Abstract
The reactivities of transition metal coordination compounds are often controlled by the environment around the coordination sphere. For ruthenium(II) complexes, differences in polypyridyl supporting ligands affect some types of reactivity despite identical coordination geometries. To evaluate the synergistic effects of (i) the supporting ligands, and (ii) the coordination geometry, a series of dicarbonyl–ruthenium(II) complexes that contain both asymmetric and symmetric bidentate polypyridyl ligands were synthesized. Molecular structures of the complexes were determined by X-ray crystallography to distinguish their steric configuration. Structural, computational, and electrochemical analysis revealed some differences between the isomers. Photo- and thermal reactions indicated that the reactivities of the complexes were significantly affected by both their structures and the ligands involved.
1. Introduction
Polypyridines with multiple covalently bonded pyridine groups exhibit unique photophysical and redox properties [1,2]. Among the polypyridines, bipyridine analogues play an important role in the formation of various transition metal complexes as bidentate ligands with two nitrogen donor atoms [3]. Bipyridine analogues function not only as supporting ligands for stabilizing metal complexes, but also as electron pool sites. For example, in addition to catalysts for the production of useful resources, such as in multi-electron reductions of carbon dioxide and water–gas shift reactions [4,5,6,7,8,9,10,11], bipyridine analogues are also utilized as photosensitizers [12] and phosphorescence materials [13]. A variety of studies that imparted selectivity to various reactions by strictly controlling the coordination sphere have been reported for many ruthenium complexes. Various molecular structures can be readily designed for a ruthenium center because ruthenium can take various oxidation states and can easily interact with the ligand. These structures include examples where differences in the coordination geometry dramatically changed both the electrical and photochemical properties of the complex, and therefore its catalysis for chemical reactions [14,15,16]. Many examples of the relationship between the supporting bipyridine ligands and the reactivity of metal complexes are known. For example, the relationship between the number of heteroatoms involved in the supporting ligand and the reactivity of the complex has been reported in a ruthenium complex containing bipyridine analogues [17].
Recently, we reported the synthesis of ruthenium complexes with 2,2’-bipyridine (bpy) and an analogue, the asymmetric bidentate ligand 2-(2-pyridyl)-1,8-naphthyridine (pynp, Chart 1). The supporting ligands affected the properties of the complexes, and diastereomers with these ligands showed different reactivities [18]. As is the case in some of the studies mentioned above, the difference in reactivities based on coordination geometry or the effect of the supporting ligand on the properties of the complex have been reported independently, but there are very few examples that investigated the synergistic effects of both. In this study, we prepared a series of Ru(II) complexes (Chart 2) containing the asymmetric bidentate pynp and a symmetric bidentate ligand N^N, [Ru(pynp)(N^N)(CO)2]2+ (N^N = bpy or 1,10-phenanthroline (phen) as a more rigid ligand, Chart 1), as a platform for investigating these synergistic effects [19]. In addition to characterizing the above complexes, we performed photo- and thermal reactions on the complexes, along with the bis–pynp complex [Ru(pynp)2(CO)2]2+ as a reference compound. We comprehensively evaluated the synergistic effects of the coordination geometry and supporting ligands on the reactivity.
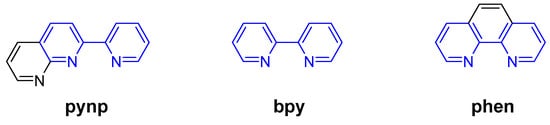
Chart 1.
Chemical structures of pynp, bpy, and phen.
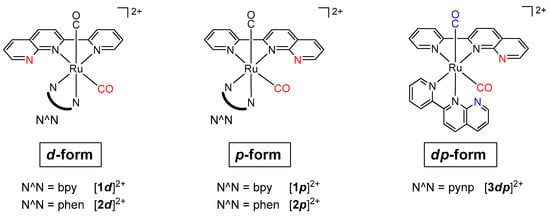
Chart 2.
[Ru(pynp)(N^N)(CO)2]2+ complexes in this study [19].
2. Results and Discussion
2.1. Diastereoselective Synthesis and Characterization of Ruthenium Complexes with the Asymmetric pynp Ligand
2.1.1. Synthesis and Structure of Complexes
Unlike unsubstituted 2,2’-bipyridine and 1,10-phenanthroline, the pynp ligand has no C2 symmetry axis (Chart 1), so metal complexes containing pynp(s) may have several diastereomers. We previously reported that for ruthenium carbonyl complexes containing both bpy and pynp, formation of the desired isomers can be controlled by the order of introduction of the two ligands [18,20]. Using this method, dicarbonylruthenium complexes containing both pynp and phen were successfully prepared. As in the previous study, the distal isomer is produced preferentially when phen is introduced first (Figure 1a). In the case of N^N = bpy, the product obtained by this method is almost all the d-form, whereas in the phen system a small quantity of the corresponding p-form (10–20%) is also formed. This may be due to energy differences between the d- and p-isomers. Density functional theory (DFT) calculations for each complex system suggest that the p-isomers are more stable, but the energy difference between the d- and p-isomers in the phen system is smaller than that of the corresponding bpy system (N^N = bpy: 4.3 kcal/mol, N^N = phen: 1.9 kcal/mol). The DFT calculations indicate that the minor formation of the p-isomer in addition to the d-isomer is due to the relatively small energy difference in the phen system.
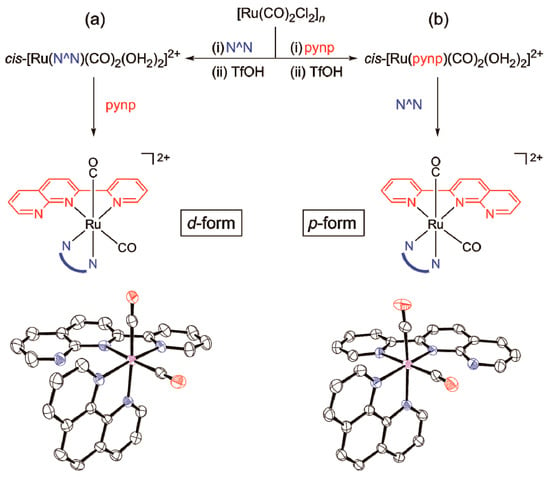
Figure 1.
Diastereoselective synthesis of [1]2+ and [2]2+. (a) synthetic route for the d-isomers and molecular structure of [2d]2+; (b) synthetic route for the p-isomers and molecular structure of [2p]2+. Counteranions, H atoms, and solvent molecules are omitted for clarity. The asymmetric unit of the [2d] crystal contains two chemically identical complex pairs, of which only one is displayed.
In contrast, initial introduction of the asymmetric pynp ligand leads to the selective formation of p-isomers (Figure 1b). Considering the molecular structure of an isolated and analyzed intermediate cis(solv)-[Ru(pynp)(CO)2(solv)2]2+; Supplementary Materials Figure S1), it is reasonable to propose that an intramolecular interaction between the non-coordinating nitrogen atom in the pynp ligand and the adjacent coordinated carbonyl promotes the formation of p-isomers [18]. In addition, only the dp-isomer was selectively isolated in the case of [Ru(pynp)2(CO)2]2+ (Figure 2), even though the bis-pynp complex could form three diastereomers (pp, dp, and dd; Chart 3). According to DFT calculations for the three diastereomers, the total energy of the experimentally formed dp-isomer was 3.79 kcal/mol higher than that of the most stable pp-isomer, which appears to contradict the experimental result. We then performed total energy calculations for the two possible intermediates in the coordination of the second pynp ligand to the ruthenium center by assuming that the aqua ligand in the trans position of the coordinated pynp is substituted earlier than the aqua ligand in the cis position (see the structure diagram in Supplementary Materials Figure S1). The results indicate that both intermediates have almost the same energy (ΔE = 0.08 kcal/mol; Figure S2). Therefore, the second pynp ligand may coordinate through a kinetically favorable pathway rather than a thermodynamic one to give the selective dp-isomer.
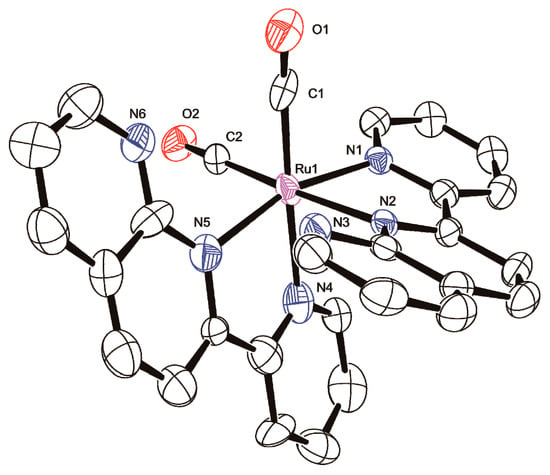
Figure 2.
Molecular structure of dp-[Ru(pynp)2(CO)2]2+ ([3dp]2+) with atom labels and displacement ellipsoids for non-H atoms drawn at the 50% probability level. Counteranions and H atoms are omitted for clarity.
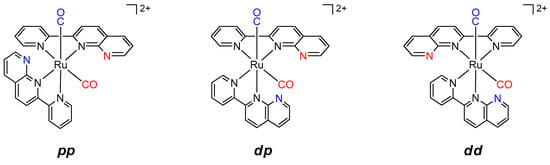
Chart 3.
Possible diastereomers in [Ru(pynp)2(CO)2]2+.
Table 1 summarizes the structural parameters of a series of [Ru(pynp)(N^N)(CO)2]2+ complexes. The data around the ruthenium center and the {Ru–CO}2+ moieties are within the typical range of dicarbonylruthenium(II) complexes that have polypyridines as supporting ligands [4,21,22]. In addition, the interatomic distances between the carbonyl carbon and the non-coordinating nitrogen of pynp (2.607(5) to 2.754(14) Å) in the three p-forms are much shorter than the sum of van der Waals radii (3.25 Å) [23]. The shortening suggests that pynp and the adjacent CO ligand interact considerably in these complexes.

Table 1.
Selected bond and interatomic distances (Å) and angles (°) for dicarbonyl complexes.
2.1.2. Characterization of Complexes
Spectroscopic and electrochemical analyses were performed on the synthesized complexes (Table 2). Two strong IR bands assignable to νCO were observed around 2100 and 2040 cm−1 in all complexes. These values were similar to those in other dicarbonyl-ruthenium(II) complexes [4,21,22], and no clear differences were observed between the isomers. Given that the complexes contain two carbonyl ligands of the highest order in the spectrochemical series, no obvious absorption was observed in the visible region (Supplementary Materials Figure S3). However, intense polypyridyl-centered π–π* intraligand transitions were observed in the UV region [24].
Table 2.
Spectral and electrochemical data for dicarbonyl complexes.
Although spectroscopic measurements did not show any marked differences between isomers, electrochemistry clearly showed different behavior. The cyclic voltammograms (CV) of all the complexes showed multiple ligand-based reduction waves in the range of −1 to −2 V vs. Fc+/Fc. Since pynp is more easily reduced than bpy or phen, the first reduction peak at ca. −1 V was attributed to the reduction of the pynp ligand [25]. When the cathodic scan was immediately reversed after the first peak potential, the coupled oxidation wave was reversible in the d-isomers (Figure 3a, dotted line); however, those of the corresponding p-isomers were either irreversible or quasi-reversible at the first reduction wave (Figure 3b). Although the first reduction wave of [2p]2+ in Figure 3b appears to be reversible, the coupled anodic current decreased as the scan rates slowed (Supplementary Materials Figure S4). The basicity of the free nitrogen atom of the 1,8-naphthyridine moiety in pynp increased due to one-electron reduction of pynp, thus making intramolecular nucleophilic attack to the adjacent carbonyl carbons possible, leading to the formation of a metallacyclic compound (Scheme 1) [26]. Due to this structural change, only the p-isomer exhibited irreversible one-electron reduction behavior.
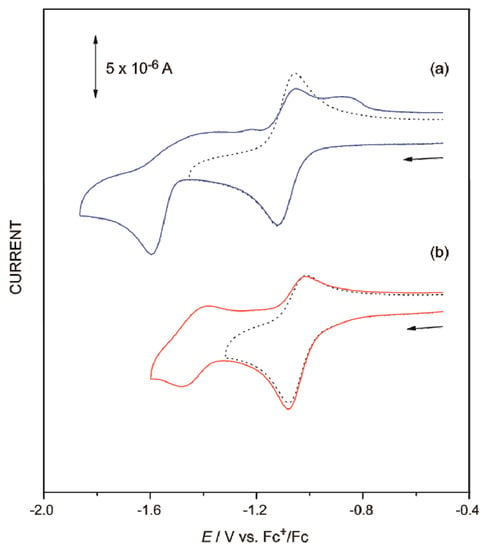
Figure 3.
Cyclic voltammograms of (a) [2d]2+ and (b) [2p]2+ in CH3CN (v = 0.1 V s−1, c = 1.0 mM).
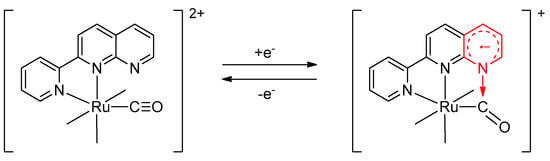
Scheme 1.
Reversible metallacyclization induced by one-electron transfer in [Ru(pynp)(CO)2(PPh3)2]2+ [26].
2.2. Reactivities of Complexes
2.2.1. Photochemical Reactions
Polypyridylruthenium complexes are generally photoreactive. When cis-[Ru(bpy)2(CO)2]2+ in acetonitrile is irradiated with light, two carbonyl ligands simultaneously dissociate and the corresponding solvent complex (cis-[Ru(bpy)2(CH3CN)2]2+) is produced [27,28]. However, when pynp is substituted for one of the two bpy ligands, the two carbonyl groups dissociate stepwise due to their electronic non-equivalence [18]. Since no comparisons between the d- and p-isomers were made in the previous report, we compared the photoreactivities of both isomers. As previously reported, there are two reaction steps for both isomers (Scheme 2) [18,29]. In the first step, one carbonyl ligand was dissociated, and, at the same time, the acetonitrile used as the solvent became coordinated (the first step in Scheme 2 and Figure 4a). Steric retention of the complex was supported by structural analysis of the isolated species at this step (Figure 5a). In the subsequent step, photoisomerization from the d- to p-isomer subsequent slow dissociation of the second CO ligand and solvent coordination occurred (the second step in Scheme 2a and Figure 4b). Reaction analysis and DFT calculations suggest that the formation of photoexcited states of the complex promote such an isomerization to a more stable p-form in similar complexes containing pynp [30,31]. In the bis-pynp complex ([3dp]2+), a similar two-step photoreaction (Scheme 2a) was confirmed from structural analyses of both products (dp-form in Figure 5b and pp-form in Supplementary Materials Figure S5). On the other hand, the final structure of the p-isomers was unchanged from spectroscopic analyses (Scheme 2b).
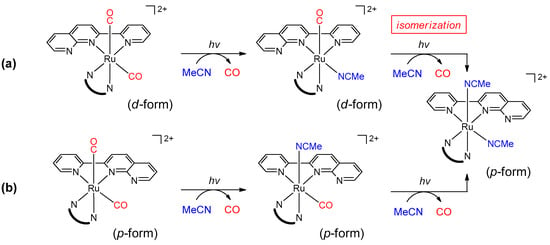
Scheme 2.
Photoreactions of [Ru(pynp)(N^N)(CO)2]2+ [29]. (a) The two-step reaction of d-isomers ([1d]2+, [2d]2+, and [3dp]2+) with isomerization; (b) The two-step reaction of p-isomers ([1p]2+ and [2p]2+) without isomerization. The structures of these species were confirmed by X-ray crystallographic analyses (Figure 5, Supplementary Materials Figure S5, and reference [18]).
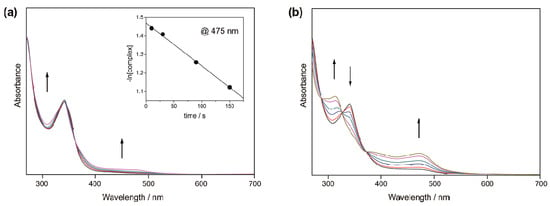
Figure 4.
Changes in the absorption spectra of [2d]2+ in CH3CN upon photoirradiation (λ = 300–400 nm). (a) 1st reaction step (t < 900 s); (b) 2nd reaction step (t > 900 s). Inset: First-order plot based on the absorbance values at 475 nm.
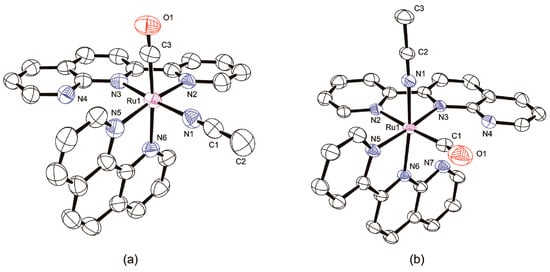
Figure 5.
Molecular structures of monosubstituted complexes with atom labels and displacement ellipsoids for non-H atoms drawn at the 50% probability level. Counteranions, H atoms, and solvent molecules are omitted for clarity. (a) d-[Ru(pynp)(phen)(CO)(CH3CN)]2+; (b) dp-[Ru(pynp)2(CO)(CH3CN)]2+.
From comparisons of the first CO-dissociation rates (Table 3), it was found that the photoreactions of the d-isomers were always faster than those of the corresponding p-isomers (ca. two times in both complexes). This difference was interpreted to be due to their geometries, based on photoreaction behavior seen in similar diastereomers [32]. The pynp ligand has a naphthyridine unit in place of the pyridine unit in bpy (or phen), which is both more delocalized and a π-acceptor. The superior charge acceptor properties of the naphthyridine unit in the d-isomers leads to better labilization of the trans-CO, and thus the d-isomers exhibit faster CO release compared with the corresponding p-isomers, despite having lower extinction coefficients between 300 and 400 nm (Table 2 and Supplementary Materials Figure S3). We also found that the overall photoreaction of the bpy system proceeded more smoothly than that of the corresponding phen system. This was probably because phen compounds were significantly more stable than their bpy analogues, based on the rigidity of phen [33]. Despite prolonged photoirradiation, incomplete dissociation of the second CO ligand in the phen system consequently prevented isolation of the single disubstituted complex ([Ru(pynp)(phen)(CH3CN)2]2+).
Table 3.
Rates of the first photoreaction step (298 K) of the dicarbonyl complexes.
2.2.2. Thermochemical Reactions
As shown in Scheme 3, in thermal reactions one carbonyl ligand of a dicarbonylruthenium(II) complex undergoes nucleophilic attack from solvent molecules [34,35]. Thus, when the d-isomers of the dicarbonyl complexes, which are expected to be more reactive (see Section 2.2.1.), were heated in water/acetonitrile or alcohol (methanol or ethanol)/acetonitrile mixtures, one of the coordinated CO moieties underwent nucleophilic attack from the solvent to the CO ligand at the trans position of pynp. As expected, molecular structures of the isolated complexes showed the formation of the hydroxycarbonyl (–C(O)OH in Figure 6a) and the methoxy- or ethoxycarbonyl (–C(O)OC2H5 in Figure 6b and Supplementary Materials Figure S6) complexes. Given that the pynp ligand is more π-acidic than bpy or phen, the CO carbon in the trans position of pynp has a more positive charge.
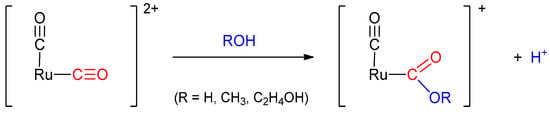
Scheme 3.
Nucleophilic attack of ROH on the coordinated CO in dicarbonyl-ruthenium(II) complexes.
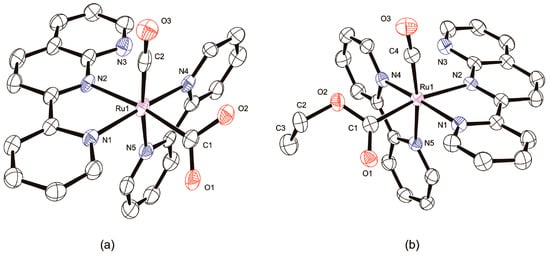
Figure 6.
Molecular structures with atom labels and displacement ellipsoids for non-H atoms drawn at the 50% probability level. Counteranions, H atoms, and solvent molecules are omitted for clarity. (a) d-[Ru(pynp)(bpy)(CO)(C(O)OH)]+; (b) d-[Ru(pynp)(bpy)(CO)(C(O)OC2H5)]+.
We next investigated other thermochemical reactions using monocarbonyl complexes that do not undergo nucleophilic attack on the coordinated carbonyl. When the monocarbonyl complex (d-[Ru(pynp)(phen)(CO)(CH3CN)]2+), which was produced by the photoreaction described in Section 2.2.1, was heated in acetone, it isomerized from the d- to the p-isomer (Figure 7). Notably, this thermal isomerization reaction was accelerated more than 70 times when water was added to the solution. A similar isomerization was observed in d-[Ru(pynp)(bpy)(CO)(CH3CN)]2+. In contrast, such thermal isomerization could not be confirmed for other monocarbonyl complexes with anionic ligands (–COR−; R− = OH, OCH3, or OC2H5). These results strongly suggest that the thermal isomerization reaction involves a donor-acceptor interaction between the solvent and the complex. In ruthenium(II) complexes, terminal CO ligands that interact with the donor solvent tend to exhibit νCO IR frequencies over 2000 cm−1 [36,37]. In this study, the CO stretching frequency of the thermally isomerized acetonitrile complexes exceeded 2000 cm−1 (2008 and 2009 cm−1), while those of complexes containing an anionic ligand were 1950–1960 cm−1. It would therefore be expected that weak interactions between the coordinated carbonyl and the solvent (acetone or water) could induce thermal isomerization. That is, the interaction between the coordinated CO and the solvent significantly changes the electronic state of the ruthenium center, resulting in lowering the activation energy for isomerization of the d-isomer to the thermally more stable p-isomer [38].
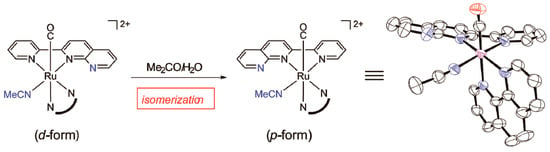
Figure 7.
Thermal isomerization of d-[Ru(pynp)(N^N)(CO)(CH3CN)]2+ and the molecular structure of the isomerized p-form (counteranions, H atoms, and solvent molecules are omitted for clarity).
3. Materials and Methods
3.1. General Remarks
All chemicals were purchased from commercial sources and used without further purification unless otherwise stated. All solvents used for the syntheses were anhydrous. Acetonitrile for electrochemical measurements was purified by passing through solvent purification columns (Glass Contour, Laguna, CA, USA). Then 2-(2-pyridyl)-1,8-naphthyridine (pynp), [Ru(CO)2Cl2]n, trans-(Cl)-[Ru(bpy)(CO)2Cl2], trans-(Cl)-[Ru(phen)(CO)2Cl2], cis(OH2)-[Ru(bpy)(CO)2(OH2)2](CF3SO3)2, cis-(OH2)-[Ru(phen)(CO)2(OH2)2](CF3SO3)2, and cis-(OH2)-[Ru(pynp)(CO)2(OH2)2](CF3SO3)2 were prepared according to previously reported procedures [18,39,40].
IR spectra were obtained using a JASCO FT–IR 4100 spectrometer (Tokyo, Japan). Electrospray ionization mass spectrometry (ESI–MS) data were obtained using a Bruker Daltonics micrOTOF spectrometer. Electronic spectra were obtained on a JASCO V-560 spectrophotometer. The 1H and 13C{1H}-NMR spectra were acquired on a JEOL JMN-AL300 spectrometer (Tokyo, Japan) operating at 1H and 13C frequencies of 300 and 75.5 MHz, respectively. Elemental analysis data were obtained on a PerkinElmer 2400II series CHN analyzer (Yokohama, Japan). Electrochemical measurements were performed on an electrochemical analyzer (ALS/CHI model 660E, Tokyo, Japan) with a solution of the complex in acetonitrile (1 mM) and n-Bu4NClO4 (0.1 M) as a supporting electrolyte in a cell consisting of a glassy carbon working electrode (ϕ = 1.6 mm), a Pt wire counter electrode, and Ag/AgNO3 (0.01 M) as the reference electrode. All potentials are reported in volts versus the ferrocenium/ferrocene couple (Fc+/Fc) under Ar at 25 °C. DFT calculations were performed using the quantum computation software Gaussian 09 [41]. The geometries of the Ru complexes were fully optimized using a restricted DFT method employing the B3LYP function [42,43], with a 6-31G(d) basis set for the light elements (H, C, N, and O) [44,45] and a LanL2DZ basis set [46] for the Ru atom. The solvent effect of acetonitrile was evaluated using an implicit solvent model and a polarizable continuum model. The vibrational analyses were performed at the same calculation level employed for geometry optimization.
3.2. Synthesis of Complexes
3.2.1. Synthesis of the Distal Isomers ([1d](PF6)2 and [2d](PF6)2)
[Ru(bpy)(CO)2(OH2)2](CF3SO3)2 (53 mg, 0.082 mmol) and pynp (20 mg, 0.098 mmol) were added to methanol (10 mL). The mixture was refluxed with stirring for 1.5 h. The reaction vessel was cooled to room temperature. The reaction mixture was condensed to 3 mL under reduced pressure. A light orange precipitate formed on addition of a saturated aqueous solution of KPF6 to the mixture. The product was collected by filtration, washed with cold water and diethyl ether, and dried in vacuo to obtain the product (56 mg, 84%). Anal. Calcd for [1d](PF6)2: C25H17N5O2F12P2Ru: C, 37.05; H, 2.11; N, 8.64. Found: C, 37.40; H, 2.03; N, 8.51. ESI–MS (CH3CN): m/z 260.5 ([M]2+), 246.5 ([M–CO]2+). IR (KBr): 2097, 2044 cm−1 (νCO). 1H-NMR (acetone-d6): δ 9.76–9.71 (m, 2H), 9.25 (d, J = 8.4 Hz, 1H), 9.09–8.99 (m, 2H), 8.77–8.71 (m, 2H), 8.64 (dd, J = 6.6, 1.8 Hz, 2H), 8.57 (td, J = 6.3, 1.5 Hz, 1H), 8.49 (dd, J = 1.8, 1.5 Hz, 1H), 8.28–8.25 (m, 2H), 8.17–8.13 (m, 1H), 7.76–7.69 (m, 2H), 7.66–7.63 (m, 1H). 13C{1H}-NMR (acetone-d6): δ 192.08 and 191.49 (CO), 158.89, 158.73, 157.02, 156.71, 156.61, 156.16, 150.19, 145.33, 143.16, 142.76, 141.93, 139.61, 131.27, 130.70, 129.41, 129.07, 128.95, 127.20, 126.18, 125.92, 125.49, 125.39, 122.23. A similar reaction between [Ru(phen)(CO)2(OH2)2](CF3SO3)2 (60 mg, 0.089 mmol) and pynp (23 mg, 0.109 mmol) gave a mixture of both [2d](PF6)2 and [2p](PF6)2. Yield: 63 mg (84%). The crude product was recrystallized from a mixture of acetonitrile, acetone, and diethyl ether. Anal. Calcd for [2d](PF6)2: C27H17N5O2F12P2Ru·CH3CN·0.5C2H6CO: C, 40.50; H, 2.56; N, 9.29. Found: C, 40.38; H, 2.29; N, 9.14. ESI–MS (CH3CN): m/z 272.6 ([M]2+), 258.6 ([M–CO]2+). IR (KBr): 2099, 2045 cm−1 (νCO). 1H-NMR (acetone-d6): δ 10.16 (dd, J = 5.1, 1.2 Hz, 1H), 9.80 (dd, J = 5.8, 0.9 Hz, 1H), 9.28 (dd, J = 7.6, 0.9 Hz, 1H), 9.18 (dd, J = 8.5, 1.2 Hz, 1H), 8.99 (s, 2H), 8.87 (dd, J = 8.3, 1.2 Hz, 1H), 8.80 (td, J = 8.1, 1.5 Hz, 1H), 8.52–8.47 (m, 2H), 8.39–8.23 (m, 4H), 8.08 (dd, J = 5.4, 1.5 Hz, 1H), 7.95 (dd, J = 8.1, 5.4 Hz, 1H), 7.60 (dd, J = 8.4, 4.5 Hz, 1H). 13C{1H}-NMR (acetone-d6): δ 192.33 and 191.47 (CO), 160.51, 159.49, 158.91, 157.29, 156.00, 155.95, 151.31, 147.34, 147.26, 145.17, 143.21, 141.68, 140.94, 139.41, 132.07, 132.06, 131.24, 129.16, 129.10, 128.46, 127.63, 127.59, 125.94, 125.83, 122.27.
3.2.2. Synthesis of the Proximal Isomers ([1p](PF6)2 and [2p](PF6)2)
Using a protocol similar to that described for the synthesis of [1d](PF6)2, the reaction between [Ru(pynp)(CO)2(OH2)2](CF3SO3)2 (106 mg, 0.161 mmol) and bpy (31 mg, 0.197 mmol) or phen (39 mg, 0.197 mmol) gave [1p](PF6)2 or [2p](PF6)2 in 81% and 86% yield, respectively. Anal. Calcd for [1p](PF6)2: C25H17N5O2F12P2Ru: C, 37.05; H, 2.11; N, 8.64. Found: C, 37.22; H, 2.01; N, 8.60. ESI–MS (CH3CN): m/z 260.5 ([M]2+), 246.5 ([M–CO]2+). IR (KBr): 2097, 2045 cm−1 (νCO). 1H-NMR (acetone-d6): δ 9.64 (d, J = 5.1 Hz, 1H), 9.49 (d, J = 3.9 Hz, 1H), 9.36 (d, J = 8.4 Hz, 1H), 9.17–9.11 (m, 2H), 9.02–8.99 (m, 2H), 8.86 (d, J = 8.1 Hz, 1H), 8.69 (t, J = 3.8 Hz, 1H), 8.50 (t, J = 4.7 Hz, 1H), 8.35 (t, J = 4.8 Hz, 1H), 8.22–8.16 (m, 2H), 8.02 (d, J = 6.3 Hz, 1H), 7.84–7.77 (m, 2H), 7.50 (t, J = 7.4 Hz, 1H). 13C{1H}-NMR (acetone-d6): δ 193.12 and 192.20 (CO), 161.36, 158.14, 157.00, 156.88, 156.71, 155.91, 153.77, 151.45, 151.15, 145.42, 143.02, 142.94, 142.87, 140.57, 130.70, 130.65, 129.66, 128.19, 127.20, 126.92, 126.76, 126.03, 122.99. Anal. Calcd for [2p](PF6)2: C27H17N5O2F12P2Ru·0.5CH3CN·0.5C2H6CO: C, 40.08; H, 2.45; N, 8.72. Found: C, 39.90; H, 2.17; N, 8.68. ESI–MS (CH3CN): m/z 272.6 ([M]2+), 258.6 ([M–CO]2+). IR (KBr): 2093, 2044 cm−1 (νCO). 1H-NMR (acetone-d6): δ 10.04 (dd, J = 5.1, 1.2 Hz, 1H), 9.48 (dd, J = 3.9, 1.8 Hz, 1H), 9.40 (d, J = 8.4 Hz, 1H), 9.30 (dd, J = 8.4, 1.2 Hz, 1H), 9.18 (d, J = 8.7 Hz, 1H), 9.10 (d, J = 7.8 Hz, 1H), 9.03 (dd, J = 7.8, 1.8 Hz, 1H), 8.96 (dd, J = 8.4, 0.9 Hz, 1H), 8.56–8.38 (m, 4H), 8.23–8.17 (m, 2H), 7.88 (d, J = 4.8 Hz, 1H), 7.82 (dd, J = 9.0, 5.4 Hz, 1H), 7.60 (dd, J = 7.5, 1.2 Hz, 1H). 13C{1H}-NMR (acetone-d6): δ 192.08 (CO), 161.61, 158.85, 157.05, 156.69, 153.96, 152.34, 151.63, 147.09, 146.35, 145.47, 142.88, 142.05, 141.81, 140.59, 132.99, 132.47, 130.39, 129.46, 129.38, 128.88, 128.13, 127.84, 127.19, 126.77, 123.01.
3.2.3. Synthesis of [3dp](PF6)2
A similar reaction between [Ru(pynp)(CO)2(OH2)2](CF3SO3)2 (58 mg, 0.083 mmol) and pynp (20 mg, 0.098 mmol) in methanol (20 mL) gave [3dp](PF6)2. Yield: 67 mg (94%). Anal. Calcd for [3dp](PF6)2: C28H18N6O2F12P2Ru·H2O: C, 38.23; H, 2.29; N, 9.56. Found: C, 37.95; H, 1.94; N, 9.32. ESI–MS (CH3CN): m/z 286.1 ([M]2+), 272.1 ([M–CO]2+). IR (KBr): 2094, 2040 cm−1 (νCO). 1H-NMR (acetone-d6): δ 9.85 (d, J = 4.8 Hz, 1H), 9.49 (dd, J = 2.2, 1.8 Hz, 1H), 9.28 (d, J = 8.4 Hz, 2H), 9.06–9.01 (m, 3H), 8.85–8.82 (m, 2H), 8.78 (t, J = 1.5 Hz, 1H), 8.53 (dd, J = 6.6, 1.8 Hz, 1H), 8.39–8.16 (m, 3H), 7.88 (d, J = 4.8 Hz, 1H), 7.75 (t, J = 1.5 Hz, 1H), 7.66 (dd, J = 2.2, 1.8 Hz, 1H), 7.54–7.49 (m, 1H). 13C{1H}-NMR (acetone-d6): δ 192.58 and 192.27 (CO), 160.76, 160.25, 158.70, 157.76, 157.19, 155.92, 155.86, 151.01, 145.17, 144.22, 142.97, 142.70, 140.27, 139.57, 139.51, 131.42, 130.27, 129.16, 127.64, 127.58, 126.57, 126.11, 125.68, 125.63, 122.23, 122.03.
3.3. Photochemical Reactions
Photochemical reactions of the complexes were conducted in the degassed CH3CN. The solution was irradiated with UV–visible light (λ = 300–400 nm) through a cutoff filter using an Asahi Spectra MAX-302 (Tokyo, Japan) with a xenon lamp (0.1 mW cm−2). In a typical reaction, an acetonitrile solution of [1d](PF6)2 (0.10 mM) was placed in a quartz flask with a stopper and irradiated with a xenon lamp for 30 min. The reaction mixture was condensed to 1 mL under reduced pressure. Addition of diethyl ether to the solution resulted in the formation of a precipitate of the mono- or disubstituted complexes in moderate yields (65–75%).
d-[Ru(pynp)(bpy)(CO)(CH3CN)](PF6)2: ESI–MS (CH3CN): m/z 246.5 ([M–CH3CN]2+). IR (KBr): 2009 cm−1 (νCO). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 250 (31,200), 312 (18,500), 341 (19,400), 480 (950). p-[Ru(pynp)(bpy)(CH3CN)2](PF6)2: ESI–MS (CH3CN): m/z 232.5 ([M–(CH3CN)2]2+). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 239 (26,400), 288 (33,600), 407 (4400), 473 (6500). d-[Ru(pynp)(phen)(CO)(CH3CN)](PF6)2: ESI–MS (CH3CN): m/z 258.5 ([M–CH3CN]2+). IR (KBr): 2008 cm−1 (νCO). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 268 (33,400), 341 (16,700), 440 (1600). dp-[Ru(pynp)2(CO)(CH3CN)](PF6)2: ESI–MS (CH3CN): m/z 272.0 ([M–CH3CN]2+), 292.5 ([M]2+). IR (KBr): 2007 cm−1 (νCO). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 241 (40,200), 340 (32,000), 420sh (2600). pp-[Ru(pynp)2(CH3CN)2](PF6)2: ESI–MS (CH3CN): m/z 258.0 ([M–(CH3CN)2]2+), 278.5 ([M–CH3CN]2+). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 315 (40,900), 496 (6400). Kinetic measurements for the photoreaction were followed at an appropriate wavelength for each complex (476 nm for [1d]2+, 478 nm for [1p]2+, 475 nm for [2d]2+ and [2p]2+, and 497 nm for [3dp]2+), and the logarithm of the complex concentration versus time plots were generated for the first step of the reaction in each measurement.
3.4. Thermochemical Reactions
3.4.1. Reaction of the Coordinated CO in [1d]2+ and [2d]2+ with Solvents
The [1d](PF6)2 (50 mg, 0.0617 mmol) was dissolved in acetonitrile (5 mL) and a suitable cosolvent (water, ethanol, or methanol) (30 mL) and refluxed for 24 h. The volume was reduced to 1 mL using a rotary evaporator. Addition of diethyl ether to the solution resulted in precipitation of the reaction products ([Ru(pynp)(bpy)(CO)(C(O)R)]PF6, R = OH, OC2H5, or OCH3) in moderate to high yields (50%–82%). [Ru(pynp)(bpy)(CO)(C(O)OH)]PF6: ESI–MS (CH3CN): m/z 538.1 ([M]+). IR (KBr): 1957 cm−1 (νCO). [Ru(pynp)(bpy)(CO)(C(O)OC2H5)]PF6: ESI–MS (CH3CN): m/z 566.1 ([M]+). IR (KBr): 1954 cm−1 (νCO). [Ru(pynp)(bpy)(CO)(C(O)OCH3)]PF6: ESI–MS (CH3CN): m/z 552.1 ([M]+). IR (KBr): 1954 cm−1 (νCO).
[Ru(pynp)(phen)(CO)(C(O)R)](PF6) complexes were also isolated in 50 to 75% yields using a similar procedure starting from [2d](PF6)2. [Ru(pynp)(phen)(CO)(C(O)OH)]PF6: ESI–MS (CH3CN): m/z 562.0 ([M]+). IR (KBr): 1960 cm−1 (νCO). [Ru(pynp)(phen)(CO)(C(O)OC2H5)]PF6: ESI–MS (CH3CN): m/z 590.2 ([M]+). IR (KBr): 1961 cm−1 (νCO). [Ru(pynp)(phen)(CO)(C(O)OCH3)]PF6: ESI–MS (CH3CN): m/z 576.1 ([M]+). IR (KBr): 1960 cm−1 (νCO).
3.4.2. Thermal-induced Isomerization Using Monocarbonyl Complexes d-[Ru(pynp)(N^N)(CO)(CH3CN)]2+ (N^N = bpy or phen) and dp-[Ru(pynp)2(CO)(CH3CN)]2+
In a typical reaction, d-[Ru(pynp)(phen)(CO)(CH3CN)](PF6)2 (4 mg, 0.005 mmol) was dissolved in acetone (20 mL) and refluxed until the solution became dark red (~72 h). The solution was condensed using a rotary evaporator, and addition of diethyl ether resulted in the formation of a precipitate. After cooling overnight, the isomerized product was collected by filtration, washed with diethyl ether, and then dried in vacuo. The yield was 3 mg (75%). A change of the solvent (acetone/water, 1:20 v/v) brought about a considerable reduction in reaction time (1 h). Similar reactions using d-[Ru(pynp)(bpy)(CO)(CH3CN)](PF6)2 or dp-[Ru(pynp)2(CO)(CH3CN)](PF6)2 gave the corresponding p- or pp-isomer, respectively. These products were characterized by spectroscopic and X-ray crystallographic analyses.
p-[Ru(pynp)(bpy)(CO)(CH3CN)](PF6)2: ESI–MS (CH3CN): m/z 246.5 ([M–CH3CN]2+). IR (KBr): 1992 cm−1 (νCO). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 251 (31,300), 312 (19,600), 343 (21,400), 383 (4700), 490 (1200). p-[Ru(pynp)(phen)(CO)(CH3CN)](PF6)2: ESI–MS (CH3CN): m/z 258.5 ([M–CH3CN]2+). IR (KBr): 1994 cm−1 (νCO). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 268 (34,500), 332sh (11,000), 521 (2200). pp-[Ru(pynp)2(CO)(CH3CN)](PF6)2: ESI–MS (CH3CN): m/z 272.0 ([M–(CH3CN)]2+), 292.5 ([M]2+). IR (KBr): 1994 cm−1 (νCO). Electronic spectrum (CH3CN): λmax/nm (ε/M−1 cm−1) 245 (31,200), 269 (26,900), 343 (28,800), 423sh (3000), 499 (1000).
3.5. X-ray Crystallographic Analyses
Single crystals of [2d]2(PF6)3(CF3SO3)·H2O, [2p](PF6)2·CH3CN, [3dp](CF3SO3)2, and d-[Ru(pynp)(phen)(CO)(CH3CN)](PF6)2 were obtained by vapor diffusion of diethyl ether into an acetonitrile/acetone solution of the complex. p-[Ru(pynp)(phen)(CO)(CH3CN)](PF6)2·CH3OH crystals were obtained by vapor diffusion of diethyl ether into an acetonitrile/acetone/methanol solution of the complex. [Ru(pynp)(CO)2(OH2)(CH3CN)](CF3SO3)2·C2H5OH crystals were obtained by vapor diffusion of diethyl ether into an acetonitrile/acetone/ethanol solution of the complex. Single crystals of dp-[Ru(pynp)2(CO)(CH3CN)](PF6)2·(CH3)2CO and [Ru(pynp)(bpy)(CO)(C(O)OH)]PF6·(CH3)2CO were obtained by vapor diffusion of diethyl ether into an acetone solution of the complex. Single crystals of pp-[Ru(pynp)2(CH3CN)2](PF6)2 were obtained by vapor diffusion of diethyl ether into an acetonitrile solution of the complex. Single crystals of d-[Ru(pynp)(bpy)(CO)(C(O)OC2H5)]PF6·(CH3)2CO and d-[Ru(pynp)(phen)(CO)(C(O)OC2H5)]CF3SO3 were obtained by layering diethyl ether over an acetone/ethanol solution of the complex. Crystal structure determination and refinement data for the complexes are given in Figure 1, Figure 2, Figure 5, Figure 6 and Figure 7, Supplementary Materials Figures S1, S5 and S6, and Table 4, Table 5 and Table 6 and Supplementary Materials Table S1. CCDC-1966877–1966887 contains the supplementary crystallographic data for this paper.
Table 4.
Crystallographic data for dicarbonyl complexes.
Table 5.
Crystallographic data for [Ru(pynp)(N^N)(CO)(CH3CN)]2+ (N^N = phen or pynp).
Table 6.
Crystallographic data for [Ru(pynp)(bpy)(CO)(COR)]+ (R = OH or OC2H5).
4. Conclusions
This study successfully formed non-equivalent CO-ligand environments by lowering the molecular symmetry of the well-known benchmark complex, [Ru(bpy)2(CO)2]2+. In addition to the diastereoselective synthesis of the desired complexes, the relationship between reaction selectivity and coordination geometry was demonstrated using redox properties. We found that the monoacetonitrile complexes undergo thermal isomerization in the d-isomers, whereas the diacetonitrile complexes undergo photoisomerization to give the corresponding p-form. This study will conduct further research on synthetic chemistry, stereochemistry, and structure–reactivity relationships in metal complexes.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/25/1/27/s1, Figure S1: Molecular structure of [Ru(pynp)(CO)2(OH2)(CH3CN)]2+, Figure S2: Optimized structures of the monosubstituted precursor of [3]2+ with the electronic energy difference, Figure S3: Electronic spectra of [1d]2+, [1p]2+, [2d]2+, [2d]2+, and [3dp]2+, Figure S4: Cyclic voltammograms of [2p]2+ at various scan rates, Figure S5: Molecular structure of [Ru(pynp)2(CH3CN)2]2+, Figure S6: Molecular structure of [Ru(pynp)(phen)(CO)(C(O)OC2H5)]+, Table S1: Crystallographic data for [Ru(pynp)(CO)2(OH2)(CH3CN)]2+, [Ru(pynp)2(CH3CN)2]2+, and [Ru(pynp)(phen)(CO)(C(O)OC2H5)]+.
Author Contributions
Syntheses and characterization of compounds, K.A. and R.A.; investigation and data analysis, K.A. and R.A.; crystallography and computational analysis, T.T.; writing—original draft preparation, K.A.; writing—review and editing, D.O.; supervision and funding acquisition, D.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI, grant number JP17K05799.
Acknowledgments
We thank Ryota Kimura at Fukushima University for his experimental assistance at an early stage of the project. We would like to thank Editage (www.editage.com) for English language editing.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Smith, A.P.; Fraser, C.L. Bipyridine ligands. In Comprehensive Coordination Chemistry II; McClevety, J.A., Meyer, T.J., Eds.; Elsevier: Oxford, UK, 2004; Volume 1, pp. 1–23. [Google Scholar]
- Winter, A.; Hoeppener, S.; Newkome, G.M.; Schubert, U.S. Terpyridine-functionalized surfaces: Redox-active, switchable, and electroactive nanoarchitectures. Adv. Mater. 2011, 23, 3484–3498. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. More hydra than Janus – Non-classical coordination modes in complexes of oligopyridine ligands. Coord. Chem. Rev. 2017, 350, 84–104. [Google Scholar] [CrossRef]
- Fukushima, T.; Ghosh, D.; Kobayashi, K.; Ohtsu, H.; Kitagawa, S.; Tanaka, K. Four-electron reduction of a new ruthenium dicarbonyl complex having two NAD model ligands through decarboxylation in water. Inorg. Chem. 2016, 55, 11613–11616. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tanaka, K. Reactivity of CO2 activated on transition metals and sulfur ligands. Inorg. Chem. 2015, 54, 5085–5095. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A molecular ruthenium electrocatalyst for the reduction of carbon dioxide to CO and formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Itabashi, J.; Fukaya, K.; Enomoto, A.; Yoshida, M.; Ishida, H. Unexpected effect of catalyst concentration on photochemical CO2 reduction by trans(Cl)-Ru(bpy)(CO)2Cl2: new mechanistic insight into the CO/HCOO– selectivity. Chem. Sci. 2015, 6, 3063–3074. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, Y.; Fukaya, K.; Yoshida, M.; Ishida, H. trans-(Cl)-[Ru(5,5’-diamide-2,2’-bipyridine)(CO)2Cl2]: Synthesis, structure, and photocatalytic CO2 reduction activity. Chem. Eur. J. 2015, 21, 10049–10060. [Google Scholar] [CrossRef] [PubMed]
- Voyame, P.; Toghill, K.E.; Méndez, M.A.; Girault, H.H. Photoreduction of CO2 using [Ru(bpy)2(CO)L]n+ catalysts in biphasic solution/supercritical CO2 systems. Inorg. Chem. 2013, 52, 10949–10957. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Majumdar, M.; Sinha, A.; Ghatak, T.; Patra, S.K.; Sadhukhan, N.; Rahaman, S.M.W.; Bera, J.K. Mapping the transformation [{RuII(CO)3Cl2}2] to [RuI2(CO)4]2+: Implications in binuclear water-gas shift chemistry. Chem. Eur. J. 2010, 16, 2574–2585. [Google Scholar] [CrossRef]
- Ishida, H.; Tanaka, K.; Morimoto, M.; Tanaka, T. Isolation of intermediates in the water gas shift reactions catalyzed by [Ru(bpy)2(CO)Cl]+ and [Ru(bpy)2(CO)2]2+. Organometallics 1986, 5, 724–730. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, P.; Aathira, M.S.; Singh, D.P.; Behera, B.; Jain, S.L. A bridged ruthenium dimer (Ru-Ru) for photoreduction of CO2 under visible light irradiation. Ind. End. Chem. 2018, 61, 381–387. [Google Scholar] [CrossRef]
- Son, A.; Kawasaki, A.; Hara, D.; Ito, T.; Tanabe, K. Phosphorescent ruthenium complexes with a nitroimidazole unit that image oxygen fluctuation in tumor tissue. Chem. Eur. J. 2014, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carrington, S.J.; Chakraborty, I.; Alvarado, J.R.; Mascharak, P.K. Differences in the CO photolability of cis- and trans-[RuCl2(azpy)(CO)2] complexes: Effect of metal-to-ligand back-bonding. Inorg. Chim. Acta. 2013, 407, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, G.; Kondo, M.; Crisalli, M.; Lee, S.K.; Shibata, A.; Ford, P.C.; Masaoka, S. Syntheses and properties of phosphine-substituted ruthenium(II) polypyridine complexes with nitrogen oxides. Dalton Trans. 2015, 44, 17189–17200. [Google Scholar] [CrossRef]
- Boyer, J.L.; Polyansky, D.E.; Szalda, D.J.; Zong, R.; Thummel, R.P.; Fujita, E. Effects of a proximal base on water oxidation and proton reduction catalyzed by geometric isomers of [Ru(tpy)(pynap)(OH2)]2+. Angew. Chem. Int. Ed. 2011, 50, 12600–12604. [Google Scholar] [CrossRef]
- Ashford, D.L.; Glasson, C.R.K.; Norris, M.R.; Concepcion, J.J.; Keinan, S.; Brennaman, M.K.; Templeton, J.L.; Meyer, T.J. Controlling ground and excited state properties through ligand changes in ruthenium polypyridyl complexes. Inorg. Chem. 2014, 53, 5637–5646. [Google Scholar] [CrossRef]
- Oyama, D.; Abe, R.; Takase, T. CO-ligand photodissociation in two Ru(II) complexes affected by different polypyridyl supporting ligands. Chem. Lett. 2017, 46, 1412–1414. [Google Scholar] [CrossRef]
- The distal and proximal isomers are defined by the relative positions of the monodentate ligand (L) and non-coordinating nitrogen atom of pynp: the d-isomer and p-isomer denote cis(L,py)- and trans(L,py)-, respectively (py: pyridyl group in pynp).
- Oyama, D.; Abe, R.; Takase, T. Coordination chemistry of mononuclear ruthenium complexes bearing versatile 1,8-naphthyridine units: Utilization of specific reaction sites constructed by the secondary coordination sphere. Coord. Chem. Rev. 2018, 375, 424–433. [Google Scholar] [CrossRef]
- Ooyama, D.; Kobayashi, T.; Shiren, K. Tanaka, K. Regulation of electron donating ability to metal center: isolation and characterization of ruthenium carbonyl complexes with N,N- and/or N,O-donor polypyridyl ligands. J. Organomet. Chem. 2003, 665, 107–113. [Google Scholar] [CrossRef]
- Tanaka, H.; Tzeng, B.-C.; Nagao, H.; Peng, S.-M.; Tanaka, K. Comparative study on crystal structures of ruthenium bipyridine carbonyl complexes [Ru(bpy)2(CO)2](PF6)2, [Ru(bpy)2(CO)(C(O)OCH3)]B(C6H5)4·CH3CN, and [Ru(bpy)2(CO)(η1-CO2)]·3H2O (bpy = 2,2’-bipyridyl). Inorg. Chem. 1993, 32, 1508–1512. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Zhao, H.C.; Fu, B.-L.; Schweinfurth, D.; Harney, J.P.; Sarkar, B.; Tsai, M.-K.; Rochford, J. Tuning oxyquinolate non-innocence at the ruthenium polypyridyl core. Eur. J. Inorg. Chem. 2013, 4410–4420. [Google Scholar] [CrossRef]
- Campos-Fernández, C.S.; Ouyang, X.; Dunbar, K.R. A homologous series of redox-active, dinuclear cations with the bridging ligand 2-(2-pyridyl)-1,8-naphthyridine. Inorg. Chem. 2000, 39, 2432–2433. [Google Scholar] [CrossRef] [PubMed]
- Oyama, D.; Ukawa, N.; Hamada, T.; Takase, T. Reversible intramolecular cyclization in ruthenium complexes induced by ligand-centered one-electron transfer on bidentate naphthyridine: An important intermediate for both metal- and organo-hydride species. Chem. Lett. 2015, 44, 533–535. [Google Scholar] [CrossRef]
- Knoll, J.D.; Albani, B.A.; Turro, C. New Ru(II) complexes for dual photoreactivity: Ligand exchange and 1O2 generation. Acc. Chem. Res. 2015, 48, 2280–2287. [Google Scholar] [CrossRef]
- Liu, Y.; Turner, D.B.; Singh, T.N.; Angeles-Boza, A.M.; Chouai, A.; Dunbar, K.R.; Turro, C. Ultrafast ligand exchange: detection of a pentacoordinate Ru(II) intermediate and product formation. J. Am. Chem. Soc. 2009, 131, 26–27. [Google Scholar] [CrossRef]
- Based on isolation and identification of the intermediates, we confirmed that the photoreaction proceeds through the two-step process as shown in Scheme 2.
- Tanaka, S.; Takahashi, K.; Hirahara, M.; Yagi, M.; Onda, K. Characterization of the excited states of distal- and proximal-[Ru(tpy)(pynp)OH2]2+ in aqueous solution using time-resolved infrared spectroscopy. J. Photochem. Photobiol. A 2015, 313, 87–98. [Google Scholar] [CrossRef]
- Hirahara, M.; Ertem, M.Z.; Komi, M.; Yamazaki, H.; Cramer, C.J.; Yagi, M. Mechanisms of photoisomerization and water-oxidation catalysis of mononuclear ruthenium(II) monoaquo complexes. Inorg. Chem. 2013, 52, 6354–6364. [Google Scholar] [CrossRef]
- Oyama, D.; Yuzuriya, K.; Naoi, R.; Hamada, T.; Takase, T. Syntheses of geometrical isomers for comparison of properties caused by steric and electronic effects in carbonylruthenium(II) complexes. Bull. Chem. Soc. Jpn. 2014, 87, 1107–1115. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. The early years of 2,2’-bipyridine – A ligand in its own lifetime. Molecules 2019, 24, 3951. [Google Scholar] [CrossRef]
- Oyama, D.; Hamada, T.; Ukawa, N.; Mochizuki, R.; Takase, T. Isolation and characterization of a metallacyclic compound by selective protection of a single CO ligand in a ruthenium complex. Bull. Chem. Soc. Jpn. 2015, 88, 1572–1574. [Google Scholar] [CrossRef]
- Haukka, M.; Kiviaho, J.; Ahlgren, M.; Pakkanen, A.T. Studies on catalytically active ruthenium carbonyl bipyridine systems. Synthesis and structural characterization of [Ru(bpy)(CO)2Cl2], [Ru(bpy)(CO)2Cl(C(O)OCH3)], [Ru(bpy)(CO)2Cl]2 and [Ru(bpy)(CO)2ClH] (bpy = 2,2’-bipyridine). Organometallics 1995, 14, 825–833. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kikuchi, T.; Kitagawa, S.; Tanaka, K. Selective generation of formamides through photocatalytic CO2 reduction catalyzed by ruthenium carbonyl compounds. Angew. Chem. Int. Ed. 2014, 53, 11813–11817. [Google Scholar] [CrossRef] [PubMed]
- Oyama, D.; Hamada, T.; Takase, T. Stereospecific synthesis and redox properties of ruthenium(II) carbonyl complexes bearing a redox-active 1,8-naphthyridine unit. J. Organomet. Chem. 2011, 696, 2263–2268. [Google Scholar] [CrossRef]
- Oyama, D.; Kainuma, S.; Akatsuka, K.; Abe, R.; Takase, T. Solvent mediated complete trans-to-cis isomerization of [Ru(polypyridine)(CO)2Cl2] complexes. J. Organomet. Chem. 2019, 900, 120883. [Google Scholar] [CrossRef]
- Campos-Fernández, C.S.; Thomson, L.M.; Galán-Mascarós, J.R.; Ouyang, X.; Dunbar, K.R. Homologous series of redox-active, dinuclear cations [M2(O2CCH3)2(pynp)2]2+ (M = Mo, Ru, Rh) with the bridging ligand 2-(2-pyridyl)-1,8-naphthyridine (pynp). Inorg. Chem. 2002, 41, 1523–1533. [Google Scholar] [CrossRef]
- Anderson, P.A.; Deacon, G.B.; Haarmann, K.H.; Keene, F.R.; Meyer, T.J.; Reitsma, D.A.; Skelton, B.W.; Strouse, G.F.; Thomas, N.C.; Treadway, J.A.; et al. Designed synthesis of mononuclear tris(heteroleptic) ruthenium complexes containing bidentate polypyridyl ligands. Inorg. Chem. 1995, 34, 6145–6157. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09W, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds[1d]2+, [1p]2+, [2d]2+, [2p]2+, and [3dp]2+ are available from the authors. |













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).