Abstract
Heteroatom-substituted alkynes have attracted a significant amount of interest in the synthetic community due to the polarized nature of these alkynes and their utility in a wide range of reactions. One specific class of heteroatom-substituted alkynes combines this utility with the presence of an azole moiety. These N-alkynyl azoles have been known for nearly 50 years, but recently there has been a tremendous increase in the number of reports detailing the synthesis and utility of this class of compound. While much of the chemistry of N-alkynyl azoles mirrors that of the more extensively studied N-alkynyl amides (ynamides), there are notable exceptions. In addition, as azoles are extremely common in natural products and pharmaceuticals, these N-alkynyl azoles have high potential for accessing biologically important compounds. In this review, the literature reports of N-alkynyl azole synthesis, reactions, and uses have been assembled. Collectively, these reports demonstrate the growth in this area and the promise of exploiting N-alkynyl azoles in synthesis.
1. Introduction
N-Alkynyl azoles, that is, alkynes containing an azole substituent through the ring nitrogen atom (Figure 1), have attracted a significant amount of interest over the last several decades, in part due to the significance of the two individual fragments: the azole and the alkyne. Alkynes are a fundamental building block and functional group that is indispensible to synthetic chemists, while azoles are extremely common in biomolecules and pharmaceuticals. The chemistry of N-alkynyl azoles represents just a fraction of the nitrogen-substituted alkyne field. Over the years, there have been many reviews of ynamines [1,2,3] and ynamides [3,4,5,6], and some of these have included N-alkynyl azoles within their content; however, the only review that primarily focused on this class of compounds was published in 2004 [7], and in the last 15 years there has been a significant amount of work published in this area.
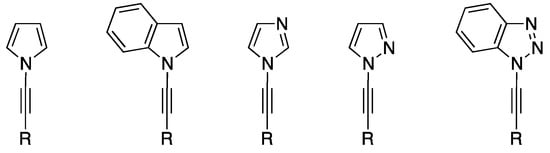
Figure 1.
Examples of N-alkynyl azoles.
Researchers were initially drawn to nitrogen-substituted alkynes for their structural relationship with enamines (nitrogen-subsituted alkenes); however, the general instability and highly reactive nature of ynamines made them undesirable research subjects and interest in them ultimately waned. Eventually, the discovery that electron-withdrawing groups on the nitrogen could stabilize these compounds led to a burgeoning interest in the synthesis and reactivity of ynamides and rejuvenated the field of nitrogen-substituted alkynes. Due to the diverse nature of the many different heterocycles that make up the azole family, there is not a single reactivity profile that defines all N-alkynyl azoles. However, in an analogous manner to ynamines being stabilized with electron-withdrawing groups, N-alkynyl azoles can be stabilized by modulating their electronic properties with functional group substitution. Over the past half century, since the first report of an N-alkynyl azole by Okamoto in 1970 [8], researchers have taken advantage of the properties of N-alkynyl azoles to develop new materials, new cytotoxic molecules, synthesize natural products, investigate new methodologies, and much more. Here we present a comprehensive review of the synthesis, reactions, and applications of N-alkynyl azoles, from Okamoto’s initial studies to the present.
2. Synthesis of N-Alkynyl Azoles
2.1. Elimination
Dehydrohalogenation and other elimination procedures with N-alkenyl azoles constitute the original strategy towards preparing N-alkynyl azoles. These strategies are convenient for researchers due to the accessibility of the starting materials and the straightforward nature of the process. Dehydrohalogenation is a classic method for preparing alkynes and was an already established method for the synthesis of related N-alkynyl amines and amides [9,10]. The dehydrochlorination of N-(α,β-dichloroethenyl)carbazole (5) in 1970 by Kundu and Okamoto to form N-ethynylcarbazole (3) represents one of the earliest examples of this procedure applied to the synthesis of an N-alkynyl azole (Scheme 1) [8]. In their work, they found that they were unable to synthesize alkyne 3 from the α,β-dihalo derivative N-(α,β-dibromoethyl)carbazole (2) and sought an alternate route. They ultimately found success in generating 3 from N-acetylcarbazole (4) by treatment with phosphorus pentachloride in refluxing benzene followed by elimination with sodium amide in liquid ammonia.
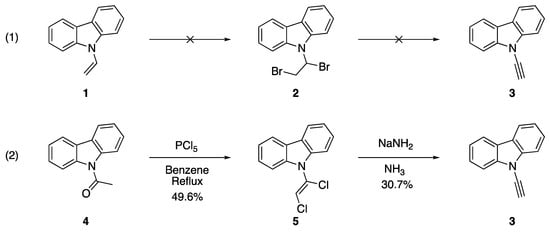
Scheme 1.
Kundu and Okamoto’s synthesis of N-ethynylcarbazole.
In 1995, researchers Pielichowski and Chrzaszcz also became interested in the synthesis of N-ethynylcarbazole (3) and sought to improve on the original preparation by Kundu and Okomoto. The researchers found success in the synthesis of dichlorovinylcarbazoles via the reaction of carbazole and carbazole derivatives with trichloroethylene (TCE) under phase-transfer conditions, using triethylbenzylammonium chloride (TEBACl) as a phase-transfer catalyst (Scheme 2) [11]. This procedure allowed them to efficiently synthesize 7 from carbazole (6). Subsequent elimination with magnesium in dry tetrahydrofuran (THF) gave good yields of N-ethynylcarbazole (3). Pielichowski and Chrzaszcz highlighted the exceptional reactivity of N-ethynylcarbazole derivatives in their work, noting these compounds should be kept in the dark at low temperature and that they readily react with water in alcoholic solvents [11].
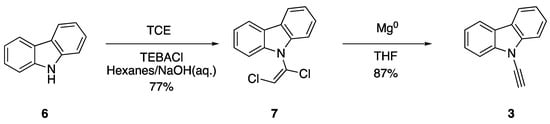
Scheme 2.
Pielichowski and Chrzaszcz’s phase-transfer synthesis of N-ethynylcarbazole.
Burger and Dreier published a rather unusual synthesis of an N-alkynyl azole in 1983 [12]. Their synthesis, although unexpected, represents just the second report of a synthesis on an N-alkynyl azole and the first reported synthesis on an N-alkynyl pyrrole (Scheme 3). The researchers were studying the reactions of nitrogen containing heteroaromatic anions with chlorocarbene, an extension of previously established ring expansions of heterocyclic compounds with halocarbenes [13]. Some of the substrates the researchers studied exhibited the anticipated reactivity. For example, compound 9, the lithium salt of 3H-pyrrolizine (8), reacted with chlorocarbene to give two compounds: the bicyclobutane derivative pyrrole-3-azabenzvalene 10 and indolizine (11), a ring-expanded product. On the other hand, they found that lithium anion 13, formed by treating dipyrrolo [1.2c:2′,1′e]2H-imidazole (12) with n-butyllithium, reacted with chlorocarbene to give N-alkynyl pyrrole 14 in a 47% yield, while generating none of the expected ring expanded product 15. Burger and Dreier proposed a mechanism for the formation of 14, suggesting that an elimination of pyrrole from the initially formed 16, followed by a dehydrohalogenation could generate the observed product [12].
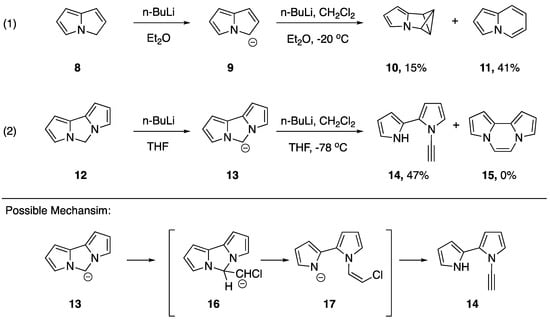
Scheme 3.
Burger and Dreier’s synthesis of an N-alkynyl pyrrole.
In 1992, Paley and coworkers published the first targeted synthesis of an N-alkynyl pyrrole [14]. The researchers were interested in studying the properties and characteristics of poly(diacetylenes) and using computer modeling to aid in both the prediction of their properties and in the screening of potentially interesting target compounds. This work led them to develop a synthesis of N-ethynyl-pyrrole (21, Scheme 4). Paley’s strategy parallels the route of Kundu and Okamoto for the synthesis of N-ethynylcarbazole. Starting with pyrrole (18) they were able to easily generate the potassium salt 19. Following decanting of the solvents the salt was refluxed in TCE overnight and N-(α,β-dichlorovinyl)pyrrole (20) was obtained in moderate yields. Subsequent elimination with methyllithium in diethyl ether gave the desired N-ethynyl pyrrole in a 90% yield.
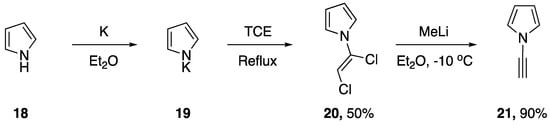
Scheme 4.
Paley and coworkers’ targeted synthesis of N-ethynylpyrrole.
In 1994, Trofimov also published a synthesis of N-ethynylpyrrole (21) [15]. Trofimov’s research program was focused on the chemistry of N-substituted pyrroles, and in an attempt to investigate the chemistry of N-ethynyl pyrrole, they found Paley and coworkers’ synthesis unsatisfactory. Trofimov was able to avoid the use of both potassium metal and TCE as a solvent by generating dichloroacetylene in situ. His procedure involves using potassium tert-butoxide in THF with TCE to generate the dichloroacetylene to which pyrrole readily adds. Trofimov also uses methyllithium for the elimination.
As part of their work studying the biological activity of various chloro-enamines and N-alkynyl amines, in 1994, Zemlicka described a synthesis of N-alkynyl purines (Scheme 5) [16]. During their investigations, they found the reaction of adenine (22) with dichloroacetylene to be disappointing, providing the corresponding N9-dichloroenamine in only a 3% yield. Ultimately, they did have success with the addition of purines 22 and 25 to tetrachloroethylene using sodium hydride as a base and hexamethylphosphoramide (HMPA) as a solvent. The authors suggest these reactions likely proceed via an addition-elimination mechanism (since tetrachloroethylene is unable to react via elimination-addition). Subsequently, the researchers were able to convert the N9-(trichlorovinyl)purine derivatives 23 and 26 into the respective N9-alkynyl purines by carefully subjecting them to n-BuLi in THF at −70 °C, providing 24 and 27 in 57% and 42% yields, respectively. In addition, the authors mention that although they were able to synthesize N9-(dichlorovinyl)purines and N9-(chlorovinyl)purines, they only managed to eliminate the trichlorovinyl derivatives to the corresponding alkynes.
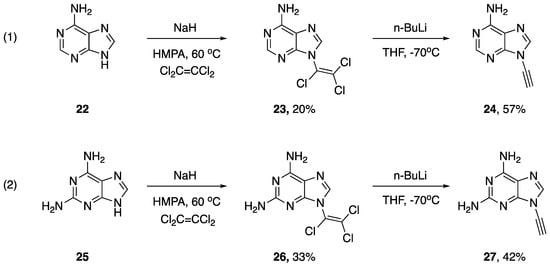
Scheme 5.
Zemlicka’s synthesis of N9-alkynyl purines.
N1-alkynyl benzotriazoles have found applications in the synthesis of disubstituted alkynes and in an alternative procedure to the Arndt-Eistert homologation of carboxylic acids [17,18]. In 2006, Katritzky published a dehydrohalogenation strategy for the rapid synthesis of various N1-alkynyl benzotriazoles (Scheme 6) [19]. This approach is appealing both for the relatively few steps involved and the accessibility of the starting materials. Starting from commercially available 28, treatment with carbon tetrachloride and triphenylphosphine in refluxing THF afforded the 1-(2,2-dichlorovinyl)-benzotriazole (29) in a 68% yield. Dehydrohalogenation of intermediate 29 followed by trapping of the transiently formed lithium acetylide with various electrophiles gave N1-alkynyl benzotriazoles 30a–e in varying yields. Katritzky observed a significant drop in yield when changing the electrophile from methyl iodide to ethyl iodide suggesting this strategy might not be suitable for synthesizing N1-alkynyl benzotriazoles with branched alkyl substitutions on the alkyne.
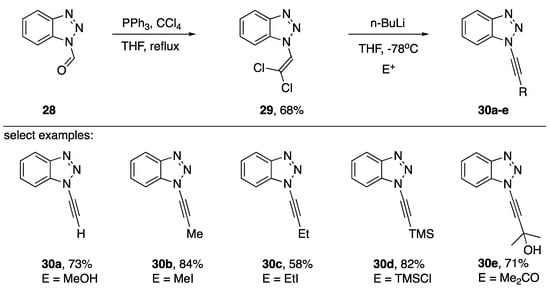
Scheme 6.
Katritzky’s ‘Corey-Fuchs’ approach to various N1-alkynyl benzotriazoles.
In 2015, Anderson published a more general approach to N-alkynyl amide synthesis, including several examples of N-alkynyl azoles (Scheme 7) [20].
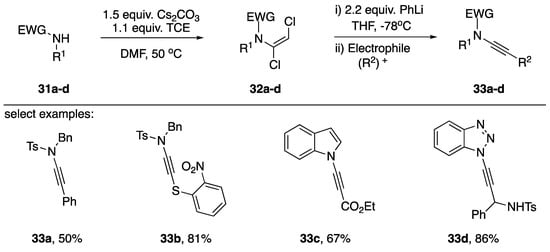
Scheme 7.
Anderson’s synthesis of N-alkynyl amides and N-alkynyl azoles.
In a similar approach as Katritzky, the researchers found they were able to trap an intermediate N-alkynyl lithium acetylide with various electrophiles. Interestingly, after transmetalation with ZnCl2 the researchers were able to perform an in situ Negishi coupling with iodobenzene to afford the N-alkynyl tosylamide 33a. Additionally, the researchers managed to generate sulfur-substituted N-alkynyl tosylamides when using the sulfur based electrophile 33b. Although Anderson reported the synthesis of many 1,2-vinyldichlorides of azoles, there were only two examples presented in which they completed the synthesis to generate the N-alkynyl azole. Using their strategy, they were able to make N-alkynyl indole 33c in a 67% yield and N1-alkynyl benzotriazole 33d in an 86% yield. The author suggests the advantages of this approach consist of a larger substrate tolerance and greater practicality to metal-catalyzed strategies; however, the use of phenyllithium precludes the use of substrates with electrophilic functional groups.
Zhao reported a transition-metal free one-step synthesis of N-alkynyl amides in 2018 (Scheme 8) [21]. In the publication, the researchers included several examples of the application of the new methodology towards the synthesis of N-alkynyl azoles. They synthesized N-alkynyl indoles 36a, 36c, and 36e in 90%, 88%, and 82% yields, respectively. Additionally, N-alkynyl carbazoles 36b and 36d were isolated in 95% and 94% yields. The methodology does have limitations: Zhao mentions their failed attempts to prepare alkyl substituted alkynes. Zhao suggests the reaction proceeds first by an elimination of the 1,1-divinylchlorides to the corresponding alkynyl-chloride followed by an addition of the nitrogen nucleophile and subsequent elimination to the N-alkynyl amide or N-alkynyl azole. In the case of aromatic substrates, the researchers explain that the nucleophile may prefer to add α to the chloride because the aromatic ring can stabilize the negatively charged intermediate. However, in the case of alkyl substrates this stabilization is not available and chloroenamides are isolated, suggesting a β-position addition of the nucleophiles is preferred in these cases.
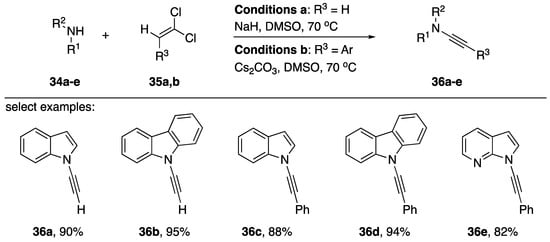
Scheme 8.
Zhao’s transition-metal free one-step synthesis of N-alkynyl amides.
In addition to dehydrohalogenation methods for the synthesis of N-alkynyl azoles there have been several publications reporting eliminations with alternative leaving groups. Interestingly, nearly all of these methods have been disclosed by Katritzky. In 1997, Katritzky reported a Shapiro-type elimination as a new method for synthesizing alkynes from esters and within this publication the author discloses the synthesis of an N-alkynyl pyrrole (Scheme 9) [22]. Starting with pyrrole-benzotriazole intermediate 37, the researchers were able to deprotonate and subsequently acylate the bridging methylene generating ketone 39 in good yields. Reaction of the ketone with (p-toluenesulfonyl)hydrazine easily converted the ketone into the corresponding hydrazone 40, and by subjecting hydrazone 40 to six equivalents of n-BuLi the authors were able to synthesize N-alkynyl pyrrole 41 in a moderate 42% yield.
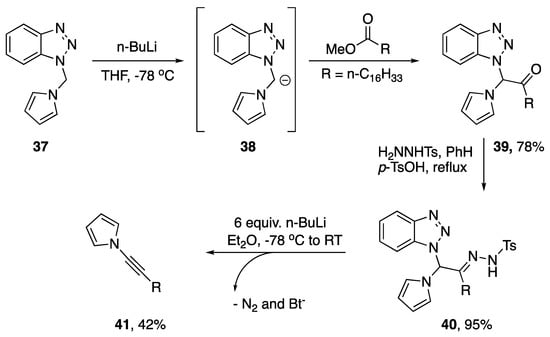
Scheme 9.
Katritzky’s Shapiro-type synthesis on an N-alkynyl pyrrole.
In 2001 and 2002, Katritzky reported on two separate transformations involving N-alkynyl benzotriazoles. In 2001, Katritzky developed an alternative protocol to the Arndt-Eistert homologation that involves an N1-alkynyl benzotriazole intermediate [17]; and in 2002, the researchers used N1-alkynyl benzotriazoles in the synthesis of disubstituted alkynes [18]. In both these publications, Katritzky used the same method to prepare the N1-alkynyl benzotriazoles (Scheme 10). Starting with (chloromethyl)trimethylsilane and benzotriazole they easily prepared the starting benzotriazole 42. This, upon treatment with an acid chloride in refluxing solvent, smoothly afforded ketones such as 43. Subsequent reaction of the ketones with triflic anhydride gave the related enol-triflates 44. Finally, elimination with aqueous NaOH gave N1-alkynyl benzotriazoles 45. The elimination of enol-phosphates has also been applied towards the synthesis of N-alkynyl azoles. In 2015, An and Dong published a one-pot enol-phosphate-elimination strategy towards the synthesis of various N-alkynyl benzimidazoles and P-alkynyl phosphonates [23].
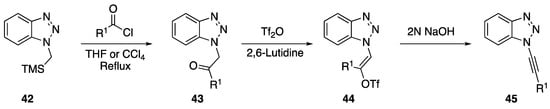
Scheme 10.
Katritzky’s N1-alkynyl benzotriazole synthesis.
2.2. Alkynyliodonium Salts
Alkynyliodonium salts have received a lot of attention from the synthetic community for their usefulness as synthons for cationic alkynes and have been used extensively as alkyne-transfer reagents [24,25]. It is widely believed that these reagents undergo a Michael-type addition followed by a rearrangement to transfer an alkyne to a nucleophile [25]. This process involves three steps: (1) the nucleophile attacks the β-carbon of the alkynyliodonium salt generating an alkenylideneiodonium ylide, (2) the ylide eliminates iodobenzene and generates an alkenylidenecarbene, and (3) the carbene rearranges into an alkyne (Scheme 11).
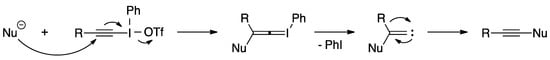
Scheme 11.
Mechanism of alkynyliodonium salts as alkyne-transfer reagents.
In 1998, Kitamura reported using alkynyl(phenyl)iodonium tosylates as alkyne transfer reagents for the synthesis of N1-alkynyl benzotriazoles (Scheme 12) [26,27]. They explored a handful of different transfer reagents (47a–d) and found that they could form the corresponding alkynes 48a–d in moderate yields (45–62%). They were able to provide support for the mechanism by trapping the intermediate carbene with tBuOH and also trapping the carbene via 1,5 C-H insertion. However, this methodology is limited by the mechanism, Kitamura suggests that the substituent on the alkynylation reagent needs to have a high migratory aptitude and that it cannot be aliphatic.
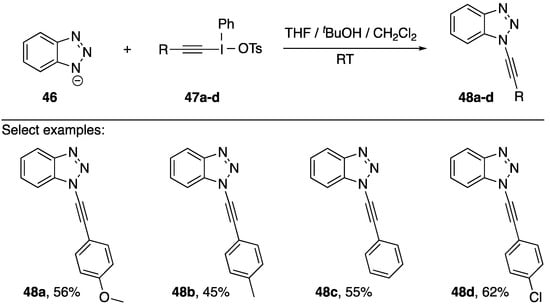
Scheme 12.
Kitamura’s synthesis of N1-alkynyl benzotriazoles.
More than a decade after their initial publication, Kitamura et al. published a follow-up synthesis of N2-alkynyl benzotriazoles (Scheme 13) [28]. Kitamura believes this is the first example of a regioselective functionalization of benzotriazole at the 2-position. The researchers found that when carrying out an alkyne transfer reaction with trimethylsilylethynyliodonium triflate (49a) under very similar reaction conditions to their previously published work, they isolated the isomeric N2-alkynyl benzotriazole 50a as the major product of the reaction. Alkyne transfer reagent 49b performed similarly; however, reagent 49c in which the alkyne has no substitution gave a near statistical ratio of the two isomers. Kitamura suggests that the differences in regioselectivity may be attributable to a steric interaction, but he does not provide any insight into the origin of the regioselective preference for the alkyne-transfer reactions with arylethynyliodonium salts.
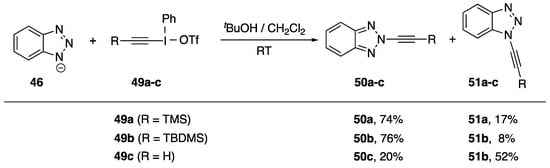
Scheme 13.
Kitamura’s synthesis of N2-alkynyl benzotriazoles.
More recently, Davydov and co-workers studied the N-alkynylation of benzotriazole with alkynyliodonium reagent 47c (R = Ph) under palladium catalysis [29]. These authors report selectivity for N1- versus N2-alkynylation ranging from 3:1 up to 21:1, with the highest regioselectivity for N1-alkynylation obtained using catalytic alumina-supported Pd in acetonitrile. Huang and Zhu have reported using a polymer-supported alkynyliodonium reagent to prepare N1-alkynylbenzotriazole 48c in 60% yield [30].
Alkynyliodonium and alkynylbenziodoxolone reagents have also been employed in alkynyl transfer to other heterocycles. Kerwin demonstrated that the iodonium reagents 47c and 49a can be used to access the N-alkynylimidazoles 53a,b (Scheme 14) by reaction with the anion of 2-iodoimidazole (52), albeit in modest yields [31,32]. As part of a study on the oxidative alkynylation of 2-oxindoles, Bisai and co-workers prepared the N-alkynylindole 56 using the ethynylbeziodoxolone 55 (Scheme 15) [33].
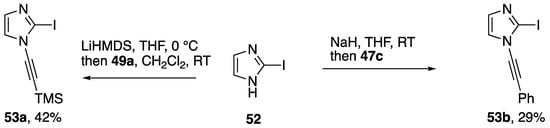
Scheme 14.
Kerwin’s synthesis of N-alkynylimidazoles using alkynyliodonium reagents.
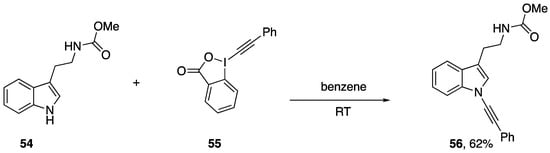
Scheme 15.
Bisai’s synthesis of an N-alkynylindole.
In 2018, Toriumi and Uchiyama published a synthesis of N-akynyl pyridinium salts as part of a research program investigating cationic nitrogen-embedded polycyclic aromatic hydrocarbons (cNe-PAHs) (Scheme 16) [34]. cNe-PAHs have many potential applications [35], but are challenging to synthesize. The researchers postulated that N-alkynyl pyridinium salts could serve as useful precursors for the synthesis of cNe-PAHs, and in the process of conducting their research they also synthesized two N-alkynyl azolium salts. Their synthesis is very similar to Kitamura’s; however, rather than using an anionic nucleophile, their nucleophile is neutral. Their strategy worked very well with pyridine, generating N-alkynyl pyridiniums 58a and 58b in 92% and 69% yields, respectively. When applying their strategy to azole nucleophiles, they were able to isolate N-alkynyl imidazolium 58c in a 50% yield and N-alkynyl pyrazolium 58d in a 72% yield, however they did have to increase the temperature for product 58d. The researchers also mention that several pyridines with electron-withdrawing groups failed to give the desired products.
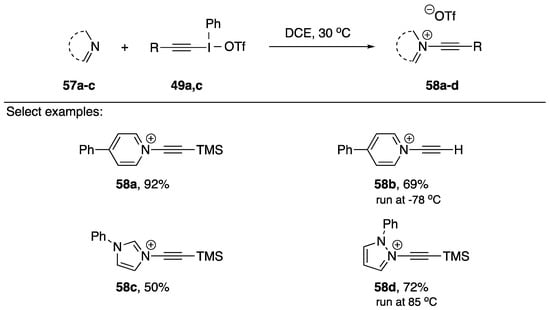
Scheme 16.
Toriumi and Uchiyama’s synthesis of N-alkynyl azolium salts.
2.3. Cross-Coupling
Metal-catalyzed cross-coupling as a strategy for synthesizing N-alkynyl azoles has become increasingly popular in recent years due to the improved functional group tolerance over elimination methods and the general ease in which these reactions can be carried out. In many publications, the synthesis of N-alkynyl azoles are often included as part of the substrate scope and are not the primary focus of the paper. The following sections cover any paper in which the synthesis of an N-alkynyl azole is reported, either when it is the main focus of the paper or as part of a screen of the substrate scope.
2.3.1. 1-Bromoalkynes
Many cross-coupling partners have been reported/evaluated for the synthesis of N-alkynyl azoles, but 1-bromoalkynes are used the most frequently. In 2003, Hsung reported the first metal-mediated synthesis of N-alkynyl amides, inspired by the work Buchwald and Hartwig. Hsung initially employed CuCN and 1-bromoalkynes [36]. The following year, in 2004, Hsung reported an improved procedure for the synthesis of N-alkynyl amides and included several N-alkynyl azoles, the first reported metal-catalyzed synthesis of N-alkynyl azoles (Scheme 17) [37]. The improved conditions involved CuSO4 as a precatalyst and 1,10-phenanthroline as a ligand and much lower temperatures. Hsung suggests these conditions are more environmentally friendly since they avoid using CuCN and copper halides.
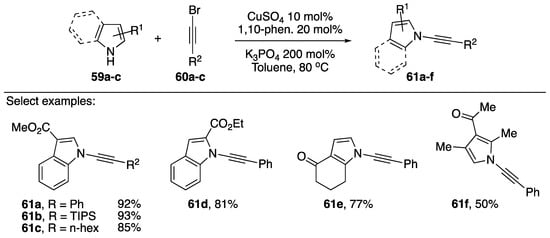
Scheme 17.
Hsung’s copper-catalyzed synthesis of various N-alkynyl azoles.
In 2006, as part of Hsung’s continued research into the chemistry of N-alkynyl amides, his group reported on their investigations into the competing alkynylation of a series of tryptamine derivatives (Scheme 18) [38]. The researchers were investigating whether N-alkynyl amides of tryptamine derivatives (62a–d) could be used in the synthesis of desbromoarborescidine A and C and were intrigued to find that these compounds also formed N-alkynyl indoles (65a–k) and bis-N-alkynylated products (66a–k) when subjected to their coupling conditions. Hsung suggests that similar pKa values between the indole and amide could be leading to the mixtures in N-alkynylated products. When testing this theory by using stronger electron-withdrawing groups on the amine they observed that the competing alkynylation of the indole nitrogen completely disappears. Remarkably, the researchers also found that when using carbamate as the protecting group that the N-alkynyl indole product was exclusively formed.
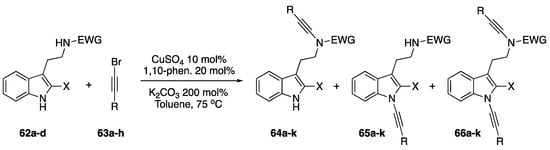
Scheme 18.
Competing alkynylation in tryptamine derivatives.
In 2008, Kerwin published a cross-coupling of imidazoles and bromoalkynes (Scheme 19) [39]. This report constitutes the first example of a dedicated procedure for the metal-catalyzed synthesis of an N-alkynyl azole.
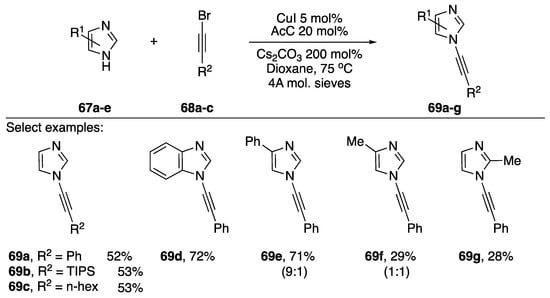
Scheme 19.
Kerwin’s copper-catalyzed synthesis of N-alkynyl imidazoles.
The researchers found that when trying to synthesize the desired N-alkynyl imidazoles using Hsung’s [37] procedure or analogous procedures for preparing N-alkynyl amides [40,41] they only isolated trace amounts of the desired compounds were obtained. However, following a screen of reaction conditions they found that they could synthesize N-alkynyl imidazoles (69a–g) in moderate to good yields (28–72%) by employing 2-acetylcyclohexanone (AcC) as ligand and CuI as copper source. A variety of bromoalkynes worked in this reaction, and the conditions worked for benzimidazoles as well. In the case of 4-substituted imidazoles, Kerwin found that the reaction gave mixtures of N-alkynylated products; 4-methyl-1H-imidazole giving a 1:1 mixture of N-alkynyl imidazoles 69f.
As part of their continued interest in the synthesis and reactivity of various N-alkynyl azoles, Kerwin has also published a synthesis of N-alkynyl pyrroles in 2017 (Scheme 20) [42]. The researchers were interested in synthesizing 2-substituted N-alkynyl pyrroles and found that the established methods for N-alkynylation were insufficient. They found that by increasing the temperature and using the more electron rich 4,7-dimethoxy-1,10-phenanthroline (DiOMePhen) ligand that they were able to synthesize various N-alkynyl pyrroles in poor to excellent yields. Kerwin found that the success of the pyrrole substrate 70 seemed to have a strong correlation with the nature of its substitution. Electron-rich (and sterically hindered) 2,5-dimethypyrrole gave only 5% of the desired N-alkynyl pyrrole 72h, while pyrroles substituted with electron-withdrawing groups often gave near quantitative conversion (compounds 72b, 72g).
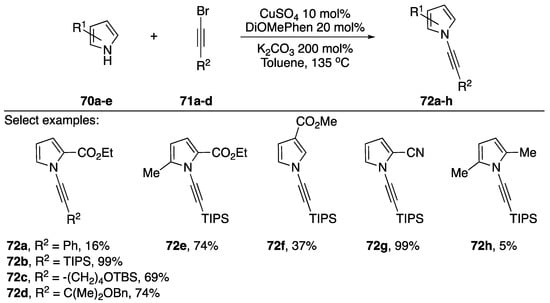
Scheme 20.
Kerwin’s copper-catalyzed synthesis of N-alkynyl pyrroles.
In 2013, as part of their research into a room temperature photo-induced Ullmann coupling, Fu and Peters reported the synthesis of an N-alkynyl carbazole derivative in conjunction with several other substituted carbazoles (Scheme 21) [43]. The researchers discuss the benefits of copper-catalyzed C-N bond formation, but suggest one of the biggest downfalls is the often-required elevated reaction temperatures. Their work circumvents elevated reaction temperatures by using 254 nm light to excite the copper catalyst. Their method was applicable to the coupling of various vinyl iodides with carbazole and indole (75a–c), and even worked for the N-alkynylation of carbazole with a 1-bromoalkyne (75d).
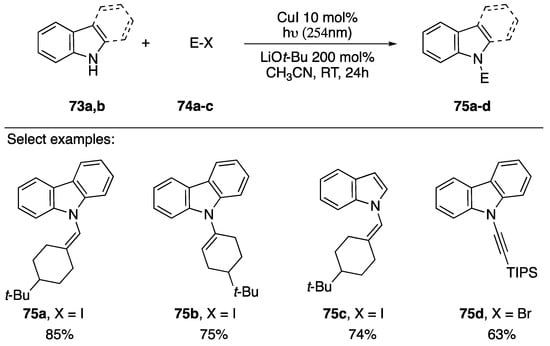
Scheme 21.
Peters and Fu’s photoinduced-copper-catalyzed synthesis of N-alkynyl carbazole.
Burley and researchers published a microwave-promoted copper-catalyzed synthesis of N-alkynyl imidazoles, benzimidazoles, pyrazoles, and indazoles in 2010 (Scheme 22) [44]. The researchers found that when using PEG-400 as a phase-transfer catalyst that the reaction times for the coupling were significantly reduced and the typical reaction was complete in only 30 min. Additionally, the researchers found that PEG-400 was also acting as a ligand for the copper catalyst and negated the need for additional ligands such as phenanthroline or acetylcyclohexanone (AcC). Using their method they synthesized various N-alkynyl imidazoles and benzimidazoles 74a–g in moderate to good yields (43–88%). They did find that a 1-bromoalkyne containing a TBS ether failed to give the desired product 74c and that their conditions failed to provide N-alkynyl purine 74g. The researchers extended their method towards the synthesis of various N-alkynyl pyrazoles and indazoles.
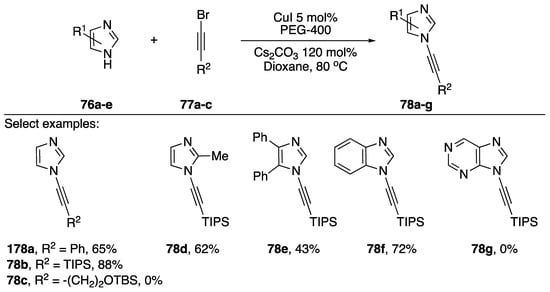
Scheme 22.
Burley’s microwave-promoted copper-catalyzed synthesis of N-alkynyl azoles.
In 2011, Das also published a “ligand-free” synthesis of N-alkynyl azoles (Scheme 23) [45]. Unlike Burley’s protocol, which employed PEG-400 as a ligand for copper, this method has no exogenous ligand; however, the substrate azoles are well known to serve as ligands for copper. Also unique to their strategy, Das uses CuO as a source of copper and KOH as their base. The researchers report isolating N-alkynyl imidazoles, benzimidazoles, and pyrazoles 81a–d in good yields (68–80%).
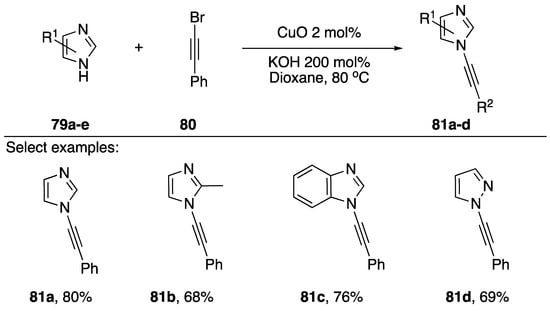
Scheme 23.
Das’s ligand-free copper-catalyzed synthesis of N-alkynyl azoles.
In 2013, Wu reported the N-alkynylation of indole from Boc-protected indoles (Scheme 24) [46]. The researchers were interested in synthesizing 2-alkynyl indoles directed by a 2-pyridyl group in the 3-position of the indole, and they were surprised when they recovered the deprotected and N-alkynylated indole product instead. Wu suggests this strategy could be synthetically useful since it is very common to employ Boc-protecting groups in the synthesis of indoles, a strategy circumventing the deprotection could improve the efficiency of the synthesis of N-alkynyl indoles derived from such compounds. They found that when there was a pyridine present on their starting indole (78a–e) that they didn’t have to use additional ligand for the synthesis to be successful (Conditions A). In other cases, they used 1,10-phenanthroline as a ligand (Conditions B). Wu’s strategy was successful at synthesizing a variety of N-alkynyl indoles both electron-rich and electron-deficient and with a variety of substitution patterns (80a–f), they also demonstrated the application of this strategy towards the synthesis on N-alkynyl pyrrole and N-alkynyl benzimidazole.
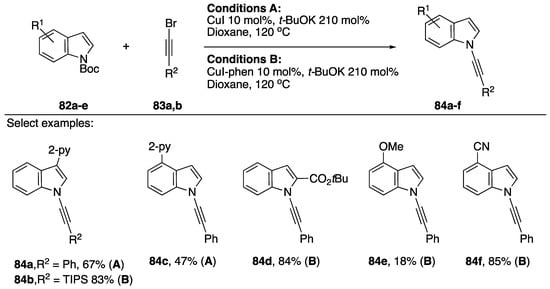
Scheme 24.
Wu’s copper-catalyzed N-alkynylation of Boc-protected indoles.
Pale and coworkers published a copper-zeolite based synthesis of N-alkynyl amides in 2014 in which they discuss its application to the synthesis of an N-alkynyl indole (Scheme 25) [47]. The researchers use a Cu(I)-ultra stable Y Zeolite (Cu(I)-USY) as their catalyst. Interestingly, by using a zeolite catalyst Pale was able to recover and recycle the catalyst, showing that after reusing the catalyst five times they only observed a marginal decline in activity (~15%). The majority of substrates screened by the researchers were amides such as 87a, however they demonstrated the use of their methodology towards the synthesis of N-alkynyl indole 87b and showed in the case of imidazole that they isolated the vinyl-bromide 87c as the only product.
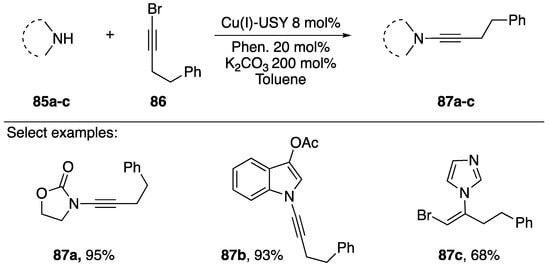
Scheme 25.
Pale’s copper-zeolite-catalyzed synthesis of N-alkynyl amides.
In 2009, Zhang reported an iron-catalyzed synthesis of N-alkynyl amides using 1-bromoalkynes as a coupling partner (Scheme 26) [48]. This is the first synthesis of N-alkynyl amides using an iron catalyst and the only instance in which a non-copper catalyst has been used in the synthesis of N-alkynyl azoles. In general, the researchers reported yields that were comparable to other published methods, synthesizing a tosyl-amide substrate 86a in a 97% yield and an N-alkynyl lactam 86b in a 57% yield. They only applied their conditions to the synthesis of one N-alkynyl azole, 86c. Interestingly, Zhang was able to recover and reuse the iron catalyst, showing that after recycling for nine runs the yields only dropped by 15%.
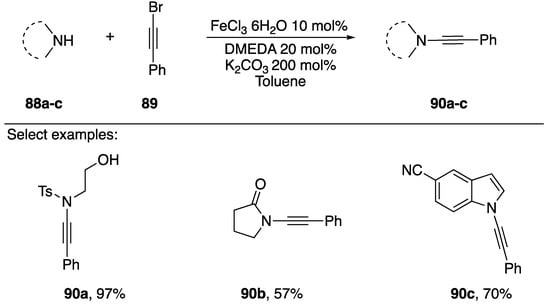
Scheme 26.
Zhang’s iron-catalyzed synthesis of N-alkynyl amides.
2.3.2. 1,1-Dibromoalkenes
In addition to 1-bromoalkynes as cross-coupling partners for the synthesis of N-alkynyl azoles, 1,1-dibromoalkenes have found use as competent coupling partners. For certain substrates, using a 1,1-dibromoalkene as a coupling partner would be advantageous due to their ease of synthesis. For example, rather than converting an aldehyde into an alkyne then subsequently the 1-bromoalkyne, researchers can simply make the analogous 1,1-dibromoalkene in one step from the same aldehyde. In 2011, Das published a paper reporting the coupling of various 1,1-dibromoalkenes with azoles (Scheme 27) [49]. This report was closely followed by two similar reports by Shang [50] and Evano [51]. Das reports that Cu(phen)(PPh3)Br in DMSO with Cs2CO3 as base provides N-alkynyl azoles 89a–e in good yields (66–78%). Interestingly this report is the only example of a phosphine ligand being used in the metal-catalyzed synthesis of an N-alkynyl azole. In their paper, the researchers suggest two possible mechanistic pathways leading to the observed alkynylated products: (1) coupling occurs with the dibromide to give an N-alkenyl azole followed by elimination to the alkyne, or (2) the 1,1-dibromoalkene first undergoes conversion to the corresponding 1-bromoalkyne and then cross-coupling occurs between the 1-bromoalkyne and the azole. Das favors the second mechanism.
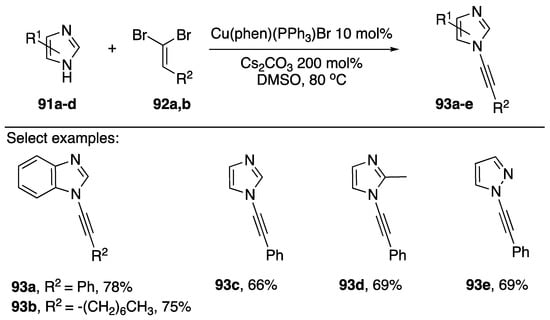
Scheme 27.
Das’s [Cu(Phen)PPh3Br]–catalyzed synthesis of N-alkynyl azoles.
2.3.3. Alternative Coupling Partners
Terminal alkynes have also been used as cross-coupling partners in the synthesis of N-alkynyl azoles. The advantage of using a terminal alkyne is avoiding the additional step to introduce the 1-bromo group and avoiding the use of halogens all together, a more atom efficient process. In 2008, Stahl reported using an oxidative N-alkynylation strategy for the synthesis of N-alkynyl amides and several N-alkynyl indoles (Scheme 28) [52]. Stahl, inspired by the Glaser-Hay coupling [53] and the Chan-Lam coupling [54] found that under the right conditions they could limit the Glaser-Hay homocoupling of the alkynes and limit the formation of 1-chloroalkynes, isolating the desired N-alkynylated products in high yields. Several indoles 94a–c were tolerated under these conditions and aromatic, alkyl, and silyl alkynes 95a–d were all tolerated as well. In 2015, Troung published a similar strategy for the synthesis of N-alkynyl oxazolidinones, however they used a copper metal-organic-framework (MOF) [55]. While their method performed well in preparing N-alkynyl oxazolidinones (31–96% yield), application to an indole substrate afforded 96f in only 32% yield.
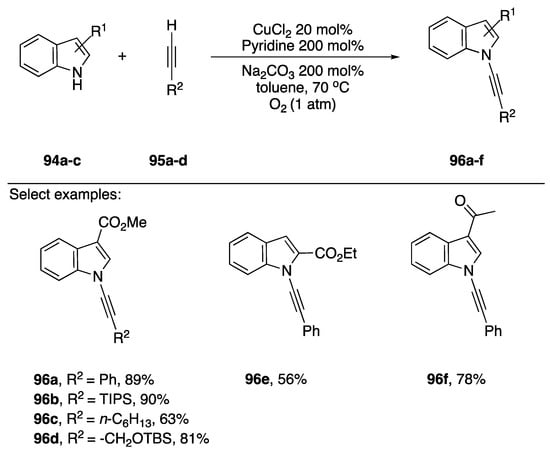
Scheme 28.
Stahl’s oxidative copper-catalyzed synthesis of N-alkynyl indoles.
In 2017, Bhattacharjee and co-workers reported the oxidative coupling of terminal acetylenes and pyrazoles (Scheme 29) [56]. They employed similar conditions as Stahl, but they used copper (II) acetate hydrate as the copper source and air as oxidant.
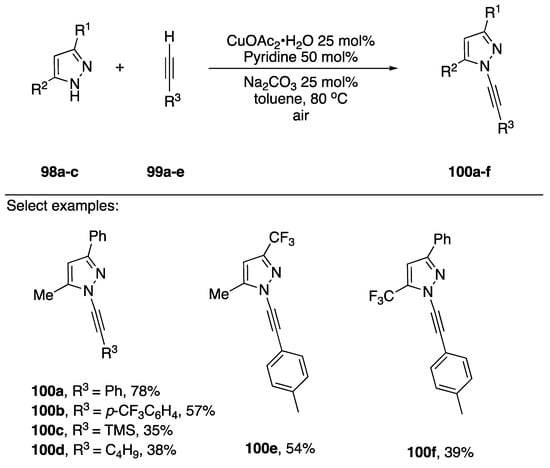
Scheme 29.
Bhattacharjee’s oxidative copper-catalyzed synthesis of N-alkynyl pyrazoles.
A series of 3,5-disubstiuted pyrazoles 94a–c were coupled to terminal acetylenes in good to modest yield to afford the regioisomerically pure N-alkynyl pyrazoles 100a–f. The authors note that yields drop when alkyl substituted acetylenes are employed.
In 2010, Jiao published a decarboxylative coupling strategy for preparing N-alkynyl amides and azoles (Scheme 30) [57]. This strategy benefits from not using halide containing starting materials, and has the added benefit that the formation of Glaser-Hay oxidative dimerization products is minimized. Additionally, many alkynyl carboxylates are readily available. The researchers showed that this strategy could be used to effectively synthesize many N-alkynyl indole substrates 103a–e, even tolerating an aldehyde. Unfortunately, when compared to other methodologies, vide supra, Jiao’s decarboxylative-coupling strategy often falls short in terms of yield, with yields ranging from 27% to 68%.
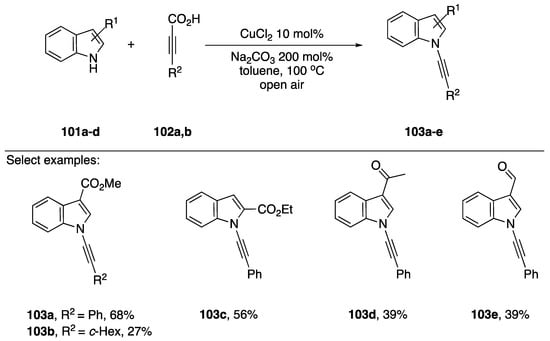
Scheme 30.
Jiao’s copper-catalyzed decarboxylative synthesis of N-alkynyl indoles.
2.4. Other Methods
One of the more unusual syntheses of an N-alkynyl azole is Brown’s synthesis of N-ethynylpyrazole (106) (Scheme 31) [58]. During the course of their research studying the flash vacuum pyrolysis of 1-alkynoyl-3-methylpyrazoles into pyrazolo[1,5-a]pyridine-5-ols, the researchers found that substrates lacking a methyl substituent in the 3-position formed N-alkynyl pyrazole in low yields. Brown proposes a mechanism that proceeds through an allene-ketene intermediate, such as 105. He suggests when a substrate lacks methyl group in the 3-position that the intermediate loses carbon monoxide and forms a vinylidene carbene that rearranges to the N-alkynyl pyrazole.
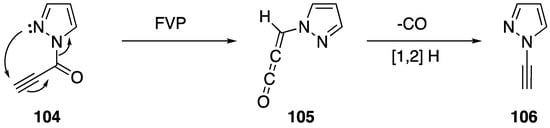
Scheme 31.
Brown’s flash vacuum pyrolysis synthesis of N-alkynyl pyrazole.
3. Reactions of N-Alkynyl Azoles
N-alkynyl azoles, in a similar manner to N-alkynyl amides, are characterized by their polarized alkyne due to the attached nitrogen, which is moderated by the participation of this nitrogen in the aromatic azole ring. This alkyne polarization, with the β-carbon becoming more nucleophilic and the α-carbon becoming more electrophilic, leads to distinct reactivity. As interest in the synthesis of N-alkynyl azoles has grown over the past decade, interest in the reactions of N-alkynyl azoles has grown as well. The following sections will discuss the many different reactions of N-alkynyl azoles that have been reported. In many cases, the reactions of N-alkynyl azoles are analogous to those of N-alkynyl amides. Publications reporting reactions of N-alkynyl amides often times include one or two examples of N-alkynyl azoles, typically N-alkynyl indoles, in sections describing the scope of the reaction. These are included in this section, with notes indicating this. In addition, there are a number of reactions specific to N-alkynyl azoles, or reactions that have only been studied in N-alkynyl azoles, which are also highlighted in this section.
3.1. Reactions of Alkyne C-H
Terminal alkynes, including N-alkynyl amides, participate in nucleophilic addition and coupling reactions [59], and similar reactions have also been reported for N-alkynyl azoles. In 1992, Paley published one of the first reactions of an N-alkynyl azole (Scheme 32, Reaction (1) [14]). In their publication they report the synthesis of diacetylene 109 from N-alkynyl pyrrole 107 and 1-bromoalkyne 108 using a Cadiot-Chodkiewicz coupling. This report provides a first glimpse into the general reactivity and stability of N-alkynyl azoles and products derived from N-alkynyl azoles. The researchers reveal that diacetylene 109 undergoes spontaneous polymerization upon formation. In 2016, Okuno reported a very similar reaction with N-alkynyl carbazole 110 (Scheme 32, Reaction (2) [60]). In their work, the analogous diacetylene 112 was more stable and Okuno was able to isolate the compound.
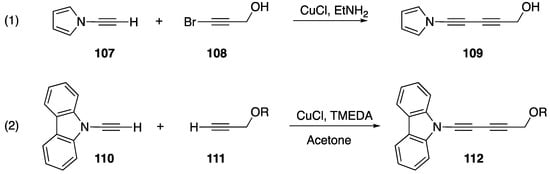
Scheme 32.
Synthesis of diacetylenes of N-alkynyl azoles.
In 1994, as part of his publication disclosing the synthesis of various N-alkynylated purines and pyrimidines, Zemlicka investigated the addition of N9-alkynyl adenine to various ketones (Scheme 33) [16].
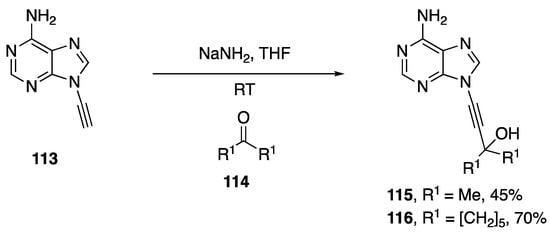
Scheme 33.
Zemlicka’s synthesis of carbinols of N9-alkynyl adenine.
The researchers discuss the importance of base on the outcome of the reaction, in the case of LiNH2 and n-BuLi the desired carbinols were not isolated. However, when NaNH2 was used as base, the carbinols of acetone (115) and of cyclohexanone (116) were isolated in 45% and 70% yield, respectively.
In 2014, Wolf reported a zinc-catalyzed enantioselective addition of N-alkynyl amides and indoles to aldehydes (Scheme 34) [61]. This publication represents the first example of a method for adding an N-alkynyl azole or N-alkynyl amide to a carbonyl electrophile without relying on a strong base to deprotonate the alkyne. Additionally, using (-)-NME as a ligand to the Zn(OTf)2 catalyst, the researchers were able to generate high levels of enantiomeric excess in the products (77–90% ee). While the scope of the indole substrate wasn’t probed extensively by Wolf, limited to 3-substituted indoles, a variety of aldehydes were tolerated in this reaction; including: benzaldehydes, furan-3-carbaldehyde, and sterically hindered aliphatic aldehydes, which afford the corresponding propargylic alcohols 119a–f in moderate to good yields (58–89%).
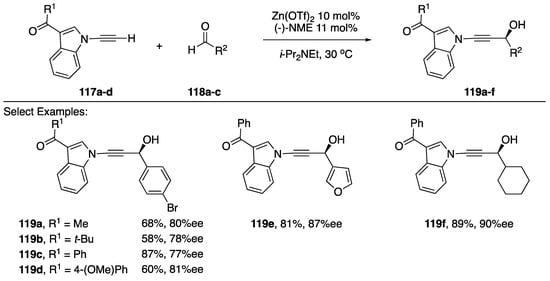
Scheme 34.
Wolf’s enantioselective addition of N-alkynyl indole to various aldehydes.
The participation of terminal N-alkynyl azoles in Sonogashira coupling reactions can be complicated by alternative, palladium-catalyzed hydroalkynylation reactions (see Section 3.2.2). However, as part of their work investigating the structure-activity relationship of ethynyl purines and analogues as BCL-ABL inhibitors, Huang and co-workers at ARIAD Pharmaceuticals prepared a series of N9-alkynyl purines 123 and 124 by Sonogoshira coupling of the N-ethynyl purine 121 with the aryl iodides 122, albeit in very low yield (Scheme 35) [62].
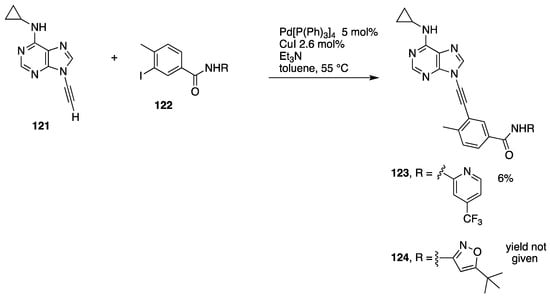
Scheme 35.
Huang’s low-yielding Sonogashira coupling of an N-alkynyl purine.
3.2. Addition Reactions
The polarization of the alkyne of N-alkynyl amides and N-alkynyl azoles (Figure 2) gives rise to regioselective addition reactions, and these reactions are one of the most explored classes of reactions of these compounds. In this section, we describe the wide variety of addition reactions, both intermolecular and intramolecular, that have been reported in the literature for N-alkynyl azoles. Cycloaddition, annulation, and other reactions involving the triple bond of N-alkynyl azoles are treated in separate sections.
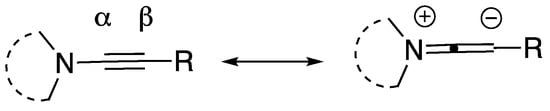
Figure 2.
Polarization of the N-alkynyl azole triple bond.
3.2.1. Carbon-Heteroatom Bond Formation
Addition reactions resulting in the formation of a new carbon-heteroatom bond are very prevalent among N-alkynyl azoles. The addition of Bronsted acids, a classic reaction of alkynes, has been reported on several occasions (Scheme 36). In 2013, Iwasawa reported a method for the stereoselective synthesis of 1-(1-halovinyl)-1H-indoles from their respective N-alkynyl indoles via the addition of in situ generated HX acids (Scheme 36, equation. 1) [63]. The researchers used TMS-X and water to generate HX in a controlled manner, and were able achieve excellent levels of stereoselectivity and isolated the vinyl-halides in good yields. In 2000, Katritzky reported the addition of p-toluenesulfonic acid to N-alkynyl benzotriazoles as part of a methodology for the homologation of carboxylic acids (Scheme 36, equation 2) [17]. The researchers isolated vinyl-tosylates, analogous compounds to those synthesized by Iwasawa, in moderate yields. In 2011, as part of a study on an aza-Bergman rearrangement of 1,2-dialkynylimidazole Kerwin reported an addition of thiophenol to an N-alkynyl imidazole (Scheme 36, equation 3) [64]. Interestingly, in this example the sulfur nucleophile adds to the β-carbon of the alkyne to give 130 in a 43% yield.
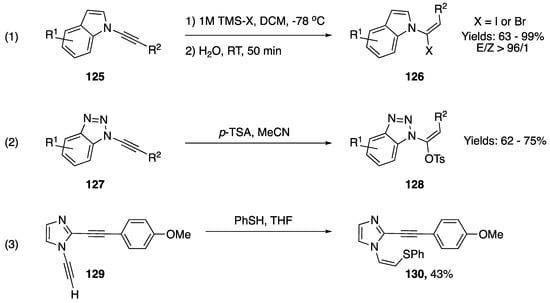
Scheme 36.
Addition of acids and thiophenol to various N-alkynyl azoles.
In 1998, while studying the rearrangement of 5-substituted 5-aminopentadienals, Neuenschwander reported an interesting transformation of an N-alkynyl pyrrole following the addition of phenol to the alkyne (Scheme 37) [65]. The researchers found that treating N-alkynyl pyrrole 131 with phenol gave the addition product 132 and that upon further treatment with HBF4, they isolated the pyrrole 133 in 70% yield.
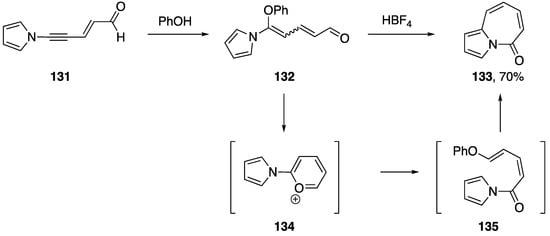
Scheme 37.
Neuenschwander’s rearrangement of 5-substituted 5-aminopentadienals.
Additions of halogens and pseudohalogens to N-alkynyl azoles can occur with high regio- and stereoselectivity due to the polarized nature of the alkyne. In 2014, Iwasawa reported iodobromination of a series on N-alkynyl amides and N-alkynylindoles 136a–c (Scheme 38) [66]. These reactions afford exclusively the (E)-1-bromo-2-iodoalkenes 137a–c in good yields from in situ generated IBr using NIS and a silylbromide.
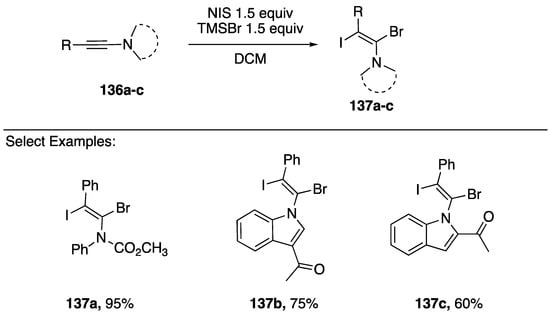
Scheme 38.
Regio- and stereoselective iodobromination of N-alkynylindoles.
In 2016, Zhu reported a sulfenylchloride-mediated addition of DMSO to N-alkynyl amides and azoles generating acrylamides with sulfur substitutions in the α-position (Scheme 39) [67]. This method worked for various N-alkynyl amides 140a,b and also worked with N-alkynyl indole 149c. The researchers suggest that initially a sulfonium ion forms between the alkyne and sulfenylchloride, which undergoes subsequent addition of DMSO to generate the observed products. In addition to sulfur, Zhu demonstrated an analogous synthesis of α-selanyl and α-tellanyl acrylamides.
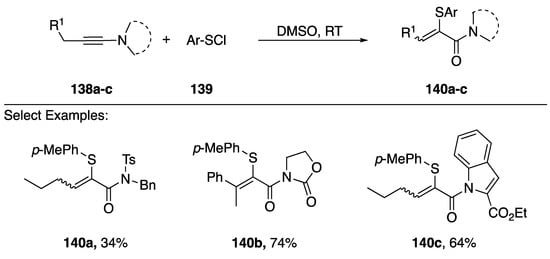
Scheme 39.
Zhu’s sulfenylchloride-mediated addition of DMSO to N-alkynyl amides.
Iodine-promoted additions of heteronucleophiles to alkenes and alkynes represent a classic method for constructing carbon-heteroatom bonds [68]. An interesting iodoamination of N-alkynyl amides and N-alkynyl azoles was reported by Zhu in 2016 (Scheme 40) [69]. The researchers found that when treating an N-alkynyl amide/azole 141a–c with a cyclic secondary amine 142a–c in the presence of iodine and tert-butyl hydrogen peroxide they could synthesize vinyl-diiodides 143a–c in good yields. Zhu suggests that under these conditions a 1-iodoalkyne initially forms, followed by an iodine-mediated addition of the nucleophile to give the observed products.
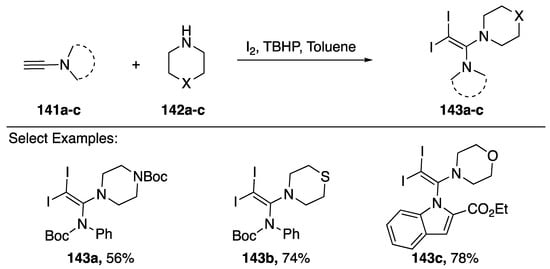
Scheme 40.
Zhu’s iodoamination of N-alkynyl amides and indole.
There have been several reports of iodocyclizations of N-alkynyl azoles published in the literature (Scheme 41). In 2017, as part of their paper investigating the intramolecular addition of hydrazide to N-alkynyl pyrrole (vide infra), Balci reported an iodocyclization from N-alkynyl pyrrole starting materials 144a–c (Scheme 41, Reaction (1) [70]). The researchers noted that all of the substrates they investigated underwent a 6-endo-dig cyclization to give the corresponding vinyl-iodides. DFT calculations performed by Balci predicted that the iodonium intermediate should carry more positive charge on the β-carbon of the alkyne, providing an explanation for the observed regioselectivity. Kerwin disclosed an iodocyclization in 2009 (Scheme 41, Reaction (2) [71]). They reported a single example of the N-iodosuccinimide mediated intramolecular addition of an alcohol to an N-alkynyl imidazole; isolating vinyl iodide 147a in a 98% yield. Interestingly, iodide 147a is also the result of a 6-endo-dig cyclization. In 2014, Okitsu published a more general iodocyclization of various N-alkynyl amides, including an example of an N-alkynyl indole (Scheme 41, Reaction (3) [72]). Okitsu uses I(coll)2PF6 as their source of iodine and ethoxyethyl ethers as the nucleophile. Amazingly, the reactions required less than 5 s to run to completion and give the substituted benzofuran product 149 in excellent yield. In 2013, Zeni published an electrophilic cyclization of a series of N-alkynyl-2-(butylselenyl)imidazoles 150a–j to imidazo[2,1-b][1,3]selenazoles 151a–j (Scheme 42, equation 4) [73]. The corresponding 2-thiomethyl imidazoles also undergo cyclization to imidazo[2,1-b][1,3]thiazoles in good yield; however, cyclization of a N-alkynyl-2-(butylselenyl)benzimidazole was lower yielding.
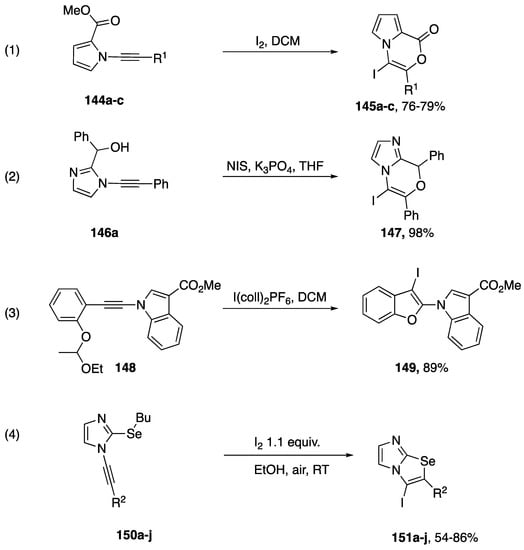
Scheme 41.
Iodocyclizations of N-alkynyl azoles.
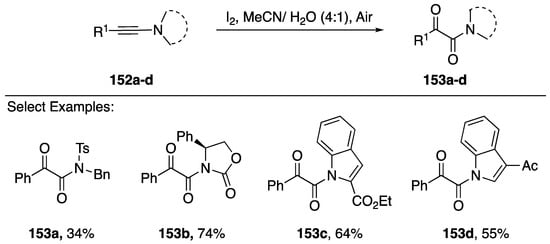
Scheme 42.
Zhu’s iodine-mediated oxidation of N-alkynyl amides and indoles.
Zhu has also reported an iodine-mediated oxidation of N-alkynyl amides/azoles (Scheme 42) [74]. Their method consists of treating the starting N-alkynes 152a–d with I2 in an acetonitrile and water mixture, open to air. Under these conditions, the researchers isolated α-ketoamides 153a–d in moderate to good yields (34–74%). Additionally, the reaction worked with both 2- and 3-substituted N-alkynyl indole starting materials. The researchers propose a reaction mechanism that proceeds initially through an iodine-promoted addition of water to the α-carbon of the alkyne, generating an α-iodo amide, and subsequently an oxidation to a ketone by air. Iwasawa also published in 2014 an NIS-mediated oxidation of N-alkynyl amides and an N-alkynyl indole, but their reported yield for 153c is lower (34%) [75].
Interestingly, an alternative route to these α-ketoamide products from N-alkynyl azoles was reported by Hwang in 2017 [76]. This method employs copper catalyst and irradiation with blue LEDs in the presence of 1 atm of O2 (Scheme 43). A series of N-alkynyl sulfonamides 155a and carbazoles 155b–d can be oxidized in good yields through a proposed photoexcitation of a Cu(I)-π complex (λmax = 460 nm) leading to formation of a Cu(II) peroxo-complex intermediate.
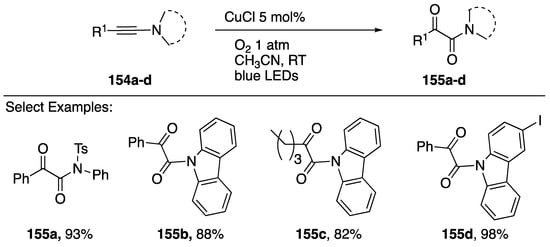
Scheme 43.
Hwang’s photochemical oxidation of N-alkynyl carbazoles.
The addition of heteronucleophiles to N-alkynyl azoles has also been reported to occur under basic conditions. Zhang, Cui and co-workers investigated a copper-free coupling of 1,1-dihaloalkenes with excess azoles in the presence of NaOH to afford dihetaryl alkenes [77]. They propose a mechanism involving the formation on N-alkynyl azoles as intermediates (see Section 2.3.2), which undergo addition under the basic reaction conditions. They provided support for this mechanism by showing that when treated under these reaction conditions in the presence of excess pyrazole, the N-alkynyl pyrrole 156 affords the same products 157 and 158, in similar ratio compared with the reaction of the corresponding 1,1,-dibromoalkene (Scheme 44, Reaction (1)). In 1994, Zemlicka reported the addition of oxygen nucleophiles to N9-alkynyl adenine (Scheme 44, Reaction (2) [16]). The heterocyclic products, 161a,b were isolated when the researchers were trying to perform the 1,2 addition of the alkyne of 159 into various ketones. The author suggests that these compounds form when the initially formed propargylic alcohol adds to another equivalent of ketone forming a hemiketal, and the hemiketal oxygen proceeds to add to the alkyne. In 2000, Katritzky reported the addition of sodium methoxide to N-alkynyl benzotriazoles (162) within the paper describing their method for the homologation of carboxylic acids, treatment of the resultant addition product with acid gave homologated esters (Scheme 44, Reaction (3) [17]). While the reaction afforded only one regioisomer, ~80:20 mixtures of (E)- and (Z)-163 were obtained. In 1995, Trofimov reported the reaction of dilithiated N-ethynyl pyrrole, 165, with elemental sulfur, selenium, and tellurium (Scheme 44, equation 4) [78]. The researchers isolate the pyrrolo[2,1-b][1,3]chalcogenazoles 166a–c, in modest yields. A similar cyclization of N-alkynyl imidazoles was reported in 2012 by Zeni [79].
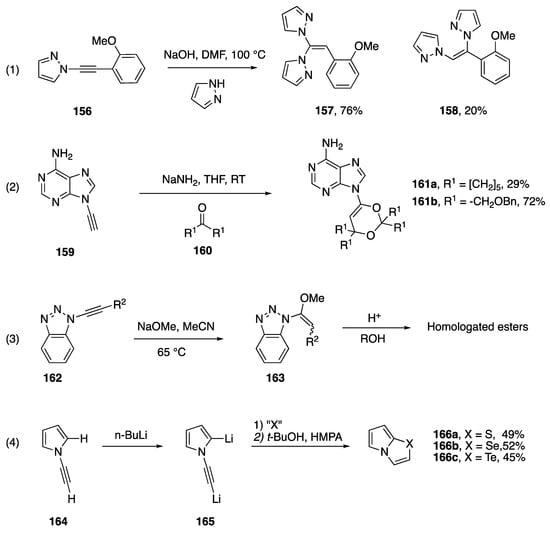
Scheme 44.
Additions to N-alkynyl azoles under basic conditions.
In 2009, Kerwin reported a method for the synthesis of imidazo[1,2-c]-oxazoles and imidazo[2,1-c][1,4]oxazines starting from N-alkynyl imidazoles (Scheme 45) [71]. Using base catalyzed conditions, the researchers reported a 5-exo-dig cyclization and when AuCl3 was used as a catalyst the reaction proceeded via a 6-endo-dig cyclization (168a and 169a). In a follow up report, Kerwin and researchers investigated the intramolecular addition of other nucleophiles; including: 5-hydroxymethyl-imidazoles, p-toluenesulfonamides, and Boc-protected hydrazine (Scheme 45) [80].
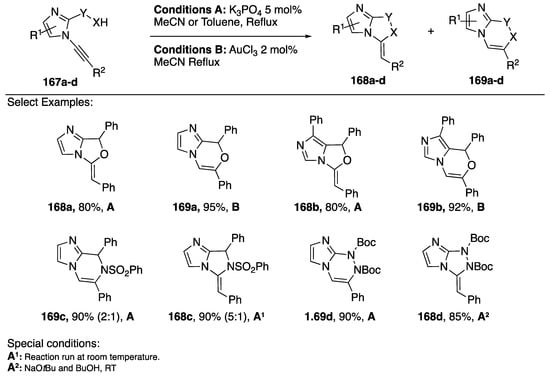
Scheme 45.
Kerwin’s regiocontrolled cyclization to imidazoazines and imidazoazoles.
In this paper, they found that the new alcohol substrates reacted analogously to the previous alcohol additions (168b and 169b); however, the nitrogen nucleophiles reacted less predictably. For both substrates containing nitrogen nucleophiles the researchers observed 6-endo-dig cyclizations when using conditions A, the same conditions that resulted in 5-exo-dig cyclizations with the oxygen nucleophiles 169c and 169d. Interestingly, gold catalysis yielded neither of the desired products. Ultimately Kerwin found that they could get the 5-exo-dig cyclization products with the nitrogen nucleophiles using base catalysis at room temperature (compounds 168c and 168d). The researchers note that the observed reaction temperature-dependent regioselectivity did not seem to be a kinetic versus thermodynamic effect.
In 2014, as part of the same paper, Kerwin reported the synthesis of several imidazo-fused heterocycles from 2-carbaldehyde-1-alkynylimidazole 170 (Scheme 46) [80]. Treatment of 170 with ammonia and catalytic Cu(OTf)2 gave imidazolopyrazine 171 in a 76% yield, while treatment with EtOH and Cu(OTf)2 gave the cyclic acetal derivative 172 nearly quantitatively. Remarkably, without the need for metal-catalysis, N-alkynyl imidazole 170 reacts with hydroxylamine to afford the cyclized product 173, an imidazolopyrazine N-oxide, in 71% yield.
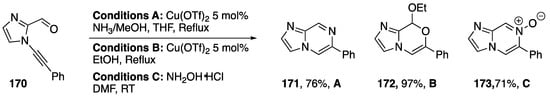
Scheme 46.
Kerwin’s synthesis of several imidazo-fused heterocycles.
In 2017, Kerwin reported the synthesis of pyrrole[2,1-c]oxazin-1-ones via the intramolecular addition of carboxylate nucleophiles to the alkyne of N-alkynyl pyrroles (Scheme 47) [42]. The researchers used trimethylsilylethyl (TMSE) esters as protected carboxylic acids to carry out the copper-catalyzed synthesis of N-alkynylazoles 176a–d. Upon deprotection of the acids with tetra-n-butylammonium fluoride (TBAF) in DMF, the substrates underwent spontaneous 6-endo-dig cyclizations to the corresponding pyrrole[2,1-c]oxazin-1-ones 177a–d. Several different substitutions on the alkyne and pyrrole were tolerated and the researchers even used this methodology to carry out the formal synthesis of peramine, a rye grass endophytic fungal alkaloid.
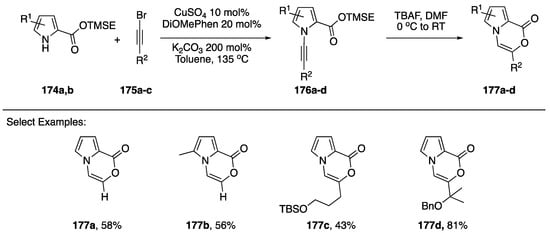
Scheme 47.
Kerwin’s synthesis of pyrrole[2,1-c]oxazin-1-ones.
Balci published an intramolecular addition of hydrazide to N-alkynyl pyrroles in 2017 (Scheme 48) [70]. Starting with N-alkynyl pyrroles 178a–d substituted with a methyl ester in the 2-position the researchers refluxed the compounds with hydrazine giving the hydroamination addition products 179a–c and 180a. Balci suggests that a hydrazide is initially formed and subsequently adds to the alkyne. The researchers observed varying regioselectivies depending on the nature of the substitution on the alkyne. If the substitution was electron withdrawing, such as nitrobenzene, they recovered the α-addition product 180a, but if the substitution was electron donating or aliphatic they observed β-addition (compounds 179b,c) In the case of phenyl substitution, they isolated a mixture of the two regioisomers, favoring addition to the β-carbon.
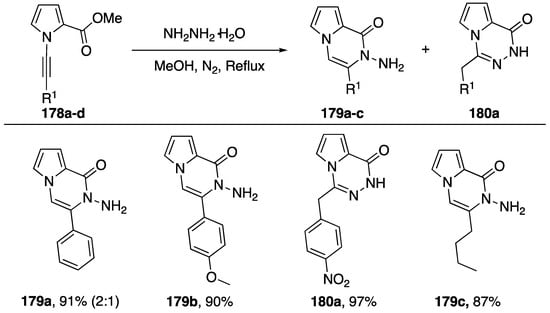
Scheme 48.
Balci’s intramolecular addition of hydrazide to N-alkynyl pyrrole.
In contrast to the predominant heteroatom additions to the β-position of N-alkynyl azoles in cyclizations under coinage metal catalysis (Scheme 45, Scheme 46 and Scheme 47), an alternative cyclization of N-alkynyl amides and azoles was reported by Gillaizeau in 2018 (Scheme 49) [81]. These workers describe the facile cyclization of ortho-ynamidyl benzoate esters 180a–c to 3-aminoisocoumarins 181a–c in the presence of an equivalent of a Bronsted acid or catalytic Lewis acid. The authors note that none of the 5-exo-dig cyclization products corresponding to addition to the β-carbon of the alkyne were observed, and that a range of ortho-ynamidyl het(aryl) esters also undergo cyclization.
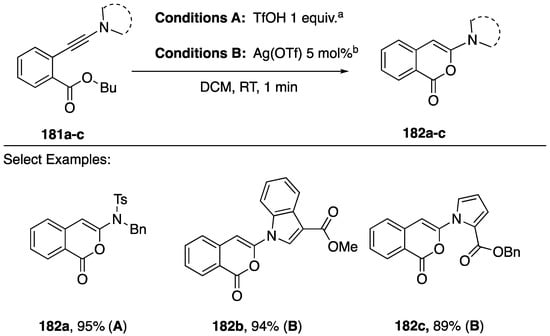
Scheme 49.
Gillaizeau’s cyclization of ortho-ynamidyl benzoate esters to 3-aminoisocoumarins.
In 2014 Clavier and Buono reported the regioselective α-addition of 1,3-diones 184a,b to N-alkynyl amides 183a,b and the N-alkynyindole 183c in the presence of the catalysis 184 to afford exclusively the products 186a–c (Scheme 50) [82]. These authors note that the indole product 186c is only formed in moderate yield.
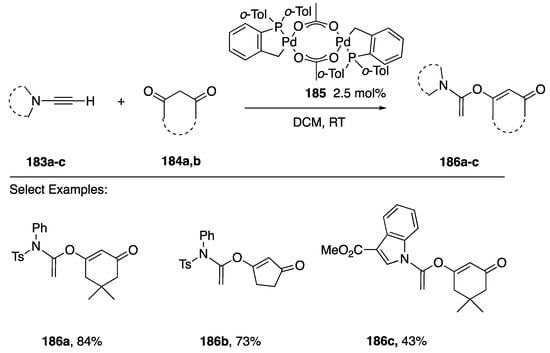
Scheme 50.
Clavier and Buono’s regioselective addition of cyclic 1,3-diones to N-alkynyl amides.
3.2.2. Carbon-Carbon Bond Formation
The addition of carbon nucleophiles to N-alkynyl azoles to generate new carbon-carbon bonds has been investigated as well. In 2002, Katritzky published an addition of Grignard and organolithium reagents to N-alkynyl benzotriazoles 187 generating disubstituted alkynes 188 in the case of aromatic nucleophiles and N-vinyl benzotriazoles 189 in the case of aliphatic carbon nucleophiles (Scheme 51) [18]. In conjunction with the researchers’ method for synthesizing N-alkynyl benznotriazoles from acid chlorides, this addition of aromatic nucleophiles represents a unique approach to synthesizing disubstituted acetylenes from carboxylic acids.
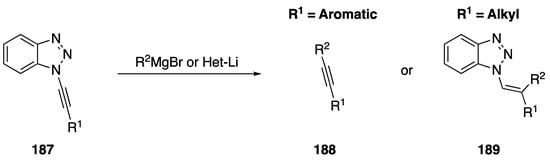
Scheme 51.
Katritzky’s synthesis of disubstituted alkynes from N-alkynyl benzotriazole.
In 2015, Reddy published a palladium-catalyzed hydroalkynylation of N-alkynyl amides (Scheme 52) [83]. Using Pd(PPh3)2Cl2 as a catalyst and triethylamine as a base the researchers were able to add terminal alkynes to N-alkynyl amides with high levels of regio- and stereoselectivity. The reaction proceeded in good yields with many different N-alkynyl amide substrates 190a–c including N-alkynyl indoles 190d. Additionally several different terminal alkynes participated in this reaction. Interestingly, in the case of N-alkynyl-p-toluenesulfonamides, such as 190c, when the substitution on the N-alkyne is aliphatic the hydroalkynylation proceeds with cis-stereoselectivity and when the substitution is aromatic the reaction proceeds with trans-stereoselectivity.
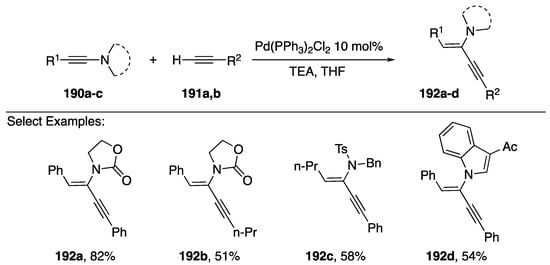
Scheme 52.
Reddy’s hydroalkynylation of N-alkynyl amides.
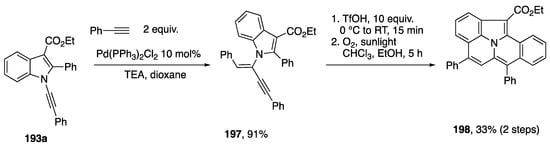
In 2017, Park published an approach to diverse fused heterocycles based on regiodivergent cyclizations and oxidative cyclodimerization of N-alknylindols (Scheme 53) [84]. On one hand, N-alkynylindols 193 were found to undergo intramolecular hydroarylation by C-H activation with Pd(OAc)2 with a phosphine ligand to afford the cyclized products 194 through 5-exo-dig cyclization, while electrophilic addition facilitated by PtCl2 led to 195 through 6-endo-dig cyclization. Interestingly, an oxidative dimerization of 193 occurs in the presence of PdCl2 and an equivalent of benzoquinone to afford the pentacycles 195. A range of aryl and alkyl substitutents on the N-alkynylindole 193 are tolerated in each of these cyclizations, although the yields of 6-endo-dig cyclization products 194 and dimers 195 are generally lower than the 5-exo-dig products 194. In this same report, Park also noted that the products of hydroalkynylation (cf. Scheme 52) of N-alkynyl-2-phenylindole can serve as substrates for cyclization to the pentacyclic benzo[7,8]indolizino[2,3,4,5-ija]quinolines (Scheme 54) [84].
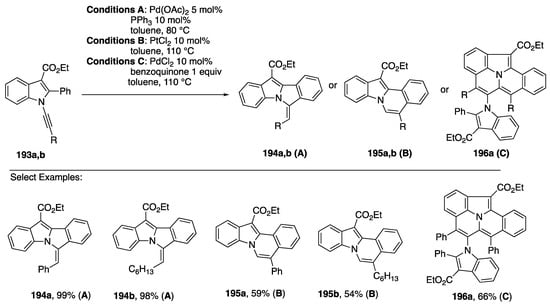
Scheme 53.
Park’s regiodivergent cyclization of N-alkynylindoles.
Scheme 54.
Park’s hydroalkynylation/cyclization approach to pentacyclic heterocycles.
In 2014, Zhu reported a stereoselective addition of boronic acids to N-alkynyl amides 199a,b and N-alkynyl azoles 199c,d to afford the (Z)-α,β-disubstituted enamines 200a–e (Scheme 55) [85]. Both aryl and alkenyl boronic acids participate in the coupling in good yields.
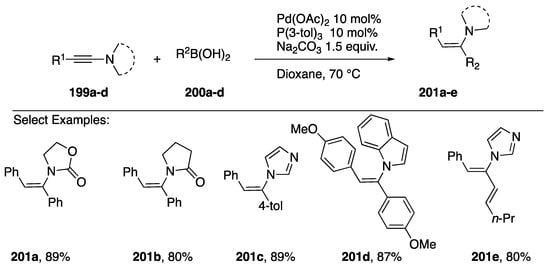
Scheme 55.
Zhu’s (Z)-stereoselective boronic acid coupling to N-alkynyl amides and azoles.
As part of an approach to access α-oxo-carbene surrogates in the absence of metal ions, Shin reported in 2017 a Bronsted acid catalyzed Friedel-Crafts-type reaction of N-alkynyl amides and the N-alkynyl indole 202 in the presence of 2-chloropyridine N-oxide (Scheme 56) [86]. These workers employed nucleophilic arenes such as pyrrole 203a, indole 203b and catechol 204 to obtain coupled products 205, 206 and 207, respectively in good yields. In support of a proposed mechanism involving SN2′ displacement on the electrophilic intermediate 209, the authors note that when reactions on N-alkynyl amides are carried out in the presence of a chiral dipyridine N,N’-dioxide affords coupled products with substantial enantioselectivity.
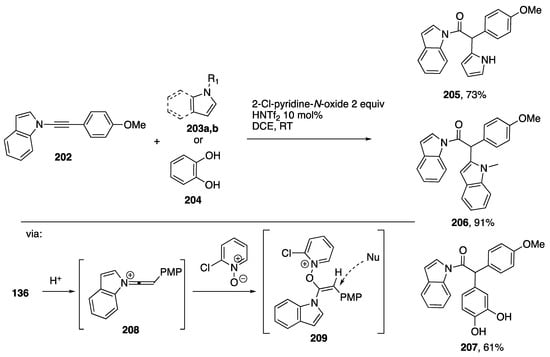
Scheme 56.
Shin’s Bronsted acid catalyzed oxygenative Friedel-Crafts coupling of an N-alkynlindole.
3.2.3. Carbon-Boron and Carbon-Hydrogen Bond Formation
The addition of hydrogen or deuterium to N-alkynyl amides and azoles is a convenient way to synthesize N-alkyl and N-vinyl azoles that may be challenging to synthesize otherwise. In 1994, Zemlicka published the reduction of N9-alkynyl adenine with hydrogen gas and palladium on carbon (Scheme 57, Reaction (1) [16]). In 2000, Bauld and Gao published the reduction of N-ethynyl carbazole with lithium aluminum deuteride forming N-(cis-2-deuteriovinyl)carbazole (213) (Scheme 57, Reaction (2) [87]). In contrast to the stereoselectivity of this transformation, Swamy reported in 2017 that the H-transfer reduction of N-alkynyl carbazole 214 affords a mixture of (E)- and (Z)-products 215 (Scheme 57, Reaction (3)), despite the highly (E)-selective reduction of N-alkynyl amides under these conditions [88].
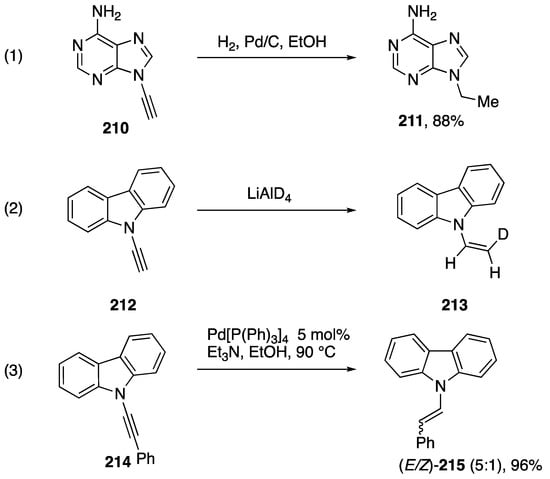
Scheme 57.
Addition of hydrogen and deuteride to N-alkynyl azoles.
In 2013, Zhu reported a copper-catalyzed reduction of N-alkynyl amides/indoles to N-alkenyl amides via a regio- and stereoselective boron addition and subsequent protonolysis (Scheme 58) [89]. The researchers found CuCl with PPh3 and NaOtBu to be the ideal conditions to carry out this reaction. A variety of N-alkynyl substrates were tolerated including N-alkynyl oxazolidin-2-one 218a, γ-lactams, and indoles 218b,c. Upon further investigation of the reaction, Zhu proposed the following mechanism: 1) a copper-boryl complex adds to the alkyne via a syn-addition and α-boron regioselectivity, 2) protonolysis of the copper-carbon bond, 3) transmetalation of the vinyl-boron with copper, 4) another protonolysis of the copper-carbon bond. In its entirety, this reaction is a formal addition of H2 to N-alkynyl amides/indoles.
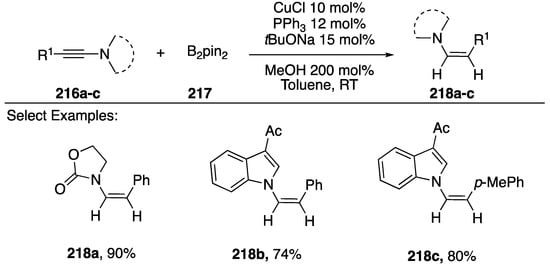
Scheme 58.
Zhu’s copper-catalyzed reduction of N-alkynyl amides.
Hydroboration of alkynes represents an important strategy for synthesizing vinylboronates, important building blocks in the synthesis of complex organic molecules as well as intermediates for alkene preparation by protonolysis [90]. In 2014, Zhu reported a copper-catalyzed hydroboration of N-alkynyl amides/indoles (Scheme 59) [91]. The researchers found that slightly modifying their previous conditions for the reduction of N-alkynyl amides/indoles (Scheme 58) by substituting PPh3 with Xantphos or LB-Phos-HBF4 changed the selectivity of the reaction from reducing the alkyne to generating N-alkenyl-vinylboronates in good yields. This reaction worked well with N-alkynyl oxazolidin-2-ones (220a) and with N-alkynyl indoles 220b,c.
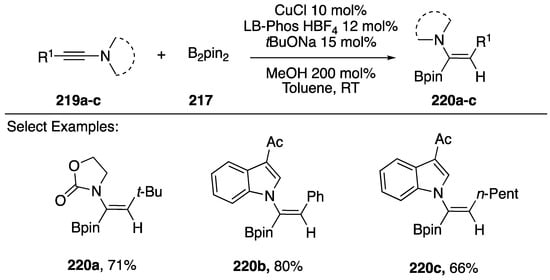
Scheme 59.
Zhu’s stereoselective hydroboration of N-alkynyl amides and indoles.
3.3. Cycloadditions and Annulations
The ability of N-alkynyl azoles to participate in a wide variety of cycloadditionas and annulations provides ready access to a variety of fused heteroaromatic ring systems. While many of these reaction types are shared with N-alkynyl amides and other alkynes, there are notable instances where the course of these cyclization is unique in the case of the N-alkynyl azoles.
3.3.1. [2 + 1] Cycloaddition
Pirrung reported a [2 + 1] cycloaddition of a rhodium carbenoid to N-alkynyl pyrrole 221 in 1994 (Scheme 60) [92]. The researchers carried out the reactions with Rh2(OAc)4 as the catalyst in hexafluorobenzene. Following formation of a rhodium carbenoid with cyclic diazodiketone 222, the carbene adds to the N-alkyne generating the short-lived cyclopropene 223; subsequent rearrangement of 223 gave tetrahydrobenzofuran 224 in moderate yield. Other terminal alkynes also afforded the tetrahydrobenzofuran products in similar yields.
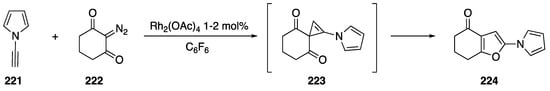
Scheme 60.
Pirrung’s [2 + 1] cycloaddition with N-ethynyl pyrrole.
In 2013, Clavier and Buono published a [2 + 1] cycloaddition of N-alkynyl amides and azoles with norbornene (Scheme 61) [93]. The researchers were interested in developing a new method for synthesizing aminomethylenecyclopropanes, a structure found in powerful antiviral agents, but with limited methods for its synthesis [94]. Ultimately Clavier and Buono found two sets of conditions (A/B) that successfully added N-alkynyl amides/azoles to norbornene. The palladium (II) catalyst used for conditions A had been previously known to promote the hydroalkynylation of alkynes to norbornadienes (Scheme 50). Interestingly, the researchers found that at lower reaction temperatures the same catalyst promoted the [2 + 1] addition of N-substituted alkynes to norbornadienes. These conditions worked well for many different norbornadienes 225a,b and substituted alkynes 226a–c to give aminomethylenecyclopropanes 227aa and 227ca in good yields (73% and 48%, respectively). Clavier and Buono had to use an alternative set of conditions (B) for the N-alkynyl indole substrate they tested, because the preferred catalyst gave an inseparable mixture of compounds.
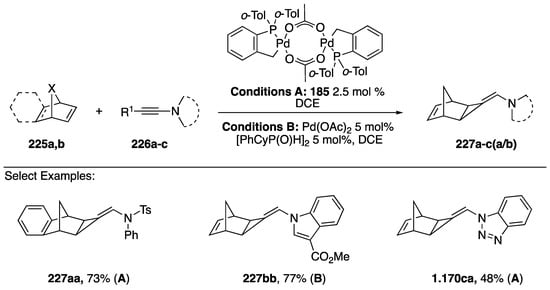
Scheme 61.
Clavier and Buono’s [2 + 1] cycloadditions of N-alkynyl amides/azoles.
3.3.2. [2 + 2] Cycloaddition
The ability of N-alkynyl amines and amides to undergo dipolar [2 + 2] cycloadditions with electron-deficient alkenes (Ficini reaction) is well established [1,95]. However, the application of the Ficini reaction with N-alkynyl azoles had not been reported until 2017 when Alcaide showed that a wide range of heterosubstituted alkynes, including N-alkynidoles, N-alkyncarbazoles, and N-alkynyindazole 228a–g undergo Ficini reaction with 1,1-bis(trifluoromethylsulfonyl)ethane, generated in situ from the zwiterionic precursor 229 to afford the cyclobutenes 230a–d or cyclobutanones 231e–g, the later formed only in the case of 228e–g bearing an electron-donating aryl substituent (Scheme 62) [96].
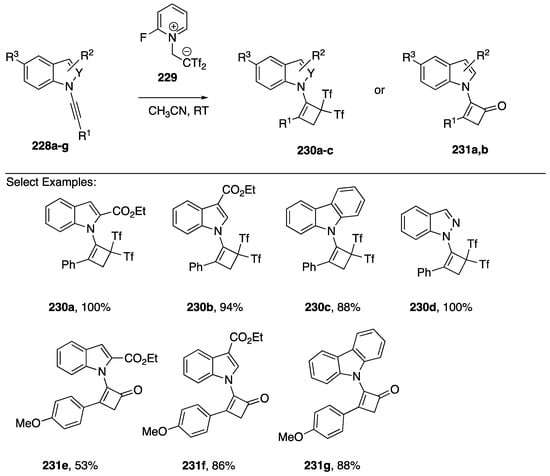
Scheme 62.
Alcaide’s Ficini cycloaddition of N-alkynylindoles, -carbazoles, and indazoles.
3.3.3. [3 + 2] Cycloaddition
In 2016, Watson and Burley disclosed a [3 + 2] copper-catalyzed “click” cycloaddition of N-alkynyl benzimidazole with assorted azides (Scheme 63) [97]. The researchers found that N-alkynyl benzimidazole 232 reacted with azides 233a–c, undergoing reaction in both MeCN and MeOH solvents to afford the triazoles 234a–c in excellent yields (83–100%). Interestingly, aliphatic alkynes gave no desired product when the cycloadditions were carried out in MeOH, and the researchers were able to exploit this characteristic to perform sequential chemoselective click reactions.
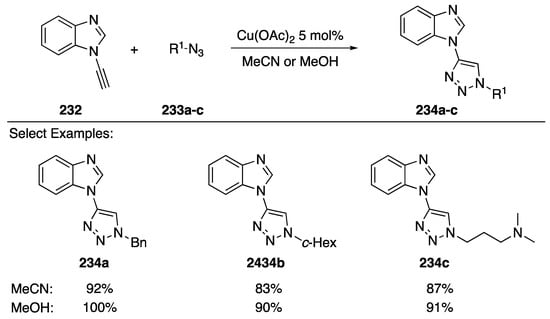
Scheme 63.
Watson and Burley’s click reaction with N-alkynyl benzimidazole.
Subsequent work by Burley and Watson revealed that there is a switch in the rate-determining step for triazole formation from copper-acetylide formation to azide-ligation in the case of 232 [98]. With this insight, these workers have developed an orthogonal scheme for click-cycloadditions based on tri(isopropyl)silyl-protected N-alkynylbenzimidazoles (Scheme 64) [99].
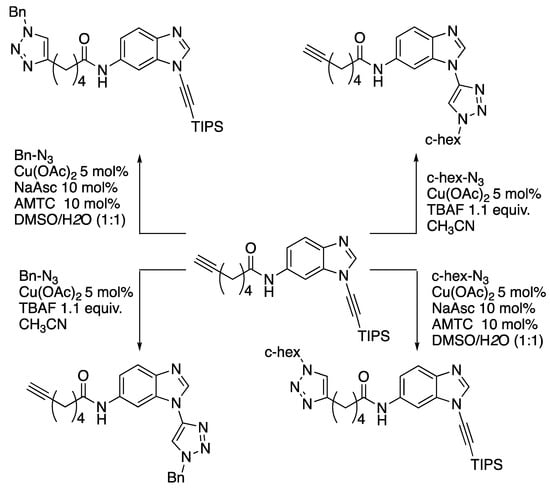
Scheme 64.
Watson and Burley’s orthogonal click-cycloaddition scheme.
A number of studies have reported metal-catalyzed formal [3 + 2] additions of N-alkynyl amides and have included N-alkynyl azole examples. Reddy reported a copper-mediated addition of 2-aminopyridine to N-alkynyl amides and the N-alkynylindoles 235a–c to afford the indole-substituted imidazolopyridines 236a–c (Scheme 65, Reaction (1) [100]). In 2017, building up their work on gold-catalyzed formal [3 + 2] cycloadditions [101,102], Davies group reported the gold-catalyzed addition of the N,N-dipole 238 to the N-alkynylindole 237 to afford the fused heterocycle 239 (Scheme 65, Reaction (2) [103]).
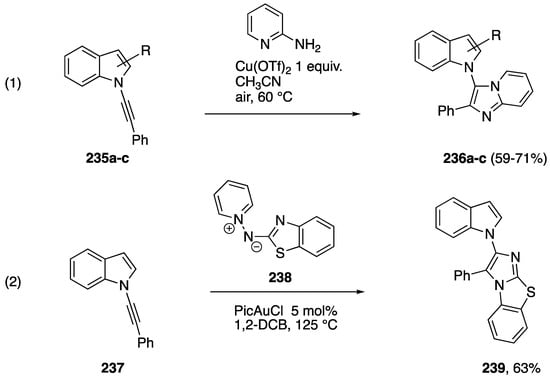
Scheme 65.
Metal-catalyzed formal [3 + 2] additions of N-alkynylindoles.
In 2017, Li published a cobalt-catalyzed synthesis of 5-aminooxazoles via a [3 + 2] cycloaddition of N-(pivaloyloxy)amides with N-alkynyl amides/azoles (Scheme 66) [104]. The researchers were interested in developing a Co(III)-catalyzed synthesis of amino-substituted isoquinolones via carbon-hydrogen (C-H) activation and were surprised when they isolated 5-aminooxazole as the sole product. Interestingly, the reaction gave the opposite regioisomer compared to previously reported metal-catalyzed syntheses of aminooxazoles [102].
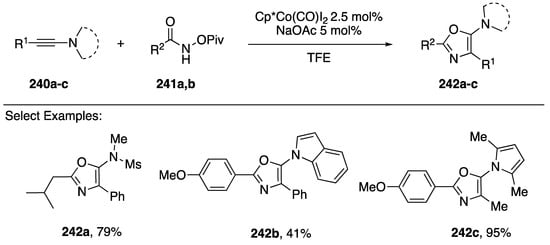
Scheme 66.
Li’s cobalt-catalyzed synthesis of 5-azole-oxazoles.
Li found that under their optimized conditions (Scheme 66) they could synthesize a large variety of 5-aminooxazoles 242a–c; the reaction tolerated both aromatic and aliphatic substitutions in the 2-position, as well as both aromatic and aliphatic substitutions in the 4-position. However, in the case of terminal N-alkynyl amides, the reaction gave significantly reduced yields. The researchers even performed the reaction using an N-alkynyl indole 242b and an N-alkynyl pyrrole 242c giving the corresponding 5-azole-oxazole products in 41% and 95% yields, respectively.
An alternative formal [3 + 2] addition to afford oxazoles was reported in 2018 by Zhou and co-workers (Scheme 67) [105]. They showed that a variety of N-alkynyl amides and N-alkynyl azoles 234a–c undergo diphenyl diselenide-catalyzed oxidative addition of solvent acetonitrile to afford 2-methyloxazoles 244a–c (Scheme 67). This process was proposed to proceed through a selenirenium ion intermediate that is trapped by acetonitrile and then undergoes oxidation and cyclization to the oxazole.
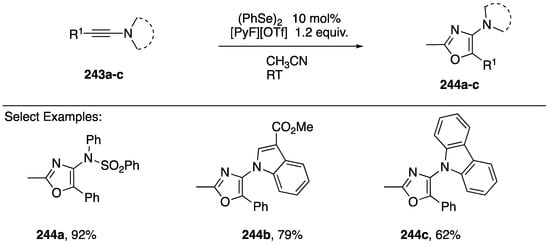
Scheme 67.
Zhou’s selenium π-acid catalyzed synthesis of 2-methylozazoles.
3.3.4. [4 + 2] Cycloaddition
In 2016, Gandon and Blanchard published an intramolecular [4 + 2] inverse electron-demand cycloaddition of N-alkynyl amides and indoles as a new method to generate unique fused pyridine bicycles (Scheme 68) [106]. Although the participation of N-substituted alkynes in cycloaddition reactions is well documented, prior to their report the reaction with pyrimidine was unknown [107]. Pyrimidines are poorly reactive as electron-deficient azadienes in [4 + 2] reactions [108], however, the researchers hypothesized that when linked with N-substituted alkynes the reaction pair should be adequately reactive.
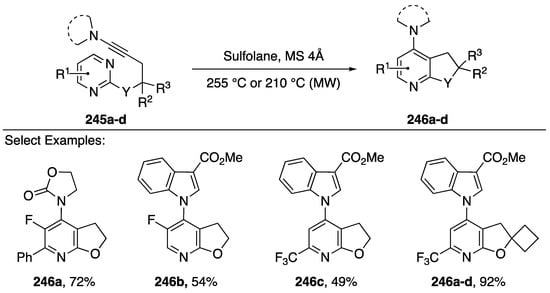
Scheme 68.
Gandon and Blanchard’s [4 + 2] cycloaddition of N-substituted alkynes.
Gandon and Blanchard found that heating the starting materials 245a–d by microwave irradiation in sulfolane afforded corresponding pyridines 246a–d in moderate to excellent yields (49–92%). The reaction tolerated many different nitrogen-substitutions on the alkyne, including oxazolidin-2-one 246a and 3-substituted indoles 246b–d. Additionally, many different substitutions were tolerated on the pyrimidine, including phenyl (compound 246a) fluorine (compounds 246a,b) and trifuoromethyl (compounds 246c,d). Following their initial report, in 2017, Gandon and Blanchard published a full paper that includes a DFT study of this inverse electron demand Diels-Alder/retro-Diels-Alder sequence [109].
3.3.5. [2 + 2 + 2] Cycloaddition
In 2015, Goswami published a study of a metal-catalyzed [2 + 2 + 2] to N-arylindoles from N-alkynylindoles [110]. After screening a number of metal catalysts, they found that N-alkynylindoles 247a–c reacted with the diynes 248a,b in the presence of catalytic FeCl2·4H2O, zinc, and ligand (E)-N-(2,6-diisopropylphenyl)-1-(pyridin-2-yl)methanimine (dipimp) in ethanol to afford the N-arylindoles 249a–d(a/b) (Scheme 69).
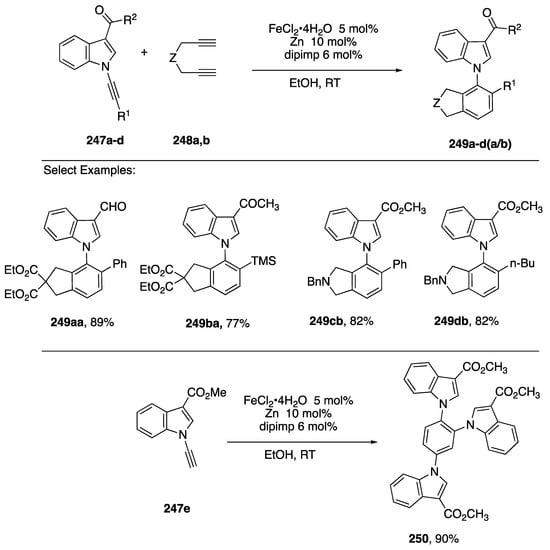
Scheme 69.
Goswami’s [2 + 2 + 2] route to N-arylindoles and tri-indolylbenzenes.
Although only 3-carbonylindole partners 247a–d were explored, a wide range of alkyne substitutents R1 were tolerated. While the reaction proceeded well with symmetrical, terminal diynes 248a,b, attempts to carry out the reaction with 1,7-octadiyne failed and reactions with non-diterminal diynes afforded mixtures of regioisomeric products. Similarly, attempts to carry out a [2 + 2 + 2] trimerization of 247a afforded a regioisomeric mixture of tri-indole-substituted benzene products; however, trimerization of the N-ethynylindole 247e gave a single product 250 in excellent yield.
3.3.6. [3 + 2 + 2] Cycloaddition
Saito reported a nickel-catalyzed [3 + 2 + 2] cycloaddition of ethyl-cyclopropylideneacetate and N-alkynyl pyrrole in 2010 (Scheme 70) [111]. The researchers were interested in the use of heteroatom-substituted alkynes as substrates in three-component cycloadditions to construct complex cycloheptadienes in one step. Saito started his investigations with O-substituted alkynes, but also documented the [3 + 2 + 2] cycloaddition of N-substituted alkynes. In their investigations, the researchers found that N-alkynyl carbamates reacted with ethylcyclopropylidene acetate (252) to give mixtures of [3 + 2 + 2] and cyclotrimerization products. However, Saito reported that in the case of N-ethynyl pyrrole substrate 251, the reaction with 251 proceeded smoothly giving only the desired cycloaddition product 252 in a 72% yield (Scheme 70, Reaction (1)). The researchers also reported a cycloaddition using diacetylenes 253a–c as substrates in the [3 + 2 + 2] reaction with N-ethynyl pyrrole 251 (Scheme 70, Reaction (2)). Due to differences in sterics and electronics Saito was able to isolate the desired products, 253a–c, in moderate yields.
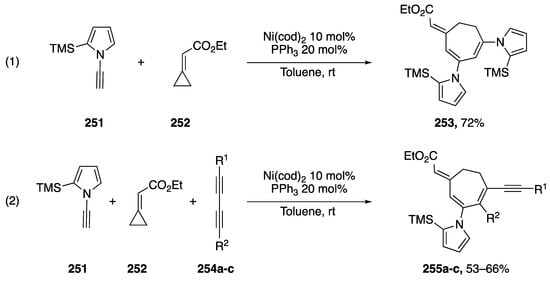
Scheme 70.
Saito’s [3 + 2 + 2] cycloaddition with N-alkynyl pyrrole.
3.3.7. [4 + 3 + 2] Cycloaddition
In 2013, Saito published a nickel-catalyzed [4 + 3 + 2] cycloaddition of ethyl-cyclopropylideneacetate 252 with a number of dienynes (Scheme 71) [112]. The researchers thoroughly investigated the scope of dienyne substrates, which included the N-alkynyl pyrrole dienyne 256. Under Saito’s optimized conditions, dienyne 256 reacted to give the polycyclic heterocycle 257 in a 69% yield. Their method represents just one of a just a few transition metal-mediated synthesis of 9-membered carbocycles.
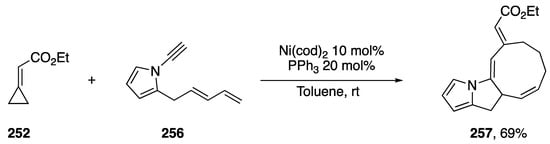
Scheme 71.
Saito’s nickel-catalyzed [4 + 3 + 2] cycloaddition with N-alkynyl pyrrole.
3.3.8. Other Annulations
There are a few examples of other annulation reactions involving N-alkynylindoles that have been reported during studies focused on N-alkynyl amide annulations. Xie and Hashmi reported in 2016 a gold-catalyzed preparation of quinolines from propargyl ethers and benzo[c]isoxazole 259 (Scheme 72, Reaction (1) [113]).
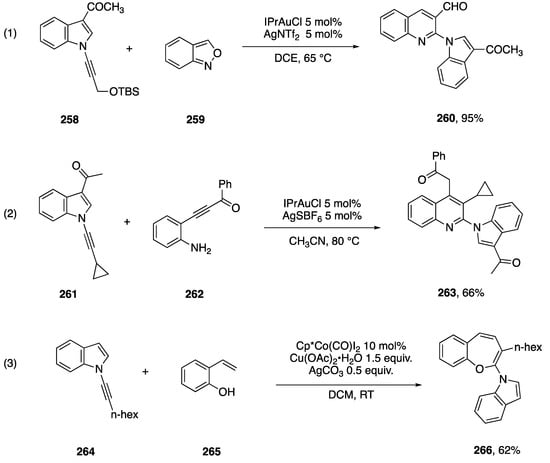
Scheme 72.
Other annulations of N-alkynylindoles.
In the course of examining the scope of propargyl ethers that participate in this reaction, they reported that N-alkynylindole 258 affords excellent yield of the quinolone 260. In 2018, Arcadi and co-workers reported an alternative route to quinolones via a gold-catalyzed annulation of N-alkynyl amides with β-(2-aminiphenyl)-α,β-ynones [114]. During this work they found that a range of N-substituted alkynes, including the N-alkynylindole 261 adds to the ynone 262 affording the quinoline 263 (Scheme 72, Reaction (2)). In 2018, Li and co-workers reported an oxidative cobalt-catalyzed [2 + 5] addition of ortho-vinylphenols with N-alkynyl amides [115]. In exploring the scope of this annulation they found that the N-alkynylindole 264 cleanly affords the benzoxepine 266 (Scheme 72, Reaction (3)). In contrast, the corresponding N-phenylethynylindole afforded mixtures of regioisomers.
3.4. Bergman Cyclizations
The Bergman cyclization has garnered the attention of many chemists due to the naturally occurring enediyne cores of many natural products that can generate diradicals via the Bergman cyclization and cleave DNA, imparting potent cytotoxicity to these compounds. As part of a research program aiming to temper the cytotoxicity and improve on the lack of selectivity of Bergman diradicals towards DNA cleavage, Kerwin reported in 2002 the surprising results from the thermolysis of the heterocyclic 3-aza-3-ene-1,5-diyne 268 (Scheme 73) [31]. The N-alkynyl imidazole 268, when heated in neat 1,4-cyclohexadiene (1,4-CDH) afforded the cyclopentapyrazines 269a,b as a mixture of stereoisomers together with the cyclopentapyrazine 270. In contrast, when the thermolysis was carried out in chlorobenzene containing 1,4-CHD, the imidazolopyridine 271 was isolated. Based upon these results and a subsequent study demonstrating the generality of these transformations [32], it was proposed that these products are the result of an unprecedented aza-Bergman/retro-aza-Bergman cascade involving initial formation of the diradical 272 which undergoes collapse to the cyclic cumulene 273. Cyclization of 273 to the carbene 275 followed by trapping with 1,4-CHD leads to the products 269a,b and 270, while an alternative cyclization to the benzyne 274 and trapping by solvent leads to 271.
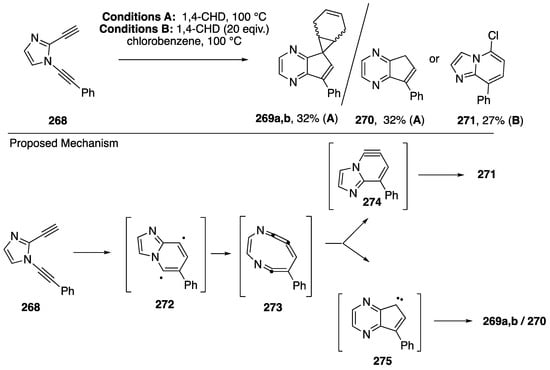
Scheme 73.
Kerwin’s aza-Bergman cyclization-based rearrangements of dialkynylimidazoles.
In 2006, Kerwin reported the synthetic application of these dialkynylimidazole rearrangements (Scheme 74) [116]. Thermolysis of the dialkynylimidazoles 276a–g in benzene affords moderate to good yields of the benzene-trapped products 277a–g. In the case of a dialkynylimidazole with a pendant alkene, thermolysis in hexafluorobenzene instead affords the tetracyclic product 278g. Additional work showed that the benzyne intermediate (cf, 274, Scheme 73) can also be intercepted with HCl to afford chloroimidazolopyridine products [117].
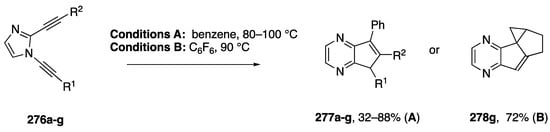
Scheme 74.
Kerwin’s dialkynylimidazole route to cyclopentapyrazines.
3.5. Sigmatropic Rearrangements
In 2012, Rabasso published a [2,3]-sigmatropic rearrangement of N-alkynyl amides [118]. The following year, 2013, during their investigations into the selective reduction of amino-allenephosphonates, Rabasso expanded the scope of their [2,3]-sigmatropic rearrangement to include N-alkynyl indole (Scheme 75) [119]. The researchers found that compound 279, underwent a [2,3]-rearrangement when treated with diethyl chlorophosphite, giving N-allenylphosphate 281 in a 78% yield. Unfortunately, Rabasso was unable to achieve a selective reduction with 281.
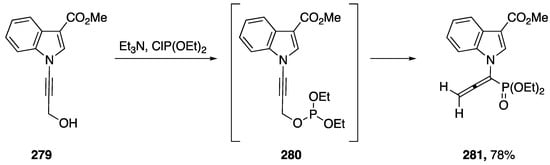
Scheme 75.
Rabasso’s [2,3]-sigmatropic rearrangement of N-alkynyl indole.
In 2017, Zhao and Gagosz published a gold-catalyzed hydride shift towards the synthesis of N-allenyl amides/azoles from N-alkynyl amides/azoles (Scheme 76) [120]. Under optimized conditions, the researchers found that a large number of N-alkynyl amides and azoles were tolerated in this reaction (282a–d): N-alkynyl indole, pyrrole, and carbazole all gave the respective allene products (283a–d), additionally a variety of R1 and R2 substitutions were tolerated. Zhao and Gagosz also discuss a cascade reaction with N-alkynyl indoles and pyrroles in which the initially formed allene reacts with the electron-rich heterocycles forming new 5-membered rings (not shown).
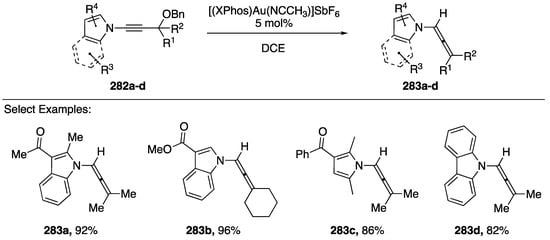
Scheme 76.
Zhao and Gagosz’s gold-catalyzed hydride shift of N-alkynyl azoles.
3.6. Other Reactions
In addition to the transformations discussed above, which focus on reactions at the alkyne of N-alkynyl azoles, there are numerous examples of reactions in which the N-alkynyl group is inert, allowing further functionalization of the azole core. Many of these are incorporated into the above section as routes to the various N-alkynyl azole substrates. In addition, there have been a number of reports the focus on these functionalization reactions. While investigating the ZnBr2 catalyzed reaction of N-alkynyl amides with benzylic alcohols Cao and Xu reported an unexpected reaction of N-alkynyl indole (Scheme 77, equation 1) [121]. While other N-substituted alkynes reacted under these conditions in a nucleophilic fashion to the benzylic alcohols, N-alkynyl indole 284 was inert, instead reacting at the 3-position with substrate 285 generating N-alkynyl indole 286 in a 45% yield. In 2018, Zeni reported the 2-functionalization of N-alkynyl indoles 287a–f by deprotonation with n-butyl lithium followed by trapping with a variety of aldehydes to afford the 2-substituted compounds 288 (Scheme 77, equation 2) [122]. No products were obtained from N-alkynyl indoles 287 bearing p-methoxyphenyl or o-chlorophenyl substituents, and the scope of aldehydes is generally limited to substituted benzaldehydes or formaldehyde; although, alternative electrophiles such as benzophenone and chlorosilanes also afforded products.
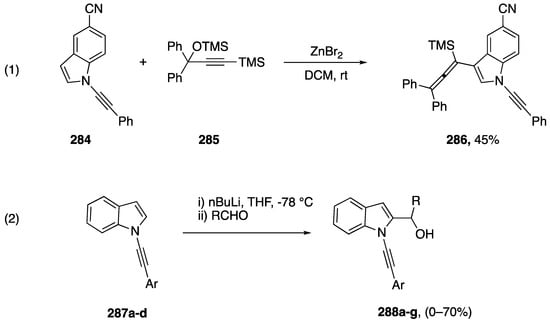
Scheme 77.
Functionalization of N-alkynyl indoles.
4. Applications of N-Alkynyl Azoles
The explosion of interest in the synthesis of N-alkynyl azoles over the last 15 years has fueled a burgeoning interest in the exploration of various applications of these compounds. N-alkynyl azoles have found applications in the total synthesis of natural products, the synthesis of polymers, and they have had their physical and biological properties evaluated.
4.1. Total Syntheses
In 2014, Beaudry published an N-alkynyl indole Diels-Alder strategy for the synthesis of the bis-indole alkaloids from Arundo donax (Scheme 78) [123]. Beaudry’s strategy involved the initial synthesis of N-alkynyl indole 291 from alkyne 290 and indole 289. Using conditions inspired by Stahl, N-alkynyl indole 291 was isolated in a 57% yield. A subsequent Diels-Alder reaction at 150 °C efficiently produced 292 in a 90% yield, completing the core of the natural products. Over several more steps the researchers were able to complete the synthesis of arundamine, arundanine, arundacine, and arundarine. Interestingly, Beaudry was able to show that these structures form atropisomers that could be resolved with a chiral HPLC. The researchers determined the half-lives of racemization to be between 1 and 7 h.
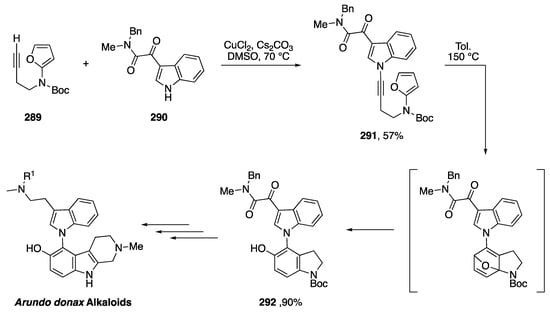
Scheme 78.
Beaudry’s synthesis of the bis-indole alkaloids from Arundo donax.
In 2017, Pale and Beneteau published a zeolite based organic synthesis of acortatarin A using an N-alkynyl pyrrole retrosynthetic strategy (Scheme 79) [124].
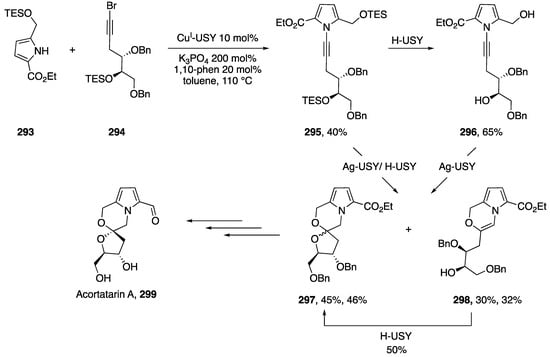
Scheme 79.
Pale and Beneteau’s zeolite strategy towards acortatarin A.
The researchers used zeolite catalysis for the key steps of their synthesis. The first instance being a CuI-USY catalyzed cross-coupling of 1-bromoalkyne 294 and pyrrole 293 to give N-alkynyl pyrrole 295 in a 40% yield. A subsequent spiroketalization of 295 was carried out with Ag-USY and H-USY to give spiroketal 297 in a 45% yield and the partially cyclized product 298 in a 30% yield. Pale and Beneteau found that separating the steps and first forming diol 296 with H-USY and then carrying out the spiroketalization did not help improve the yields. Additionally, the researchers were able to finish the spirocyclization of 298 using a H-USY zeolite. Following a few finishing steps, acortatarin A was synthesized with a 3:1 dr.
4.2. Polymer Synthesis
Some of the earliest reports of N-alkynyl azoles in the literature involve their synthesis and subsequent polymerization. One of the more studied examples is the polymerization of the diacetylenes of N-alkynyl pyrrole and carbazole 300 (Scheme 80, Reaction (1) [14,60,125]).
Scheme 80.
Application of N-alkynyl azoles in polymerization reactions.
The diacetylene polymers 301 were often studied for their optical properties. Polyacetylene polymers of N-alkynyl azoles have been investigated as well. In 1970, Okamoto published the synthesis of poly-N-ethynyl carbazole 303 and studied the conductivity of the polymer (Scheme 80, Reaction (2) [8]). Subsequent studies on this and related polymers have appeared [125,126,127]. Additionally, polymerizations in which the alkyne remains intact have been reported. Yamamoto published the co-polymerization of N-alkynyl pyrroles 304a,b with the di(stanyl)thiophenes 305a,b (Scheme 80, Reaction (3) [128,129,130]). Yamamoto also reported the oxidative polymerization of 307 with TBAPF6 in 2012 (Scheme 80, Reaction (4) [131]). Okamoto also published a paper in 1973 investigating the photoconductive properties of arylethynylcopper polymers including the copper polymer of N-ethynyl carbazole [132].
4.3. Other Applications
N-alkynyl azoles have been studied for their physical properties. In 1998, Dellepiane published a paper investigating the vibrational properties of several diacetylenes, including a diacetylene of N-alkynyl carbazole [133]. Additionally, in 2013, Sambri, Tonelli, and Armaroli studied donor-acceptor luminophores made from N-alkynyl carbazole [134]. Trolez has reported on the optoelectronic properties of N-alkynylcarbazole- and indole derivatives [135]. Burley has studied the metal ion coordination of triazoles derived from N-alkynyl benzimidazoles [136].
In addition to studies of their physical properties, there have been several reports investigating the biological properties of N-alkynyl azoles. Zemlicka disclosed their investigations into the biological activity of N9-alkynyl adenine in 1994 [16]. They found N9-alkynyl adenine to be a substrate of moderate activity for adenosine deaminase, in addition it inhibited the growth of murine leukemia L1210, and suppressed growth of mouse and human colon tumors C38, H8 or H116. In 2009, Kerwin published a paper disclosing their biological evaluation of dialkynylimidazoles specifically designed to target p38α kinase [137]. Using this scaffold, these workers developed a molecular probe that led to the discovery of a novel class of p-38α docking-recognition site inhibitors [138]. In future studies, Kerwin investigated the ability of dialkynylimidazoles to induce apoptosis in A549 cancer cells, and the researchers also investigated whether aza-Bergman rearrangement rates could be used to predict cytotoxicity [64]. Bhattacharjee has also investigated the cytotoxicity of N-alkynyl pyrazoles [56].
5. Conclusions
This review has covered the synthesis and reactions of N-alkynyl azoles, a small subclass of nitrogen-substituted alkynes, which have received a lot of attention over the last several decades. Initially researchers synthesized these molecules using classical dehydrohalogenation chemistry that limited the scope of accessible substrates; however, with the discovery of milder catalytic protocols for the synthesis of N-alkynyl amides and azoles the synthesis of more complicated structures containing sensitive functional groups has become possible. Additionally, these catalytic protocols have brought more attention to the reactions of N-alkynyl azoles and the various potential applications of the compounds. N-alkynyl azoles have been used in metal-catalyzed addition reactions, they have found tremendous use in cycloaddition reactions, they have been applied to the total synthesis of several natural products, and have been investigated for their biological activity and use in polymers.
We believe the importance of both azoles and alkynes to the organic community will ensure a continued interest in N-alkynyl azoles. Notably, although there has been significant improvement in the procedures for their synthesis, we believe that continued efforts towards developing even more efficient methods would be of value to the synthetic community. Additionally, further work still needs to be done towards understanding the distinct reactivity of the many different N-alkynyl azoles. On a final note, due to the importance of azoles within the pharmaceutical industry we believe the application of N-alkynyl azoles for the synthesis of natural products and designed molecules will continue to grow in the future.
Author Contributions
Writing—original draft preparation, B.R.; writing—review and editing, and funding acquisition, S.M.K.
Funding
We gratefully acknowledge the William and Ella Owens Medical Research Foundation, the Robert Welch Foundation, and Texas State University for support.
Acknowledgments
We would like to thank all of the authors whose work cited here has advanced the chemistry of N-alkynyl azoles, and we apologize to any authors whose work we unknowingly overlooked.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ficini, J. Ynamine—Versatile Tool in Organic-Synthesis. Tetrahedron 1976, 32, 1449–1486. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Paine, S.W.; Popik, V.V. Reactive Species: Ynols and Ynamines. J. Phys. Org. Chem. 1996, 9, 361–370. [Google Scholar] [CrossRef]
- Zificsak, C.A.; Mulder, J.A.; Hsung, R.P.; Rameshkumar, C.; Wei, L.L. Recent advances in the chemistry of ynamines and ynamides. Tetrahedron 2001, 57, 7575–7606. [Google Scholar] [CrossRef]
- Pan, F.; Shu, C.; Ye, L.W. Recent progress towards gold-catalyzed synthesis of N-containing tricyclic compounds based on ynamides. Org. Biomol. Chem. 2016, 14, 9456–9465. [Google Scholar] [CrossRef] [PubMed]
- DeKorver, K.A.; Li, H.; Lohse, A.G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R.P. Ynamides: A Modern Functional Group for the New Millennium. Chem. Rev. 2010, 110, 5064–5106. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Blanchard, N.; Compain, G.; Coste, A.; Demmer, C.S.; Gati, W.; Guissart, C.; Heimburger, J.; Henry, N.; Jouvin, K.; et al. A Journey in the Chemistry of Ynamides: From Synthesis to Applications. Chem. Lett. 2016, 45, 574–585. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Jiang, R.; Singh, S.K. Synthesis and reactions of N-ethynyl-heterocycles. Heterocycles 2004, 63, 1455–1475. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kundu, S.K. Synthesis and Properties of N-Ethynylcarbazoleand Poly-N-ethynylcarbazole. J. Org. Chem. 1970, 35, 4250–4252. [Google Scholar] [CrossRef]
- Viehe, H.G. Synthesis and Reactions of Alkynylamines. Angew. Chem. Int. Ed. 1967, 6, 767–778. [Google Scholar] [CrossRef]
- Pielichowski, J.; Chrzaszcz, R. A New Method of Synthesis of 9-Ethynylcarbazole and Its Derivatives. Bull. Soc. Chim. Belg. 1995, 104, 117–118. [Google Scholar] [CrossRef]
- Burger, U.; Dreier, F. Reactions of Nitrogen Containing Aromatic Anions with Chlorocarbene. Tetrahedron 1983, 39, 2065–2071. [Google Scholar] [CrossRef]
- Deangelis, F.; Gambacorta, A.; Nicoletti, R. Improved Route for Ring Expansion of 5-Membered Heterocyclic-Compounds by Use of Phase Transfer Catalysts. Synthesis 1976, 798–800. [Google Scholar] [CrossRef]
- Paley, M.S.; Frazier, D.O.; Abeledeyem, H.; McManus, S.P.; Zutaut, S.E. Synthesis, Vapor Growth, Polymerization, and Characterization of Thin-Films of Novel Diacetylene Derivatives of Pyrrole—The Use of Computer Modeling to Predict Chemical and Optical-Properties of these Diacetylenes and Poly(Diacetylenes). J. Am. Chem. Soc. 1992, 114, 3247–3251. [Google Scholar] [CrossRef]
- Brandsma, L.; Mal’kina, A.G.; Trofimov, B.A. An Improved Procedure for N-Ethynylpyrrole. Synth. Commun. 1994, 24, 2721–2724. [Google Scholar] [CrossRef]
- Joshi, R.V.; Xu, Z.-Q.; Ksebati, M.B.; Kessel, D.; Corbett, T.H.; Drach, J.C.; Zemlicka, J. Synthesis, Transformations and Biological Activity of Chloro Enamines and Ynamines Derived from Chloroalkenyl- and Alkynyl-N-substituted Purine and Pyrimidine Bases of Nucleic Acids. J. Chem. Soc. Perkin Trans. 1 1994, 1089–1098. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Zhang, S.M.; Hussein, A.H.M.; Fang, Y.F.; Steel, P.J. One-carbon homologation of carboxylic acids via BtCH(2)TMS: A safe alternative to the Arndt-Eistert reaction. J. Org. Chem. 2001, 66, 5606–5612. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Abdel-Fattah, A.A.A.; Wang, M. A Novel Access to Disubstituted Acetylenes. J. Org. Chem. 2002, 67, 7526–7529. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Singh, S.K.; Jiang, R. A convenient synthesis of functionalized N-(ethynyl)benzotriazoles. Tetrahedron 2006, 62, 3794–3797. [Google Scholar] [CrossRef]
- Mansfield, S.J.; Campbell, C.D.; Jones, M.W.; Anderson, E.A. A robust and modular synthesis of ynamides. Chem. Commun. 2015, 51, 3316–3319. [Google Scholar] [CrossRef]
- Tu, Y.L.; Zeng, X.Z.; Wang, H.; Zhao, J.F. A Robust One-Step Approach to Ynamides. Org. Lett. 2018, 20, 280–283. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Wang, J.; Karodia, N.; Li, J. A Novel Transformation of Esters to Alkynes with 1-Substituted Benzotriazoles. J. Org. Chem. 1997, 62, 4142–4147. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Xiao, J.; Peng, Z.; Dong, W.; An, D. Base-Induced One-Pot Preparation of N- or P-Substituted Alkynes. Eur. J. Org. Chem. 2015, 2015, 7806–7815. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Alkynyliodonium salts in organic synthesis. Tetrahedron 1998, 54, 10927–10966. [Google Scholar] [CrossRef]
- Brand, J.P.; Waser, J. Electrophilic alkynylation: The dark side of acetylene chemistry. Chem. Soc. Rev. 2012, 41, 4165–4179. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Tashi, N.; Tsuda, K.; Chen, H.Y.; Fujiwara, Y. A convenient synthesis of new 1-alkynyl-1H-benzotriazoles by reaction of alkynyl(phenyl)iodonium salts with benzotriazole ion. Heterocycles 2000, 52, 303–312. [Google Scholar] [CrossRef]
- Kitamura, T.; Tashi, N.; Tsuda, K.; Fujiwara, Y. Alkynylbenzotriazoles by direct alkynylation of benzotriazole using alkynyliodonium salts. Tetrahedron Lett. 1998, 39, 3787–3790. [Google Scholar] [CrossRef]
- Kitamura, T.; Morshed, M.H.; Tsukada, S.; Miyazaki, Y.; Iguchi, N.; Inoue, D. Alkynylation of benzotriazole with silylethynyliodonium triflates. Regioselective synthesis of 2-ethynyl-2H-benzotriazole derivatives. J. Org. Chem. 2011, 76, 8117–8120. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.Y.; Oprunenko, Y.F.; Beletskaya, I.P. Pd/Al2O3-catalysed regioselective N-1-modification of benzotriazoles using iodonium salts. Tetrahedron Lett. 2017, 58, 4465–4467. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, Q. Preparation of resin-bound alkynyl iodonium salts and their application in organic synthesis as alkynyl transfer reagents. Tetrahedron Lett. 2001, 42, 6373–6375. [Google Scholar] [CrossRef]
- Nadipuram, A.N.; David, W.M.; Kumar, D.; Kerwin, S.M. Synthesis and Thermolysis of Heterocyclic 3-Aza-3-ene-1,5-diynes. Org. Lett. 2002, 4, 4543–4546. [Google Scholar] [CrossRef]
- Kerwin, S.M.; Nadipuram, A. 5H-Cyclopentapyrazines from 1,2-Dialkynylimidazoles. Synlett 2004, 1404–1408. [Google Scholar] [CrossRef]
- Roy, A.; Das, M.K.; Chaudhuri, S.; Bisai, A. Transition-Metal Free Oxidative Alkynylation of 2-Oxindoles with Ethynylbenziodoxolone (EBX) Reagents. J. Org. Chem. 2018, 83, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Toriumi, N.; Asano, N.; Miyamoto, K.; Muranaka, A.; Uchiyama, M. N-Alkynylpyridinium Salts: Highly Electrophilic Alkyne-Pyridine Conjugates as Precursors of Cationic Nitrogen-Embedded Polycyclic Aromatic Hydrocarbons. J. Am. Chem. Soc. 2018, 140, 3858–3862. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications. ACS Catal. 2012, 2, 781–794. [Google Scholar] [CrossRef]
- Frederick, M.O.; Mulder, J.A.; Tracey, M.R.; Hsung, R.P.; Huang, J.; Kurtz, K.C.M.; Shen, L.C.; Douglas, C.J. A copper-catalyzed C-N bond formation involving sp-hybridized carbons. A direct entry to chiral ynamides via N-alkynylation of amides. J. Am. Chem. Soc. 2003, 125, 2368–2369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Hsung, R.P.; Tracey, M.R.; Kurtz, K.C.M.; Vera, E.L. Copper sulfate-pentahydrate-1,10-phenanthroline catalyzed amidations of alkynyl bromides. Synthesis of heteroaromatic amine substituted ynamides. Org. Lett. 2004, 6, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Zhang, Y.S.; Huang, J.; Hsung, R.P.; Kurtz, K.C.M.; Oppenheimer, J.; Petersen, M.E.; Sagamanova, I.K.; Shen, L.C.; Tracey, M.R. Copper(II)-catalyzed amidations of alkynyl bromides as a general synthesis of ynamides and Z-enamides. An intramolecular amidation for the synthesis of macrocyclic ynamides. J. Org. Chem. 2006, 71, 4170–4177. [Google Scholar] [CrossRef] [PubMed]
- Laroche, C.; Li, J.; Freyer, M.W.; Kerwin, S.M. Coupling reactions of bromoalkynes with imidazoles mediated by copper salts: Synthesis of novel N-alkynylimidazoles. J. Org. Chem. 2008, 73, 6462–6465. [Google Scholar] [CrossRef]
- Hirano, S.; Tanaka, R.; Urabe, H.; Sato, F. Practical preparation of N-(1-alkynyl)sulfonamides and their remote diastereoselective addition to aldehydes via titanation. Org. Lett. 2004, 6, 727–729. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Danheiser, R.L. Copper-mediated N-alkynylation of carbamates, ureas, and sulfonamides. A general method for the synthesis of ynamides. Org. Lett. 2003, 5, 4011–4014. [Google Scholar] [CrossRef]
- Reinus, B.J.; Kerwin, S.M. A Copper-Catalyzed N-Alkynylation Route to 2-Substituted N-Alkynyl Pyrroles and Their Cyclization into Pyrrolo 2,1-c oxazin-1-ones: A Formal Total Synthesis of Peramine. Synthesis 2017, 49, 2544–2554. [Google Scholar]
- Ziegler, D.T.; Choi, J.; Munoz-Molina, J.M.; Bissember, A.C.; Peters, J.C.; Fu, G.C. A Versatile Approach to Ullmann C-N Couplings at Room Temperature: New Families of Nucleophiles and Electrophiles for Photoinduced, Copper-Catalyzed Processes. J. Am. Chem. Soc. 2013, 135, 13107–13112. [Google Scholar] [CrossRef] [PubMed]
- Burley, G.A.; Davies, D.L.; Griffith, G.A.; Lee, M.; Singh, K. Cu-Catalyzed N-Alkynylation of Imidazoles, Benzimidazoles, Indazoles, and Pyrazoles Using PEG as Solvent Medium. J. Org. Chem. 2010, 75, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Reddy, G.C.; Balasubramanyam, P.; Salvanna, N. Copper(II) Oxide Catalyzed Ligand-Free Coupling Reaction of Heteroarenes with Bromoalkynes. Synthesis 2011, 816–820. [Google Scholar] [CrossRef]
- Song, J.H.; Wang, Q.B.; Fu, H.X.; Fu, S.L.; Li, S.B.; Shi, F.; Wu, C.R. Copper-Catalyzed N-Alkynylation of N-tert-Butyloxycarbonyl (Boc)-Protected Indoles. Asian J. Org. Chem. 2013, 2, 877–881. [Google Scholar] [CrossRef]
- Harkat, H.; Borghèse, S.; Nigris, M.D.; Kiselev, S.; Bénéteau, V.; Pale, P. Zeo-Click Synthesis: Copper-Zeolite-Catalyzed Synthesis of Ynamides. Adv. Synth. Catal. 2014, 356, 3842–3848. [Google Scholar] [CrossRef]
- Yao, B.B.; Liang, Z.J.; Niu, T.M.; Zhang, Y. Iron-Catalyzed Amidation of Alkynyl Bromides: A Facile Route for the Preparation of Ynamides. J. Org. Chem. 2009, 74, 4630–4633. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Salvanna, N.; Reddy, G.C.; Balasubramanyam, P. Copper-catalyzed coupling of imidazoles and s with 1,1-dibromo-1-alkenes: A distinct approach for direct N-alkynylation of heteroarenes. Tetrahedron Lett. 2011, 52, 6497–6500. [Google Scholar] [CrossRef]
- Wang, M.G.; Wu, J.; Shang, Z.C. A Simple and Efficient Copper-Catalyzed Synthesis of N-Alkynylimidazoles. Synlett 2012, 23, 589–594. [Google Scholar]
- Jouvin, K.; Coste, A.; Bayle, A.; Legrand, F.; Karthikeyan, G.; Tadiparthi, K.; Evano, G. Copper-Mediated Selective Cross-Coupling of 1,1-Dibromo-1-alkenes and Heteronucleophiles: Development of General Routes to Heterosubstituted Alkynes and Alkenes. Organometallics 2012, 31, 7933–7947. [Google Scholar] [CrossRef]
- Hamada, T.; Ye, X.; Stahl, S.S. Copper-catalyzed aerobic oxidative amidation of terminal alkynes: Efficient synthesis of ynamides. J. Am. Chem. Soc. 2008, 130, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Siemsen, P.; Livingston, R.C.; Diederich, F. Acetylenic coupling: A powerful tool in molecular construction. Angew. Chem. Int. Ed. 2000, 39, 2633–2657. [Google Scholar] [CrossRef]
- Chan, D.M.T.; Lam, P.Y.S. Boronic Acids in Organic Synthesis and Chemical Biology; Wiley-VCH: New York, NY, USA, 2005; pp. 205–240. [Google Scholar]
- Le, H.T.N.; Tran, T.V.; Phan, N.T.S.; Truong, T. Efficient and recyclable Cu-2(BDC)(2)(BPY)-catalyzed oxidative amidation of terminal alkynes: Role of bipyridine ligand. Catal. Sci. Technol. 2015, 5, 851–859. [Google Scholar] [CrossRef]
- Sau, M.C.; Rajesh, Y.; Mandal, M.; Bhattacharjee, M. Copper Catalyzed Regioselective N-Alkynylation of Pyrazoles and Evaluation of the Anticancer Activity of Ethynyl-Pyrazoles. ChemistrySelect 2018, 3, 3511–3515. [Google Scholar] [CrossRef]
- Jia, W.; Jiao, N. Cu-Catalyzed Oxidative Amidation of Propiolic Acids Under Air via Decarboxylative Coupling. Org. Lett. 2010, 12, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.F.C.; Eastwood, F.W.; Fallon, G.D.; Lee, S.C.; McGeary, R.P. The Pyrolytic Rearrangement of 1-Alkynoyl-3-methylpyrazoles: Synthesis of Pyrazolo[1,5-a]pyridin-5-ols and Related Compounds. Aust. J. Chem. 1994, 47, 991–1007. [Google Scholar] [CrossRef]
- Cook, A.M.; Wolf, C. Terminal ynamides: Synthesis, coupling reactions, and additions to common electrophiles. Tetrahedron Lett. 2015, 56, 2377–2392. [Google Scholar] [CrossRef]
- Tabata, H.; Kuwamoto, K.; Okuno, T. Conformational polymorphs and solid-state polymerization of 9-(1,3-butadiynyl)carbazole derivatives. J. Mol. Struct. 2016, 1106, 452–459. [Google Scholar] [CrossRef]
- Cook, A.M.; Wolf, C. Catalytic enantioselective nucleophilic addition of ynamides to aldehydes. Chem. Commun. 2014, 50, 3151–3154. [Google Scholar] [CrossRef]
- Huang, W.-S.; Metcalf, C.A.; Sundaramoorthi, R.; Wang, Y.; Zou, D.; Thomas, R.M.; Zhu, X.; Cai, L.; Wen, D.; Liu, S.; et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-[4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl]benzamide (AP24534), a Potent, Orally Active Pan-Inhibitor of Breakpoint Cluster Region-Abelson (BCR-ABL) Kinase Including the T315I Gatekeeper Mutant. J. Med. Chem. 2010, 53, 4701–4719. [Google Scholar]
- Sato, A.H.; Ohashi, K.; Ito, K.; Iwasawa, T. Regio- and stereoselective synthesis of 1-(1-halovinyl)-1H-indoles from 1-ethynyl-1H-indoles with in situ generated HX. Tetrahedron Lett. 2013, 54, 2878–2881. [Google Scholar] [CrossRef]
- Laroche, C.; Li, J.; Kerwin, S.M. Cytotoxic 1,2-Dialkynylimidazole-Based Aza-Enediynes: Aza-Bergman Rearrangement Rates Do Not Predict Cytotoxicity. J. Med. Chem. 2011, 54, 5059–5069. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Berger, D.; Neuenschwander, M. Rearrangement of 5-Substituted 5-Aminopentadienals. Angew. Chem. Int. Ed. 1998, 37, 2138–2140. [Google Scholar] [CrossRef]
- Ide, M.; Yauchi, Y.; Iwasawa, T. Regio-, and stereoselective iodobromination of ynamides for synthesis of (E)-1-bromo-2-iodoenamides. Eur. J. Org. Chem. 2014, 2014, 3262–3267. [Google Scholar] [CrossRef]
- Huang, H.; Tang, L.; Liu, Q.; Xi, Y.; He, G.; Zhu, H. Formation of α-chalcogenyl acrylamides through unprecedented chalcogen-mediated metal-free oxyfunctionalization of ynamides with DMSO as an oxidant. Chem. Commun. (Camb. UK) 2016, 52, 5605–5608. [Google Scholar] [CrossRef] [PubMed]
- Mphahlele, M.J. Molecular Iodine-Mediated Cyclization of Tethered Heteroatom-Containing Alkenyl or Alkynyl Systems. Molecules 2009, 14, 4814–4837. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tang, L.; Han, X.; He, G.; Xi, Y.; Zhu, H. Regioselective iodoamination of terminal ynamides for the synthesis of α-amino-β,β-diiodo-enamides. Chem. Commun. 2016, 52, 4321–4324. [Google Scholar] [CrossRef]
- Yenice, I.; Basceken, S.; Balci, M. Nucleophilic and electrophilic cyclization of N-alkyne-substituted pyrrole derivatives: Synthesis of pyrrolopyrazinone, pyrrolotriazinone, and pyrrolooxazinone moieties. Beilstein J. Org. Chem. 2017, 13, 825–834. [Google Scholar] [CrossRef]
- Laroche, C.; Kerwin, S.M. Efficient, Regioselective Access to Bicyclic Imidazo 1,2-x-Heterocycles via Gold- and Base-Promoted Cyclization of 1-Alkynylimidazoles. J. Org. Chem. 2009, 74, 9229–9232. [Google Scholar] [CrossRef]
- Okitsu, T.; Nakata, K.; Nishigaki, K.; Michioka, N.; Karatani, M.; Wada, A. Iodocyclization of Ethoxyethyl Ethers to Ynamides: An Immediate Construction to Benzo b furans. J. Org. Chem. 2014, 79, 5914–5920. [Google Scholar] [CrossRef]
- Grimaldi, T.B.; Godoi, B.; Roehrs, J.A.; Speranca, A.; Zeni, G. Electrophilic Cyclization of N-Alkynyl-2-(organochalcogen)imidazoles: An Alternative Access to Imidazo[2,1-b]chalcogenazoles. Eur. J. Org. Chem. 2013, 2013, 2646–2652. [Google Scholar] [CrossRef]
- Huang, H.; He, G.; Zhu, X.; Jin, X.; Qiu, S.; Zhu, H. Iodine-Mediated Oxidation of Ynamides: A Facile Access to N-Monosubstituted α-Ketoamides and α-Ketoimides. Eur. J. Org. Chem. 2014, 2014, 7174–7183. [Google Scholar] [CrossRef]
- Chikugo, T.; Yauchi, Y.; Ide, M.; Iwasawa, T. Transition metal-free oxidation of ynamides for synthesis of α-keto-imides. Tetrahedron 2014, 70, 3988–3993. [Google Scholar] [CrossRef]
- Ragupathi, A.; Charpe, V.P.; Sagadevan, A.; Hwang, K.C. Visible Light-Mediated Copper(I)-catalyzed aerobic oxidation of ynamides/ynamines at room temperature: A sustainable approach to the synthesis of α-ketoimides/α-ketoamides. Adv. Synth. Catal. 2017, 359, 1138–1143. [Google Scholar] [CrossRef]
- Zhang, C.; Shi, Y.-L.; Zhang, L.-Y.; Yuan, D.-P.; Ban, M.-T.; Zheng, J.-Y.; Liu, D.-H.; Guo, S.-N.; Cui, D.-M. NaOH-promoted reaction of 1,1-dihaloalkenes and 1H-azoles: Synthesis of dihetaryl substituted alkenes. New J. Chem. 2018, 42, 17732–17739. [Google Scholar] [CrossRef]
- Mal’kina, A.G.; den Besten, R.; van der Kerk, A.C.H.T.M.; Brandsma, L.; Trofimov, B.A. Dimetallation of N-ethynylpyrrole and subsequent regiospecific derivatization. J. Organomet. Chem. 1995, 493, 271–273. [Google Scholar] [CrossRef]
- Roehrs, J.A.; Pistoia, R.P.; Back, D.F.; Zeni, G. Three-Step One-Pot Synthesis of Imidazo[2,1-b]chalcogenazoles via Intramolecular Cyclization of N-Alkynylimidazoles. Adv. Synth. Catal. 2012, 354, 1791–1796. [Google Scholar] [CrossRef]
- Laroche, C.; Gilbreath, B.; Kerwin, S.M. Exploring the synthetic utility of 1-alkynylimidazoles: Regiocontrolled cyclization to diverse imidazoazines and imidazoazoles. Tetrahedron 2014, 70, 4534–4539. [Google Scholar] [CrossRef]
- Habert, L.; Retailleau, P.; Gillaizeau, I. Rapid synthesis of 3-aminoisocoumarin derivatives from ynamides. Org. Biomol. Chem. 2018, 16, 7351–7355. [Google Scholar] [CrossRef]
- Graux, L.V.; Clavier, H.; Buono, G. Palladium-Catalyzed Addition of 1,3-Diones to Ynamides: An Entry to Alkoxy-Substituted Enamides. ChemCatChem 2014, 6, 2544–2548. [Google Scholar] [CrossRef]
- Dwivedi, V.; Hari Babu, M.; Kant, R.; Sridhar Reddy, M. N-Substitution dependent stereoselectivity switch in palladium catalyzed hydroalkynylation of ynamides: A regio and stereoselective synthesis of ynenamides. Chem. Commun. 2015, 51, 14996–14999. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Hong, S.W.; Oh, K.H.; Park, J.K. Divergent C-H Annulation for Multifused N-Heterocycles: Regio- and Stereospecific Cyclizations of N-Alkynylindoles. Angew. Chem. Int. Ed. 2017, 56, 13387–13391. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Zhang, F.; Zhu, G. Preparation of (Z)-α,β-Disubstituted Enamides via Palladium-Catalyzed Addition of Boronic Acids to Ynamides. J. Org. Chem. 2014, 79, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.V.; Kim, S.W.; Nguyen, Q.H.; Kim, H.; Wang, S.; Hoang, T.; Shin, S. Bronsted Acid-Catalyzed Oxygenative Bimolecular Friedel-Crafts-type Coupling of Ynamides. Angew. Chem. Int. Ed. 2017, 56, 3670–3674. [Google Scholar] [CrossRef] [PubMed]
- Bauld, N.L.; Gao, D.X. The mechanism of the prototype cation radical cycloaddition reaction: The cyclodimerization of N-vinylcarbazole. J. Chem. Soc. Perkin Trans. 2 2000, 191–192. [Google Scholar] [CrossRef]
- Siva Reddy, A.; Kumara Swamy, K.C. Ethanol as a Hydrogenating Agent: Palladium-Catalyzed Stereoselective Hydrogenation of Ynamides to Give Enamides. Angew. Chem. Int. Ed. 2017, 56, 6984–6988. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhang, Q.; Huang, H.; Chen, S.; Wang, Q.; Zhang, D.; Zhang, R.; Zhu, H. Copper(I)-Catalyzed Highly Regio- and Stereoselective Boron Addition-Protonolysis of Alkynamides to give Alkenamides. Eur. J. Org. Chem. 2013, 2013, 6979–6989. [Google Scholar] [CrossRef]
- Itami, K.; Yoshida, J. Multisubstituted olefins: Platform synthesis and applications to materials science and pharmaceutical chemistry. B Chem Soc Jpn. 2006, 79, 811–824. [Google Scholar] [CrossRef]
- He, G.; Chen, S.; Wang, Q.; Huang, H.; Zhang, Q.; Zhang, D.; Zhang, R.; Zhu, H. Studies on copper(I)-catalyzed highly regio- and stereo-selective hydroboration of alkynamides. Org. Biomol. Chem. 2014, 12, 5945–5953. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Zhang, J.C.; Morehead, A.T. Dipolar Cycloaddition of Cyclic Rhodium Carbenoids to Digonal Carbon—Synthesis of Isoeuparin. Tetrahedron Lett. 1994, 35, 6229–6230. [Google Scholar] [CrossRef]
- Clavier, H.; Lepronier, A.; Bengobesse-Mintsa, N.; Gatineau, D.; Pellissier, H.; Giordano, L.; Tenaglia, A.; Buono, G. Palladium-Mediated [2+1] Cycloaddition of Norbornene Derivatives with Ynamides. Adv. Synth. Catal. 2013, 355, 403–408. [Google Scholar] [CrossRef]
- Audran, G.; Pellissier, H. Synthesis of Methylene- and Alkylidenecyclopropane Derivatives. Adv. Synth. Catal. 2010, 352, 575–608. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Lazaro-Milla, C. Direct Metal-Free Entry to Aminocyclobutenes or Aminocyclobutenols from Ynamides: Synthetic Applications. Chem. Eur. J. 2016, 22, 8998–9005. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Lazaro-Milla, C. Regioselective Synthesis of Heteroatom-Functionalized Cyclobutene-triflones and Cyclobutenones. Adv. Synth. Catal. 2017, 359, 2630–2639. [Google Scholar] [CrossRef]
- Hatit, M.Z.C.; Sadler, J.C.; McLean, L.A.; Whitehurst, B.C.; Seath, C.P.; Humphreys, L.D.; Young, R.J.; Watson, A.J.B.; Burley, G.A. Chemoselective Sequential Click Ligations Directed by Enhanced Reactivity of an Aromatic Ynamine. Org. Lett. 2016, 18, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Seath, C.P.; Burley, G.A.; Watson, A.J.B. Determining the Origin of Rate-Independent Chemoselectivity in CuAAC Reactions: An Alkyne-Specific Shift in Rate-Determining Step. Angew. Chem. Int. Ed. 2017, 56, 3314–3318. [Google Scholar] [CrossRef] [PubMed]
- Hatit, M.Z.C.; Seath, C.P.; Watson, A.J.B.; Burley, G.A. Strategy for Conditional Orthogonal Sequential CuAAC Reactions Using a Protected Aromatic Ynamine. J. Org. Chem. 2017, 82, 5461–5468. [Google Scholar] [CrossRef]
- Dwivedi, V.; Kumar, R.; Sharma, K.; Sridhar, B.; Reddy, M.S. Copper-Promoted Regioselective Intermolecular Diamination of Ynamides: Synthesis of Imidazo[1,2-a]pyridines. ACS Omega 2017, 2, 2770–2777. [Google Scholar] [CrossRef]
- Davies, P.W.; Cremonesi, A.; Dumitrescu, L. Intermolecular and Selective Synthesis of 2,4,5-Trisubstituted Oxazoles by a Gold-Catalyzed Formal [3+2] Cycloaddition. Angew. Chem. Int. Ed. 2011, 50, 8931–8935. [Google Scholar] [CrossRef]
- Gillie, A.D.; Jannapu Reddy, R.; Davies, P.W. Efficient and Flexible Synthesis of Highly Functionalised 4-Aminooxazoles by a Gold-Catalysed Intermolecular Formal [3+2] Dipolar Cycloaddition. Adv. Synth. Catal. 2016, 358, 226–239. [Google Scholar] [CrossRef]
- Garzon, M.; Arce, E.M.; Reddy, R.J.; Davies, P.W. General Entry into o-,o’-Heteroatom-Linked N-(Hetero)aryl-Imidazole Motifs by Gold-Catalysed Formal [3+2]-Dipolar Cycloaddition. Adv. Synth. Catal. 2017, 359, 1837–1843. [Google Scholar] [CrossRef]
- Han, X.-L.; Zhou, C.-J.; Liu, X.-G.; Zhang, S.-S.; Wang, H.; Li, Q. Regioselective Synthesis of 5-Aminooxazoles via Cp*Co(III)-Catalyzed Formal [3 + 2] Cycloaddition of N-(Pivaloyloxy)amides with Ynamides. Org. Lett. 2017, 19, 6108–6111. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhang, H.; Zhao, X. Selenium-π-Acid Catalyzed Oxidative Functionalization of Alkynes: Facile Access to Ynones and Multisubstituted Oxazoles. ACS Catal. 2018, 8, 6745–6750. [Google Scholar] [CrossRef]
- Duret, G.; Quinlan, R.; Martin, R.E.; Bisseret, P.; Neuburger, M.; Gandon, V.; Blanchard, N. Inverse Electron-Demand 4+2 -Cycloadditions of Ynamides: Access to Novel Pyridine Scaffolds. Org. Lett. 2016, 18, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Yeom, H.S.; Fang, L.C.; He, S.H.; Ma, Z.X.; Kedrowski, B.L.; Hsung, R.P. Ynamides in Ring Forming Transformations. Acc. Chem. Res. 2014, 47, 560–578. [Google Scholar] [CrossRef]
- Foster, R.A.A.; Willis, M.C. Tandem inverse-electron-demand hetero-/retro-Diels-Alder reactions for aromatic nitrogen heterocycle synthesis. Chem. Soc. Rev. 2013, 42, 63–76. [Google Scholar] [CrossRef]
- Duret, G.; Quinlan, R.; Yin, B.; Martin, R.E.; Bisseret, P.; Neuburger, M.; Gandon, V.; Blanchard, N. Intramolecular Inverse Electron-Demand [4 + 2] Cycloadditions of Ynamides with Pyrimidines: Scope and Density Functional Theory Insights. J. Org. Chem. 2017, 82, 1726–1742. [Google Scholar] [CrossRef]
- Chowdhury, H.; Chatterjee, N.; Goswami, A. An Eco-Friendly Route to N-Arylindoles by Iron-Catalyzed [2+2+2] Cycloaddition of Diynes with (Indol-1-yl)alkynes. Eur. J. Org. Chem. 2015, 2015, 7735–7742. [Google Scholar] [CrossRef]
- Amasaki, R.; Terashima, N.; Sotome, I.; Komagawa, S.; Saito, S. Nickel-Catalyzed [3+2+2] Cycloaddition of Ethyl Cyclopropylideneacetate and Heteroatom-Substituted Alkynes: Application to Selective Three-Component Reaction with 1,3-Diynes. J. Org. Chem. 2010, 75, 480–483. [Google Scholar] [CrossRef]
- Yamasaki, R.; Ohashi, M.; Maeda, K.; Kitamura, T.; Nakagawa, M.; Kato, K.; Fujita, T.; Kamura, R.; Kinoshita, K.; Masu, H.; et al. Ni-Catalyzed [4+3+2] Cycloaddition of Ethyl Cyclopropylideneacetate and Dienynes: Scope and Mechanistic Insights. Chem. Eur. J. 2013, 19, 3415–3425. [Google Scholar] [CrossRef]
- Jin, H.; Tian, B.; Song, X.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Gold-Catalyzed Synthesis of Quinolines from Propargyl Silyl Ethers and Anthranils through the Umpolung of a Gold Carbene Carbon. Angew. Chem. Int. Ed. 2016, 55, 12688–12692. [Google Scholar] [CrossRef] [PubMed]
- Rode, N.D.; Arcadi, A.; Di Nicola, A.; Marinelli, F.; Michelet, V. Gold-Catalyzed Cascade Reaction of β-(2-Aminophenyl)-α,β-ynones with Ynamides: A Sequential Route to Polysubstituted 2-Aminoquinolines. Org. Lett. 2018, 20, 5103–5106. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-L.; Liu, X.-G.; Lin, E.; Chen, Y.; Chen, Z.; Wang, H.; Li, Q. Cp*Co(III)-Catalyzed oxidative [5+2] annulation: Regioselective synthesis of 2-aminobenzoxepines via C-H/O-H functionalization of 2-vinylphenols with ynamides. Chem. Commun. (Camb. UK) 2018, 54, 11562–11565. [Google Scholar] [CrossRef] [PubMed]
- Nadipuram, A.K.; Kerwin, S.M. Intra- and intermolecular trapping of cyclopentapyrazine carbenes derived from 1,2-dialkynylimidazoles. Tetrahedron Lett. 2006, 47, 353–356. [Google Scholar] [CrossRef]
- Nadipuram, A.K.; Kerwin, S.M. Thermal cyclization of 1,2-dialkynylimidazoles to imidazo 1,2-a pyridines. Tetrahedron 2006, 62, 3798–3808. [Google Scholar] [CrossRef]
- Gomes, F.; Fadel, A.; Rabasso, N. 2,3-Sigmatropic Rearrangement of Ynamides: Preparation of alpha-Amino Allenephosphonates. J. Org. Chem. 2012, 77, 5439–5444. [Google Scholar] [CrossRef]
- Adler, P.; Gomes, F.; Fadel, A.; Rabasso, N. Selective Reduction of Amino Allenephosphonates: Preparation of α-Amino Vinylphosphonates. Eur. J. Org. Chem. 2013, 2013, 7546–7555. [Google Scholar] [CrossRef]
- Zhao, Q.; Gagosz, F. Synthesis of Allenamides and Structurally Related Compounds by a Gold-Catalyzed Hydride Shift Process. Adv. Synth. Catal. 2017, 359, 3108–3113. [Google Scholar] [CrossRef]
- Chen, L.; Yu, L.; Deng, Y.; Zheng, Z.-J.; Xu, Z.; Cao, J.; Xu, L.-W. C-H Functionalization/C-O Bond Cleavage of Benzyl Silyl Ethers with Ynamides for the Chemoselective Synthesis of Skeletally Diverse Compounds. Adv. Synth. Catal. 2016, 358, 480–485. [Google Scholar] [CrossRef]
- Prochnow, T.; Maroneze, A.; Back, D.F.; Jardim, N.S.; Nogueira, C.W.; Zeni, G. Synthesis and anticholinesterase activity of 2-substituted-N-alkynylindoles. Org. Biomol. Chem. 2018, 16, 7926–7934. [Google Scholar] [CrossRef]
- Chen, J.J.; Ferreira, A.J.; Beaudry, C.M. Synthesis of Bis(indole) Alkaloids from Arundo donax: The Ynindole Diels-Alder Reaction, Conformational Chirality, and Absolute Stereochemistry. Angew. Chem. Int. Ed. 2014, 53, 11931–11934. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, E.; Borghese, S.; Blanc, A.; Beneteau, V.; Pale, P. Zeolite-Based Organic Synthesis (ZeoBOS) of Acortatarin A: First Total Synthesis Based on Native and Metal-Doped Zeolite-Catalyzed Steps. Chem. Eur. J. 2017, 23, 1484–1489. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Nakanishi, H.; Hosomi, T.; Kato, M. Synthesis and Solid-State Polymerization of a New Diacetylene—1-(N-carbazolyl)penta-1,3-diyn-5-ol. Macromolecules 1988, 21, 1238–1240. [Google Scholar] [CrossRef]
- Dumitrescu, S.; Percec, V.; Simionescu, C.I. Polymerization of Acetylenic Derivatives. 27. Synthesis and Properties of Isomeric Poly-N-Ethynylcarbazole. J. Polym. Sci. Part A Polym. Chem. 1977, 15, 2893–2907. [Google Scholar] [CrossRef]
- Sata, T.; Nomura, R.; Wada, T.; Sasabe, H.; Masuda, T. Polymerization of N-carbazolylacetylene by various transition metal catalysts and polymer properties. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 2489–2492. [Google Scholar] [CrossRef]
- Yamamoto, T.; Mahmut, A.; Abe, M.; Kuroda, S.I.; Imase, T.; Sasaki, S. Alternating copolymer of thiophene and N-(phenylethynyl)pyrrole. New pi-conjugated alternating five-membered ring copolymer and its packing structure. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2219–2224. [Google Scholar] [CrossRef]
- Yamashita, R.; Koizumi, T.; Sasaki, S.; Yamamoto, T. Preparation of Soluble Polypyrrole with -C C-p-C6H4-hexyl Side Chains at the N-Position and Its Self-assembling Behavior. Polym. J. 2007, 39, 1202–1206. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamashita, R. Preparation of New pi-Conjugated Thiophene-Pyrrole Copolymers Having Ethynyl Substituents at the N-Position of Pyrrole. Polym. J. 2008, 40, 775–778. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamagata, Y.; Yamashita, R.; Abla, M.; Fukumoto, H.; Koizumi, T.-A. Copolymers of pyrrole with N-alkynylpyrroles. Synth. Met. 2012, 162, 2406–2413. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kundu, S.K. Photoconductive Properties of Arylethynylcopper Polymers—Effects of Structure and Oxygen. J. Phys. Chem. 1973, 77, 2677–2680. [Google Scholar] [CrossRef]
- Moggio, I.; Alloisio, M.; Cravino, A.; Comoretto, D.; Piaggio, P.; Musso, G.F.; Garbarino, G.; Cuniberti, C.; Dell’Erba, C.; Dellepiane, G. Vibrational properties of novel diacetylenic monomers. J. Chem. Soc. Perkin Trans. 2 1998, 2249–2254. [Google Scholar] [CrossRef]
- Baschieri, A.; Sambri, L.; Gualandi, I.; Tonelli, D.; Monti, F.; Esposti, A.D.; Armaroli, N. Carbazole-terpyridine donor-acceptor luminophores. RSC Adv. 2013, 3, 6507–6517. [Google Scholar] [CrossRef]
- Betou, M.; Durand, R.J.; Sallustrau, A.; Gousset, C.; Le Coz, E.; Leroux, Y.R.; Toupet, L.; Trzop, E.; Roisnel, T.; Trolez, Y. Reactivity of Functionalized Ynamides with Tetracyanoethylene: Scope, Limitations and Optoelectronic Properties of the Adducts. Chem. Asian J. 2017, 12, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Burley, G.A.; Boutadla, Y.; Davies, D.L.; Singh, K. Triazoles from N-Alkynylheterocycles and Their Coordination to Iridium. Organometallics 2012, 31, 1112–1117. [Google Scholar] [CrossRef]
- Li, J.; Kaoud, T.S.; Laroche, C.; Dalby, K.N.; Kerwin, S.M. Synthesis and biological evaluation of p38α kinase-targeting dialkynylimidazoles. Bioorg. Med. Chem. Lett. 2009, 19, 6293–6297. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kaoud, T.S.; LeVieux, J.; Gilbreath, B.; Moharana, S.; Dalby, K.N.; Kerwin, S.M. A Fluorescence-Based Assay for p38 alpha Recruitment Site Binders: Identification of Rooperol as a Novel p38 alpha Kinase Inhibitor. ChemBioChem 2013, 14, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Laroche, C.; Li, J.; Gonzales, C.; David, W.M.; Kerwin, S.M. Cyclization kinetics and biological evaluation of an anticancer 1,2-dialkynylimidazole. Org. Biomol. Chem. 2010, 8, 1535–1539. [Google Scholar] [CrossRef] [PubMed]


















































































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).