
Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages

 , ,
, ,  and
and 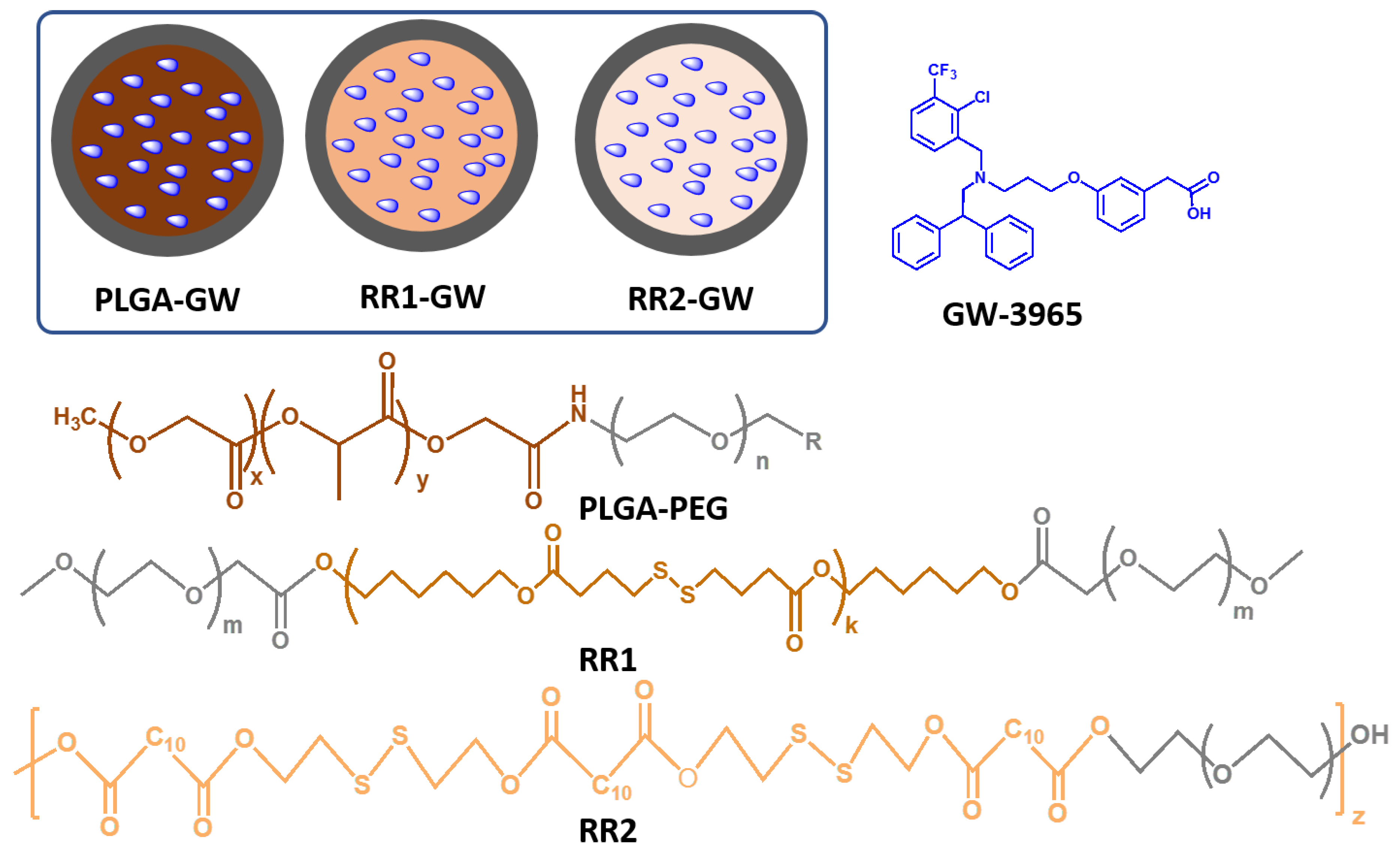{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
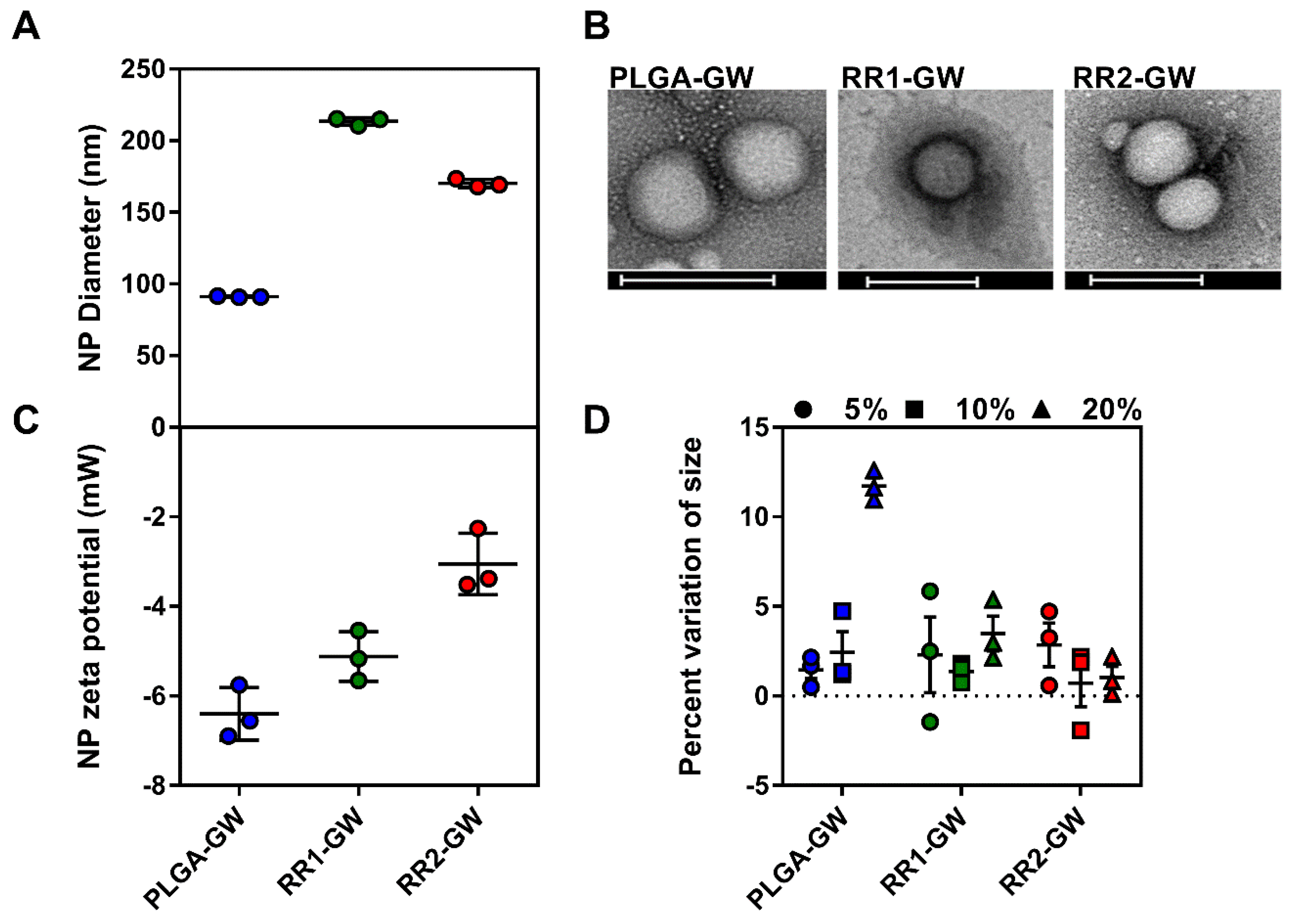
2.1. Synthesis and Characterization of Polymers and Nanoparticles
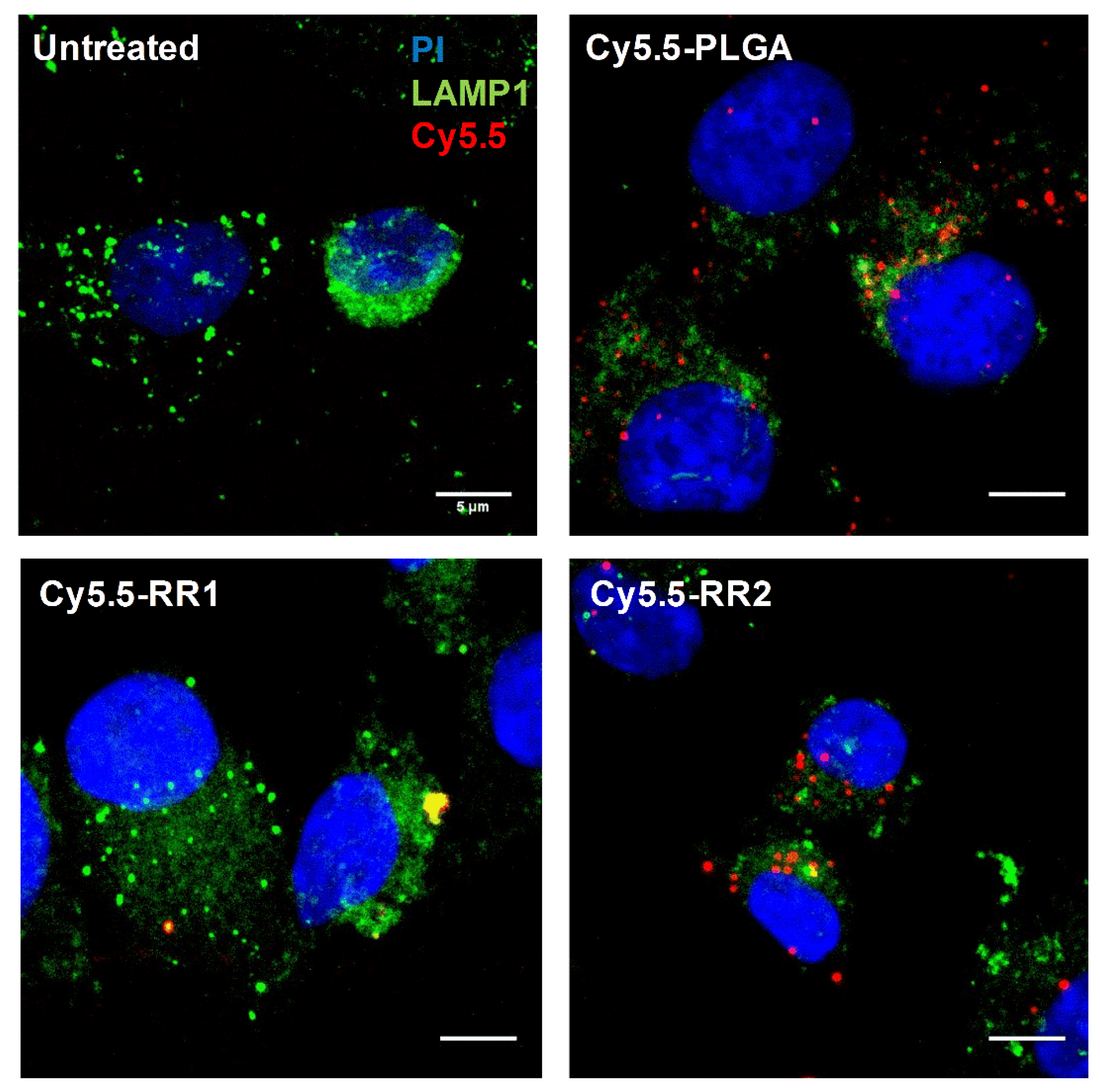
2.2. Cellular Uptake Experiments
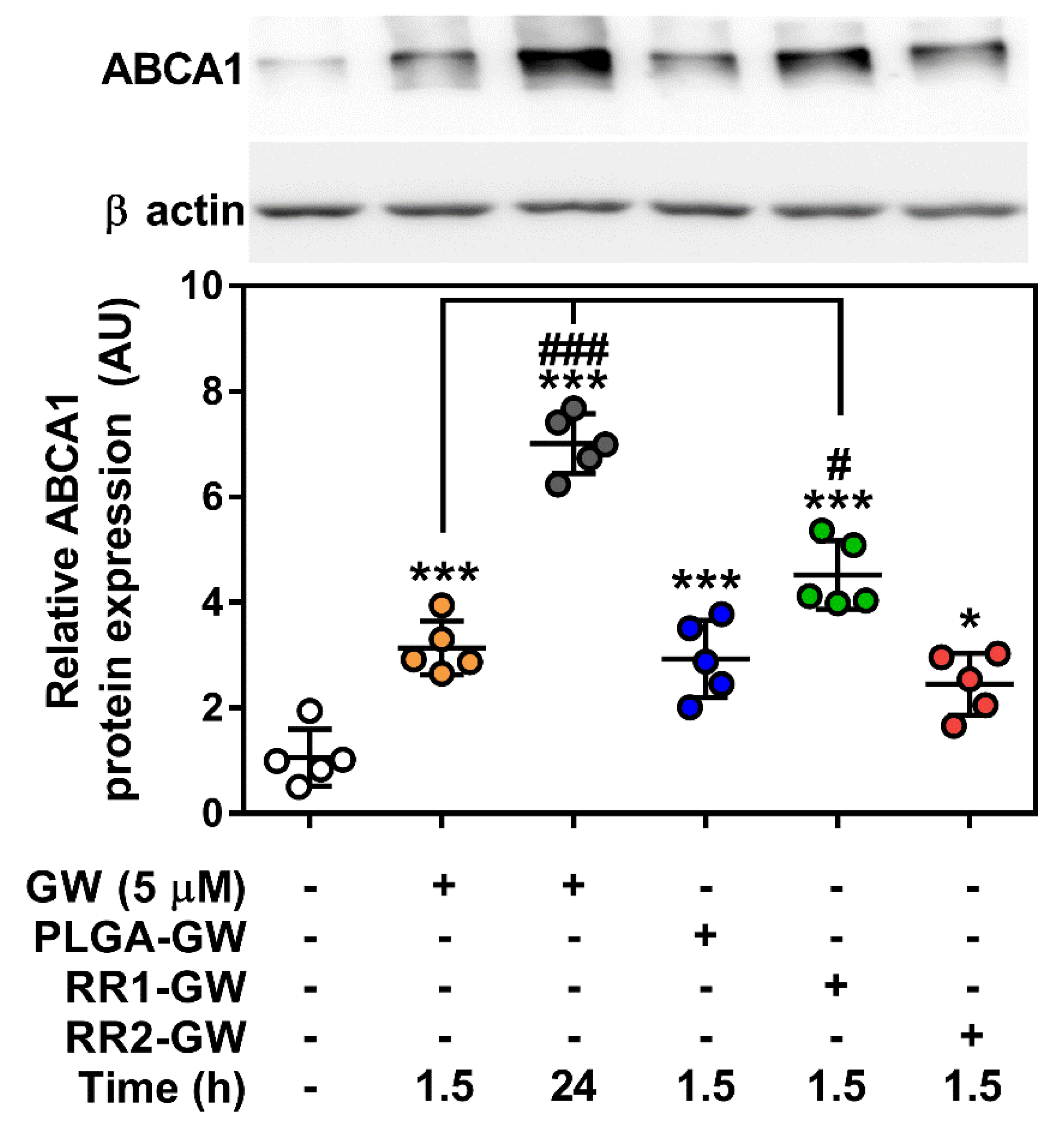
2.3. In vitro Functional Assays
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Polymers Synthesis
4.3. Synthesis of DBA-16HD-PEG2K (RR1)
4.4. Synthesis of DDA-2HDS-PEG4K (RR2)
4.5. GW-NPs Synthesis and Characterization
4.6. Mice
4.7. Cell Culture
4.8. GW Treatment
4.9. Western Blotting
4.10. RNA Isolation, cDNA Synthesis, and Quantitative PCR
4.11. Flow Cytometry
4.12. Microscopy
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Pineda-Torra, I.; Sanson, M.; Bradley, M.N.; Vengrenyuk, Y.; Bogunovic, D.; Gautier, E.L.; Rubinstein, D.; Hong, C.; Liu, J.; et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J. Clin. Investig. 2010, 120, 4415–4424. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Bradley, M.N.; Joseph, S.B.; Zelcer, N.; Janssen, E.M.; Hausner, M.A.; Shih, R.; Parks, J.S.; Edwards, P.A.; Jamieson, B.D.; et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 2008, 134, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. United States Am. 2007, 104, 10601–10606. [Google Scholar] [CrossRef]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Fowler, A.J.; Sheu, M.Y.; Schmuth, M.; Kao, J.; Fluhr, J.W.; Rhein, L.; Collins, J.L.; Willson, T.M.; Mangelsdorf, D.J.; Elias, P.M.; et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: Liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J. Invest. Derm. 2003, 120, 246–255. [Google Scholar] [CrossRef]
- Lund, E.G.; Menke, J.G.; Sparrow, C.P. Liver X receptor agonists as potential therapeutic agents for dyslipidemia and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1169–1177. [Google Scholar] [CrossRef]
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. United States Am. 2002, 99, 7604–7609. [Google Scholar] [CrossRef]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Gadde, S.; Even-Or, O.; Kamaly, N.; Hasija, A.; Gagnon, P.G.; Adusumilli, K.H.; Erakovic, A.; Pal, A.K.; Zhang, X.Q.; Kolishetti, N.; et al. Development of therapeutic polymeric nanoparticles for the resolution of inflammation. Adv. Healthc Mater. 2014, 3, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Amengual, J.; Menon, A.; Kamaly, N.; Zhou, F.; Xu, X.; Saw, P.E.; Lee, S.J.; Si, K.; Ortega, C.A.; et al. Targeted nanotherapeutics encapsulating liver x receptor agonist GW3965 enhance antiatherogenic effects without adverse effects on hepatic lipid metabolism in Ldlr(-/-) mice. Adv. Healthc Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Even-Or, O.; Xu, X.; van Rosmalen, M.; Lim, L.; Gadde, S.; Farokhzad, O.C.; Fisher, E.A. Nanoparticles containing a liver X receptor agonist inhibit inflammation and atherosclerosis. Adv. Healthc Mater. 2015, 4, 228–236. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef]
- Gadde, S. Multi-drug delivery nanocarriers for combination therapy. Medchemcomm 2015, 6, 1916–1929. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; El-Sahli, S.; Li, L.; Chambers, J.; Phan, A.; Cote, M.; Cron, G.O.; Alain, T.; Le, Y.; et al. Co-targeting bulk tumor and cscs in clinically translatable TNBC patient-derived xenografts via combination nanotherapy. Mol. Cancer Ther. 2019, 18, 1755–1764. [Google Scholar] [CrossRef]
- Nguyen, M.A.; Wyatt, H.; Susser, L.; Geoffrion, M.; Rasheed, A.; Duchez, A.C.; Cottee, M.L.; Afolayan, E.; Farah, E.; Kahiel, Z.; et al. delivery of micrornas by chitosan nanoparticles to functionally alter macrophage cholesterol efflux in vitro and in vivo. Acs Nano 2019, 13, 6491–6505. [Google Scholar] [CrossRef]
- Binderup, T.; Duivenvoorden, R.; Fay, F.; van Leent, M.M.T.; Malkus, J.; Baxter, S.; Ishino, S.; Zhao, Y.; Sanchez-Gaytan, B.; Teunissen, A.J.P.; et al. Imaging-assisted nanoimmunotherapy for atherosclerosis in multiple species. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Mitchell, S.L.; Carlson, E.E. Tiny things with enormous impact: Nanotechnology in the fight against infectious disease. Acs Infect. Dis 2018, 4, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Dehaini, D.; Zhang, Y.; Zhou, J.; Chen, X.; Zhang, L.; Fang, R.H.; Gao, W.; Zhang, L. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol 2018, 13, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Kamaly, N.; Spolitu, S.; Milton, J.; Ghorpade, D.; Chiasson, R.; Kuriakose, G.; Perretti, M.; Farokzhad, O.; Tabas, I. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 2015, 7, 275ra220. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Fredman, G.; Subramanian, M.; Gadde, S.; Pesic, A.; Cheung, L.; Fayad, Z.A.; Langer, R.; Tabas, I.; Farokhzad, O.C. Development and in vivo efficacy of targeted polymeric inflammation-resolving nanoparticles. Proc. Natl. Acad. Sci. United States Am. 2013, 110, 6506–6511. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Fredman, G.; Fojas, J.J.; Subramanian, M.; Choi, W.I.; Zepeda, K.; Vilos, C.; Yu, M.; Gadde, S.; Wu, J.; et al. targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. Acs Nano 2016, 10, 5280–5292. [Google Scholar] [CrossRef] [PubMed]
- Nakashiro, S.; Matoba, T.; Umezu, R.; Koga, J.; Tokutome, M.; Katsuki, S.; Nakano, K.; Sunagawa, K.; Egashira, K. Pioglitazone-incorporated nanoparticles prevent plaque destabilization and rupture by regulating monocyte/macrophage differentiation in ApoE-/- mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 491–500. [Google Scholar] [CrossRef]
- Ling, X.; Chen, X.; Riddell, I.A.; Tao, W.; Wang, J.; Hollett, G.; Lippard, S.J.; Farokhzad, O.C.; Shi, J.; Wu, J. Glutathione-scavenging poly(disulfide amide) nanoparticles for the effective delivery of Pt(IV) Prodrugs and reversal of cisplatin resistance. Nano Lett 2018, 18, 4618–4625. [Google Scholar] [CrossRef]
- Chen, J.; Qiu, X.; Ouyang, J.; Kong, J.; Zhong, W.; Xing, M.M. pH and reduction dual-sensitive copolymeric micelles for intracellular doxorubicin delivery. Biomacromolecules 2011, 12, 3601–3611. [Google Scholar] [CrossRef]
- Quinn, J.F.; Whittaker, M.R.; Davis, T.P. Glutathione responsive polymers and their application in drug delivery systems. Polym Chem-Uk 2017, 8, 97–126. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, K.; Zhu, W.; Deng, W.; Lam, K.S. Stimuli-responsive cross-linked micelles for on-demand drug delivery against cancers. Adv. Drug Deliv Rev. 2014, 66, 58–73. [Google Scholar] [CrossRef]
- Su, Y.; Hu, Y.; Du, Y.; Huang, X.; He, J.; You, J.; Yuan, H.; Hu, F. Redox-responsive polymer-drug conjugates based on doxorubicin and chitosan oligosaccharide-g-stearic acid for cancer therapy. Mol. Pharm 2015, 12, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Gadde, S.; Pfirschke, C.; Engblom, C.; Sprachman, M.M.; Kohler, R.H.; Yang, K.S.; Laughney, A.M.; Wojtkiewicz, G.; Kamaly, N.; et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci. Transl. Med. 2015, 7, 314ra183. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Wellington, C.L.; Walker, E.K.; Suarez, A.; Kwok, A.; Bissada, N.; Singaraja, R.; Yang, Y.Z.; Zhang, L.H.; James, E.; Wilson, J.E.; et al. ABCA1 mRNA and protein distribution patterns predict multiple different roles and levels of regulation. Lab. Invest. 2002, 82, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, X.; Song, X.S.; Bai, R.; Yang, H.Y.; Yang, Z.M.; Xiao, C.S.; Bian, Y.F. MicroRNA-20a/b regulates cholesterol efflux through post-transcriptional repression of ATP-binding cassette transporter A1. Biochim. Et Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 929–938. [Google Scholar] [CrossRef]
- Bea, F.; Hudson, F.N.; Chait, A.; Kavanagh, T.J.; Rosenfeld, M.E. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ. Res. 2003, 92, 386–393. [Google Scholar] [CrossRef]
- Duan, J.; Kodali, V.K.; Gaffrey, M.J.; Guo, J.; Chu, R.K.; Camp, D.G.; Smith, R.D.; Thrall, B.D.; Qian, W.J. Quantitative profiling of protein s-glutathionylation reveals redox-dependent regulation of macrophage function during nanoparticle-induced oxidative stress. Acs Nano 2016, 10, 524–538. [Google Scholar] [CrossRef]
- Giacalone, G.; Tsapis, N.; Mousnier, L.; Chacun, H.; Fattal, E. PLA-PEG nanoparticles improve the anti-inflammatory effect of rosiglitazone on macrophages by enhancing drug uptake compared to free rosiglitazone. Materials 2018, 11. [Google Scholar] [CrossRef]
- Cobitz, A.; Zambanini, A.; Sowell, M.; Heise, M.; Louridas, B.; McMorn, S.; Semigran, M.; Koch, G. A retrospective evaluation of congestive heart failure and myocardial ischemia events in 14,237 patients with type 2 diabetes mellitus enrolled in 42 short-term, double-blind, randomized clinical studies with rosiglitazone. Pharm. Drug Saf 2008, 17, 769–781. [Google Scholar]
- Beaven, S.W.; Matveyenko, A.; Wroblewski, K.; Chao, L.; Wilpitz, D.; Hsu, T.W.; Lentz, J.; Drew, B.; Hevener, A.L.; Tontonoz, P. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 2013, 18, 106–117. [Google Scholar] [CrossRef]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.P.; van Denderen, B.J.; et al. Hematopoietic AMPK beta 1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available, but the authors are happy to provide more information upon reasonable request. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, T.K.T.; Kahiel, Z.; LeBlond, N.D.; Ghorbani, P.; Farah, E.; Al-Awosi, R.; Cote, M.; Gadde, S.; Fullerton, M.D. Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages. Molecules 2019, 24, 3751. https://doi.org/10.3390/molecules24203751
Smith TKT, Kahiel Z, LeBlond ND, Ghorbani P, Farah E, Al-Awosi R, Cote M, Gadde S, Fullerton MD. Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages. Molecules. 2019; 24(20):3751. https://doi.org/10.3390/molecules24203751
Chicago/Turabian StyleSmith, Tyler K. T., Zaina Kahiel, Nicholas D. LeBlond, Peyman Ghorbani, Eliya Farah, Refel Al-Awosi, Marceline Cote, Suresh Gadde, and Morgan D. Fullerton. 2019. "Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages" Molecules 24, no. 20: 3751. https://doi.org/10.3390/molecules24203751
APA StyleSmith, T. K. T., Kahiel, Z., LeBlond, N. D., Ghorbani, P., Farah, E., Al-Awosi, R., Cote, M., Gadde, S., & Fullerton, M. D. (2019). Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages. Molecules, 24(20), 3751. https://doi.org/10.3390/molecules24203751