Analysis and Comparison of Vector Space and Metric Space Representations in QSAR Modeling


Abstract
:1. Introduction
1.1. Molecular Similarity and Metric Space Representation
1.2. Metric Spaces vs. Vector Spaces
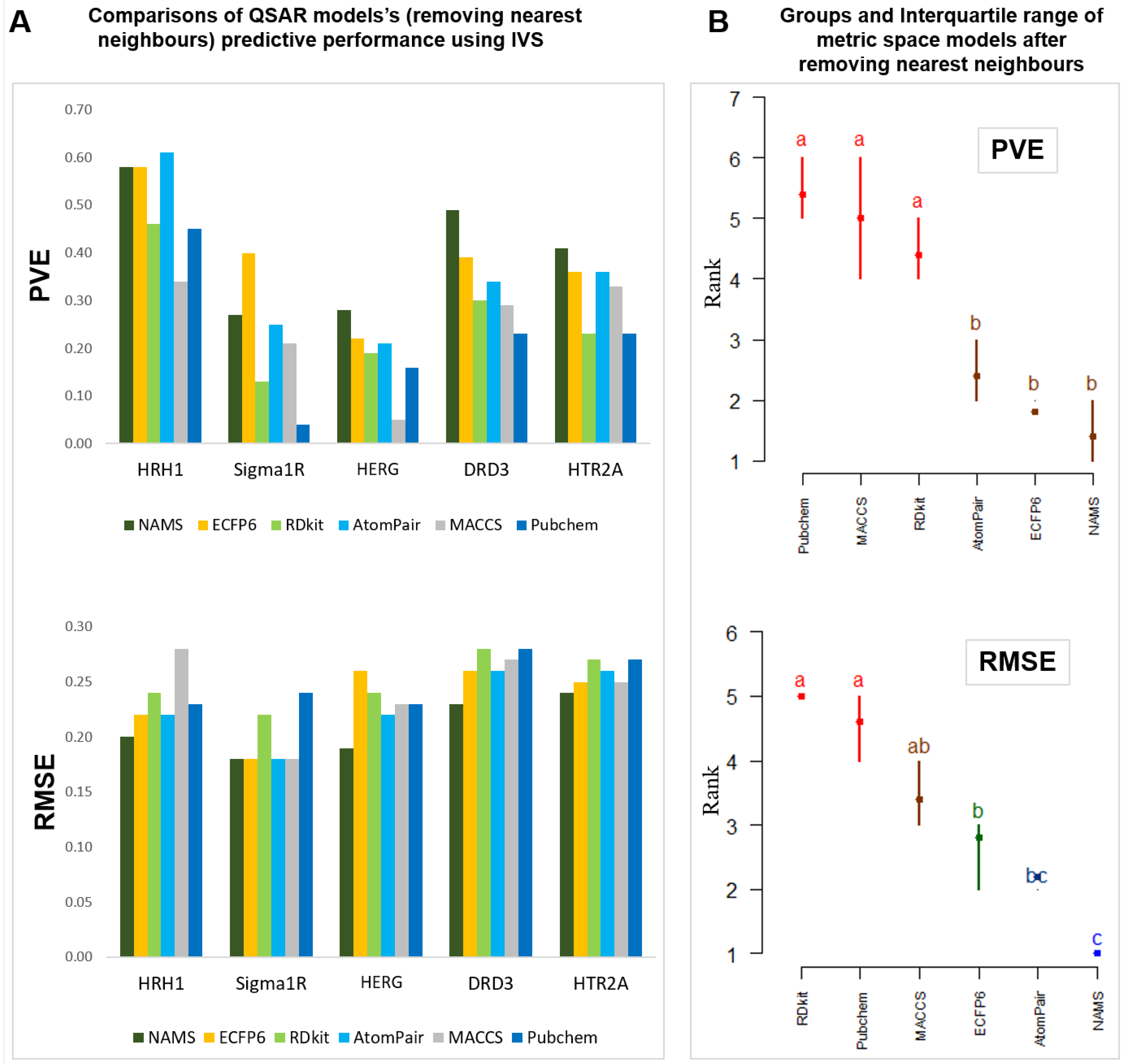
- Is metric space representation as good as the most common vector space-based approaches?
- Which similarity representation carries the maximum chemical/structural information content to establish the best relationship between structural similarity and activity?
- How effective is the reduction of dimensionality of the feature space with principal components by the replacement of explicit descriptors/fingerprints in QSAR modeling?
- Is there any one molecular structure representation method that is generally better than the others?
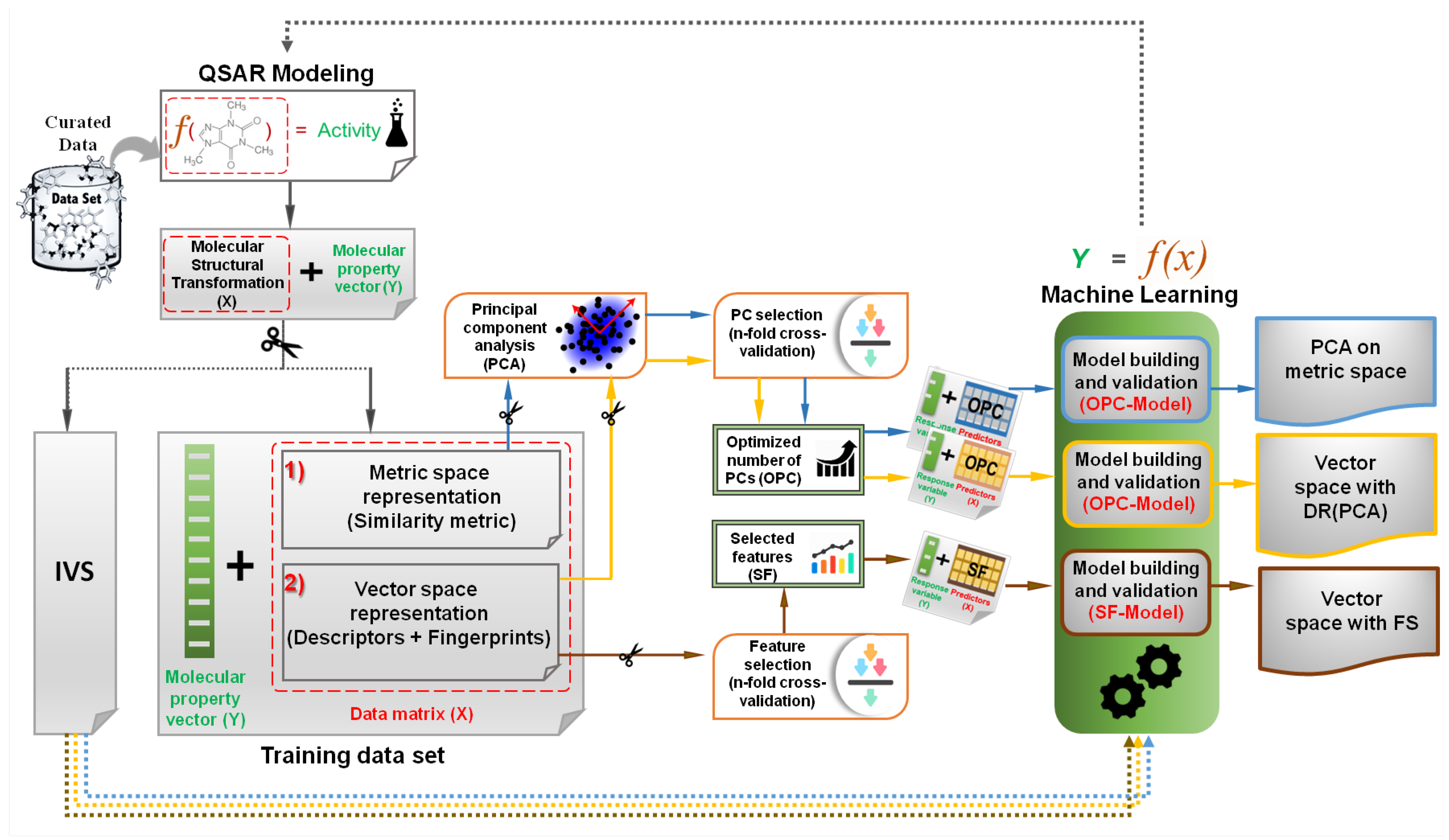
2. Methodology
2.1. Overview of the Methodology
2.2. Vector Space Representation
2.2.1. Descriptor-Based Representations
2.2.2. Fingerprint-Based Representations
- Topological/path-based fingerprints (e.g., Daylight-like RDkit [27,60] and Atom Pair [61]) capture the paths between atom types by describing their different combinations and always assign the same bit’s position to the same substructures within the compared molecules, which sometimes results in bit collisions but is also useful for clustering compounds.
- Circular fingerprints (e.g., ECFP [62]) record circular atom environments that grow radially from the central atom connections. In topological and circular fingerprints, an individual bit has no definite meaning.
2.3. Metric Space Representation
2.3.1. Fingerprint-Based Similarity
2.3.2. NAMS-Based Similarity
2.4. Model Building
2.4.1. Feature Reduction with PCA
2.4.2. Feature Selection with Random Forests
2.4.3. Support Vector Machine
2.4.4. Model Evaluation and External Validation
3. Data
Data Preparation for Vector and Metric Space Representations
4. Results
4.1. Implementation of Analysis
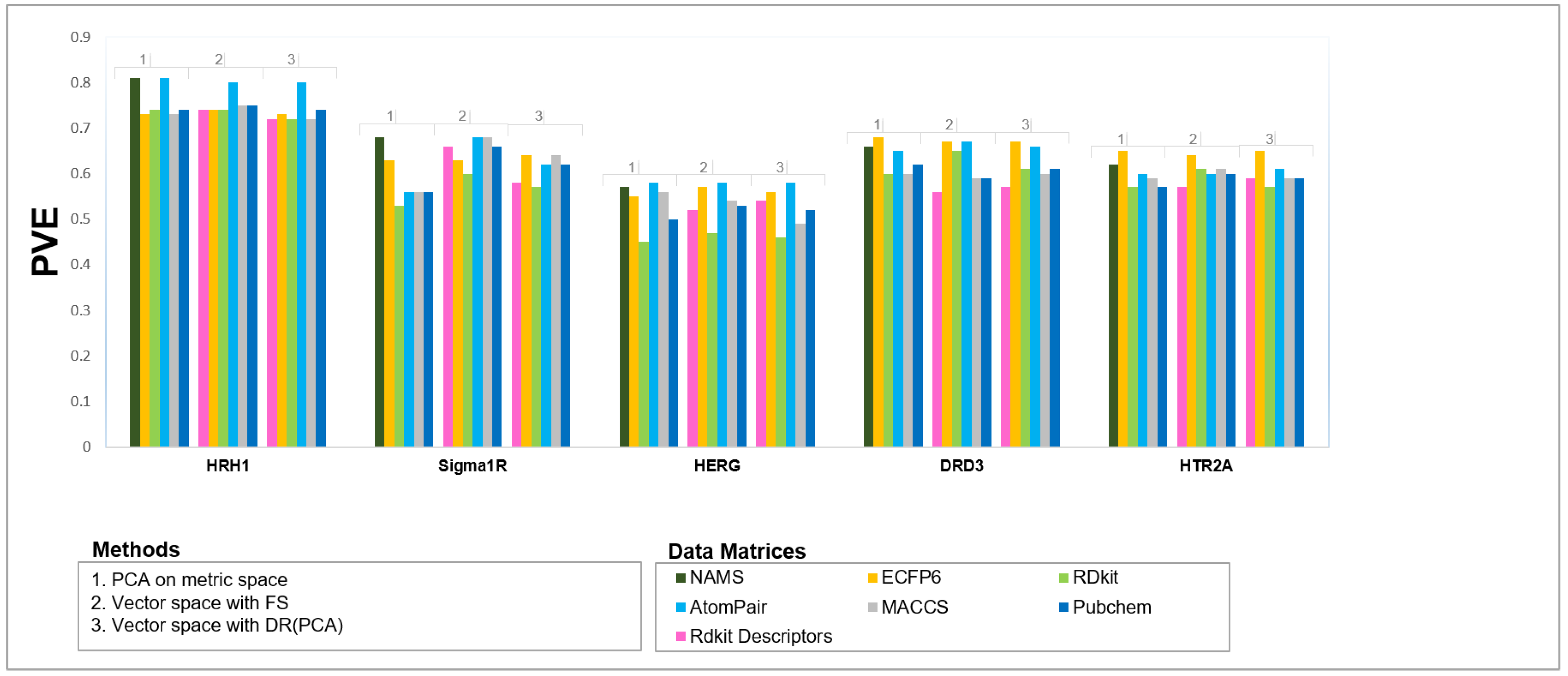
4.2. Results of Generated Models
4.2.1. Is Metric Space Representation as Good as the Most Common Vector Space-Based Approaches?
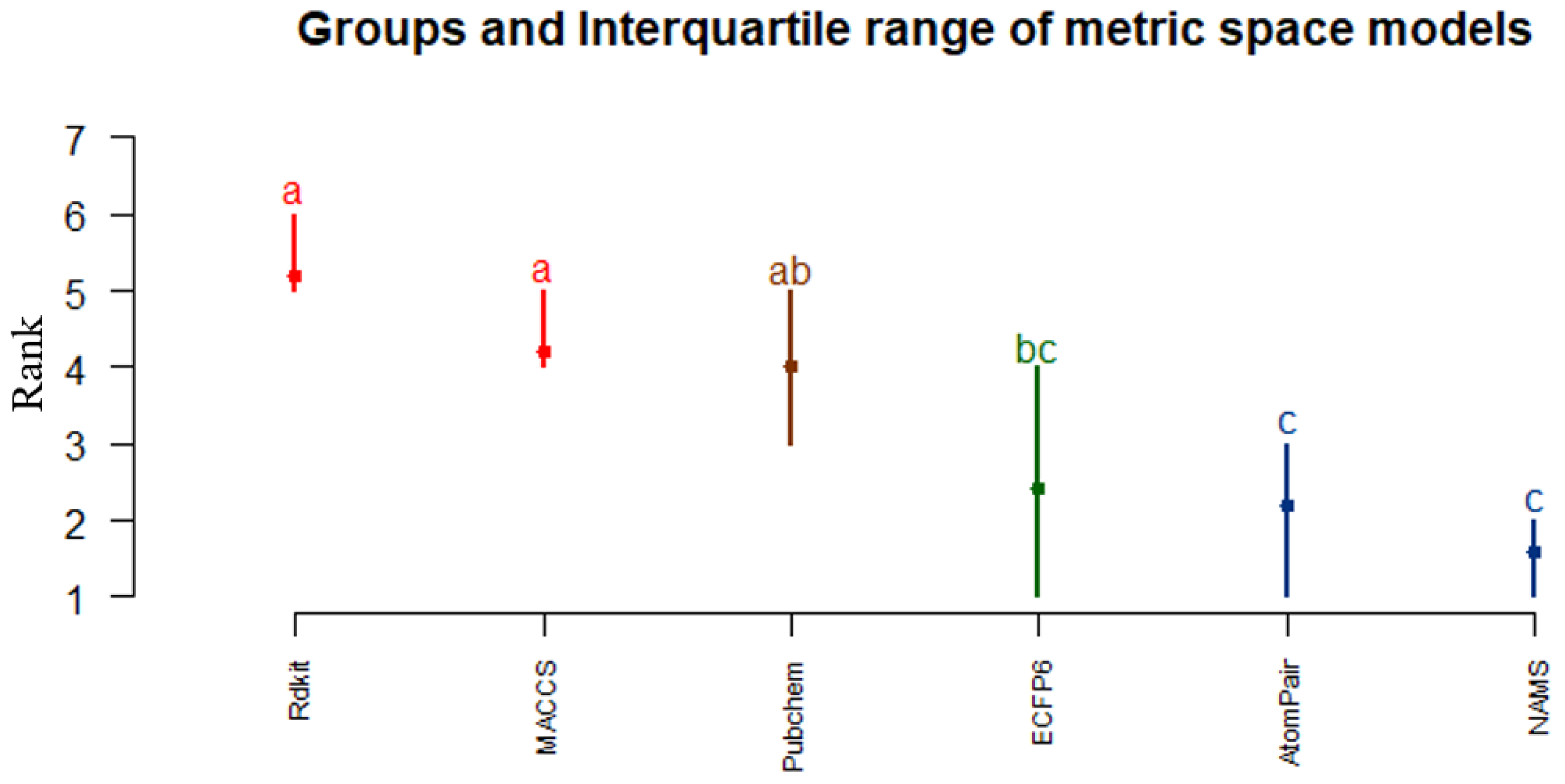
4.2.2. Which Similarity Representation Carries the Maximum Chemical/Structural Information Content to Establish the Best Relationship between Local Similarities and Activity?
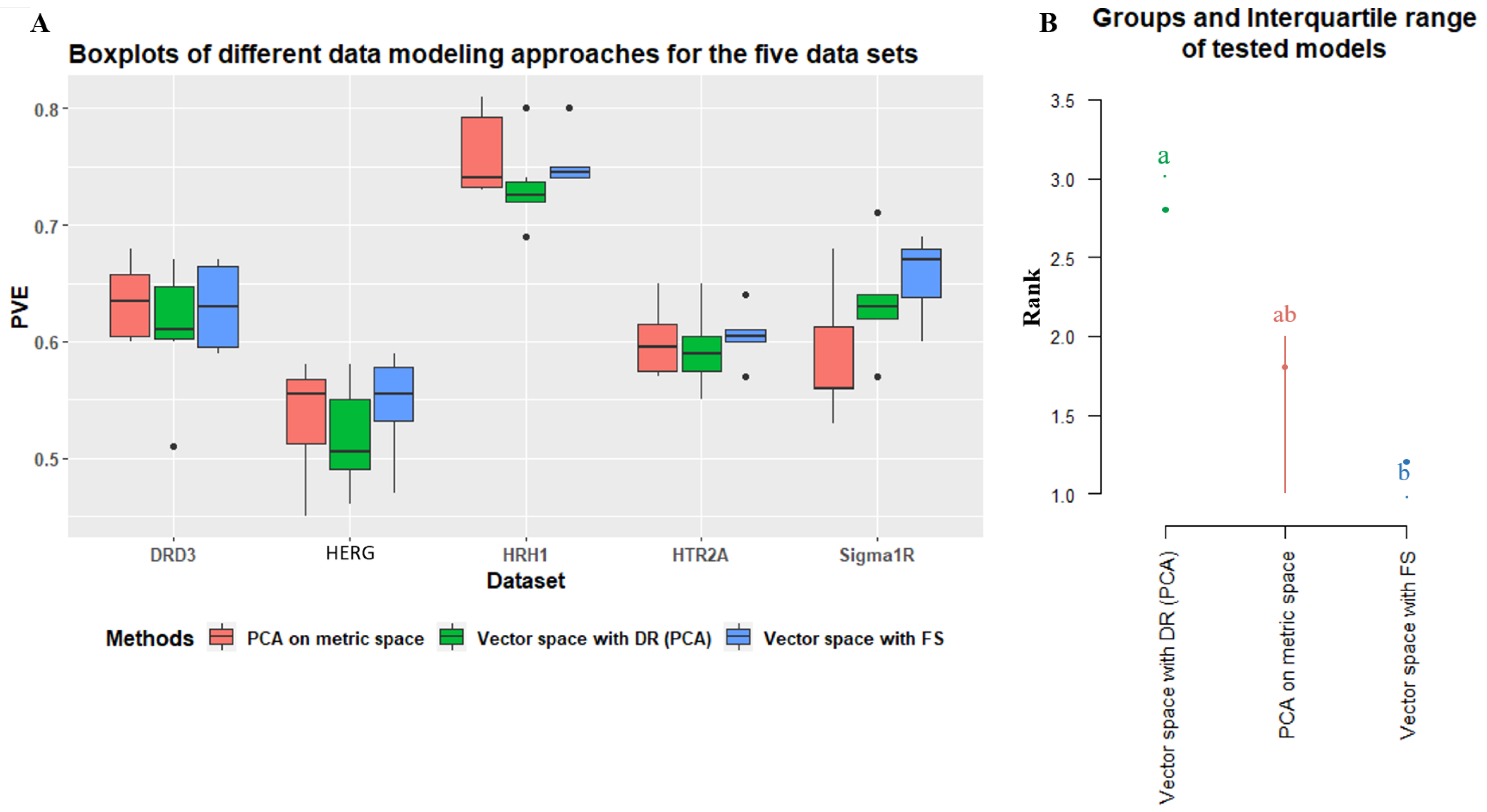
4.2.3. How Effective Is Using a Reduced Dimensionality of the Metric/Vector Space with Principal Components, Replacing Explicit Descriptors/Fingerprints, in QSAR Modeling?
4.2.4. Is There Any Solution That Is Globally Better on a Variety of Difficult Problems?
5. Discussion
Computation Time
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAMS | Non-contiguous atom matching structure similarity |
| DR | Dimensionality reduction |
| PC | Principal component |
| OPC-models | Optimized number of PC-based models |
| SF-models | Selected number of features-based model |
| RF | Random forest |
| SVM | Support vector machines |
| IVS | Independent validation sets |
References
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Arodz, T.; Galvez, J. Computational methods in developing quantitative structure-activity relationships (QSAR): A review. Comb. Chem. High Throughput Screen. 2006, 9, 213–228. [Google Scholar] [CrossRef]
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar] [CrossRef]
- Yoo, C.; Shahlaei, M. The applications of PCA in QSAR studies: A case study on CCR5 antagonists. Chem. Biol. Drug Des. 2017. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, Volume 11; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; p. 688. [Google Scholar]
- Chávez, E.; Navarro, G.; Baeza-Yates, R.; Marroquín, J.L. Searching in Metric Spaces. ACM Comput. Surv. 2001, 33, 273–321. [Google Scholar] [CrossRef]
- Gasteiger, J. Handbook of Chemoinformatics: From Data to Knowledge, Volumes 1–4; Wiley-VCH: Weinheim, Germany, 2008; pp. 1–1870. [Google Scholar]
- O’Boyle, N.M.; Sayle, R.A. Comparing structural fingerprints using a literature-based similarity benchmark. J. Cheminform. 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Yasri, A.; Hartsough, D. Toward an Optimal Procedure for Variable Selection and QSAR Model Building. J. Chem. Inf. Comput. Sci. 2001, 41, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Puzyn, T.; Leszczynski, J.; Cronin, M.T. Recent Advances in QSAR Studies: Methods and Applications (Challenges and Advances in Computational Chemistry and Physics); Springer: Berlin, Germany, 2009. [Google Scholar]
- Dearden, J.C.; Cronin, M.T.D.; Kaiser, K.L.E. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). SAR QSAR Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Golbraikh, A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Lesk, A.M. Introduction to Bioinformatics, 4th ed.; Oxford University Press: Oxford, UK, 2014; p. 400. [Google Scholar]
- Orengo, C.A.; Bateman, A. Protein Families: Relating Protein Sequence, Structure, and Function; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; p. 552. [Google Scholar]
- Teixeira, A.L.; Falcao, A.O. Structural similarity based kriging for quantitative structure activity and property relationship modeling. J. Chem. Inf. Model. 2014, 54, 1833–1849. [Google Scholar] [CrossRef]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do Structurally Similar Molecules Have Similar Biological Activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Nikolova, N.; Jaworska, J. Approaches to Measure Chemical Similarity—A Review. QSAR Comb. Sci. 2003, 22, 1006–1026. [Google Scholar] [CrossRef]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Eckert, H.; Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov. Today 2007, 12, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Stumpfe, D.; Bajorath, J. Similarity searching. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular Similarity Measures. In Methods in Molecular Biology; Springer: Clifton, NJ, USA, 2004; pp. 1–50. [Google Scholar]
- Bajorath, J. Molecular Similarity Concepts for Informatics Applications. In Bioinformatics: Volume II: Structure, Function, and Applications; Keith, J.M., Ed.; Springer: New York, NY, USA, 2017; pp. 231–245. [Google Scholar]
- James, C.; Weininger, D.; Delaney, J. Daylight Theory Manual Version 4.9; Daylight Chemical Information Systems, Inc.: Laguna Niguel, CA, USA, 2011. [Google Scholar]
- Teixeira, A.L.; Falcao, A.O. Noncontiguous atom matching structural similarity function. J. Chem. Inf. Model. 2013, 53, 2511–2524. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.C.; Rarey, M. Maximum common subgraph isomorphism algorithms and their applications in molecular science: A review. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 68–79. [Google Scholar] [CrossRef]
- Raymond, J.W.; Willett, P. Maximum common subgraph isomorphism algorithms for the matching of chemical structures. J. Comput.-Aided Mol. Des. 2002, 16, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.M. Substructure searching methods: Old and new. J. Chem. Inf. Model. 1993, 33, 532–538. [Google Scholar] [CrossRef]
- Flower, D. On the Properties of Bit String-Based Measures of Chemical Similarity. J. Chem. Inf. Model. 1998, 38, 379–386. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 1–13. [Google Scholar] [CrossRef]
- Tversky, A. Features of similarity. Psychol. Rev. 1977, 84, 327–352. [Google Scholar] [CrossRef]
- Leskovec, J.; Rajaraman, A.; Ullman, J.D. Mining of Massive Datasets, 2nd ed.; Cambridge University Press: New York, NY, USA, 2014. [Google Scholar]
- Benigni, R.; Cotta-Ramusino, M.; Giorgi, F.; Gallo, G. Molecular similarity matrices and quantitative structure-activity relationships: A case study with methodological implications. J. Med. Chem. 1995, 38, 629–635. [Google Scholar] [CrossRef] [PubMed]
- So, S.S.; Karplus, M. Three-dimensional quantitative structure-activity relationships from molecular similarity matrices and genetic neural networks. 2. Applications. J. Med. Chem. 1997, 40, 4360–4371. [Google Scholar] [CrossRef]
- Robert, D.; Amat, L.; Carbó-Dorca, R. Quantum similarity QSAR: Study of inhibitors binding to thrombin, trypsin, and factor Xa, including a comparison with CoMFA and CoMSIA methods. Int. J. Quantum Chem. 2000, 80, 265–282. [Google Scholar] [CrossRef]
- Gironés, X.; Carbó-Dorca, R. Molecular quantum similarity-based QSARs for binding affinities of several steroid sets. J. Chem. Inf. Comput. Sci. 2002, 42, 1185–1193. [Google Scholar] [CrossRef]
- Besalú, E.; Gironés, X.; Amat, L.; Carbó-Dorca, R. Molecular quantum similarity and the fundamentals of QSAR. Acc. Chem. Res. 2002, 35, 289–295. [Google Scholar] [CrossRef]
- Carbó-Dorca, R. About the prediction of molecular properties using the fundamental Quantum QSPR (QQSPR) equation †. SAR QSAR Environ. Res. 2007, 18, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Carbó-Dorca, R.; Mezey, P.G. Advances in Molecular Similarity; Number v. 2 in Advances in Molecular Similarity; Elsevier Science: Amsterdam, The Netherlands, 1999; p. 296. [Google Scholar]
- Urbano Cuadrado, M.; Luque Ruiz, I.; Gómez-Nieto, M.Á. A Steroids QSAR Approach Based on Approximate Similarity Measurements. J. Chem. Inf. Model. 2006, 46, 1678–1686. [Google Scholar] [CrossRef]
- Girschick, T.; Almeida, P.R.; Kramer, S.; Staìšlring, J. Similarity boosted quantitative structure-activity relationship—A systematic study of enhancing structural descriptors by molecular similarity. J. Chem. Inf. Model. 2013. [Google Scholar] [CrossRef]
- Luque Ruiz, I.; Gómez-Nieto, M.Á. QSAR classification and regression models for β-secretase inhibitors using relative distance matrices. SAR QSAR Environ. Res. 2018, 29, 355–383. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Kausar, S.; Falcao, A.O. An automated framework for QSAR model building. J. Cheminform. 2018, 10, 1. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Methods and Principles in Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Katritzky, A.R.; Lobanov, V.S.; Karelson, M. QSPR: The correlation and quantitative prediction of chemical and physical properties from structure. Chem. Soc. Rev. 1995, 24, 279–287. [Google Scholar] [CrossRef]
- Gasteiger, J. Handbook of Chemoinformatics; Volumes 1–4; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003; pp. 1–1870. [Google Scholar]
- Bajorath, J. Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery, Volume 275; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Roy, K.; Kar, S.; Das, R.N. Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Varnek, A.; Baskin, I.I. Chemoinformatics as a theoretical chemistry discipline. Mol. Inform. 2011, 30, 20–32. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D. Comparison of Topological, Shape, and Docking Methods in Virtual Screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. Synergies of Virtual Screening Approaches. Mini-Rev. Med. Chem. 2008, 8, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.P.; Kearsley, S.K. Why do we need so many chemical similarity search methods? Drug Discov. Today 2002, 7, 903–911. [Google Scholar] [CrossRef]
- Zhang, Q.; Muegge, I. Scaffold Hopping through Virtual Screening Using 2D and 3D Similarity Descriptors: Ranking, Voting, and Consensus Scoring. J. Med. Chem. 2006, 49, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit Documentation. Release 2018, 1, 1–79. [Google Scholar]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: Definition and applications. J. Chem. Inf. Model. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. PubChem Substructure Fingerprint; U.S. National Library of Medicine: Bethesda, MD, USA, 2009.
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Willett, P. The Calculation of Molecular Structural Similarity: Principles and Practice. Mol. Inform. 2014, 33, 403–413. [Google Scholar] [CrossRef]
- Jasial, S.; Hu, Y.; Vogt, M.; Bajorath, J. Activity-relevant similarity values for fingerprints and implications for similarity searching. F1000Research 2016, 5, 591. [Google Scholar] [CrossRef]
- Han, J.; Kamber, M.; Pei, J. Data Mining: Concepts and Techniques; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer Series in Statistics; Springer: New York, NY, USA, 2009. [Google Scholar]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef]
- Vogt, M.; Stumpfe, D.; Geppert, H.; Bajorath, J. Scaffold Hopping Using Two-Dimensional Fingerprints: True Potential, Black Magic, or a Hopeless Endeavor? Guidelines for Virtual Screening. J. Med. Chem. 2010, 53, 5707–5715. [Google Scholar] [CrossRef]
- Willett, P. Similarity-based approaches to virtual screening. Biochem. Soc. Trans. 2003, 31, 603–606. [Google Scholar] [CrossRef]
- Liu, P.; Long, W. Current mathematical methods used in QSAR/QSPR studies. Int. J. Mol. Sci. 2009, 10, 1978–1998. [Google Scholar] [CrossRef]
- Lima, A.N.; Philot, E.A.; Goulart Trossini, G.H.; Barbour Scott, L.P.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 225–239. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Leal, J.P.; Falcao, A.O. Random forests for feature selection in QSPR models—An application for predicting standard enthalpy of formation of hydrocarbons. J. Cheminform. 2013, 5, 1. [Google Scholar] [CrossRef]
- Statnikov, A.; Wang, L.; Aliferis, C. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinform. 2008, 9, 319. [Google Scholar] [CrossRef]
- Yee, L.C.; Wei, Y.C. Current Modeling Methods Used in QSAR/QSPR. In Statistical Modelling of Molecular Descriptors in QSAR/QSPR; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 1–31. [Google Scholar]
- Varnek, A.; Baskin, I. Machine Learning Methods for Property Prediction in Chemoinformatics. J. Chem. Inf. Model. 2012, 52, 1413–1437. [Google Scholar] [CrossRef]
- Gertrudes, J.C.; Maltarollo, V.G.; Silva, R.A.; Oliveira, P.R.; Honório, K.M.; da Silva, A.B.F. Machine learning techniques and drug design. Curr. Med. Chem. 2012, 19, 4289–4297. [Google Scholar] [CrossRef]
- Dobchev, D.; Pillai, G.; Karelson, M. In silico machine learning methods in drug development. Curr. Top. Med. Chem. 2014, 14, 1913–1922. [Google Scholar] [CrossRef]
- González, M.P.; Terán, C.; Saíz-Urra, L.; Teijeira, M. Variable selection methods in QSAR: An overview. Curr. Top. Med. Chem. 2008, 8, 1606–1627. [Google Scholar] [CrossRef]
- Dehmer, M.; Varmuza, K.; Bonchev, D.; Emmert-Streib, F. Statistical Modelling of Molecular Descriptors in QSAR/QSPR; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2012; p. 32434. [Google Scholar]
- Genuer, R.; Poggi, J.M.; Tuleau-Malot, C. Variable selection using Random Forests. Pattern Recognit. Lett. 2012, 31, 2225–2236. [Google Scholar] [CrossRef]
- Zaki, J.M.; Meira, W. Data Mining and Analysis: Fundamental Concepts and Algorithms; Cambridge University Press: New York, NY, USA, 2014. [Google Scholar]
- Lee, J.A.; Verleysen, M. Nonlinear Dimensionality Reduction; Information Science and Statistics; Springer: New York, NY, USA, 2007. [Google Scholar]
- Eriksson, L.; Andersson, P.L.; Johansson, E.; Tysklind, M. Megavariate analysis of environmental QSAR data. Part I—A basic framework founded on principal component analysis (PCA), partial least squares (PLS), and statistical molecular design (SMD). Mol. Divers. 2006, 10, 169–186. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Petrukhin, R.; Tatham, D.; Basak, S.; Benfenati, E.; Karelson, M.; Maran, U. Interpretation of Quantitative Structure-Property and -Activity Relationships. J. Chem. Inf. Comput. Sci. 2001, 41, 679–685. [Google Scholar] [CrossRef]
- Genuer, R.; Poggi, J.M.; Tuleau, C. Random Forests: Some methodological insights. Inria 2008, 6729, 32. [Google Scholar]
- Biau, G. Analysis of a Random Forests Model. J. Mach. Learn. Res. 2012, 13, 1063–1095. [Google Scholar]
- Spiess, A.N.; Neumeyer, N. An evaluation of R2 as an inadequate measure for nonlinear models in pharmacological and biochemical research: A Monte Carlo approach. BMC Pharmacol. 2010, 10, 6. [Google Scholar] [CrossRef]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz Information Miner. SIGKDD Explor. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2011. [Google Scholar] [CrossRef]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. Misc Functions of the Department of Statistics (e1071), TU Wien; R Development Core Team: Vienna, Austria, 2014. [Google Scholar]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Kassambara, A.; Mundt, F. Package ‘Factoextra’ for R: Extract and Visualize the Results of Multivariate Data Analyses; R Development Core Team: Vienna, Austria, 2017. [Google Scholar]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2006, 46, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust but verify: On the importance of chemical structure curation in chemoinformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Fourches, D.; Tropsha, A. Using graph indices for the analysis and comparison of chemical datasets. Mol. Inform. 2013, 32, 827–842. [Google Scholar] [CrossRef]
- Young, D.; Martin, T.; Venkatapathy, R.; Harten, P. Are the chemical structures in your QSAR correct? QSAR Comb. Sci. 2008, 27, 1337–1345. [Google Scholar] [CrossRef]
- Golbraikh, A.; Muratov, E.; Fourches, D.; Tropsha, A. Data set modelability by QSAR. J. Chem. Inf. Model. 2014, 54, 1–4. [Google Scholar] [CrossRef]
- Golbraikh, A.; Fourches, D.; Sedykh, A.; Muratov, E.; Liepina, I.; Tropsha, A. Modelability Criteria: Statistical Characteristics Estimating Feasibility to Build Predictive QSAR Models for a Dataset; Springer: Boston, MA, USA, 2014; pp. 187–230. [Google Scholar]
- Marcou, G.; Horvath, D.; Varnek, A. Kernel Target Alignment Parameter: A New Modelability Measure for Regression Tasks. J. Chem. Inf. Model. 2016, 56, 6–11. [Google Scholar] [CrossRef]
- Hollander, M.; Wolfe, D.; Chicken, E. Nonparametric Statistical Methods, 3rd ed.; Wiley Series in Probability and Statistics; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Mendiburu, F.D. Agricolae: Statistical Procedures for Agricultural Research; R Package Version 1.2-8; R Package Team: Vienna, Austria, 2017. [Google Scholar]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Öberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical assessment of QSAR models of environmental toxicity against tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef]
- Zhu, H.; Tropsha, A.; Fourches, D.; Varnek, A.; Papa, E.; Gramatical, P.; Öberg, T.; Dao, P.; Cherkasov, A.; Tetko, I.V. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J. Chem. Inf. Model. 2008, 48, 766–784. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot ID. | Gene Name | Target Protein Name | Associated Bioactivities (Y) | Total Number of Observations (N-Processed) |
|---|---|---|---|---|
| P35367 | HRH1 | Histamine H1 receptor | Ki | 1222 |
| Q99720 | SIGMAR1 | Sigma non-opioid intracellular receptor 1 | Ki | 226 |
| Q12809 | HERG | Potassium voltage-gated channel subfamily H member 2 | Ki | 1481 |
| P35462 | DRD3 | D(3) dopamine receptor | Ki | 2902 |
| P28223 | HTR2A | 5-hydroxytryptamine receptor 2A | Ki | 2088 |
| Target Protein Name | Data Size without Removing Nearest Neighbors | NAMS | ECFP6 | RDkit | Atom Pair | MACCS | Pubchem | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thr | N | Thr | N | Thr | N | Thr | N | Thr | N | Thr | N | ||
| Histamine H1 receptor (HRH1) | 1222 | 0.80 | 379 | 0.55 | 378 | 0.80 | 371 | 0.67 | 376 | 0.84 | 379 | 0.87 | 391 |
| Sigma non-opioid intracellular receptor 1 (Sigma1R) | 226 | 0.87 | 312 | 0.61 | 310 | 0.89 | 305 | 0.75 | 309 | 0.92 | 311 | 0.94 | 321 |
| Potassium voltage-gated channel subfamily H member 2 (HERG) | 1481 | 0.80 | 397 | 0.54 | 394 | 0.82 | 392 | 0.69 | 395 | 0.83 | 395 | 0.86 | 403 |
| D(3) dopamine receptor (DRD3) | 2902 | 0.80 | 478 | 0.52 | 481 | 0.77 | 470 | 0.67 | 480 | 0.87 | 484 | 0.86 | 484 |
| 5-hydroxytryptamine receptor 2A (HTR2A) | 2088 | 0.80 | 432 | 0.47 | 432 | 0.78 | 424 | 0.63 | 426 | 0.83 | 429 | 0.85 | 437 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kausar, S.; Falcao, A.O. Analysis and Comparison of Vector Space and Metric Space Representations in QSAR Modeling. Molecules 2019, 24, 1698. https://doi.org/10.3390/molecules24091698
Kausar S, Falcao AO. Analysis and Comparison of Vector Space and Metric Space Representations in QSAR Modeling. Molecules. 2019; 24(9):1698. https://doi.org/10.3390/molecules24091698
Chicago/Turabian StyleKausar, Samina, and Andre O. Falcao. 2019. "Analysis and Comparison of Vector Space and Metric Space Representations in QSAR Modeling" Molecules 24, no. 9: 1698. https://doi.org/10.3390/molecules24091698
APA StyleKausar, S., & Falcao, A. O. (2019). Analysis and Comparison of Vector Space and Metric Space Representations in QSAR Modeling. Molecules, 24(9), 1698. https://doi.org/10.3390/molecules24091698