuzumab ozogamicin [12] | CD33 | Calicheamicin | DNA damage | Acute myeloid leukemia |
| Inotuzumab ozogamicin [13] | CD22 | Calicheamicin | DNA damage | Acute lymphoblastic leukemia |
| Moxetumomab pasudotox [14] | CD22 | PE38 | Apoptosis induction | Hairy cell leukemia |
| Polatuzumab vedotin [15] | CD79b | Auristatin | Microtubule disruptor | B-cell lymphoma |
| Enfortumab vedotin [16] | Nectin-4 | Auristatin | Microtubule disruptor | Bladder Cancer |
| Trastuzumab deruxtecan [4] | HER2 | Deruxtecan | Topoisomerase I inhibitor | HER2 positive breast cancer |
| Trastuzumab emtansine [3] | HER2 | Maytansine | Microtubule disruptor | HER2 positive breast cancer |
| Sacituzumab govitecan [17] | Trop-2 | SN38 | Topoisomerase inhibitor | Triple-negative breast cancer |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlgren, D.; Lennernäs, H. Antibody-Drug Conjugates and Targeted Treatment Strategies for Hepatocellular Carcinoma: A Drug-Delivery Perspective. Molecules 2020, 25, 2861. https://doi.org/10.3390/molecules25122861
Dahlgren D, Lennernäs H. Antibody-Drug Conjugates and Targeted Treatment Strategies for Hepatocellular Carcinoma: A Drug-Delivery Perspective. Molecules. 2020; 25(12):2861. https://doi.org/10.3390/molecules25122861
Chicago/Turabian StyleDahlgren, David, and Hans Lennernäs. 2020. "Antibody-Drug Conjugates and Targeted Treatment Strategies for Hepatocellular Carcinoma: A Drug-Delivery Perspective" Molecules 25, no. 12: 2861. https://doi.org/10.3390/molecules25122861
APA StyleDahlgren, D., & Lennernäs, H. (2020). Antibody-Drug Conjugates and Targeted Treatment Strategies for Hepatocellular Carcinoma: A Drug-Delivery Perspective. Molecules, 25(12), 2861. https://doi.org/10.3390/molecules25122861