Synthesis and PI3 Kinase Inhibition Activity of Some Novel Trisubstituted Morpholinopyrimidines

Abstract
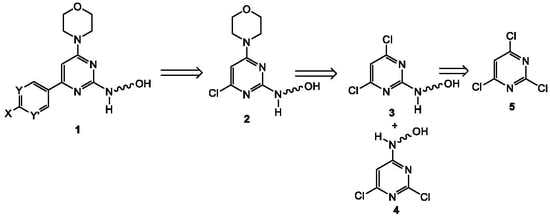
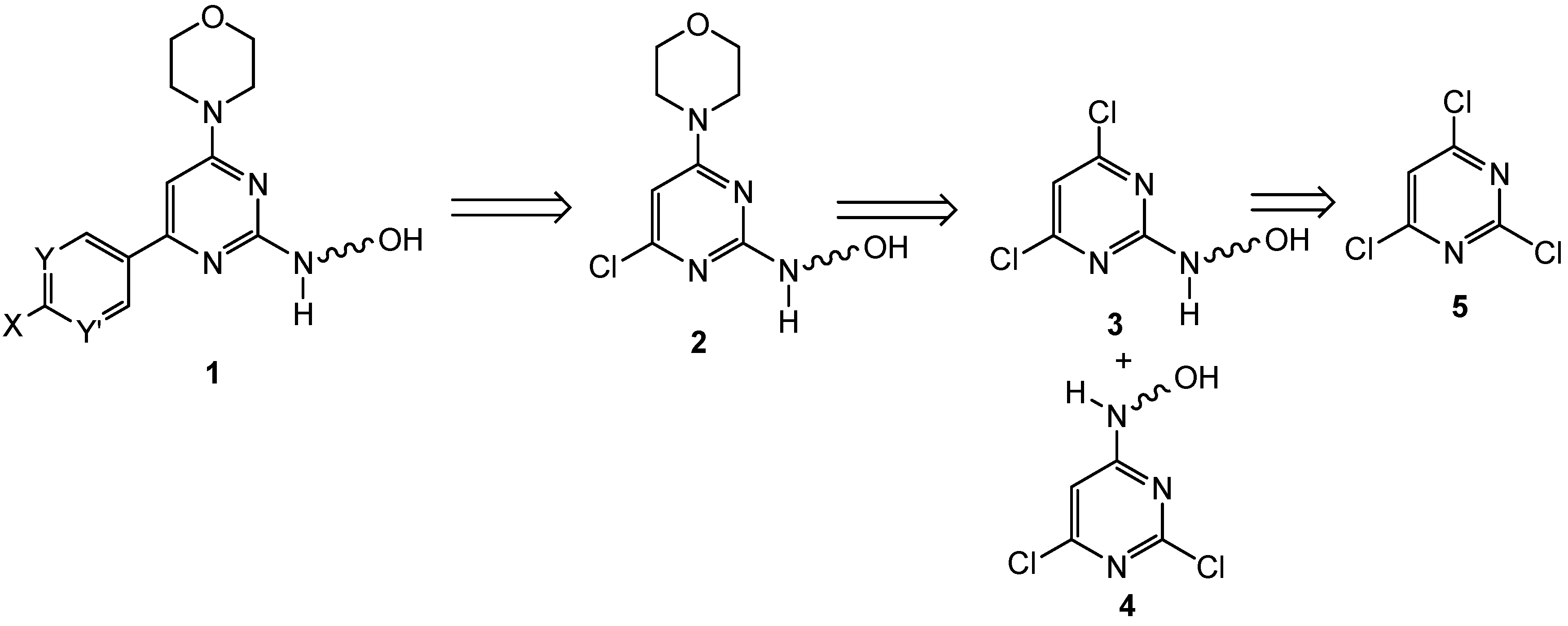
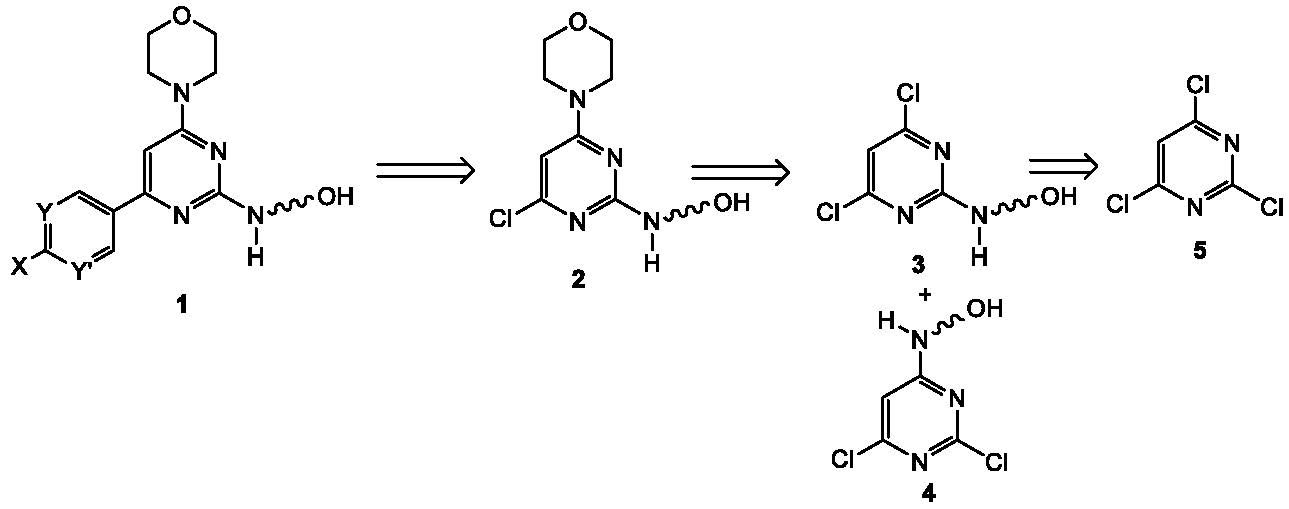{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
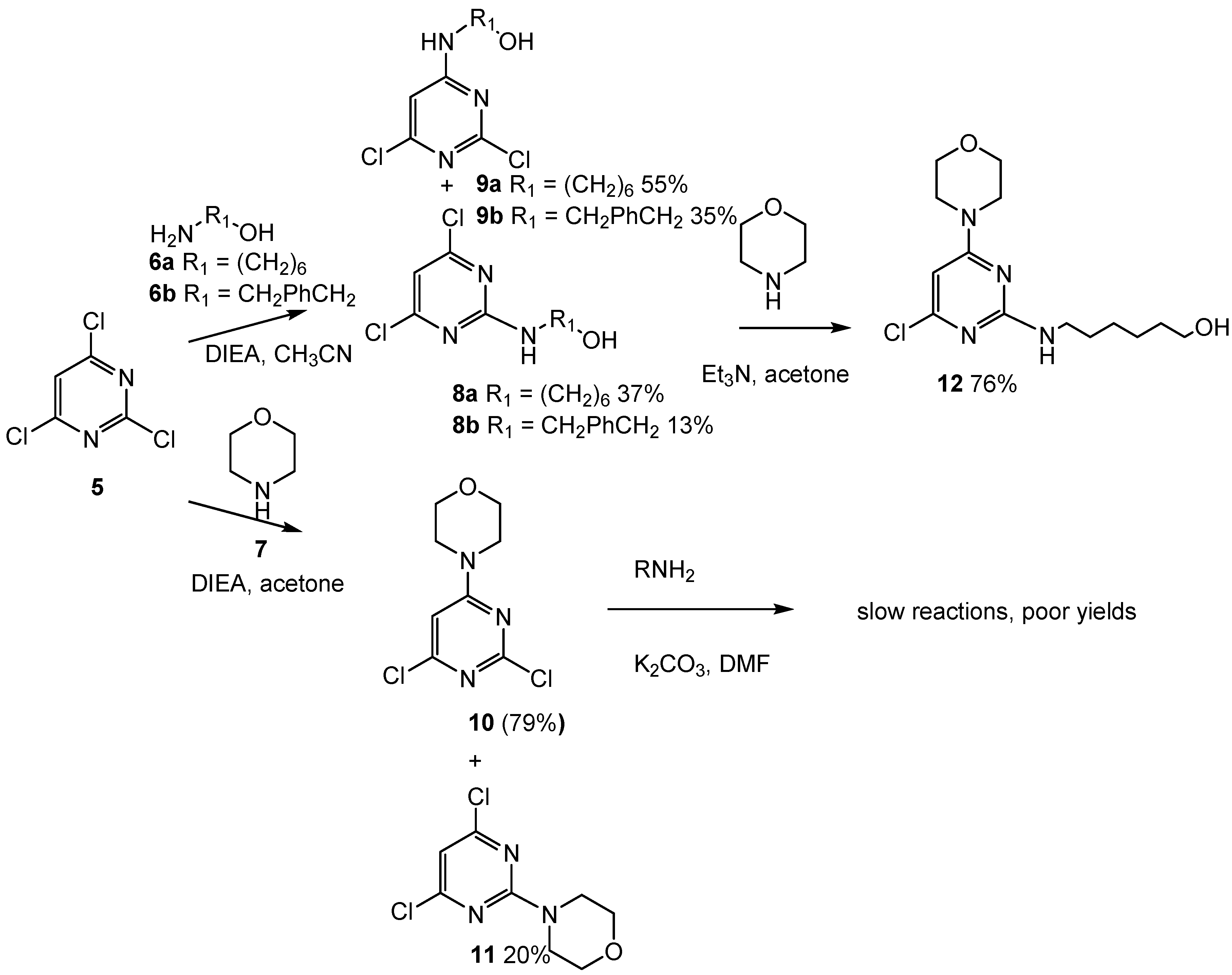
2.1. Chemistry
2.2. First Addition Reaction (Scheme 2)
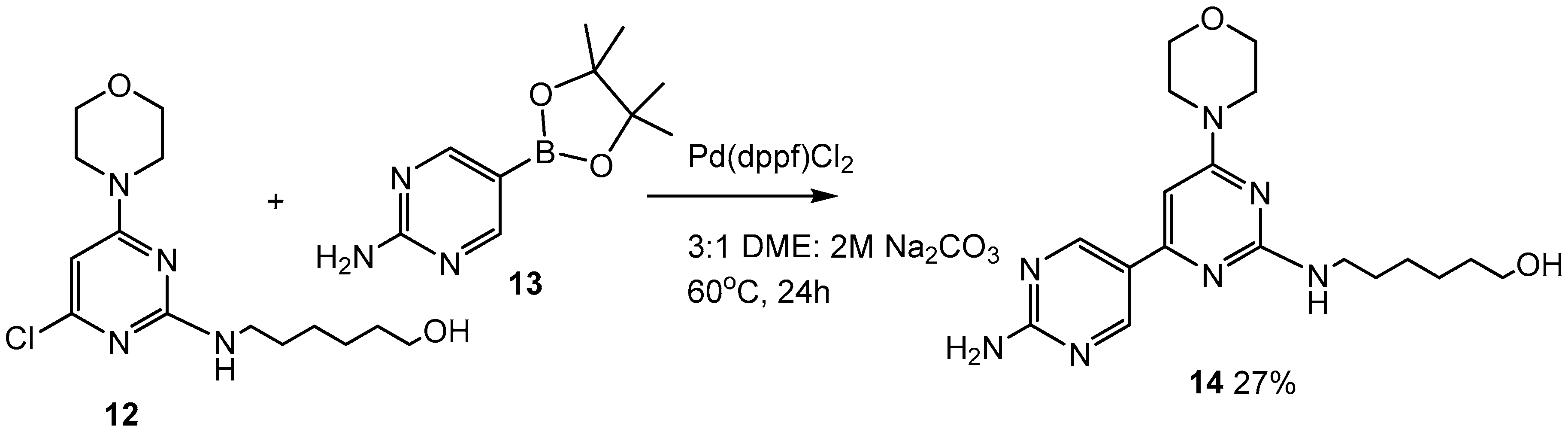
2.3. Third Addition Reaction (Scheme 3)
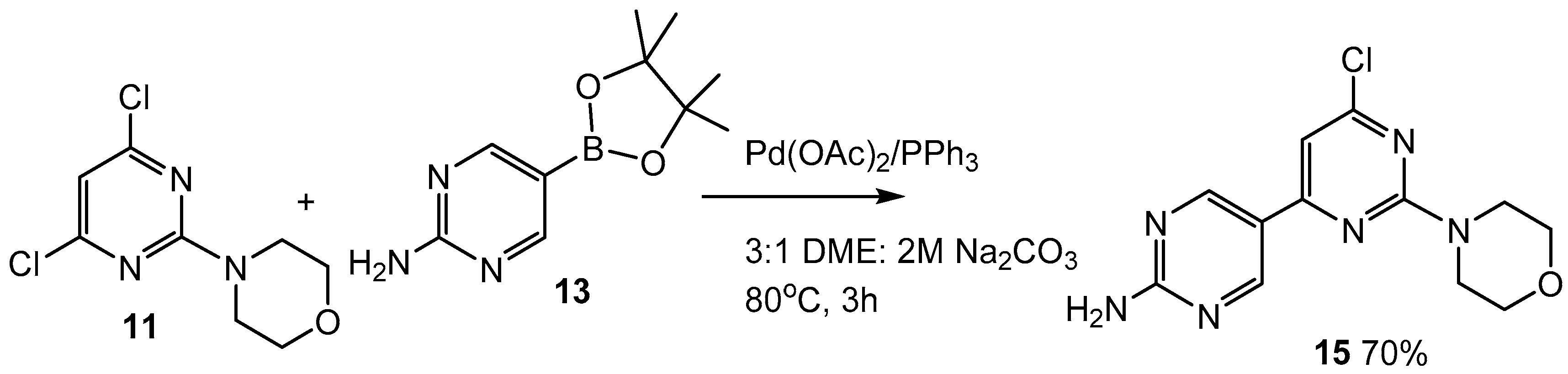
2.4. Attempt to Prepare an Isomer of 14
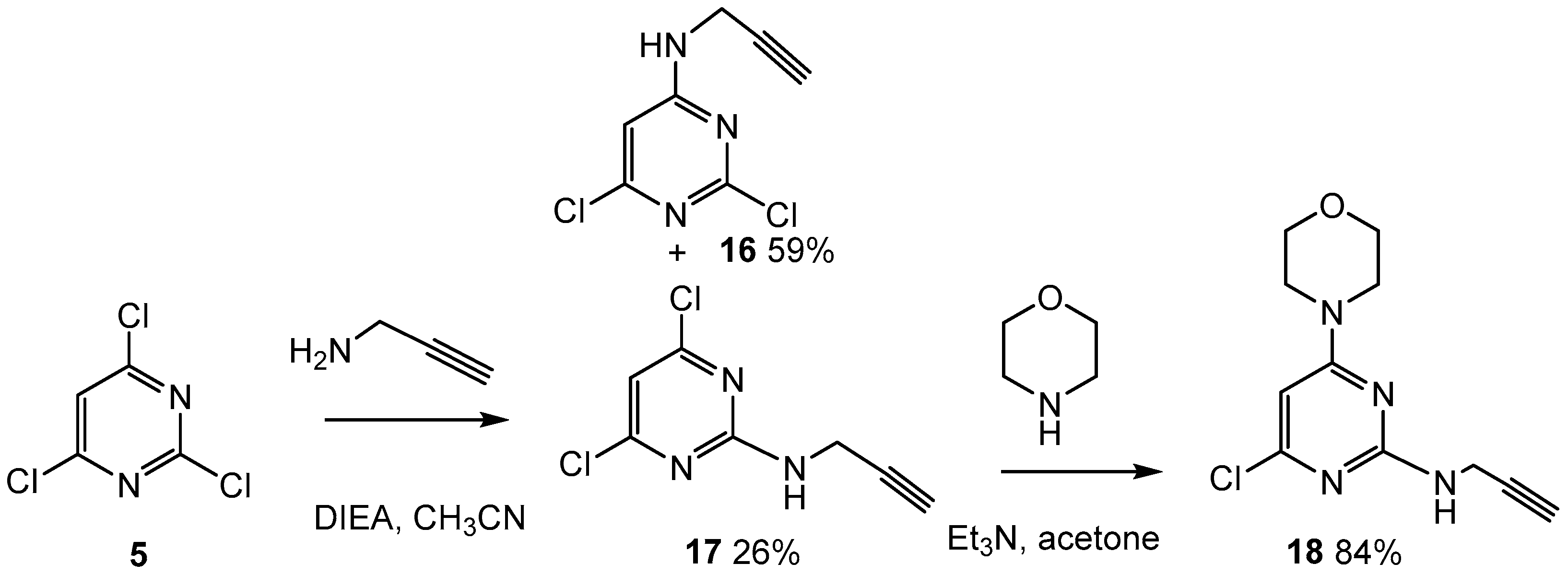
2.5. Attempts to Prepare an Analog of 14 with a Terminal Alkyne Rather than Primary OH as Peptide Link Point
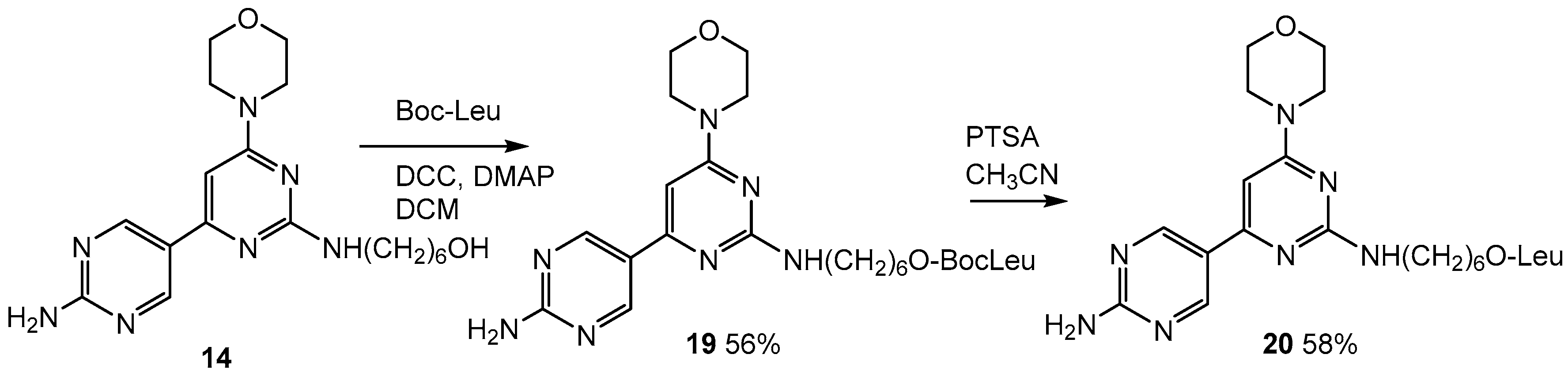
2.6. Addition of Leucine to the Lead Compound (14)
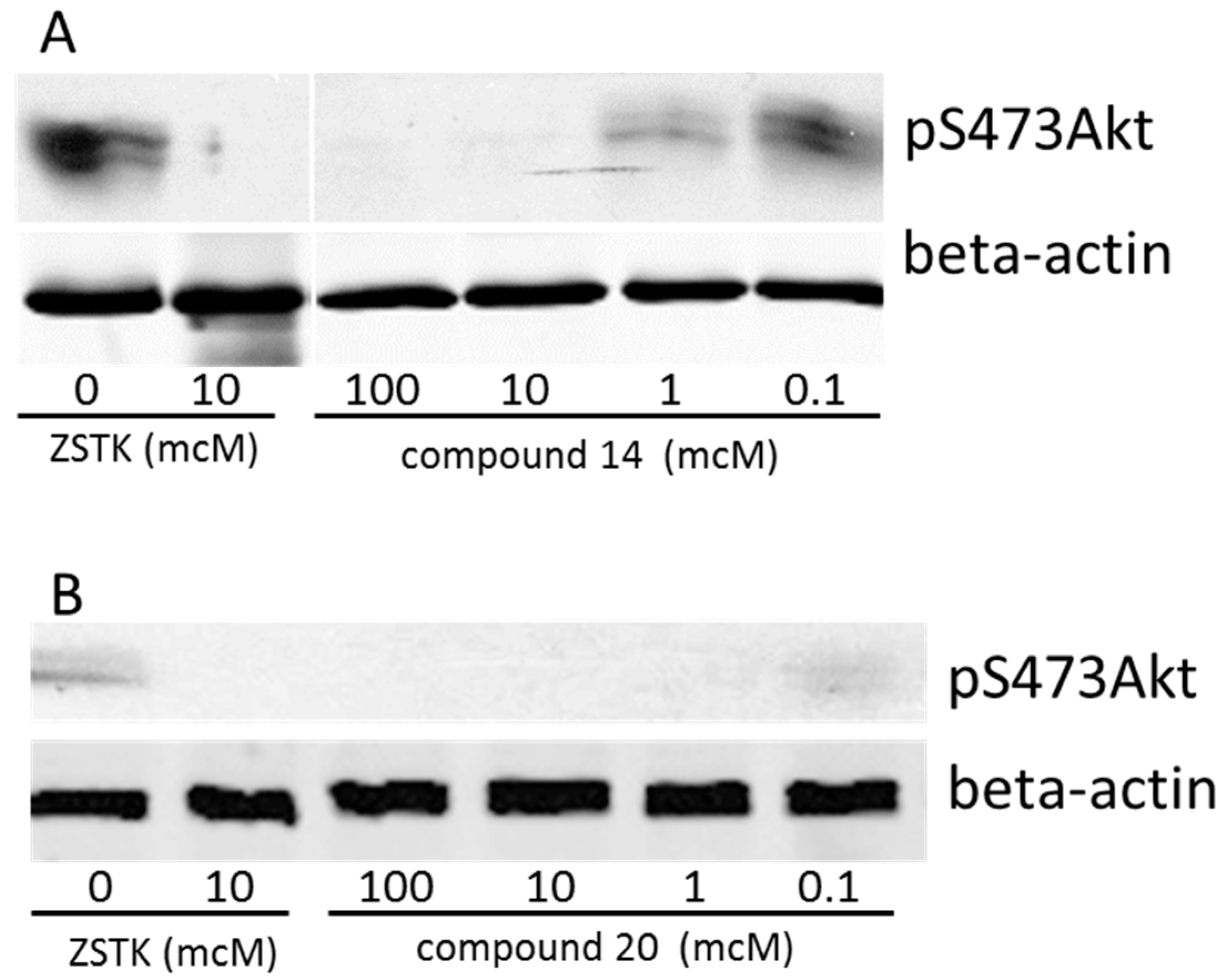
2.7. Biological Activity
3. Materials and Methods
3.1. Compound Synthesis
3.1.1. General Methods
3.1.2. 6-((4,6-Dichloropyrimidin-2-yl)amino)hexan-1-ol (8a) and 6-((2,6-dichloropyrimidin-4-yl)amino)hexan-1-ol (9a)
3.1.3. (4-(((2,6-Dichloropyrimidin-4-yl)amino)methyl)phenyl)methanol, (8b) (4-(((4,6-dichloropyrimidin-2-yl)amino)methyl)phenyl)methanol, (9b)
3.1.4. 4-(2,6-Dichloropyrimidin-4-yl)morpholine, (10) and 4-(4,6-dichloropyrimidin-2-yl)morpholine, (11)
3.1.5. 6-((4-Chloro-6-morpholinopyrimidin-2-yl)amino)hexan-1-ol (12)
3.1.6. 6-((2′-Amino-6-morpholino-[4,5′-bipyrimidin]-2-yl)amino)hexan-1-ol (14)
3.1.7. 6-Chloro-2-morpholino-[4,5′-bipyrimidin]-2′-amine (15)
3.1.8. 2-(But-3-yn-1-yl)-4,6-dichloropyrimidine (17) and 4-(but-3-yn-1-yl)-2,6-dichloropyrimidine (16)
3.1.9. 4-(2-(But-3-yn-1-yl)-6-chloropyrimidin-4-yl)morpholine (18)
3.1.10. 6-((2′-Amino-6-morpholino-[4,5′-bipyrimidin]-2-yl)amino)hexyl (tert-butoxycarbonyl)-l-leucinate (19)
3.1.11. 6-((2′-Amino-6-morpholino-[4,5′-bipyrimidin]-2-yl)amino)hexyl-l-leucinate (20)
3.2. Western Blot Analysis on Synthesized PI3K Inhibitors
General Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuhn, J.-M.; Billebaud, T.; Navratil, H.; Moulonguet, A.; Fiet, J.; Grise, P.; Louis, J.-F.; Costa, P.; Husson, J.-M.; Dahan, R.; et al. Prevention of the Transient Adverse Effects of a Gonadotropin-Releasing Hormone Analogue (Buserelin) in Metastatic Prostatic Carcinoma by Administration of an Antiandrogen (Nilutamide). N. Engl. J. Med. 1989, 321, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Small, E.J. Hormone-Refractory prostate cancer. Curr. Treat. Options Oncol. 2002, 3, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Denis, L.; Murphy, G.P. Overview of Phase-III Trials on Combined Androgen Treatment in Patients with Metastatic Prostate-Cancer. Cancer 1993, 72, 3888–3895. [Google Scholar] [CrossRef]
- Dorkin, T.J.; Neal, D.E. Basic science aspects of prostate cancer. Semin. Cancer Biol. 1997, 8, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G.; Porter, A.T. Target to apoptosis: A hopeful weapon for prostate cancer. Prostate 1997, 32, 284–293. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Kyprianou, N.; Isaacs, J.T. Activation of Programmed Cell-Death in the Rat Ventral Prostate after Castration. Endocrinology 1988, 122, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.D.; Ling, M.T. Targeting Drug-Resistant Prostate Cancer with Dual PI3K/mTOR Inhibition. Curr. Med. Chem. 2014, 21, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Hu, Y. Targeting PI3K/AKT/mTOR Cascade: The Medicinal Potential, Updated Research Highlights and Challenges Ahead. Curr. Med. Chem. 2013, 20, 2991–3010. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.B.; Rokhlin, O.W. Mechanisms of prostate cancer cell survival after inhibition of AR expression. J. Cell. Biochem. 2009, 106, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Khurshid Qazi, A.; Hussain, A.; Hamid, A.; Qurishi, Y.; Majeed, R.; Ahmad, M.; Ahmad Najar, R.; Ahmad Bhat, J.; Kumar Singh, S.; Afzal Zargar, M.; et al. Recent Development in Targeting PI3K-AKT-mTOR Signaling for Anticancer Therapeutic Strategies. Anticancer Agents Med. Chem. 2013, 13, 1552–1564. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Pten Inactivation and the Emergence of Androgen-Independent Prostate Cancer. Cancer Res. 2007, 67, 6535–6538. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Wu, H.; Nelson, C.C. PTEN and GSK3β: Key regulators of progression to androgen-independent prostate cancer. Oncogene 2006, 25, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Uzoh, C.C.; Perks, C.M.; Bahl, A.; Holly, J.M.P.; Sugiono, M.; Persad, R.A. PTEN-mediated pathways and their association with treatment-resistant prostate cancer. BJU Int. 2009, 104, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Whang, Y.E.; Wu, X.Y.; Suzuki, H.; Reiter, R.E.; Tran, C.; Vessella, R.L.; Said, J.W.; Isaacs, W.B.; Sawyers, C.L. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. USA 1998, 95, 5246–5250. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.; Peacock, J.W.; Fazli, L.; Mui, A.L.-F.; Chung, S.W.; Cox, M.E.; Monia, B.; Gleave, M.E.; Ong, C.J. Loss of PTEN is associated with progression to androgen independence. Prostate 2006, 66, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Cairns, P.; Okami, K.; Halachmi, S.; Halachmi, N.; Esteller, M.; Herman, J.G.; Jen, J.; Isaacs, W.B.; Bova, G.S.; Sidransky, D. Frequent Inactivation of PTEN/MMAC1 in Primary Prostate Cancer. Cancer Res. 1997, 57, 4997–5000. [Google Scholar] [PubMed]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal Heterogeneity of PTEN/MMAC1 Gene Alterations in Multiple Metastatic Prostate Cancer Tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar] [PubMed]
- Robinson, D.; van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Sarker, D.; Reid, A.H.M.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT Pathway for the Treatment of Prostate Cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Q.; Adam, R.M.; Santiestevan, E.; Freeman, M.R. The phosphatidylinositol 3’-kinase pathway is a dominant growth factor-activated cell survival pathway in LNCaP human prostate carcinoma cells. Cancer Res. 1999, 59, 2891–2897. [Google Scholar] [PubMed]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; Ahmed, M.S.; Bakanauskas, V.J.; Muschel, R.J.; McKenna, W.G. Radiation sensitization of human cancer cells in vivo by inhibiting the activity of PI3K using LY294002. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 846–853. [Google Scholar] [CrossRef]
- Wadhwa, B.; Makhdoomi, U.; Vishwakarma, R.; Malik, F. Protein kinase B: Emerging mechanisms of isoform-specific regulation of cellular signaling in cancer. Anticancer Drugs 2017, 28, 569–580. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.; Urbano, T.G. New drug development in head and neck squamous cell carcinoma: The PI3-K inhibitors. Oral Oncol. 2017, 67, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Hermida, M.A.; Leslie, N.R. Prostate cancer, PI3K, PTEN and prognosis. Clin. Sci. 2017, 131, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wan, W.Z.; Li, Y.; Zhou, G.L.; Liu, X.G. Recent development of ATP-competitive small molecule phosphatidylinostitol-3-kinase inhibitors as anticancer agents. Oncotarget 2017, 8, 7181–7200. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Baiz, D.; Pinder, T.A.; Hassan, S.; Karpova, Y.; Salsbury, F.; Welker, M.E.; Kulik, G. Synthesis and Characterization of a Novel Prostate Cancer-Targeted Phosphatidylinositol-3-kinase Inhibitor Prodrug. J. Med. Chem. 2012, 55, 8038–8046. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Shukla, R.S.; Qin, B.; Li, B.; Cheng, K. Development of a Peptide–Drug Conjugate for Prostate Cancer Therapy. Mol. Pharm. 2011, 8, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Coombs, G.S.; Bergstrom, R.C.; Pellequer, J.-L.; Baker, S.I.; Navre, M.; Smith, M.M.; Tainer, J.A.; Madison, E.L.; Corey, D.R. Substrate specificity of prostate-specific antigen (PSA). Chem. Biol. 1998, 5, 475–488. [Google Scholar] [CrossRef]
- Welker, M.E.; Kulik, G. Recent syntheses of PI3K/AKT/mTOR signaling pathway inhibitors. Bioorg. Med. Chem. 2013, 21, 4063–4091. [Google Scholar] [CrossRef] [PubMed]
- Elshemy, H.A.H.; Abdelall, E.K.A.; Azouz, A.A.; Moawad, A.; Ali, W.A.M.; Safwat, N.M. Synthesis, anti-inflammatory, cyclooxygenases inhibitions assays and histopathological study of poly-substituted 1,3,5-triazines: Confirmation of regiospecific pyrazole cyclization by HMBC. Eur. J. Med. Chem. 2017, 127, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Plebanek, E.; Chevrier, F.; Roy, V.; Garenne, T.; Lecaille, F.; Warszycki, D.; Bojarski, A.J.; Lalmanach, G.; Agrofoglio, L.A. Straightforward synthesis of 2,4,6-trisubstituted 1,3,5-triazine compounds targeting cysteine cathepsins K and S. Eur. J. Med. Chem. 2016, 121, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Latacz, G.; Kechagioglou, P.; Papi, R.; Lazewska, D.; Wiecek, M.; Kaminska, K.; Wencel, P.; Karcz, T.; Schwed, J.S.; Stark, H.; et al. The Synthesis of 1,3,5-triazine Derivatives and JNJ7777120 Analogues with Histamine H-4 Receptor Affinity and Their Interaction with PTEN Promoter. Chem. Biol. Drug Des. 2016, 88, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Luxami, V.; Paul, K. Synthesis and in vitro evaluation of novel triazine analogues as anticancer agents and their interaction studies with bovine serum albumin. Eur. J. Med. Chem. 2016, 117, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Luxami, V.; Paul, K. Synthesis, in vitro antitumor activity, dihydrofolate reductase inhibition, DNA intercalation and structure-activity relationship studies of 1,3,5-triazine analogues. Bioorg. Med. Chem. Lett. 2016, 26, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.R.; Singh, U.P.; Gahtori, P.; Ghosh, S.K.; Gogoi, K.; Prakash, A.; Singh, R.K. Synthesis, Docking, In Vitro and In Vivo Antimalarial Activity of Hybrid 4-aminoquinoline-1,3,5-triazine Derivatives against Wild and Mutant Malaria Parasites. Chem. Biol. Drug Des. 2015, 86, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Xu, J.; Xin, M.H.; Lu, S.M.; Zhang, S.Q. Design, synthesis and antiproliferative activity evaluation of m-(4-morpholinyl-1,3,5-triazin-2-yl)benzamides in vitro. Bioorg. Med. Chem. Lett. 2015, 25, 1730–1735. [Google Scholar] [CrossRef] [PubMed]
- Andrs, M.; Korabecny, J.; Jun, D.; Hodny, Z.; Bartek, J.; Kuca, K. Phosphatidylinositol 3-Kinase (PI3K) and Phosphatidylinositol 3-Kinase-Related Kinase (PIKK) Inhibitors: Importance of the Morpholine Ring. J. Med. Chem. 2015, 58, 41–71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Tan, Q.; Zhang, Z.T.; Zhao, Y. 1,3,5-Triazine inhibitors of histone deacetylases: Synthesis and biological activity. Med. Chem. Res. 2014, 23, 5188–5196. [Google Scholar] [CrossRef]
- Burger, M.T.; Knapp, M.; Wagman, A.; Ni, Z.J.; Hendrickson, T.; Atallah, G.; Zhang, Y.C.; Frazier, K.; Verhagen, J.; Pfister, K.; et al. Synthesis and in Vitro and in Vivo Evaluation of Phosphoinositide-3-kinase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.-J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; de Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Chen, M.; Yue, P.; Tao, H.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R.; Sun, S.-Y. The combination of RAD001 and NVP-BKM120 synergistically inhibits the growth of lung cancer in vitro and in vivo. Cancer Lett. 2012, 325, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Nanni, P.; Nicoletti, G.; Palladini, A.; Croci, S.; Murgo, A.; Ianzano, M.L.; Grosso, V.; Stivani, V.; Antognoli, A.; Lamolinara, A.; et al. Multiorgan Metastasis of Human HER-2+ Breast Cancer in Rag2−/−; Il2rg−/− Mice and Treatment with PI3K Inhibitor. PLoS ONE 2012, 7, e39626. [Google Scholar] [CrossRef] [PubMed]
- Sutherlin, D.P.; Bao, L.; Berry, M.; Castanedo, G.; Chuckowree, I.; Dotson, J.; Folks, A.; Friedman, L.; Goldsmith, R.; Gunzner, J.; et al. Discovery of a Potent, Selective, and Orally Available Class I Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Kinase Inhibitor (GDC-0980) for the Treatment of Cancer. J. Med. Chem. 2011, 54, 7579–7587. [Google Scholar] [CrossRef] [PubMed]
- Safina, B.S.; Baker, S.; Baumgardner, M.; Blaney, P.M.; Chan, B.K.; Chen, Y.-H.; Cartwright, M.W.; Castanedo, G.; Chabot, C.; Cheguillaume, A.J.; et al. Discovery of Novel PI3-Kinase δ Specific Inhibitors for the Treatment of Rheumatoid Arthritis: Taming CYP3A4 Time-Dependent Inhibition. J. Med. Chem. 2012, 55, 5887–5900. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhai, X.; Fu, Q.; Guo, F.; Bai, M.; Wang, J.; Wang, H.; Gong, P. Design, Synthesis and Anticancer Activity of 4-Morpholinothieno[3,2-d]pyrimidine Derivatives Bearing Arylmethylene Hydrazine Moiety. Chem. Pharm. Bull. 2012, 60, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Sutherlin, D.P.; Baker, S.; Bisconte, A.; Blaney, P.M.; Brown, A.; Chan, B.K.; Chantry, D.; Castanedo, G.; DePledge, P.; Goldsmith, P.; et al. Potent and selective inhibitors of PI3Kδ: Obtaining isoform selectivity from the affinity pocket and tryptophan shelf. Bioorg. Med. Chem. Lett. 2012, 22, 4296–4302. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.K.-C.; Bailey, S.; Dinh, D.M.; Lam, H.; Li, C.; Wells, P.A.; Yin, M.-J.; Zou, A. Conformationally-restricted cyclic sulfones as potent and selective mTOR kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5114–5117. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P.; Salphati, L.; Alicke, B.; Cheong, J.; Dotson, J.; Edgar, K.; Goldsmith, R.; Gould, S.E.; Lee, L.B.; Lesnick, J.D.; et al. The Design and Identification of Brain Penetrant Inhibitors of Phosphoinositide 3-Kinase α. J. Med. Chem. 2012, 55, 8007–8020. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R.; Alessi, D.R. Regulation of AKT/PKB Ser473 Phosphorylation. Mol. Cell 2005, 18, 143–145. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, E.W.; Nelson, R.A., Jr.; Karpova, Y.; Kulik, G.; Welker, M.E. Synthesis and PI3 Kinase Inhibition Activity of Some Novel Trisubstituted Morpholinopyrimidines. Molecules 2018, 23, 1675. https://doi.org/10.3390/molecules23071675
Wright EW, Nelson RA Jr., Karpova Y, Kulik G, Welker ME. Synthesis and PI3 Kinase Inhibition Activity of Some Novel Trisubstituted Morpholinopyrimidines. Molecules. 2018; 23(7):1675. https://doi.org/10.3390/molecules23071675
Chicago/Turabian StyleWright, Emily W., Ronald A. Nelson, Jr., Yelena Karpova, George Kulik, and Mark E. Welker. 2018. "Synthesis and PI3 Kinase Inhibition Activity of Some Novel Trisubstituted Morpholinopyrimidines" Molecules 23, no. 7: 1675. https://doi.org/10.3390/molecules23071675
APA StyleWright, E. W., Nelson, R. A., Jr., Karpova, Y., Kulik, G., & Welker, M. E. (2018). Synthesis and PI3 Kinase Inhibition Activity of Some Novel Trisubstituted Morpholinopyrimidines. Molecules, 23(7), 1675. https://doi.org/10.3390/molecules23071675