Recognition Selectivities of Lasso-Type Pseudo[1]rotaxane Based on a Mono-Ester-Functionalized Pillar[5]arene

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
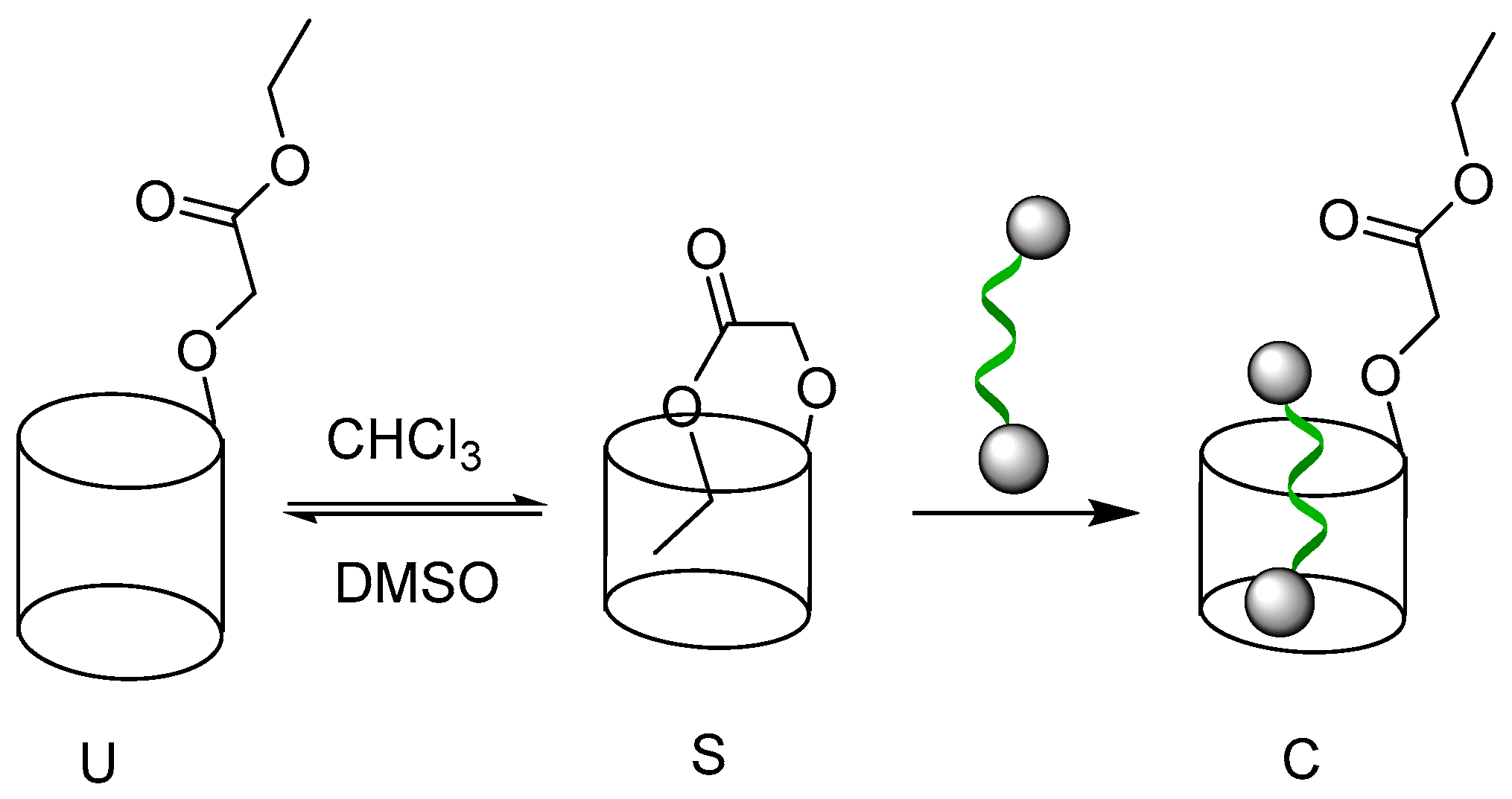
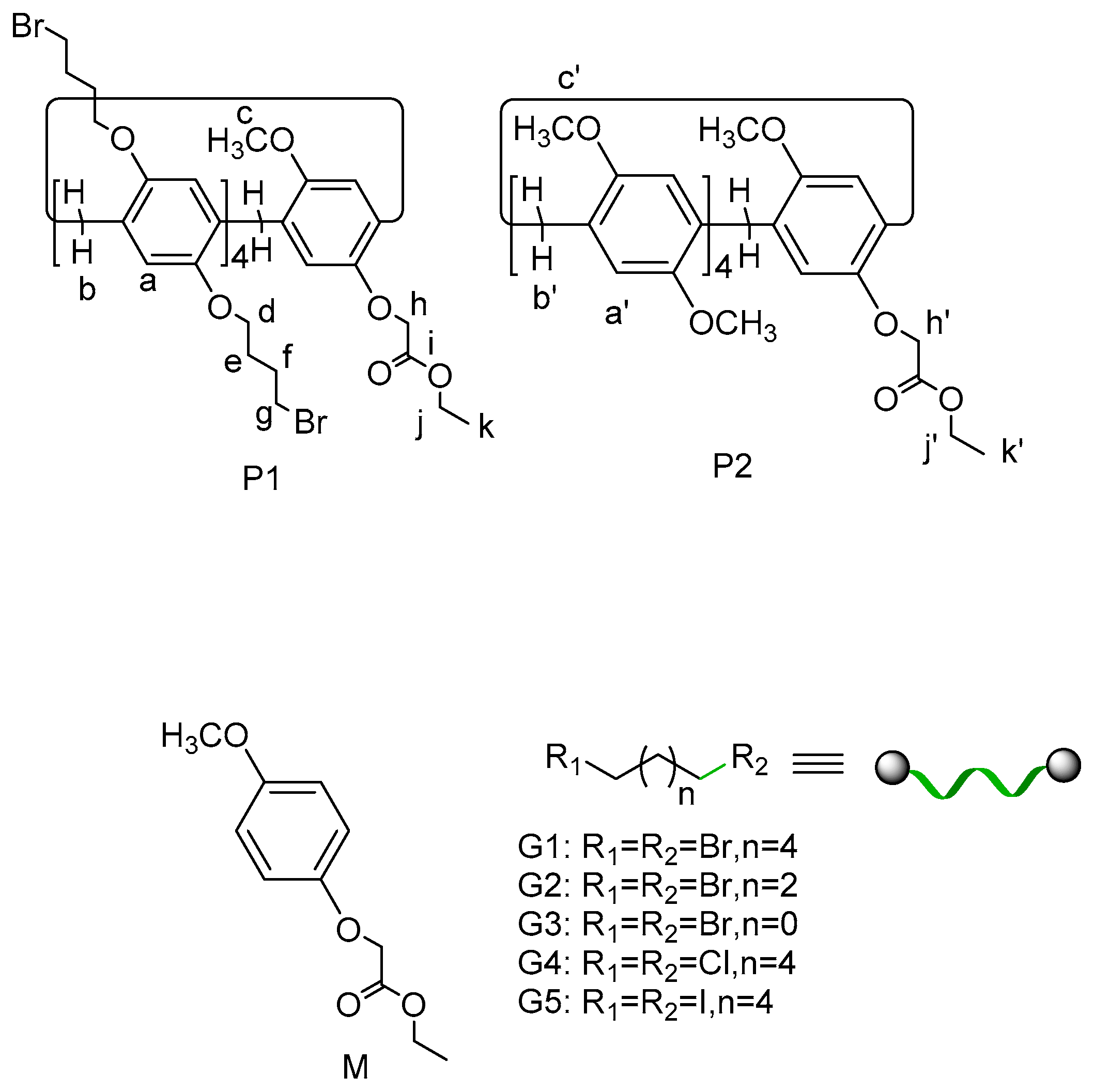
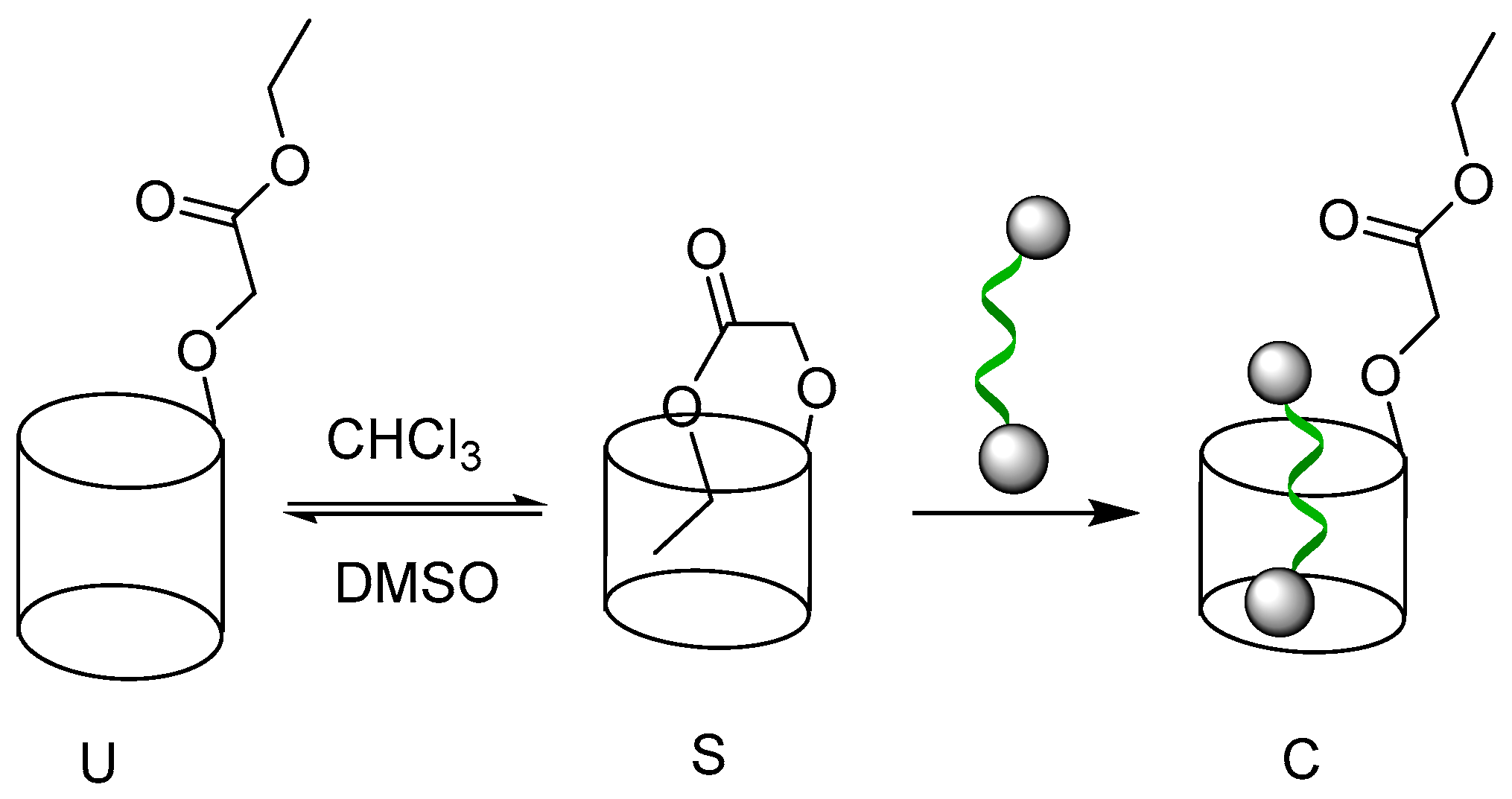
2.1. Self-Assembly Behavior of P1 and P2
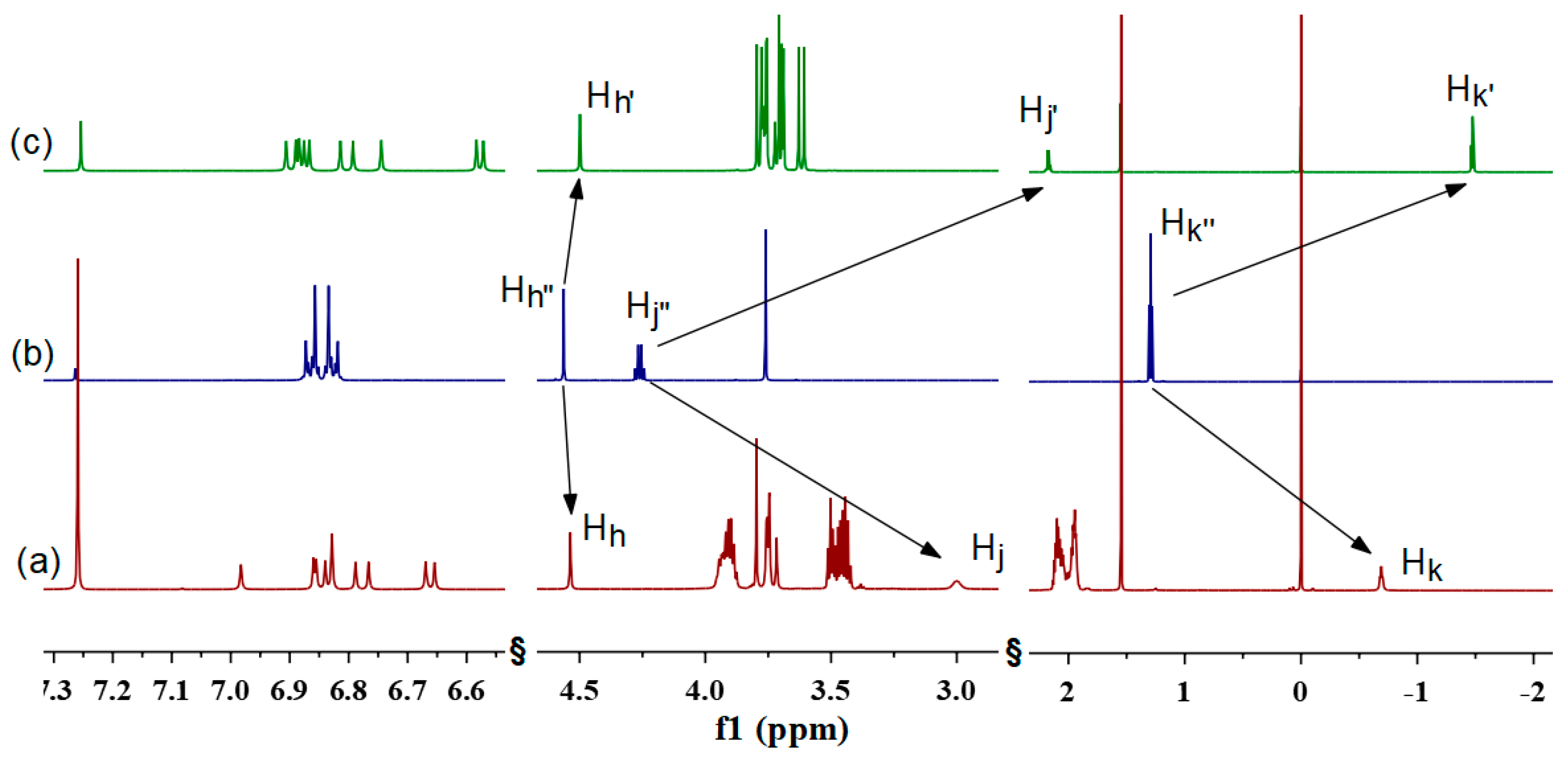
2.2. Host–Guest Properties
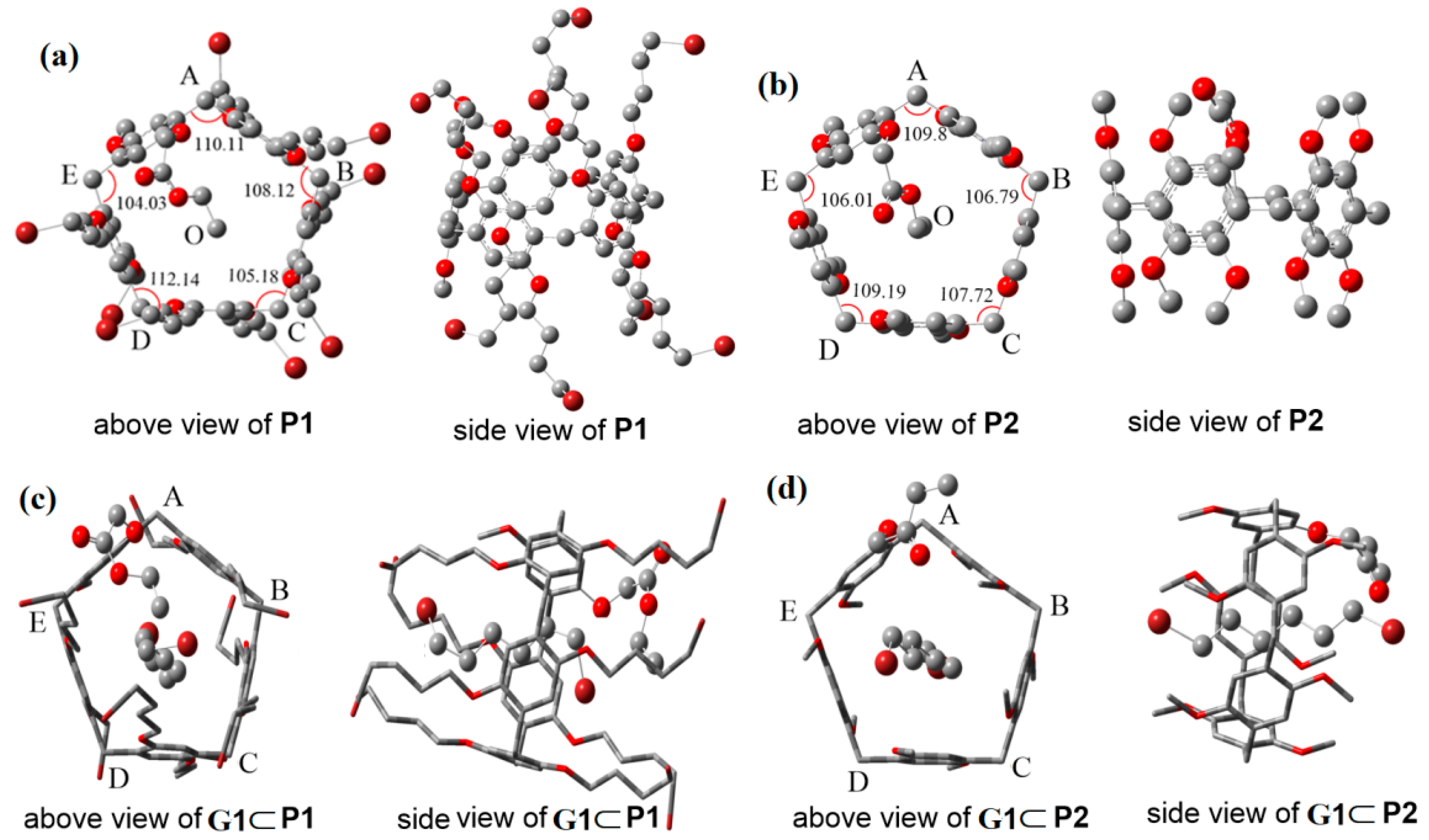
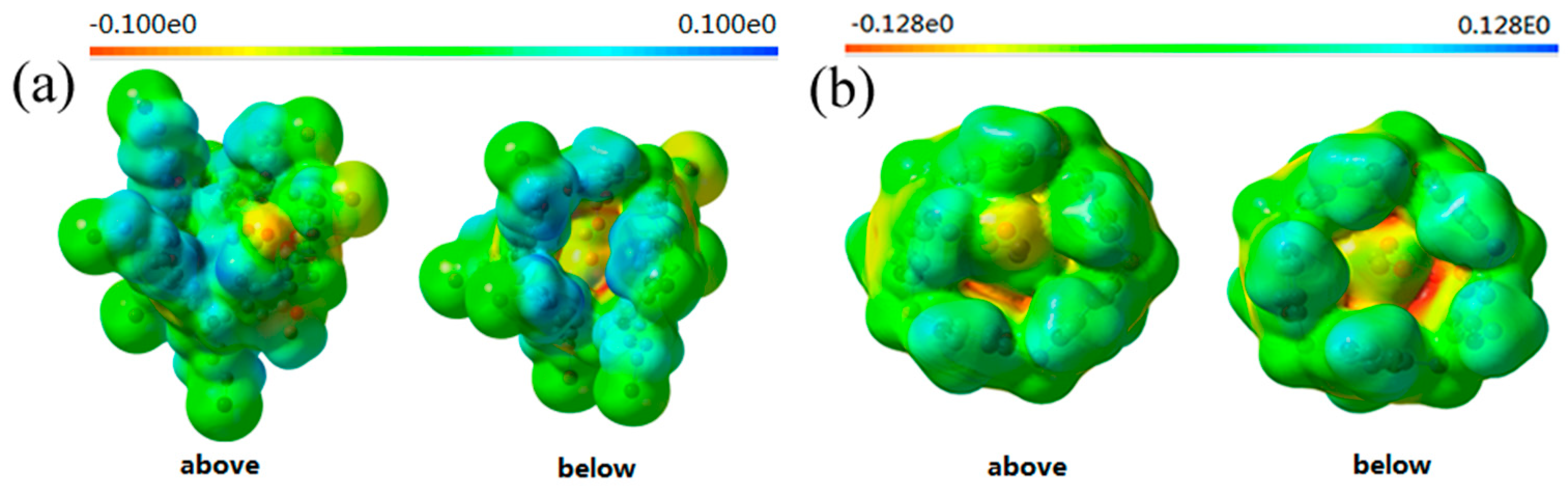
2.3. Quantum Chemical Calculations
3. Experimental Section
3.1. General
3.2. Synthesis of P1 and P2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Amirjalayer, S.; Martinez-Cuezva, A.; Berna, J.; Woutersen, S.; Buma, W.J. Photoinduced pedalo-type motion in an azodicarboxamide-based molecular switch. Angew. Chem. Int. Ed. 2018, 57, 1792–1796. [Google Scholar] [CrossRef]
- Pazos, E.; Novo, P.; Peinador, C.; Kaifer, A.E.; Garcia, M.D. Cucurbit[8]uril (CB[8])-based supramolecular switches. Angew. Chem. Int. Ed. 2019, 58, 403–416. [Google Scholar] [CrossRef]
- Ren, J.M.; Knight, A.S.; van Ravensteijn, B.G.P.; Kohl, P.; Bou Zerdan, R.; Li, Y.L.; Lunn, D.J.; Abdilla, A.; Qiao, G.G.; Hawker, C.J. DNA-inspired strand-exchange for switchable pmma-based supramolecular morphologies. J. Am. Chem. Soc. 2019, 141, 2630–2635. [Google Scholar] [CrossRef]
- Goujon, A.; Du, G.Y.; Moulin, E.; Fuks, G.; Maaloum, M.; Buhler, E.; Giuseppone, N. Hierarchical self-assembly of supramolecular muscle-like fibers. Angew. Chem. Int. Ed. 2016, 55, 703–707. [Google Scholar] [CrossRef]
- Chang, J.C.; Tseng, S.H.; Lai, C.C.; Liu, Y.H.; Peng, S.M.; Chiu, S.H. Mechanically interlocked daisy-chain-like structures as multidimensional molecular muscles. Nature Chemistry 2016, 9, 128–134. [Google Scholar] [CrossRef]
- Chen, J.W.; Leung, F.K.C.; Stuart, M.C.A.; Kajitani, T.; Fukushima, T.; van der Giessen, E.; Feringa, B.L. Artificial muscle-like function from hierarchical supramolecular assembly of photoresponsive molecular motors. Nature Chemistry 2017, 10, 132–138. [Google Scholar] [CrossRef]
- Schröder, H.V.; Wollschlager, J.M.; Schalley, C.A. Redox-controlled self-inclusion of a lasso-type pseudo[1]rotaxane. Chem. Commun. 2017, 53, 9218–9221. [Google Scholar] [CrossRef]
- Madhu, V.; Das, S.K. Diverse supramolecular architectures having well-defined void spaces formed from a pseudorotaxane cation: Influential role of metal dithiolate coordination complex anions. Cryst. Growth Des. 2014, 14, 2343–2356. [Google Scholar] [CrossRef]
- Zanichelli, V.; Ragazzon, G.; Orlandini, G.; Venturi, M.; Credi, A.; Silvi, S.; Arduini, A.; Secchi, A. Efficient active-template synthesis of calix[6]arene-based oriented pseudorotaxanes and rotaxanes. Cryst. Growth Des. 2017, 15, 6753–6763. [Google Scholar] [CrossRef]
- Talotta, C.; De Simone, N.A.; Gaeta, C.; Neri, P. Calix[6]arene threading with weakly interacting tertiary ammonium axles: Generation of chiral pseudorotaxane architectures. Org. Lett. 2015, 17, 1006–1009. [Google Scholar] [CrossRef]
- Wang, Y.; Du, J.W.; Wang, Y.X.; Jin, Q.; Ji, J. Pillar[5]arene based supramolecular prodrug micelles with pH induced aggregate behavior for intracellular drug delivery. Chem. Commun. 2015, 51, 2999–3002. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, K.K.; Wang, Y.C.; Shao, W.; Yao, C.H.; Peng, L.M.; Zhang, D.M.; Hu, X.Y.; Wang, L.Y. Dual-Responsive bola-type supra-amphiphile constructed from water-soluble pillar[5]arene and naphthalimide-containing amphiphile for intracellular drug delivery. ACS Appl. Mater. Interfaces 2017, 9, 4843–4850. [Google Scholar] [CrossRef] [PubMed]
- He, J.P.; Chen, J.Z.; Lin, S.L.; Niu, D.C.; Hao, J.N.; Jia, X.B.; Li, N.; Gu, J.L.; Li, Y.S.; Shi, J.L. Synthesis of a Pillar[5]arene-based polyrotaxane for enhancing the drug loading capacity of pcl-based supramolecular amphiphile as an excellent drug delivery platform. Biomacromolecules 2018, 19, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Wei, R.Y.; Yan, G.H.; Sun, J.; Li, C.J.; Wang, H.F.; Shi, L.Y.; Capobianco, J.A.; Sun, L.N. Smart self-assembled nanosystem based on water-soluble pillararene and rare-earth-doped upconversion nanoparticles for ph-responsive drug delivery. ACS Appl. Mater. Interfaces 2018, 10, 4910–4920. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Lin, Q.; Zhang, Y.M.; Yao, H.; Wei, T.B. Pillararene-based fluorescent chemosensors: Recent advances and perspectives. Chem. Commun. 2017, 53, 13296–13311. [Google Scholar] [CrossRef]
- Yao, Q.F.; Lü, B.Z.; Ji, C.D.; Cai, Y.; Yin, M.Z. Supramolecular host-guest system as ratiometric fe3+ ion sensor based on water-soluble pillar[5]arene. ACS Appl. Mater. Interfaces 2017, 9, 36320–36326. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Z.; Yang, Y.W. Tetraphenylethylene-interweaving conjugated macrocycle polymer materials as two-photon fluorescence sensors for metal ions and organic molecules. Adv. Mater. 2018. [Google Scholar] [CrossRef]
- Lin, Q.; Jiang, X.M.; Liu, L.; Chen, J.F.; Zhang, Y.M.; Yao, H.; Wei, T.B. A novel supramolecular organogel based on acylhydrazone functionalized pillar[5]arene acts as an I– responsive smart material. Soft Matter 2017, 13, 7222–7226. [Google Scholar] [CrossRef]
- Cui, W.; Tang, H.; Xu, L.X.; Wang, L.Y.; Meier, H.; Cao, D.R. Pillar[5]arene-diketopyrrolopyrrole fluorescent copolymer: A promising recognition and adsorption material for adiponitrile by selective formation of a conjugated polypseudorotaxane. Macromol. Rapid Commun. 2017. [Google Scholar] [CrossRef]
- Song, N.; Kakuta, T.; Yamagishi, T.A.; Yang, Y.W.; Ogoshi, T. Molecular-scale porous materials based on pillar[n]arenes. Chem 2018, 4, 2029–2053. [Google Scholar] [CrossRef]
- Li, C. Pillararene-based supramolecular polymers: From molecular recognition to polymeric aggregates. Chem. Commun. 2014, 50, 12420–12433. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Q.; Wu, K.; Zhang, Q.; Li, K.Q.; Li, Y.Y.; Xin, P.Y.; Zhang, W.W.; Guo, H.M. A dual-responsive hyperbranched supramolecular polymer constructed by cooperative host-guest recognition and hydrogen-bond interactions. Chem. Commun. 2018, 54, 13821–13824. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Xu, F.F.; Liang, T.X.; Wen, H.R.; Tian, W. Pillararene-based supramolecular polymers. Chem. Commun. 2019, 55, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cao, D.R.; Wang, L.Y.; He, M.Q.; Zhou, L.X.; Schollmeyer, D.; Meier, H. Monoester copillar[5]arenes: Synthesis, unusual self-inclusion behavior, and molecular recognition. Chem. Eur. J. 2013, 19, 7064–7070. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Z.M.; Chen, Z.X.; Hou, J.L. Pillar[5]arenes with an introverted amino group: A hydrogen bonding tuning effect. Org. Biomol. Chem. 2013, 11, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.F.; Hu, X.Y.; Jiang, J.L.; Wang, L.Y. The self-complexation of mono-urea-functionalized pillar[5]arenes with abnormal urea behaviors. Chem. Commun. 2014, 50, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.F.; Liu, P.Y.; Deng, C.; Ni, M.F.; Xiong, S.H.; Lin, C.; Hu, X.Y.; Ma, J.; Wang, L.Y. Dynamic self-inclusion behavior of pillar[5]arene-based pseudo[1]rotaxanes. Org. Biomol. Chem. 2014, 12, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ni, M.F.; Xia, W.; Hu, X.Y.; Wang, L.Y. A novel dynamic pseudo[1]rotaxane based on a mono-biotin-functionalized pillar[5]arene. Org. Chem. Front. 2015, 2, 1013–1017. [Google Scholar] [CrossRef]
- Wu, X.; Gao, L.; Sun, J.Z.; Hu, X.Y.; Wang, L.Y. Stable pillar[5]arene-based pseudo[1]rotaxanes formed in polar solution. Chinese Chem. Lett. 2016, 27, 1655–1660. [Google Scholar] [CrossRef]
- Liu, L.Z.; Wang, L.Y.; Liu, C.C.; Fu, Z.Y.; Meier, H.; Cao, D.R. Dimerization control in the self-assembly behavior of copillar[5]arenes bearing omega-hydroxyalkoxy groups. J. Org. Chem. 2012, 77, 9413–9417. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Cao, D.R.; Jin, Y.; Tao, H.Q.; Kou, Y.H.; Meier, H. Efficient synthesis of copillar[5]arenes and their host-guest properties with dibromoalkanes. Org. Biomol. Chem. 2011, 9, 7007–7010. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Hua, Z.Z.; Duan, W.G.; Huang, H.F.; Huang, Y.; Lin, G.S.; Cen, B. Selective and effective rotation mode of copillar[5]arene by mono-functionalizing bulky substituent. Tetrahedron Letters 2016, 57, 2969–2971. [Google Scholar] [CrossRef]
- Liu, L.Z.; Duan, W.G.; Kou, Y.H.; Wang, L.Y.; Meier, H.; Cao, D.R. Crystal Structure and Host-Guest Binding Ability of Three Types of Pillar[5]arenes. Chin. J. Chem. 2015, 33, 346–350. [Google Scholar] [CrossRef]
- Huang, H.F.; Liu, L.Z.; Duan, W.G.; Huang, Y.; Lin, G.S. Synthesis of copillar[5]arenes and their host-guest complexation with two types of guests. Chin. J. Chem. 2015, 33, 384–388. [Google Scholar] [CrossRef]
- Li, C.J.; Shu, X.Y.; Li, J.; Fan, J.Z.; Chen, Z.X.; Weng, L.H.; Jia, X.S. Selective and effective binding of pillar[5,6]arenes toward secondary ammonium salts with a weakly coordinating counteranion. Org. Lett. 2012, 14, 4126–4129. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision, A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Samples of the compounds P1 and P2 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P1 | P2 | |
|---|---|---|
| Bond lengths (Å) | ||
| O–A | 5.91 | 5.13 |
| O–B | 5.53 | 5.04 |
| O–C | 5.27 | 4.81 |
| O–D | 5.62 | 4.83 |
| O–E | 6.33 | 5.20 |
| Inner angles (°) | ||
| E–A–B | 110.11 | 109.80 |
| A–B–C | 108.12 | 106.79 |
| B–C–D | 105.18 | 107.72 |
| C–D–E | 112.14 | 109.20 |
| D–E–A | 104.03 | 106.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.-X.; Liu, L.-Z.; Duan, W.-G.; Zhou, Q.-Q.; Ma, C.-G.; Huang, Y. Recognition Selectivities of Lasso-Type Pseudo[1]rotaxane Based on a Mono-Ester-Functionalized Pillar[5]arene. Molecules 2019, 24, 2693. https://doi.org/10.3390/molecules24152693
Zhang W-X, Liu L-Z, Duan W-G, Zhou Q-Q, Ma C-G, Huang Y. Recognition Selectivities of Lasso-Type Pseudo[1]rotaxane Based on a Mono-Ester-Functionalized Pillar[5]arene. Molecules. 2019; 24(15):2693. https://doi.org/10.3390/molecules24152693
Chicago/Turabian StyleZhang, Wen-Xue, Lu-Zhi Liu, Wen-Gui Duan, Qing-Qing Zhou, Cui-Guang Ma, and Yan Huang. 2019. "Recognition Selectivities of Lasso-Type Pseudo[1]rotaxane Based on a Mono-Ester-Functionalized Pillar[5]arene" Molecules 24, no. 15: 2693. https://doi.org/10.3390/molecules24152693
APA StyleZhang, W.-X., Liu, L.-Z., Duan, W.-G., Zhou, Q.-Q., Ma, C.-G., & Huang, Y. (2019). Recognition Selectivities of Lasso-Type Pseudo[1]rotaxane Based on a Mono-Ester-Functionalized Pillar[5]arene. Molecules, 24(15), 2693. https://doi.org/10.3390/molecules24152693