Design of a Novel and Selective IRAK4 Inhibitor Using Topological Water Network Analysis and Molecular Modeling Approaches

Abstract
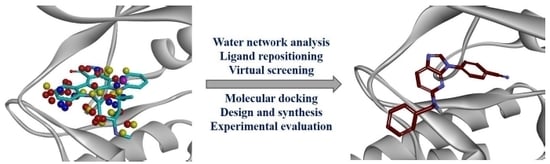
1. Introduction
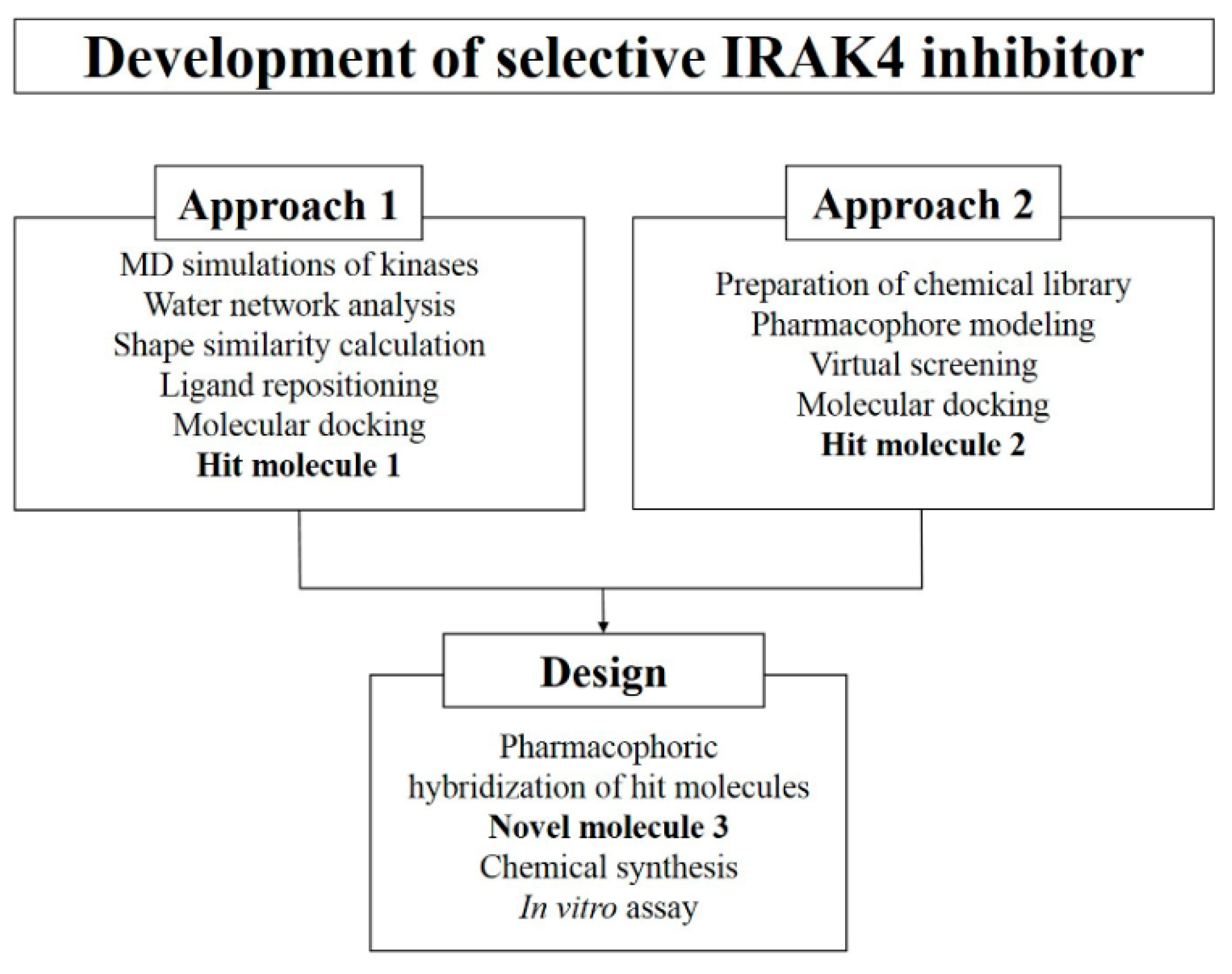
2. Results and Discussion
2.1. TWN Analysis and Staurosporine-Based Repositioning
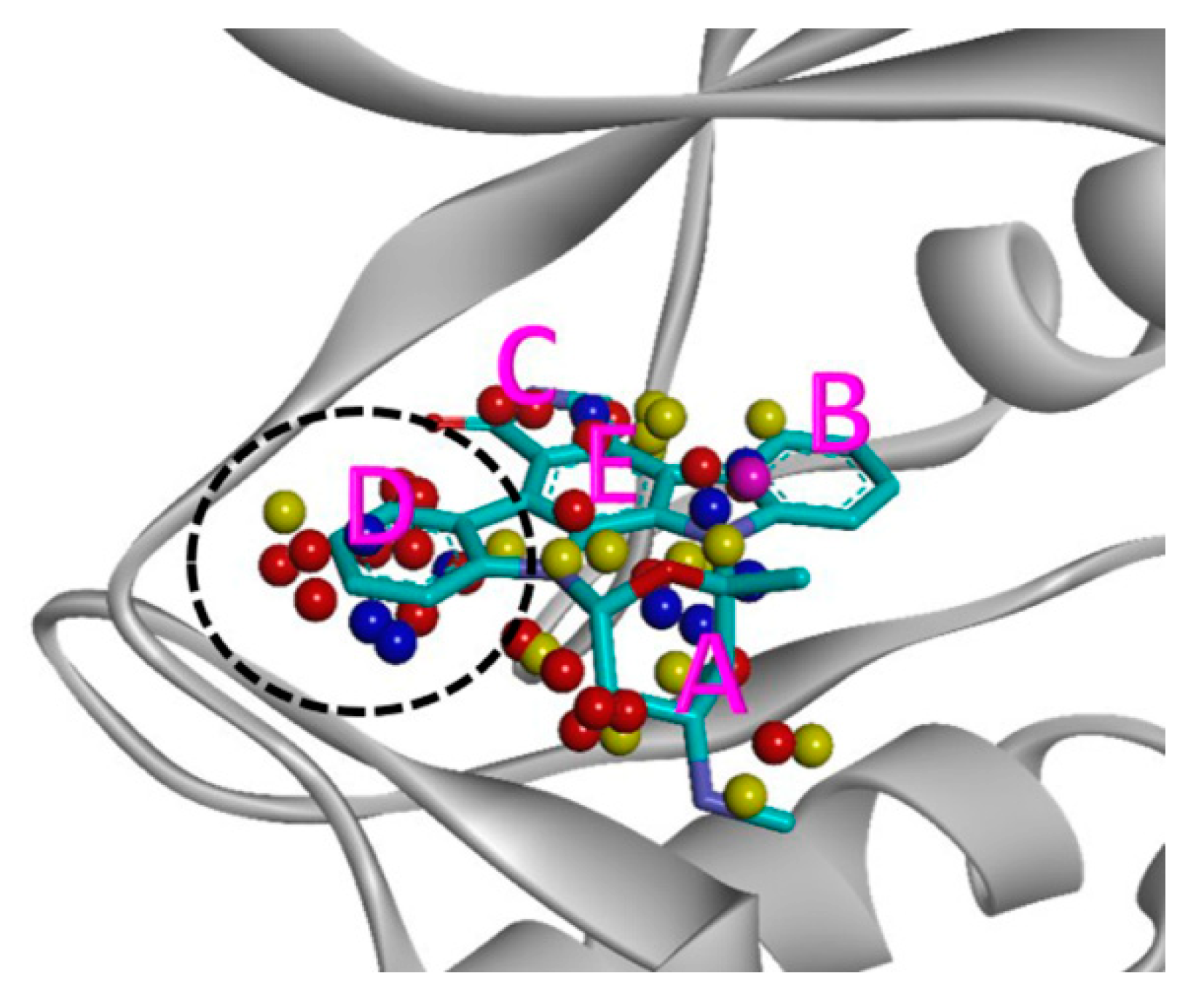
2.2. Selectivity Analysis by Shape Similarity
2.3. Pharmacophore-Based Virtual Screening and Molecular Docking
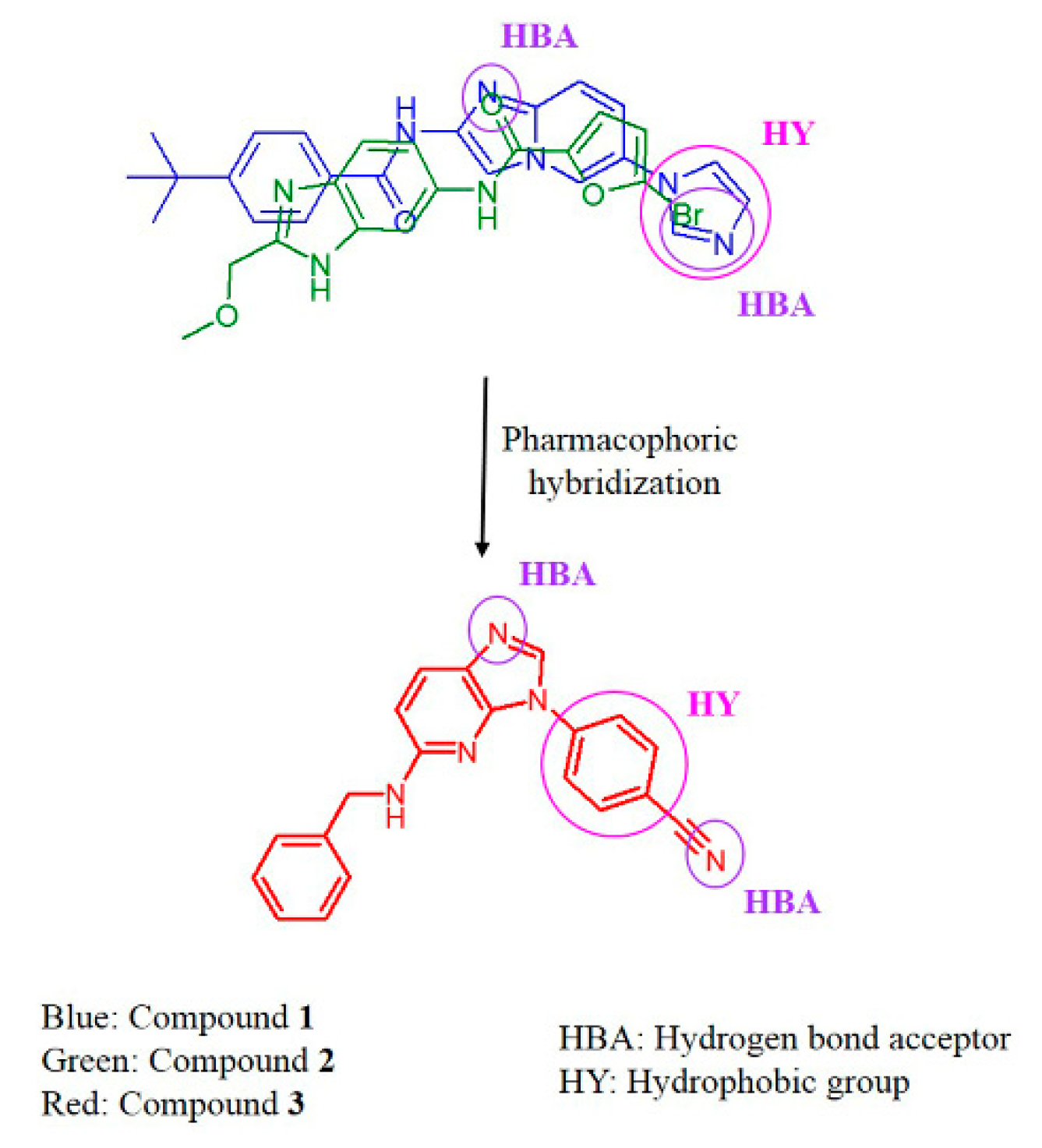
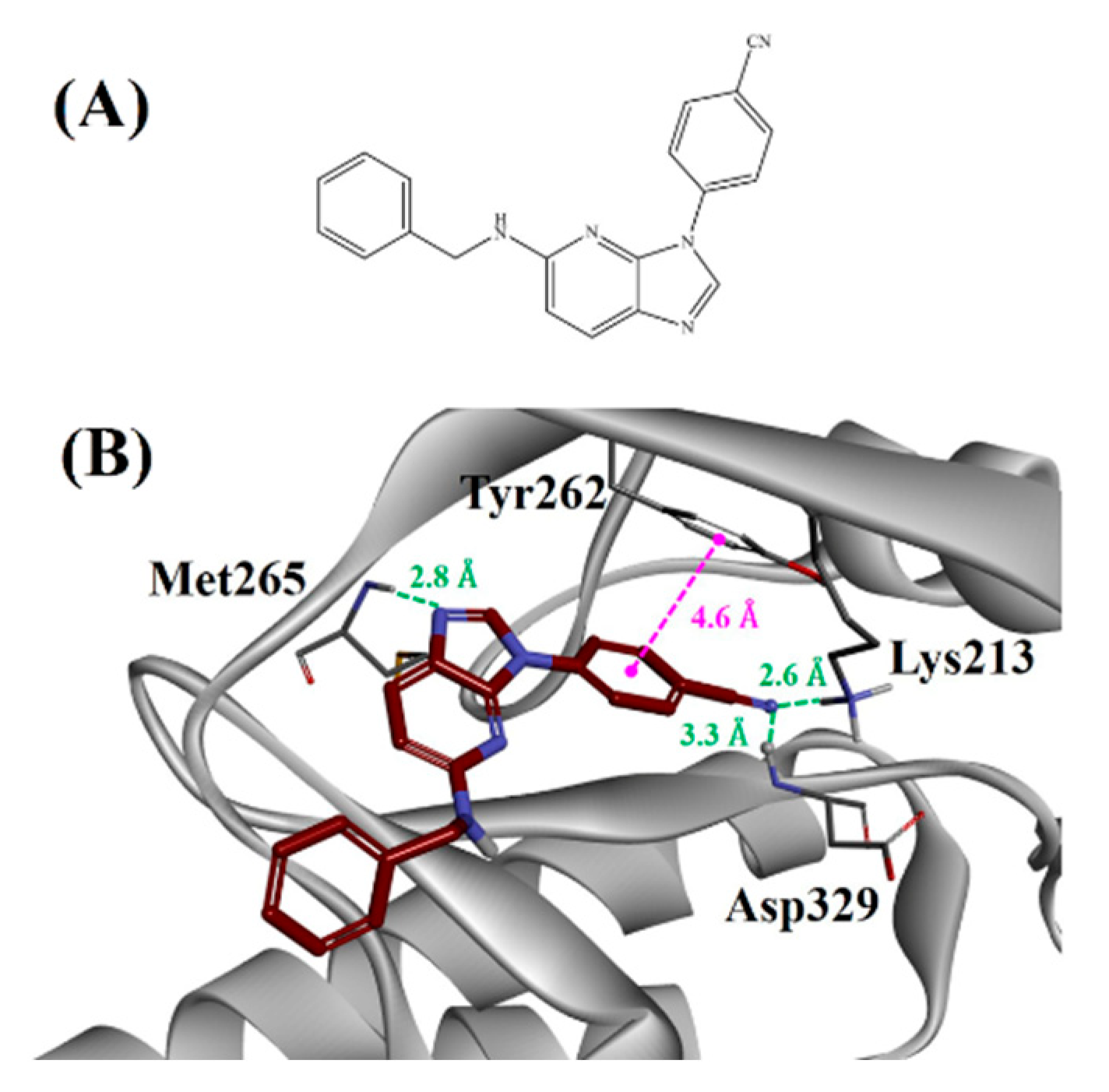
2.4. Design, Synthesis and Testing of IRAK4 Inhibitor
3. Materials and Methods
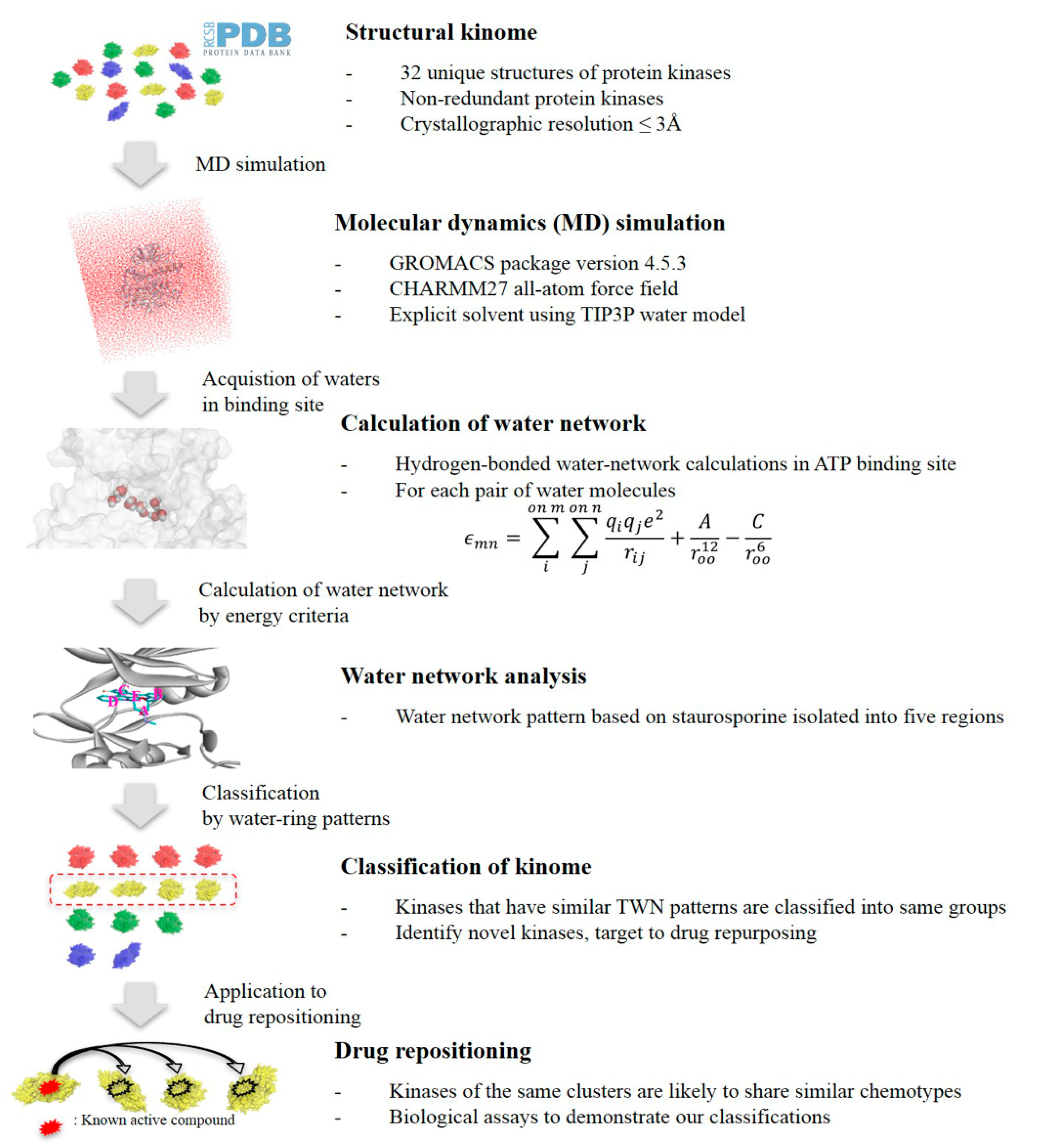
3.1. Protein Preparation
3.2. MD Simulation
3.3. TWN Analysis
3.4. Chemical Library Preparation for Virtual Screening
3.5. Pharmacophore Model Generation
3.6. Molecular Docking
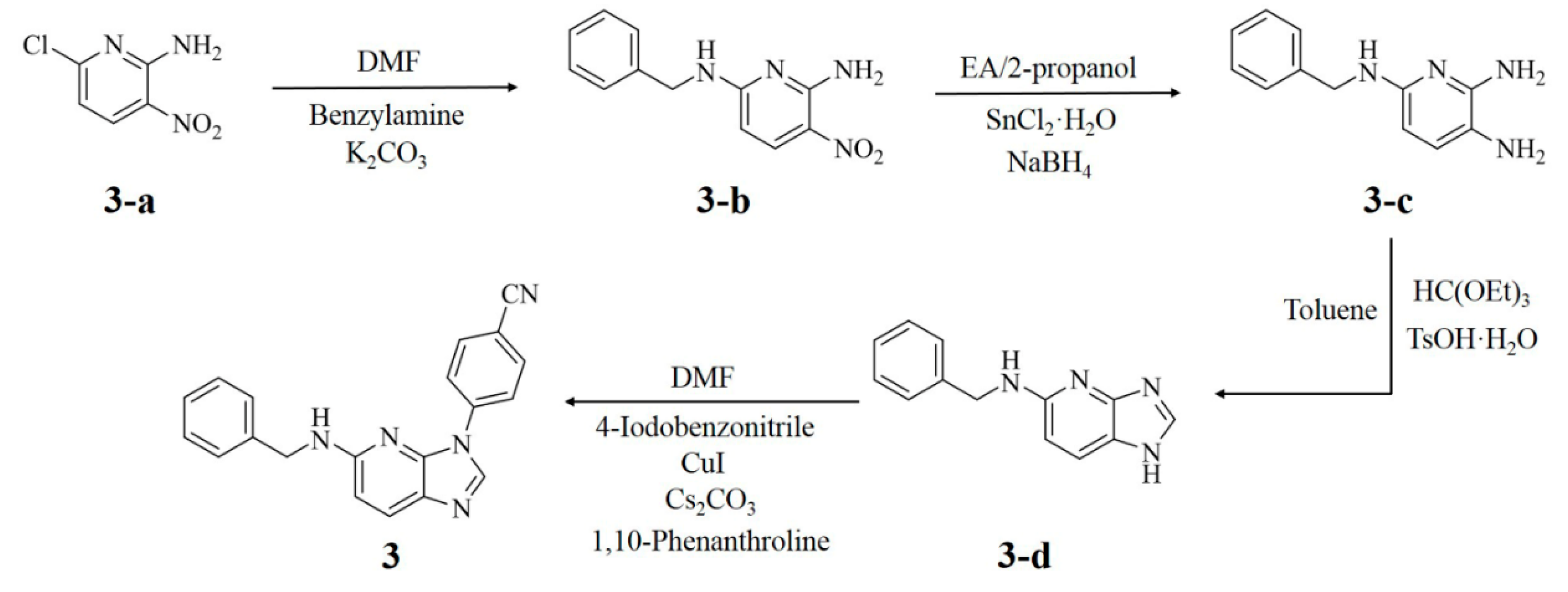
3.7. Synthesis and Characterization
3.7.1. N6-Benzyl-3-nitro-pyridine-2,6-diamine (3-b)
3.7.2. N6-Benzyl-pyridine-2,3,6-triaine (3-c)
3.7.3. Benzyl-(1H-imidazo[4,5-b]pyridine-5-yl)-amine (3-d)
3.7.4. 4-(5-Benzylamino-imidazo[4,5-b]pyridine-1-yl)-benzonitrile (3)
3.8. In Vitro Assay
3.8.1. Enzymatic Assay for IRAK4
3.8.2. Enzymatic Assay for ASK1
3.8.3. Enzymatic Assay for ITK
3.8.4. Enzymatic Assay for LYN
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stout, T.; Foster, P.; Matthews, D. High-throughput structural biology in drug discovery: Protein kinases. Curr. Pharm. Des. 2004, 10, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.; Scheraga, H.A. Insufficiently dehydrated hydrogen bonds as determinants of protein interactions. Proc. Natl. Acad. Sci. USA 2003, 100, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Abel, R.; Kim, B.; Berne, B.J.; Friesner, R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Nagata, N.; Nakanishi, I.; Kitaura, K. Ligand shape emerges in solvent dipole ordering region at ligand binding site of protein. J. Comput. Chem. 2010, 31, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.D.; Lee, M.H.; Kang, N.S. Quantitative assessment of kinase selectivity based the water-ring network in protein binding sites using molecular dynamics simulations. J. Mol. Liq. 2016, 221, 316–322. [Google Scholar] [CrossRef]
- Jang, W.D.; Kim, J.-T.; Kang, N.S. The analysis of water network for kinase selectivity based on the MD simulations. J. Mol. Liq. 2014, 191, 37–41. [Google Scholar] [CrossRef]
- Hynes Jr, J.; Nair, S.K. Advances in the discovery of small-molecule IRAK4 inhibitors. In Annual Reports in Medicinal Chemistry, 1st ed.; Weinstein, D.S., Desai, M.C., Eds.; Elsevier: Foster, CA, USA, 2014; Volume 49, pp. 117–133. ISBN 978-0-12-800167-7. [Google Scholar]
- Gosu, V.; Choi, S. Structural dynamic analysis of apo and ATP-bound IRAK4 kinase. Sci. Rep. 2014, 4, 5748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wesche, H.; Stevens, T.; Walker, N.; Yeh, W.-C. IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem. 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Kuglstatter, A.; Villaseñor, A.G.; Shaw, D.; Lee, S.W.; Tsing, S.; Niu, L.; Song, K.W.; Barnett, J.W.; Browner, M.F. Cutting edge: IL-1 receptor-associated kinase 4 structures reveal novel features and multiple conformations. J. Immunol. 2007, 178, 2641–2645. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Zhou, H.; Shan, Y.; Liu, Q.; Li, Q.; Shaw, D.E.; Li, X.; Wu, H. IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly. Mol. Cell 2014, 55, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Altman, M.D.; Baker, J.; Brubaker, J.D.; Chen, H.; Chen, Y.; Fischmann, T.; Gibeau, C.; Kleinschek, M.A.; Leccese, E. Discovery of 5-Amino-N-(1H-pyrazol-4-yl) pyrazolo [1, 5-a] pyrimidine-3-carboxamide inhibitors of IRAK4. ACS Med. Chem. Lett. 2015, 6, 683–688. [Google Scholar] [CrossRef] [PubMed]
- McElroy, W.T.; Seganish, W.M.; Herr, R.J.; Harding, J.; Yang, J.; Yet, L.; Komanduri, V.; Prakash, K.C.; Lavey, B.; Tulshian, D. Discovery and hit-to-lead optimization of 2, 6-diaminopyrimidine inhibitors of interleukin-1 receptor-associated kinase 4. Bioorg. Med. Chem. Lett. 2015, 25, 1836–1841. [Google Scholar] [CrossRef] [PubMed]
- McElroy, W.T.; Tan, Z.; Ho, G.; Paliwal, S.; Li, G.; Seganish, W.M.; Tulshian, D.; Tata, J.; Fischmann, T.O.; Sondey, C. Potent and selective amidopyrazole inhibitors of IRAK4 that are efficacious in a rodent model of inflammation. ACS Med. Chem. Lett. 2015, 6, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Seganish, W.M.; Fischmann, T.O.; Sherborne, B.; Matasi, J.; Lavey, B.; McElroy, W.T.; Tulshian, D.; Tata, J.; Sondey, C.; Garlisi, C.G. Discovery and structure enabled synthesis of 2, 6-diaminopyrimidin-4-one IRAK4 inhibitors. ACS Med. Chem. Lett. 2015, 6, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Hanisak, J.; Seganish, W.M.; McElroy, W.T.; Tang, H.; Zhang, R.; Tsui, H.-C.; Fischmann, T.; Tulshian, D.; Tata, J.; Sondey, C. Efforts towards the optimization of a bi-aryl class of potent IRAK4 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4250–4255. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-Rongen, M.; Godlewska, E.; Brunn, A.; Wiestler, O.D.; Siebert, R.; Deckert, M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011, 122, 791–792. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Terao, Y.; Suzuki, H.; Yoshikawa, M.; Yashiro, H.; Takekawa, S.; Fujitani, Y.; Okada, K.; Inoue, Y.; Yamamoto, Y.; Nakagawa, H. Design and biological evaluation of imidazo [1, 2-a] pyridines as novel and potent ASK1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7326–7329. [Google Scholar] [CrossRef] [PubMed]
- Kinnings, S.L.; Jackson, R.M. Binding site similarity analysis for the functional classification of the protein kinase family. J. Chem. Inf. Model. 2009, 49, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, D.; Robinson, S.; Romero, D.L. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders: Miniperspective. J. Med. Chem. 2014, 58, 96–110. [Google Scholar] [CrossRef] [PubMed]
- OTAVA chemicals. Available online: www.otavachemicals.com (accessed on 8 April 2014).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2009; p. 1257. ISBN 978-3-527-31852-0. [Google Scholar]
- Guner, O.F. Pharmacophore Perception, Development, and Use in Drug Design, Illustrated ed.; International University Line: La Jolla, CA, USA, 2000; p. 537. ISBN 0-9636817-6-1. [Google Scholar]
- Wang, Z.; Liu, J.; Sudom, A.; Ayres, M.; Li, S.; Wesche, H.; Powers, J.P.; Walker, N.P. Crystal structures of IRAK-4 kinase in complex with inhibitors: A serine/threonine kinase with tyrosine as a gatekeeper. Structure 2006, 14, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Long, J.M.; Vial, S.C.; Dedi, N.; Dunster, N.J.; Renwick, S.B.; Tanner, A.J.; Frantz, J.D.; Fleming, M.A.; Cheetham, G.M. Crystal structures of interleukin-2 tyrosine kinase and their implications for the design of selective inhibitors. J. Biol. Chem. 2004, 279, 18727–18732. [Google Scholar] [CrossRef] [PubMed]
- Miyano, N.; Kinoshita, T.; Nakai, R.; Kirii, Y.; Yokota, K.; Tada, T. Structural basis for the inhibitor recognition of human Lyn kinase domain. Bioorg. Med. Chem. Lett. 2009, 19, 6557–6560. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | A (%) | B (%) | C (%) | D (%) | E (%) |
|---|---|---|---|---|---|
| IRAK4 | 25.0 | 7.1 | 17.9 | 35.7 | 14.3 |
| ASK1 | 18.5 | 0.0 | 29.6 | 33.3 | 18.5 |
| LYN | 6.3 | 3.1 | 9.4 | 31.3 | 50.0 |
| ITK | 36.0 | 6.0 | 16.0 | 26.0 | 16.0 |
| Total Sequence Similarity (%)/Binding Site Similarity (%) | ||||
|---|---|---|---|---|
| Kinase | IRAK4 | ASK1 | LYN | ITK |
| IRAK4 | 100.0/100.0 | 44.0/40.6 | 47.9/35.6 | 44.6/39.6 |
| ASK1 | 44.0/40.6 | 100.0/100.0 | 45.8/46.8 | 46.8/49.5 |
| LYN | 47.9/35.6 | 45.8/46.8 | 100.0/100.0 | 64.0/42.7 |
| ITK | 44.6/39.6 | 46.8/49.5 | 64.0/42.7 | 100.0/100.0 |
| Kinase | Full Ligand (%) | Around D-Site (%) |
|---|---|---|
| IRAK4 | 22.7 | 67.6 |
| ASK1 | 24.2 | 60.7 |
| LYN | 18.2 | 49.2 |
| ITK | 17.4 | 41.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.H.; Balupuri, A.; Jung, Y.-r.; Choi, S.; Lee, A.; Cho, Y.S.; Kang, N.S. Design of a Novel and Selective IRAK4 Inhibitor Using Topological Water Network Analysis and Molecular Modeling Approaches. Molecules 2018, 23, 3136. https://doi.org/10.3390/molecules23123136
Lee MH, Balupuri A, Jung Y-r, Choi S, Lee A, Cho YS, Kang NS. Design of a Novel and Selective IRAK4 Inhibitor Using Topological Water Network Analysis and Molecular Modeling Approaches. Molecules. 2018; 23(12):3136. https://doi.org/10.3390/molecules23123136
Chicago/Turabian StyleLee, Myeong Hwi, Anand Balupuri, Ye-rim Jung, Sungwook Choi, Areum Lee, Young Sik Cho, and Nam Sook Kang. 2018. "Design of a Novel and Selective IRAK4 Inhibitor Using Topological Water Network Analysis and Molecular Modeling Approaches" Molecules 23, no. 12: 3136. https://doi.org/10.3390/molecules23123136
APA StyleLee, M. H., Balupuri, A., Jung, Y.-r., Choi, S., Lee, A., Cho, Y. S., & Kang, N. S. (2018). Design of a Novel and Selective IRAK4 Inhibitor Using Topological Water Network Analysis and Molecular Modeling Approaches. Molecules, 23(12), 3136. https://doi.org/10.3390/molecules23123136