A Critical View of the Application of the APEX Software (Aqueous Photochemistry of Environmentally-Occurring Xenobiotics) to Predict Photoreaction Kinetics in Surface Freshwaters


Abstract

1. Introduction
- The main manuscript provides a general introduction to the software, its capabilities, and the way to use it. It does not replace the user’s guide, but it gives an initial taste of how the software works. Attention is also given to the experimental procedures that can be used to obtain the input data. These procedures are described in detail, as are the tips and tricks that enable modeling to be extended to stratified lakes and to rivers.
- Readme.pdf is the user’s guide, which explains how to install the software (which is provided in the .zip file of the Supplementary Materials), lists the equations of the photochemical model, and provides all the needed details on how to use APEX.
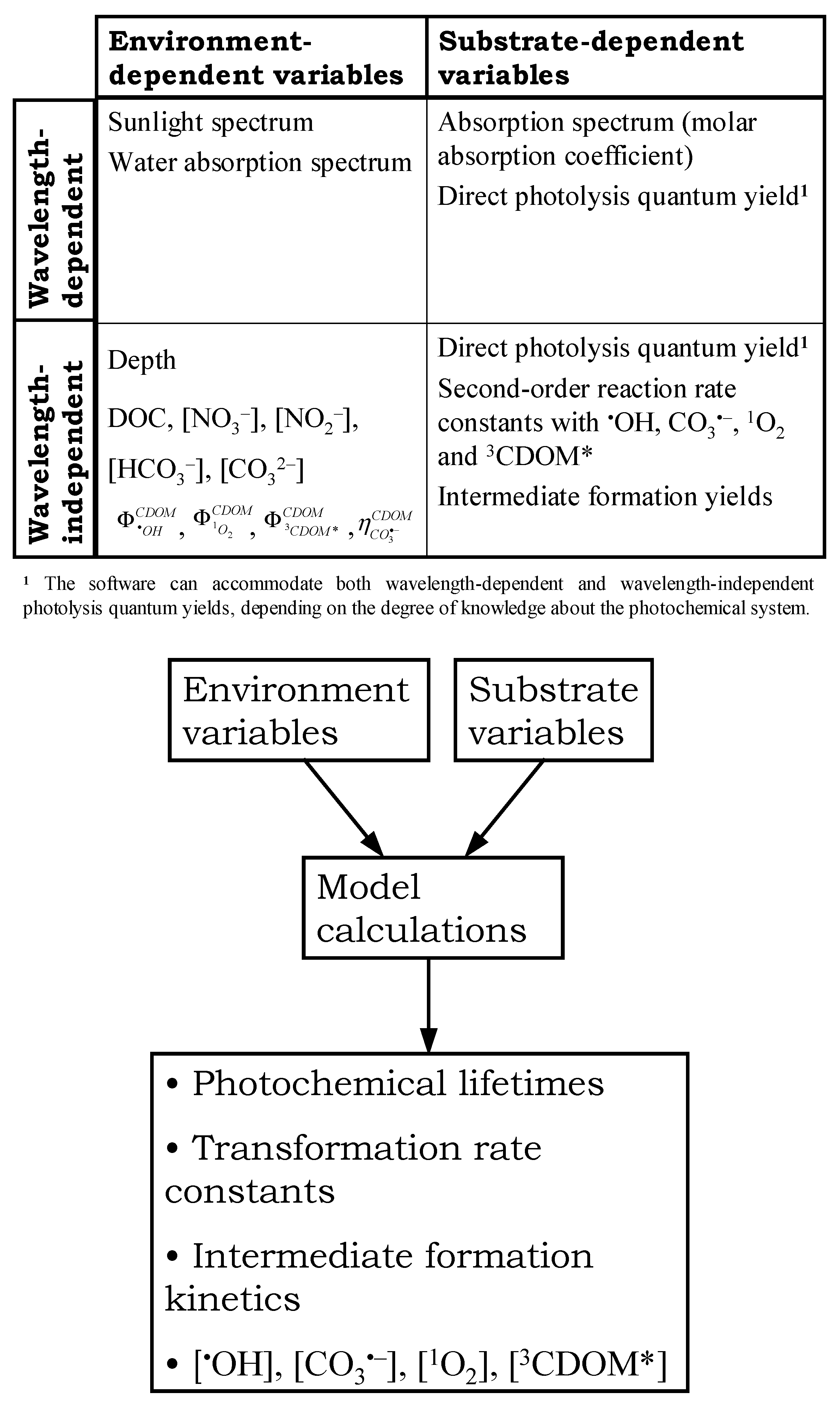
2. What APEX Is, What APEX Does
3. Input Data
3.1. Environment-Dependent Variables
3.1.1. Savetable/Plotgraph
3.1.2. Molecule.csv
3.2. Substrate-Related Variables
3.2.1. Savetable/Plotgraph
- Insert the known wavelength trend in molecule.csv (column phi); in this case, the software will ignore the value of fi_P provided in Savetable/Plotgraph;
- Insert the known constant value of fi_P (suppose it is 0.33, as for ibuprofen) in Savetable/Plotgraph; to make the software read this value with priority, the phi column in molecule.csv should be made equal to −1; note that the same result can be obtained by inserting the above constant value (0.33) in the whole phi column of molecule.csv, which will be read with priority;
- Define the quantum yield as a variable (X, “fi_P = −1”, or Y, “fi_P = −2”) within Savetable/Plotgraph. In this case, it is mandatory to have −1 in the whole phi column of molecule.csv.
3.2.2. Molecule.csv
3.2.3. Final Considerations Concerning the Input Data
4. Experimental Measurement of Photoreaction Parameters
4.1. Direct Photolysis Quantum Yield
4.2. Measurement of Second-Order Reaction Rate Constants without Using Reference Compounds
4.2.1. Reaction with •OH
4.2.2. Reaction with CO3•−
- Irradiation of the substrate (e.g., at 20 μmol L−1) in the presence of nitrate (e.g., 10−2 mol·L−1) and different HCO3− values (e.g., 0 to 10−2 mol·L−1).
- Irradiation of the substrate with nitrate at the same concentrations as before, but with a phosphate buffer (H2PO4− + HPO42−) instead of bicarbonate. The phosphate buffer should have the same total concentration as the bicarbonate salt, and the same pH within 0.2–0.3 units.
- Irradiation of the substrate at the usual concentration without nitrate, but with the same bicarbonate concentrations as in series (1) (direct photolysis check).
4.2.3. Reaction with 1O2
4.2.4. Reaction with 3CDOM*

4.3. Measurement of Second-Order Reaction Rate Constants by Using Reference Compounds (Competition Kinetics)
4.4. QSAR Approaches to Determining the Second-Order Reaction Rate Constants
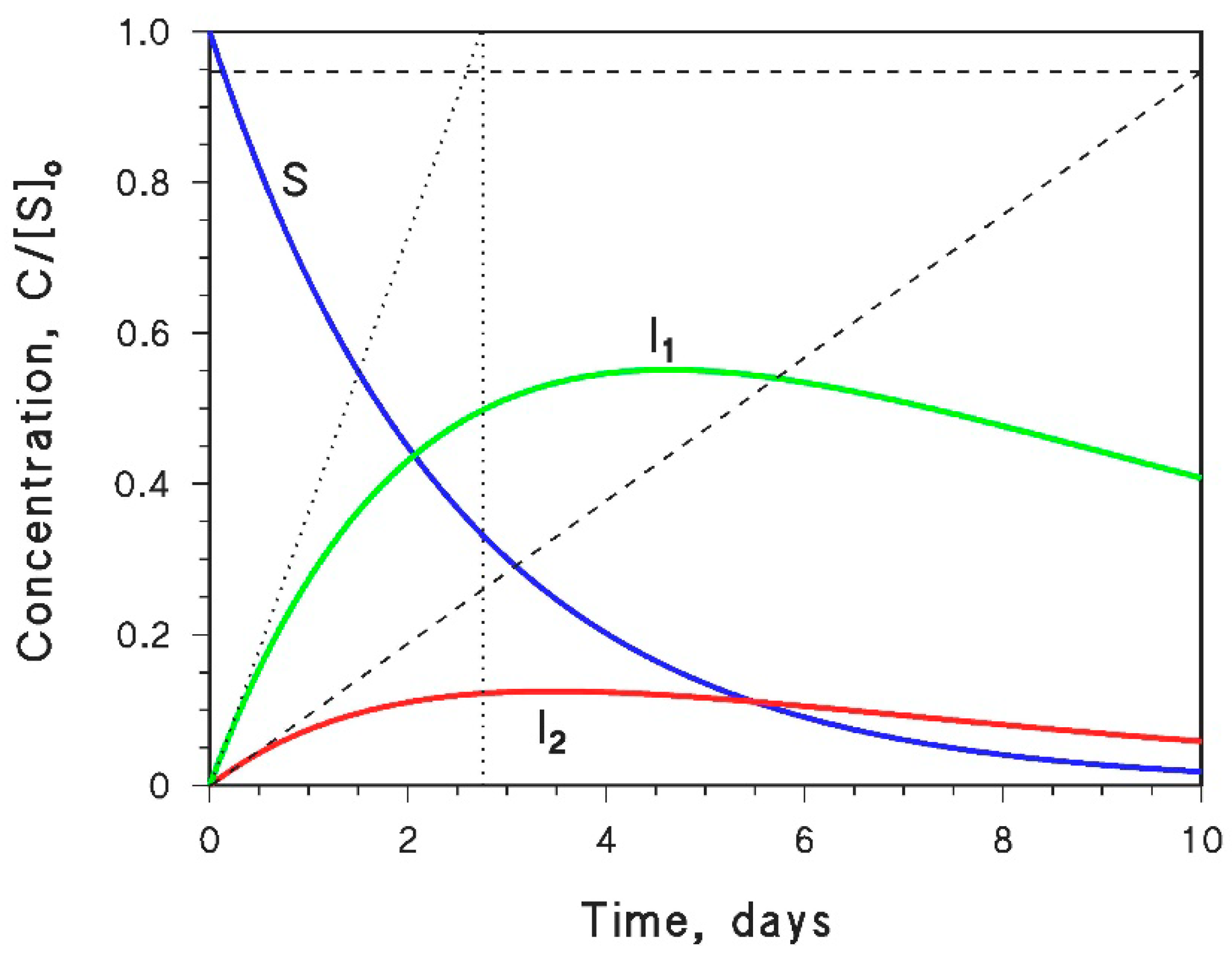
4.5. Measurement of Intermediate Formation Yields

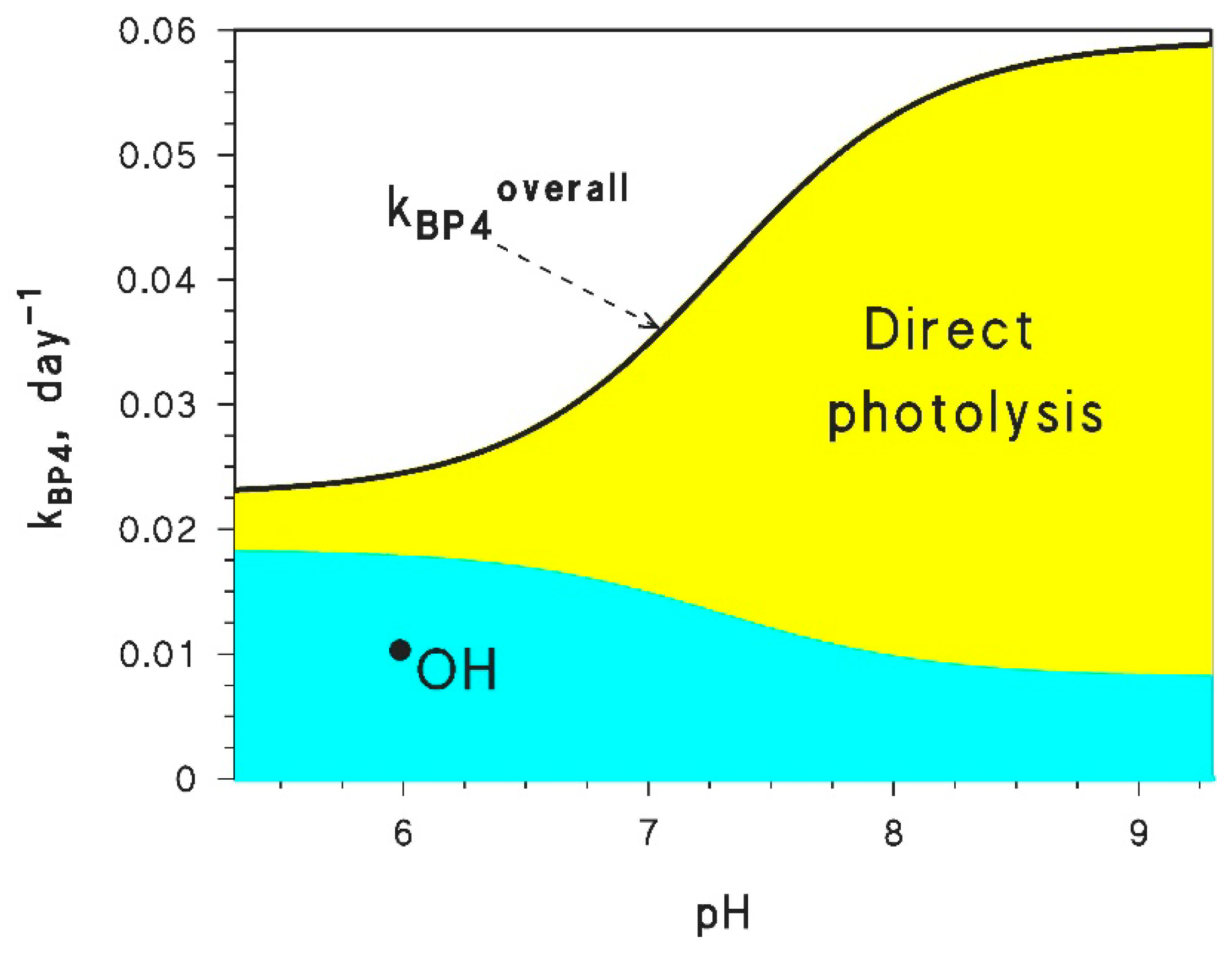
5. How to Quantify the pH Effect
6. Model Treatment of Non-Thoroughly Mixed or Flowing Systems
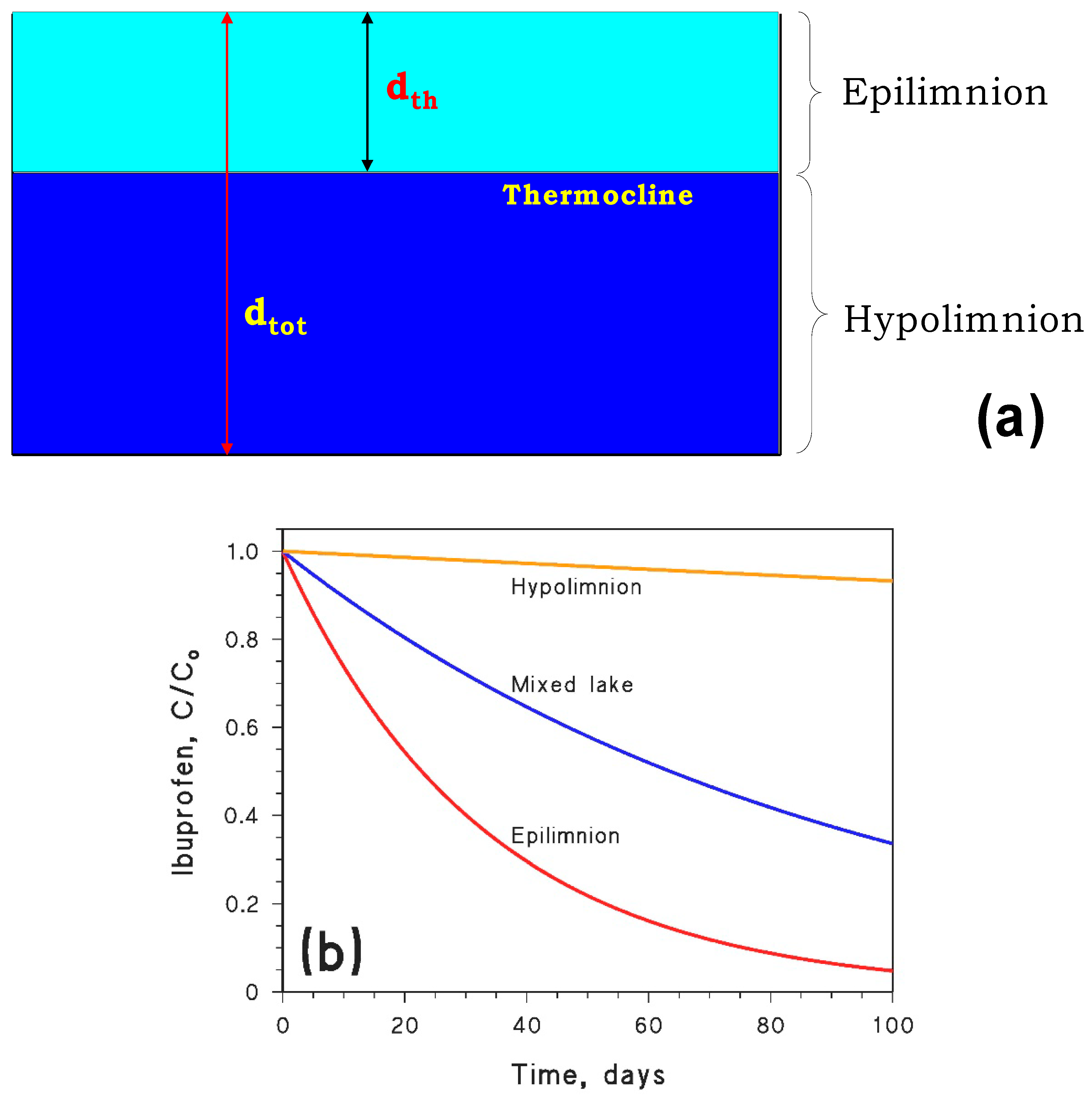
6.1. Stratified Lakes
6.2. Rivers
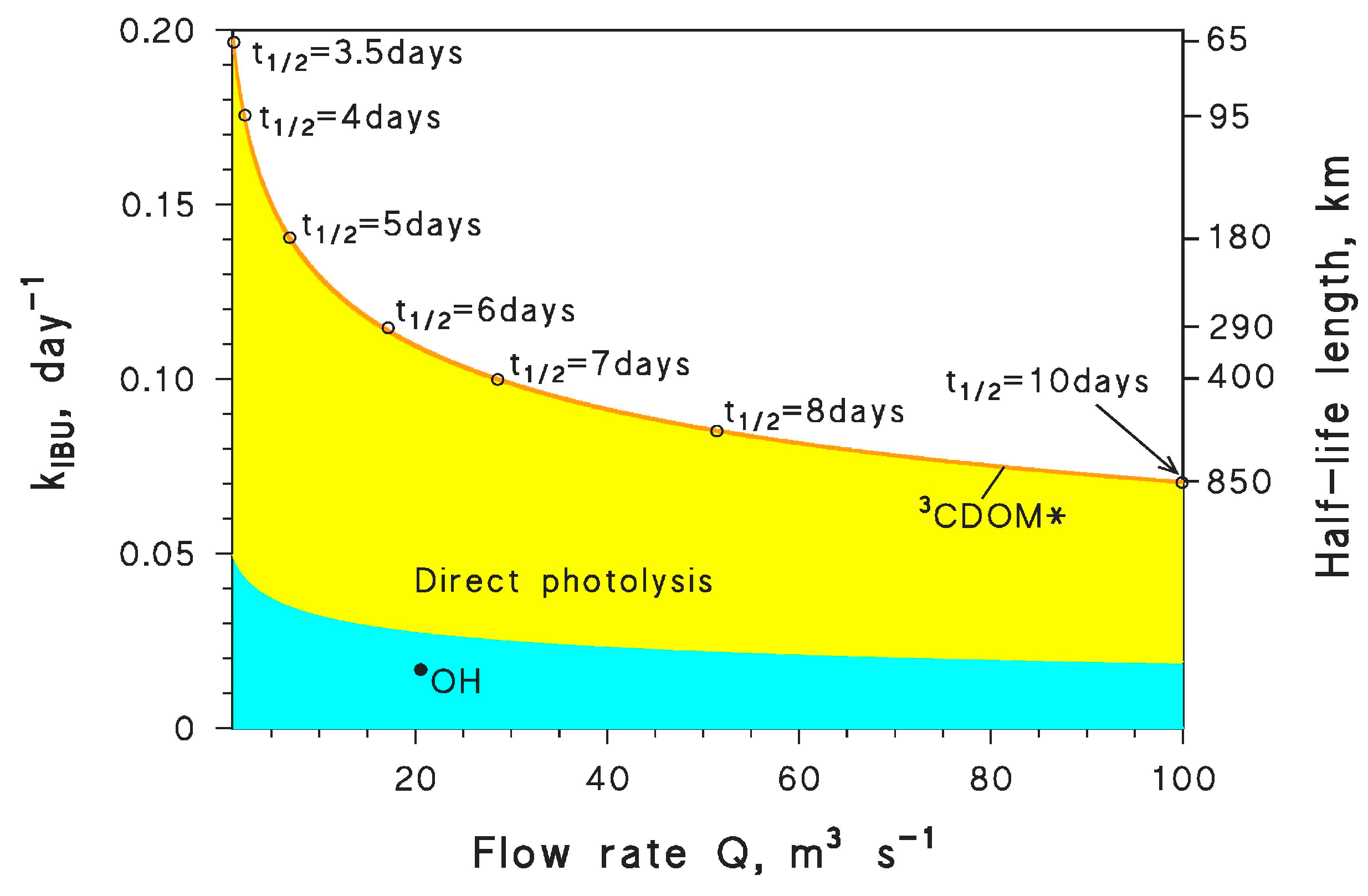
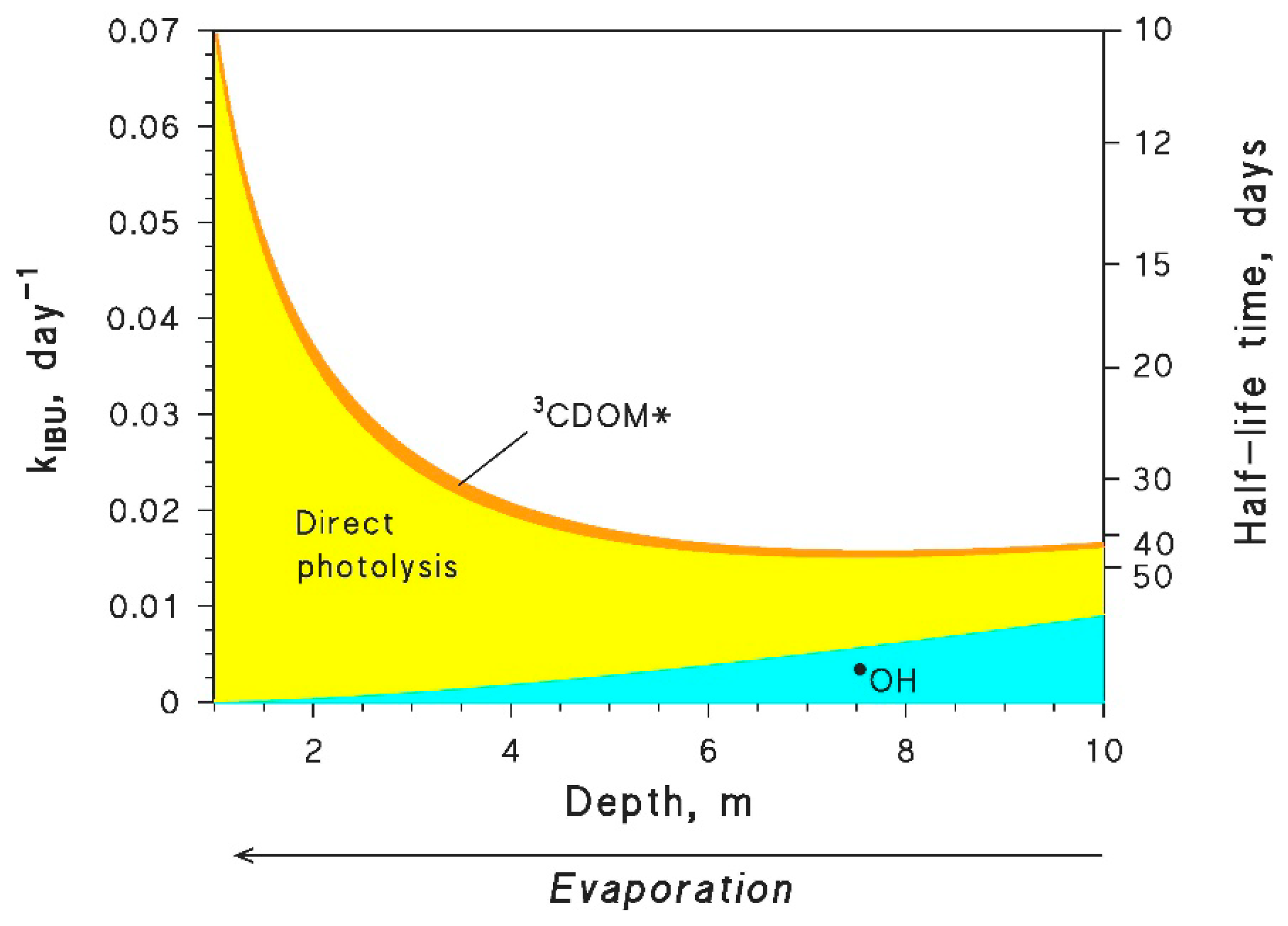
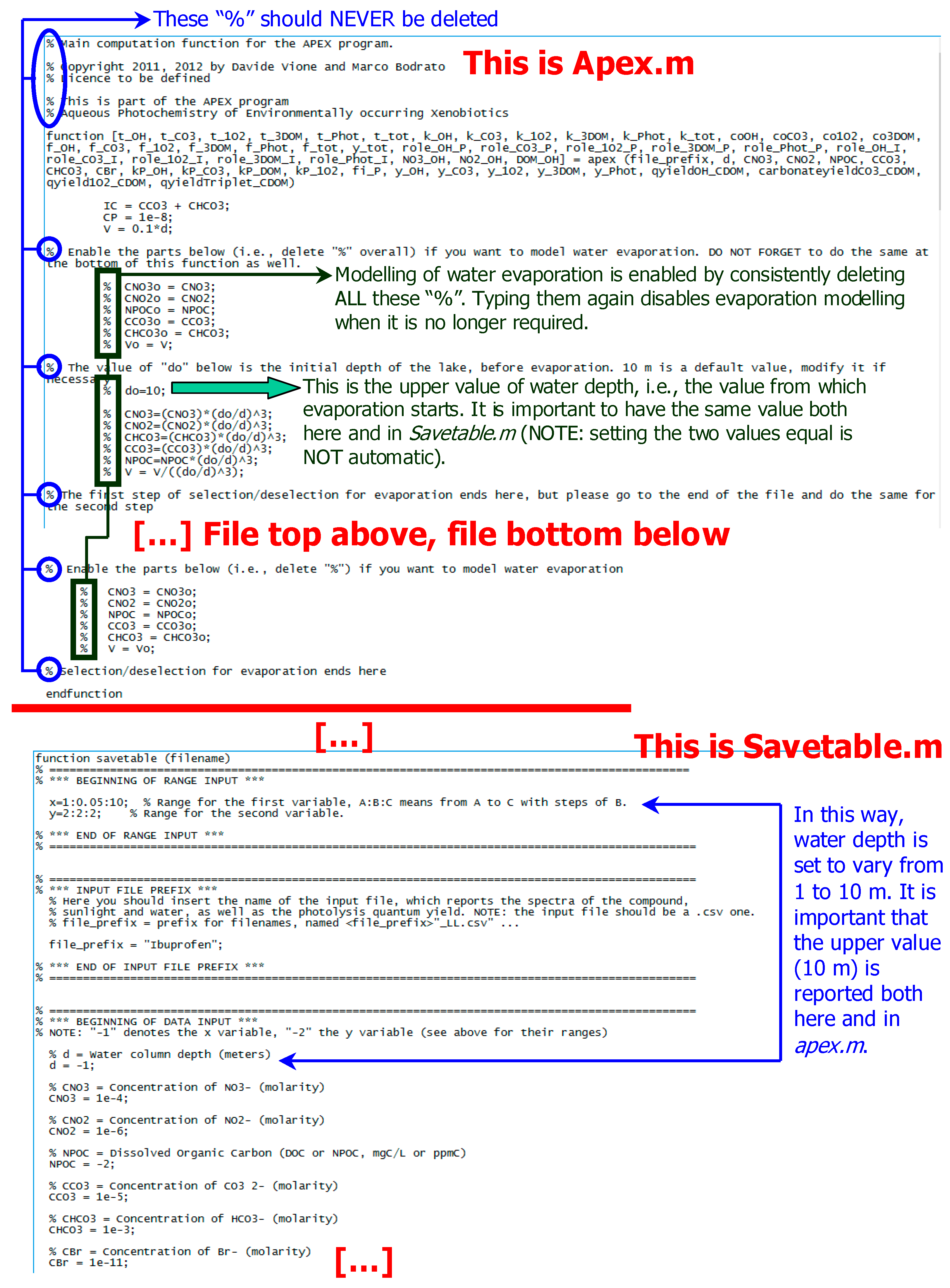
6.3. Aquatic Systems under Evaporative Concentration
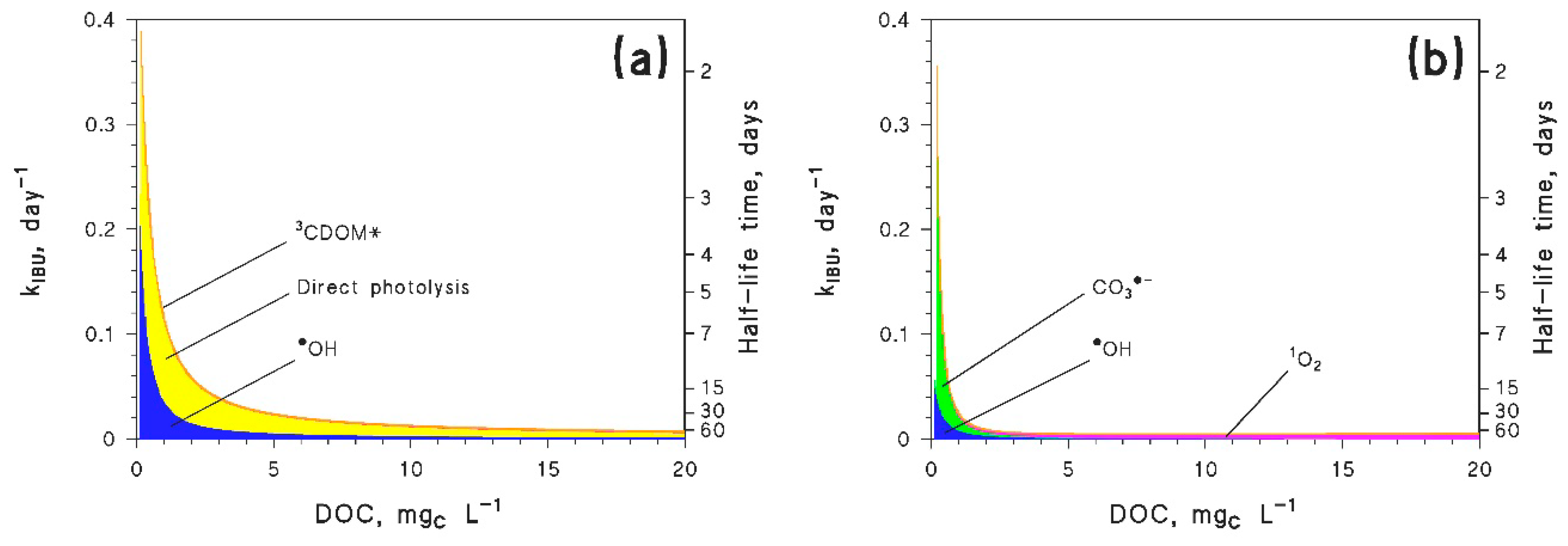
7. Ibuprofen as a Case Study
8. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Fenner, K.; Canonica, S.; Wackett, L.P.; Elsner, M. Evaluating pesticide degradation in the environment: Blind spots and emerging opportunities. Science 2013, 341, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.D.; Ternes, T.A. Water analysis: Emerging contaminants and current issues. Anal. Chem. 2014, 86, 2813–2848. [Google Scholar] [CrossRef] [PubMed]
- Mc Neill, K.; Canonica, S. Triplet state dissolved organic matter in aquatic photochemistry: Reaction mechanisms, substrate scope, and photophysical properties. Environ. Sci.-Process Impacts 2016, 18, 1381–1399. [Google Scholar] [CrossRef] [PubMed]
- Rosario-Ortiz, F.L.; Canonica, S. Probe compounds to assess the photochemical activity of dissolved organic matter. Environ. Sci. Technol. 2016, 50, 12532–12547. [Google Scholar] [CrossRef]
- Canonica, S.; Kohn, T.; Mac, M.; Real, F.J.; Wirz, J.; Von Gunten, U. Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds. Environ. Sci. Technol. 2005, 39, 9182–9188. [Google Scholar] [CrossRef]
- Canonica, S.; Freiburghaus, M. Electron-rich phenols for probing the photochemical reactivity of freshwaters. Environ. Sci. Technol. 2001, 35, 690–695. [Google Scholar] [CrossRef]
- Vione, D.; Minella, M.; Maurino, V.; Minero, C. Indirect photochemistry in sunlit surface waters: Photoinduced production of reactive transient species. Chem.-Eur. J. 2014, 20, 10590–10606. [Google Scholar] [CrossRef]
- Gligorovski, S.; Strekowski, R.; Barbati, S.; Vione, D. Environmental implications of hydroxyl radicals (•OH). Chem. Rev. 2015, 115, 13051–13092. [Google Scholar] [CrossRef]
- Koehler, B.; Barsotti, F.; Minella, M.; Landelius, T.; Minero, C.; Tranvik, L.J.; Vione, D. Simulation of photoreactive transients and of photochemical transformation of organic pollutants in sunlit boreal lakes across 14 degrees of latitude: A photochemical mapping of Sweden. Water Res. 2018, 129, 94–104. [Google Scholar] [CrossRef]
- Trawinski, J.; Skibinski, R. Studies on photodegradation process of psychotropic drugs: A review. Environ. Sci. Pollut. Res. 2017, 24, 1152–1199. [Google Scholar] [CrossRef]
- Calza, P.; Noé, G.; Fabbri, D.; Santoro, V.; Minero, C.; Vione, D.; Medana, C. Photoinduced transformation of pyridinium-based ionic liquids, and implications for their photochemical behavior in surface waters. Water Res. 2017, 122, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Zepp, R.G.; Cline, D.M. Rates of direct photolysis in aquatic environment. Environ. Sci. Technol. 1977, 11, 359–366. [Google Scholar] [CrossRef]
- Williamson, C.E.; Zepp, R.G.; Lucas, R.M.; Madronich, S.; Austin, A.T.; Ballaré, C.L.; Norval, M.; Sulzberger, B.; Bais, A.F.; McKenzie, R.L.; et al. Solar ultraviolet radiation in a changing climate. Nature Clim. Change 2014, 4, 434–441. [Google Scholar] [CrossRef]
- Nelson, K.L.; Boehm, A.B.; Davies-Colley, R.J.; Dodd, M.C.; Kohn, T.; Linden, K.G.; Liu, Y.Y.; Maraccini, P.A.; McNeill, K.; Mitch, W.A.; et al. Sunlight-mediated inactivation of health-relevant microorganisms in water: A review of mechanisms and modeling approaches. Environ. Sci.: Processes Impacts 2018, 20, 1089–1122. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Z.; Chen, J.W.; Xie, H.J.; Zhang, Y.N.; Li, Y.J.; Wang, Y.; Xie, Q.; Zhang, S.Y. Modeling photodegradation kinetics of organic micropollutants in water bodies: A case of the Yellow River estuary. J. Haz. Mat. 2018, 349, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Bintou, A.T.; Bianco, A.; Mailhot, G.; Brigante, M. A new insight into ethoxyquin fate in surface waters: Stability, direct and indirect photochemical behaviour and the identification of main products. J. Photochem. Photobiol. A: Chem. 2015, 311, 118–126. [Google Scholar] [CrossRef]
- Bodrato, M.; Vione, D. APEX (Aqueous Photochemistry of Environmentally occurring Xenobiotics): A free software tool to predict the kinetics of photochemical processes in surface waters. Environ. Sci.-Process Impacts 2014, 16, 732–740. [Google Scholar] [CrossRef]
- De Laurentiis, E.; Chiron, S.; Kouras-Hadef, S.; Richard, C.; Minella, M.; Maurino, V.; Minero, C.; Vione, D. Photochemical fate of carbamazepine in surface freshwaters: Laboratory measures and modeling. Environ. Sci. Technol. 2012, 46, 8164–8173. [Google Scholar] [CrossRef]
- Vione, D.; Maddigapu, P.R.; De Laurentiis, E.; Minella, M.; Pazzi, M.; Maurino, V.; Minero, C.; Kouras, S.; Richard, C. Modelling the photochemical fate of ibuprofen in surface waters. Water Res. 2011, 45, 6725–6736. [Google Scholar] [CrossRef]
- Avetta, P.; Fabbri, D.; Minella, M.; Brigante, M.; Maurino, V.; Minero, C.; Pazzi, M.; Vione, D. Assessing the phototransformation of diclofenac, clofibric acid and naproxen in surface waters: Model predictions and comparison with field data. Water Res. 2016, 105, 383–394. [Google Scholar] [CrossRef]
- Marchetti, G.; Minella, M.; Maurino, V.; Minero, C.; Vione, D. Photochemical transformation of atrazine and formation of photointermediates under conditions relevant to sunlit surface waters: Laboratory measures and modelling. Water Res. 2013, 47, 6211–6222. [Google Scholar] [CrossRef] [PubMed]
- De Laurentiis, E.; Minella, M.; Bodrato, M.; Maurino, V.; Minero, C.; Vione, D. Modelling the photochemical generation kinetics of 2-methyl-4-chlorophenol, an intermediate of the herbicide MCPA (2-methyl-4-chlorophenoxyacetic acid) in surface waters. Aquat. Ecosys. Health Manag. 2013, 16, 216–221. [Google Scholar] [CrossRef]
- Maddigapu, P.R.; Minella, M.; Vione, D.; Maurino, V.; Minero, C. Modeling phototransformation reactions in surface water bodies: 2,4-Dichloro-6-nitrophenol as a case study. Environ. Sci. Technol. 2011, 45, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Sur, B.; De Laurentiis, E.; Minella, M.; Maurino, V.; Minero, C.; Vione, D. Photochemical transformation of anionic 2-nitro-4-chlorophenol in surface waters: Laboratory and model assessment of the degradation kinetics, and comparison with field data. Sci. Total Environ. 2012, 426, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Carena, L.; Proto, M.; Minella, M.; Ghigo, G.; Giovannoli, C.; Brigante, M.; Mailhot, G.; Maurino, V.; Minero, C.; Vione, D. Evidence of an important role of photochemistry in the attenuation of the secondary contaminant 3,4-dichloroaniline in paddy water. Environ. Sci. Technol. 2018, 52, 6334–6342. [Google Scholar] [CrossRef] [PubMed]
- Minella, M.; Leoni, B.; Salmaso, N.; Savoye, L.; Sommaruga, R.; Vione, D. Long-term trends of chemical and modelled photochemical parameters in four Alpine lakes. Sci. Total Environ. 2016, 541, 247–256. [Google Scholar] [CrossRef]
- Vione, D.; Scozzaro, A. Photochemistry of surface fresh waters in the framework of climate change. Environ. Sci. Technol. 2019, 53, 7945–7963. [Google Scholar] [CrossRef]
- Silva, M.P.; Mostafa, S.; McKay, G.; Rosario-Ortiz, F.L.; Teixeira, A.C.S.C. Photochemical fate of amicarbazone in aqueous media: Laboratory measurement and simulations. Environ. Engin. Sci. 2015, 32, 730–740. [Google Scholar] [CrossRef]
- Lastre-Acosta, A.M.; Barberato, B.; Parizi, M.P.S.; Teixeira, A.C.S.C. Direct and indirect photolysis of the antibiotic enoxacin: Kinetics of oxidation by reactive photo-induced species and simulations. Environ. Sci. Pollut. Res. 2019, 26, 4337–4347. [Google Scholar] [CrossRef]
- Parizi, M.P.S.; Acosta, A.M.L.; Ishiki, H.M.; Rossi, R.C.; Mafra, R.C.; Teixeira, A.C.S.C. Environmental photochemical fate and UVC degradation of sodium levothyroxine in aqueous medium. Environ. Sci. Pollut. Res. 2019, 26, 4393–4403. [Google Scholar] [CrossRef]
- Ge, L.; Dong, Q.; Halsall, C.; Chen, C.-E.L.; Li, J.; Wang, D.; Zhang, P.; Yao, Z. Aqueous multivariate phototransformation kinetics of dissociated tetracycline: Implications for the photochemical fate in surface waters. Environ. Sci. Pollut. Res. 2018, 25, 15726–15732. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, P.; Halsall, C.; Li, Y.; Chen, C.-E.; Li, J.; Sun, H.; Yao, Z. The importance of reactive oxygen species on the aqueous phototransformation of sulfonamide antibiotics: Kinetics, pathways, and comparisons with direct photolysis. Water Res. 2019, 149, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Carena, L.; Minella, M.; Barsotti, F.; Brigante, M.; Milan, M.; Ferrero, A.; Berto, S.; Minero, C.; Vione, D. Phototransformation of the herbicide propanil in paddy field water. Environ. Sci. Technol. 2017, 51, 2695–2704. [Google Scholar] [CrossRef] [PubMed]
- Vione, D.; Das, R.; Rubertelli, F.; Maurino, V.; Minero, C.; Barbati, S.; Chiron, S. Modelling the occurrence and reactivity of hydroxyl radicals in surface waters: Implications for the fate of selected pesticides. Intern. J. Environ. Anal. Chem. 2010, 90, 260–275. [Google Scholar] [CrossRef]
- Page, S.E.; Arnold, W.A.; McNeill, K. Assessing the contribution of free hydroxyl radical in organic matter-sensitized photohydroxylation reactions. Environ. Sci. Technol. 2011, 45, 2818–2825. [Google Scholar] [CrossRef]
- Minella, M.; Romeo, F.; Vione, D.; Maurino, V.; Minero, C. Low to negligible photoactivity of lake-water matter in the size range from 0.1 to 5 µm. Chemosphere 2011, 83, 1480–1485. [Google Scholar] [CrossRef]
- Kistiakowsky, G.B. The temperature coefficients of some photochemical reactions. Proc. Natl. Acad. Sci. USA 1929, 15, 194–197. [Google Scholar] [CrossRef]
- Bianco, A.; Fabbri, D.; Minella, M.; Brigante, M.; Mailhot, G.; Maurino, V.; Minero, C.; Vione, D. New insights into the environmental photochemistry of 5-chloro-2-(2,4-dichlorophenoxy)phenol (triclosan): Reconsidering the importance of indirect photoreactions. Water Res. 2015, 72, 271–280. [Google Scholar] [CrossRef]
- Tremblay, L.; Caparros, J.; Leblanc, K.; Obernosterer, I. Origin and fate of particulate and dissolved organic matter in a naturally iron-fertilized region of the Southern Ocean. Biogeosciences 2015, 12, 607–621. [Google Scholar] [CrossRef]
- Minella, M.; Merlo, M.P.; Maurino, V.; Minero, C.; Vione, D. Transformation of 2,4,6-trimethylphenol and furfuryl alcohol, photosensitised by Aldrich humic acids subject to different filtration procedures. Chemosphere 2013, 90, 306–311. [Google Scholar] [CrossRef]
- Millero, F.J.; Pierrot, D. A chemical equilibrium model for natural waters. Aquatic Geochem. 1998, 4, 153–199. [Google Scholar] [CrossRef]
- Carena, L.; Terrenzio, D.; Mosley, L.M.; Toldo, M.; Minella, M.; Vione, D. Photochemical consequences of prolonged hydrological drought: A model assessment of the Lower Lakes of the Murray-Darling Basin (Southern Australia). Chemosphere 2019, 236, 124356. [Google Scholar] [CrossRef] [PubMed]
- Kohn, T.; Mattle, M.J.; Minella, M.; Vione, D. A modeling approach to estimate the solar disinfection of viral indicator organisms in waste stabilization ponds and surface waters. Water Res. 2016, 88, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Serna-Galvis, E.A.; Troyon, J.A.; Giannakis, S.; Torres-Palma, R.A.; Minero, C.; Vione, D.; Pulgarin, C. Photoinduced disinfection in sunlit natural waters: Measurement of the second order inactivation rate constants between E. coli and photogenerated transient species. Water Res. 2018, 147, 242–253. [Google Scholar] [CrossRef]
- Minero, C.; Pellizzari, P.; Maurino, V.; Pelizzetti, E.; Vione, D. Enhancement of dye sonochemical degradation by some inorganic anions present in natural waters. Appl. Catal. B: Environ. 2008, 77, 308–316. [Google Scholar] [CrossRef]
- Braslavsky, S.E. Glossary of terms used in photochemistry. third edition. Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar] [CrossRef]
- Galbavy, E.S.; Ram, K.; Anastasio, C. 2-Nitrobenzaldehyde as a chemical actinometer for solution and ice photochemistry. J. Photochem. Photobiol. A: Chem. 2010, 209, 186–192. [Google Scholar] [CrossRef]
- Marchisio, A.; Minella, M.; Maurino, V.; Minero, C.; Vione, D. Photogeneration of reactive transient species upon irradiation of natural water samples: Formation quantum yields in different spectral intervals, and implications for the photochemistry of surface waters. Water Res. 2015, 73, 145–156. [Google Scholar] [CrossRef]
- Kuhn, H.J.; Braslavsky, S.E.; Schmidt, R. Chemical actinometry. Pure Appl. Chem. 2004, 76, 2105–2146. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O− in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Vione, D.; Khanra, S.; Cucu Man, S.; Maddigapu, P.R.; Das, R.; Arsene, C.; Olariu, R.I.; Maurino, V.; Minero, C. Inhibition vs. enhancement of the nitrate-induced phototransformation of organic substrates by the •OH scavengers bicarbonate and carbonate. Water Res. 2009, 43, 4718–4728. [Google Scholar] [CrossRef] [PubMed]
- Vione, D.; Sur, B.; Dutta, B.K.; Maurino, V.; Minero, C. On the effect of 2-propanol on phenol photonitration upon nitrate photolysis. J. Photochem. Photobiol. A: Chem. 2011, 224, 68–70. [Google Scholar] [CrossRef]
- Nissenson, P.; Dabdub, D.; Das, R.; Maurino, V.; Minero, C.; Vione, D. Evidence of the water-cage effect on the photolysis of NO3− and FeOH2+. Implications of this effect and of H2O2 surface accumulation on photochemistry at the air–water interface of atmospheric droplets. Atmos. Environ. 2010, 44, 4859–4866. [Google Scholar] [CrossRef]
- Rodgers, M.A.J.; Snowden, P.T. Lifetime of 1O2 in liquid water as determined by time-resolved infrared luminescence measurements. J. Am. Chem. Soc. 1982, 104, 5541–5543. [Google Scholar] [CrossRef]
- Appiani, E.; Ossola, R.; Latch, D.E.; Erickson, P.R.; McNeill, K. Aqueous singlet oxygen reaction kinetics of furfuryl alcohol: Effect of temperature, pH, and salt content. Environ. Sci.: Processes Impacts 2017, 19, 507–516. [Google Scholar] [CrossRef]
- Schmitt, M.; Erickson, P.R.; McNeill, K. Triplet-state dissolved organic matter quantum yields and lifetimes from direct observation of aromatic amine oxidation. Environ. Sci. Technol. 2017, 51, 13151–13160. [Google Scholar] [CrossRef]
- Erickson, P.R.; Moor, K.J.; Werner, J.J.; Latch, D.E.; Arnold, W.A.; McNeill, K. Singlet oxygen phosphorescence as a probe for triplet-state dissolved organic matter reactivity. Environ. Sci. Technol. 2018, 52, 9170–9178. [Google Scholar] [CrossRef]
- Bedini, A.; De Laurentiis, E.; Sur, B.; Maurino, V.; Minero, C.; Brigante, M.; Mailhot, G.; Vione, D. Phototransformation of anthraquinone-2-sulphonate in aqueous solution. Photochem. Photobiol. Sci. 2012, 11, 1445–1453. [Google Scholar] [CrossRef]
- Carena, L.; Puscasu, C.G.; Comis, S.; Sarakha, M.; Vione, D. Environmental photodegradation of emerging contaminants: A re-examination of the importance of triplet-sensitised processes, based on the use of 4-carboxybenzophenone as proxy for the chromophoric dissolved organic matter. Chemosphere 2019, 237, 124476. [Google Scholar] [CrossRef]
- Minella, M.; Rapa, L.; Carena, L.; Pazzi, M.; Maurino, V.; Minero, C.; Brigante, M.; Vione, D. An experimental methodology to measure the reaction rate constants of processes sensitised by the triplet state of 4-carboxybenzophenone as a proxy of the triplet states of chromophoric dissolved organic matter, under steady-state irradiation conditions. Environ. Sci.: Processes Impacts 2018, 20, 1007–1019. [Google Scholar] [CrossRef]
- Canonica, S.; Laubscher, H.-U. Inhibitory effect of dissolved organic matter on triplet-induced oxidation of aquatic contaminants. Photochem. Photobiol. Sci. 2008, 7, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Wenk, J.; von Gunten, U.; Canonica, S. Effect of dissolved organic matter on the transformation of contaminants induced by excited triplet states and the hydroxyl radical. Environ. Sci. Technol. 2011, 45, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Wenk, J.; Canonica, S. Phenolic antioxidants inhibit the triplet-induced transformation of anilines and sulfonamide antibiotics in aqueous solution. Environ. Sci. Technol. 2012, 46, 5455–5462. [Google Scholar] [CrossRef] [PubMed]
- Vione, D.; Fabbri, D.; Minella, M.; Canonica, S. Effects of the antioxidant moieties of dissolved organic matter on triplet-sensitized phototransformation processes: Implications for the photochemical modeling of sulfadiazine. Water Res. 2018, 128, 38–48. [Google Scholar] [CrossRef]
- De Laurentiis, E.; Prasse, C.; Ternes, T.A.; Minella, M.; Maurino, V.; Minero, C.; Sarakha, M.; Brigante, M.; Vione, D. Assessing the photochemical transformation pathways of acetaminophen relevant to surface waters: Transformation kinetics, intermediates, and modelling. Water Res. 2014, 53, 235–248. [Google Scholar] [CrossRef]
- Ervens, B.; Gligorovski, S.; Herrmann, H. Temperature-dependent rate constants for hydroxyl radical reactions with organic compounds in aqueous solutions. Phys. Chem. Chem. Phys. 2003, 5, 1811–1824. [Google Scholar] [CrossRef]
- Mattle, M.J.; Vione, D.; Kohn, T. Conceptual model and experimental framework to determine the contributions of direct and indirect photoreactions to the solar disinfection of MS2, phiX174, and adenovirus. Environ. Sci. Technol. 2015, 49, 334–342. [Google Scholar] [CrossRef]
- Minella, M.; Giannakis, S.; Mazzavillani, A.; Maurino, V.; Minero, C.; Vione, D. Phototransformation of Acesulfame K in surface waters: Comparison of two techniques for the measurement of the second-order rate constants of indirect photodegradation, and modelling of photoreaction kinetics. Chemosphere 2017, 186, 185–192. [Google Scholar] [CrossRef]
- Wols, B.; Hofman-Caris, C. Review of photochemical reaction constants of organic micropollutants required for UV advanced oxidation processes in water. Water Res. 2012, 46, 2815–2827. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. An expanded and revised compilation. J. Phys. Chem. Ref. Data 1995, 24, 663–1021. [Google Scholar] [CrossRef]
- Chen, J.; Peijnenburg, W.J.G.M.; Quan, X.; Yang, F. Quantitative structure–property relationships for direct photolysis quantum yields of selected polycyclic aromatic hydrocarbons. Sci. Total Environ. 2000, 246, 11–20. [Google Scholar] [CrossRef]
- Chen, J.; Quan, X.; Peijnenburg, W.J.G.M.; Yang, F. Quantitative structure–property relationships (QSPRs) on direct photolysis quantum yields of PCDDs. Chemosphere 2001, 43, 235–241. [Google Scholar] [CrossRef]
- US EPA. Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.11; United States Environmental Protection Agency: Washington, DC, USA, 2019.
- Arnold, W.A.; Oueis, Y.; O’Connor, M.; Rinaman, J.E.; Taggart, M.G.; McCarthy, R.E.; Foster, K.A.; Latch, D.E. QSARs for phenols and phenolates: Oxidation potential as a predictor of reaction rate constants with photochemically produced oxidants. Environ. Sci.: Processes Impacts 2017, 19, 324–338. [Google Scholar] [CrossRef]
- Lima, A.B.; Faria, E.O.; Montes, R.H.O.; Cunha, R.R.; Richter, E.M.; Munoz, R.A.A.; dos Santos, W.T.P. Electrochemical oxidation of ibuprofen and its voltammetric determination at a boron---doped diamond electrode. Electroanalysis 2013, 25, 1585–1588. [Google Scholar] [CrossRef]
- Ruggeri, G.; Ghigo, G.; Maurino, V.; Minero, C.; Vione, D. Photochemical transformation of ibuprofen into harmful 4-isobutylacetophenone: Pathways, kinetics, and significance for surface waters. Water Res. 2013, 47, 6109–6121. [Google Scholar] [CrossRef]
- De Laurentiis, E.; Minella, M.; Sarakha, M.; Marrese, A.; Minero, C.; Mailhot, G.; Brigante, M.; Vione, D. Photochemical processes involving the UV absorber benzophenone-4 (2-hydroxy-4-methoxybenzophenone-5-sulphonic acid) in aqueous solution: Reaction pathways and implications for surface waters. Water Res. 2013, 47, 5943–5953. [Google Scholar] [CrossRef]
- Solomon, C.T.; Jones, S.E.; Weidel, B.C.; Buffam, I.; Fork, M.L.; Karlsson, J.; Larsen, S.; Lennon, J.T.; Read, J.S.; Sadro, S.; et al. Ecosystem consequences of changing inputs of terrestrial dissolved organic matter to lakes: Current knowledge and future challenges. Ecosystems 2015, 18, 376–389. [Google Scholar] [CrossRef]
- Garibaldi, L.; Mezzanotte, V.; Brizzio, M.C.; Rogora, M.; Mosello, R. The trophic evolution of Lake Iseo as related to its holomixis. J. Limnol. 1999, 62, 10–19. [Google Scholar] [CrossRef][Green Version]
- Worrall, F.; Howden, N.J.K.; Burt, T.P. A method of estimating in-stream residence time of water in rivers. J. Hydrol. 2014, 512, 274–284. [Google Scholar] [CrossRef]
- Osorio, V.; Proia, L.; Ricart, M.; Pérez, S.; Ginebreda, A.; Cortina, J.L.; Sabater, S.; Barceló, D. Hydrological variation modulates pharmaceutical levels and biofilm responses in a Mediterranean river. Sci. Total Environ. 2014, 472, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Gökbulak, F.; Özhan, S. Water loss through evaporation from water surfaces of lakes and reservoirs in Turkey. E-Water 2006. Available online: http://www.ewa-online.eu/tl_files/_media/content/documents_pdf/Publications/E-WAter/documents/40_2006_07.pdf (accessed on 10 November 2019).
- Majidi, M.; Alizadeh, A.; Farid, A.; Vazifedoust, M. Estimating evaporation from lakes and reservoirs under limited data condition in a semi-arid region. Water Res. Manag. 2015, 29, 3711–3733. [Google Scholar] [CrossRef]
- Zuccato, E.; Castiglioni, S.; Fanelli, R. Identification of the pharmaceuticals for human use contaminating the Italian aquatic environment. J. Hazard. Mater. 2005, 122, 205–209. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
M−1 s−1 | M−1 s−1 | M−1 s−1 | M−1 s−1 | Direct Photolysis | |
|---|---|---|---|---|---|
| Acesulfame K | 5.9 × 109 | n/a | 2.8 × 104 | n/a | No |
| Acetaminophen | 1.9 × 109 | 3.8 × 108 | 3.7 × 107 | 1.6 × 109 | Yes (UVB & UVA) |
| Atrazine | 2.7 × 109 | 4 × 106 | n/a | 7.2 × 108 | Yes (UVB & UVA) |
| Tyrosine | 1.3 × 1010 | 4.5 × 107 | 8 × 106 | n/a | Yes (UVB & UVA) |
| Direct Photolysis | •OH | 1O2 | 3CDOM* | |
|---|---|---|---|---|
| Ibuprofen [19] | Φ = 0.33 | k = 1.0 × 1010 M−1 s−1 | k = 6 × 104 M−1 s−1 | k = 4.5 × 107 M−1 s−1 |
| IBAP [77] | y = 0.25 | y = 0.023 | y ~ 0 | y = 0.31 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vione, D. A Critical View of the Application of the APEX Software (Aqueous Photochemistry of Environmentally-Occurring Xenobiotics) to Predict Photoreaction Kinetics in Surface Freshwaters. Molecules 2020, 25, 9. https://doi.org/10.3390/molecules25010009
Vione D. A Critical View of the Application of the APEX Software (Aqueous Photochemistry of Environmentally-Occurring Xenobiotics) to Predict Photoreaction Kinetics in Surface Freshwaters. Molecules. 2020; 25(1):9. https://doi.org/10.3390/molecules25010009
Chicago/Turabian StyleVione, Davide. 2020. "A Critical View of the Application of the APEX Software (Aqueous Photochemistry of Environmentally-Occurring Xenobiotics) to Predict Photoreaction Kinetics in Surface Freshwaters" Molecules 25, no. 1: 9. https://doi.org/10.3390/molecules25010009
APA StyleVione, D. (2020). A Critical View of the Application of the APEX Software (Aqueous Photochemistry of Environmentally-Occurring Xenobiotics) to Predict Photoreaction Kinetics in Surface Freshwaters. Molecules, 25(1), 9. https://doi.org/10.3390/molecules25010009