A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
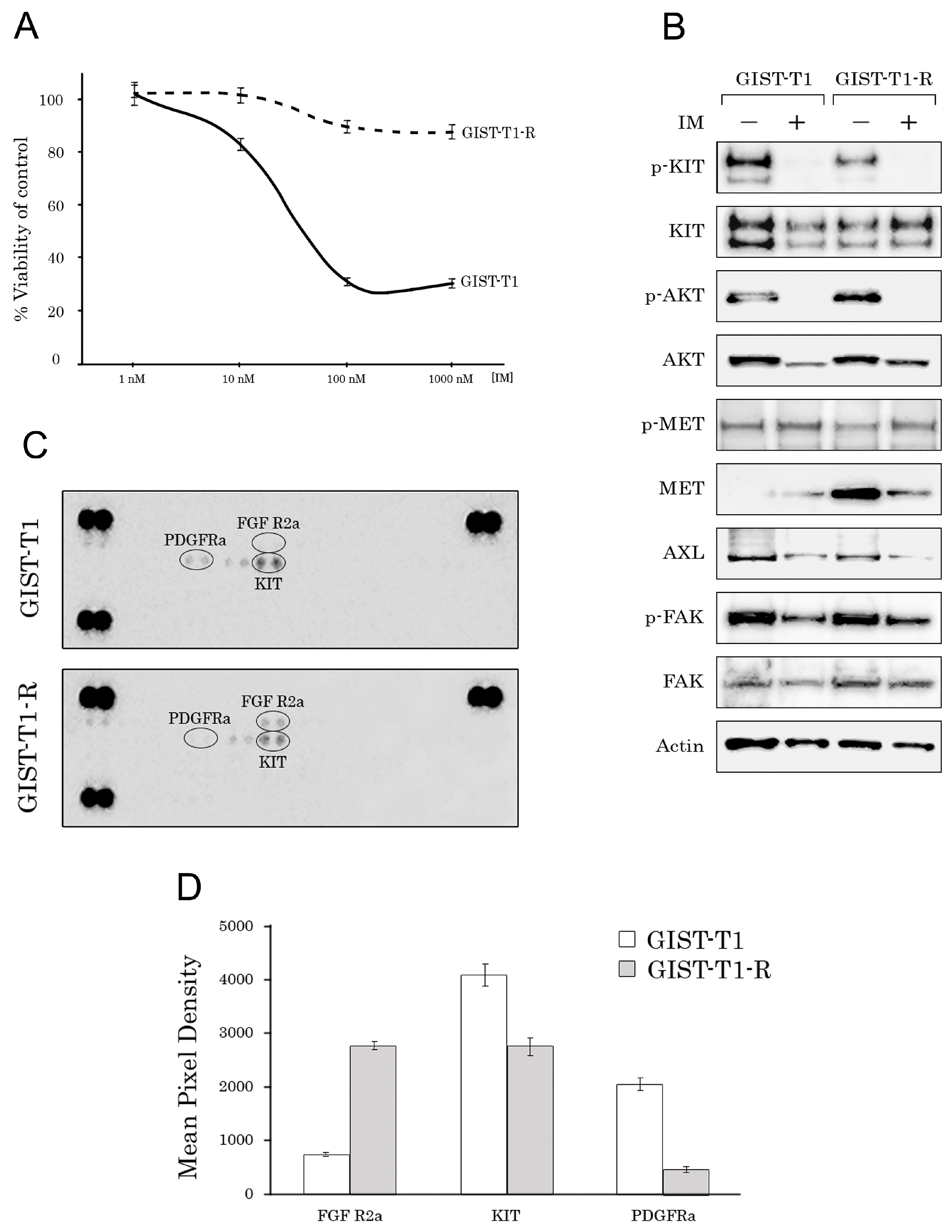
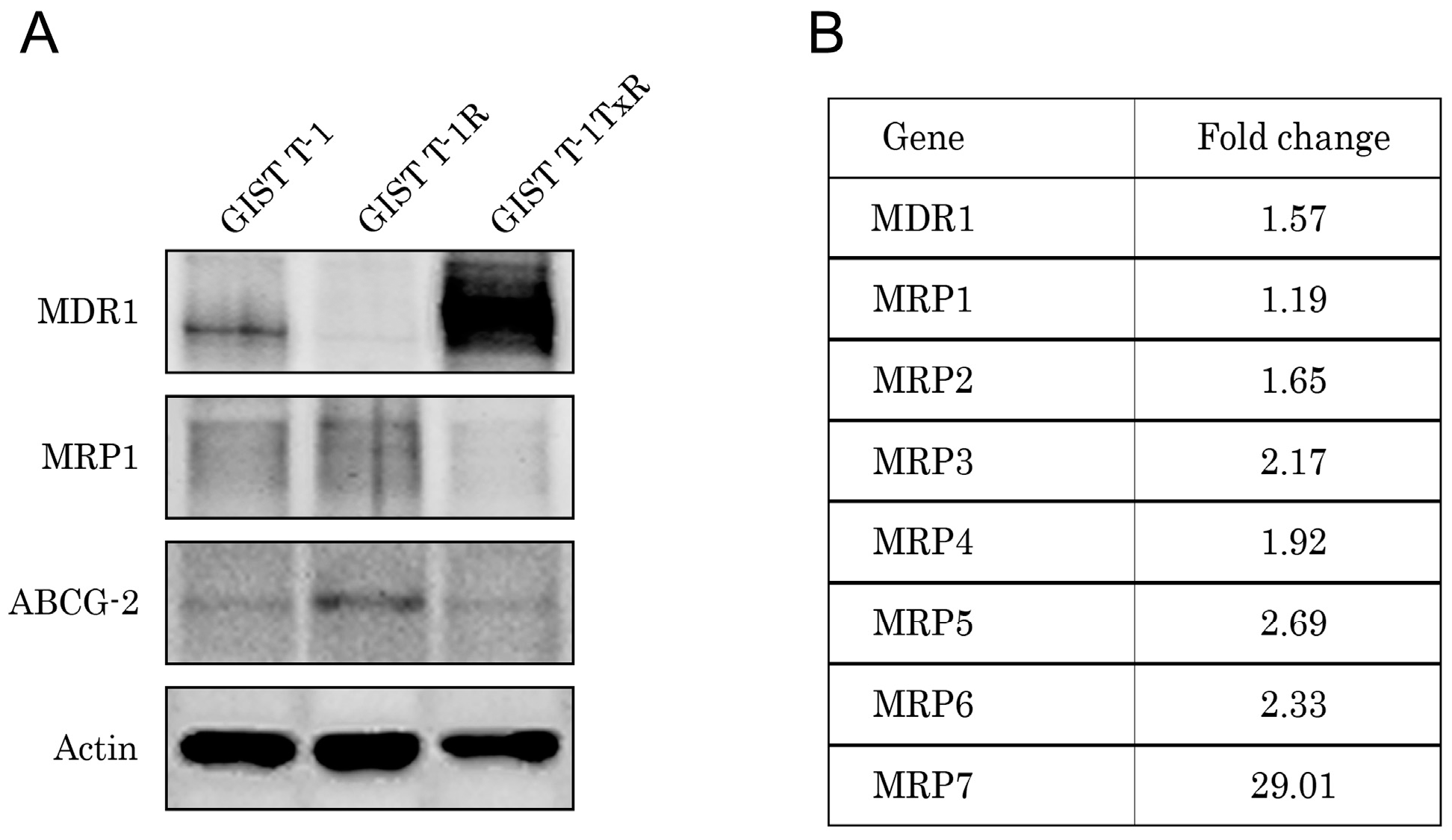
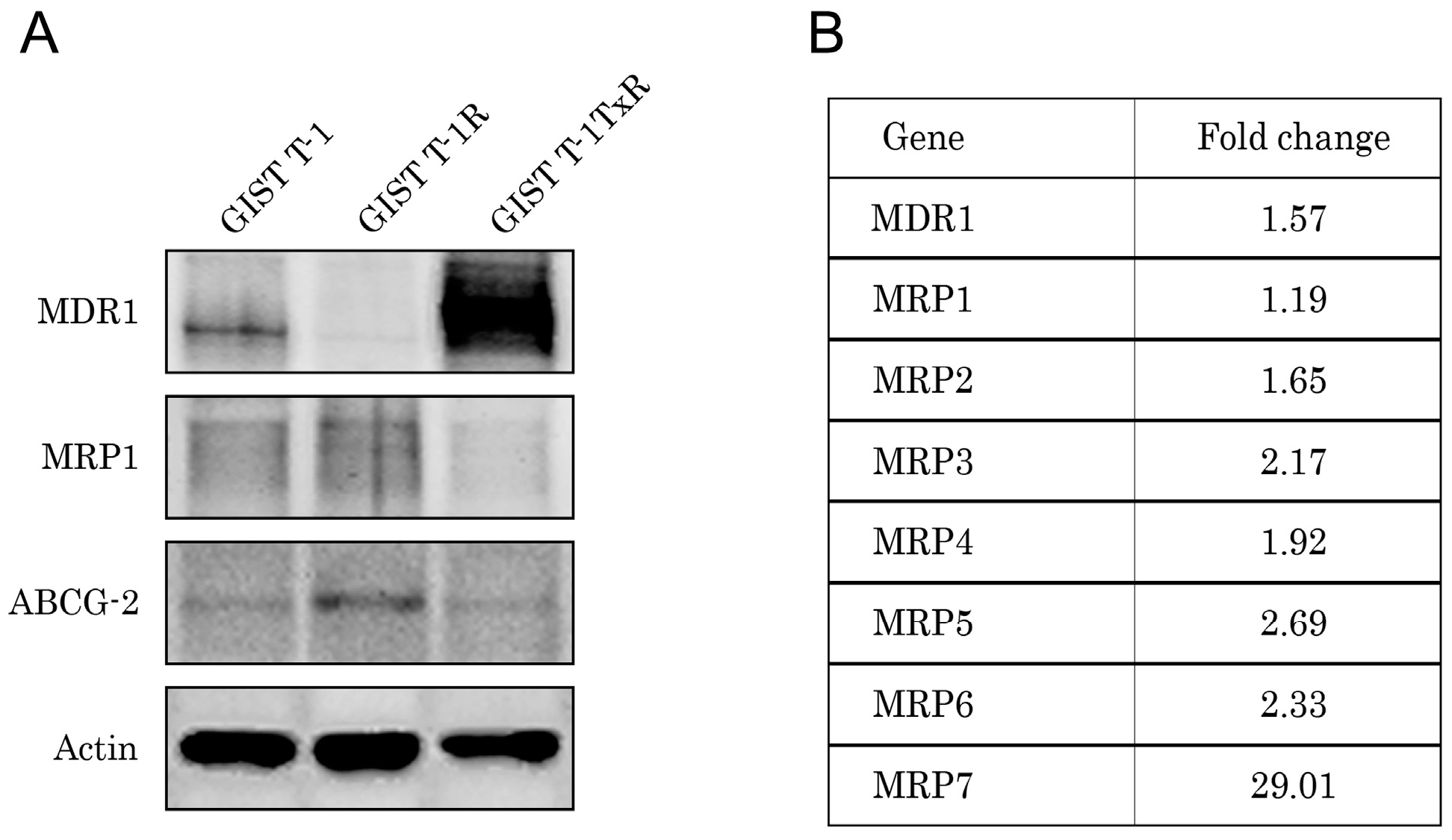
2.1. Establishment and Characterization of the IM-Resistant GIST T-1R Cell Subline
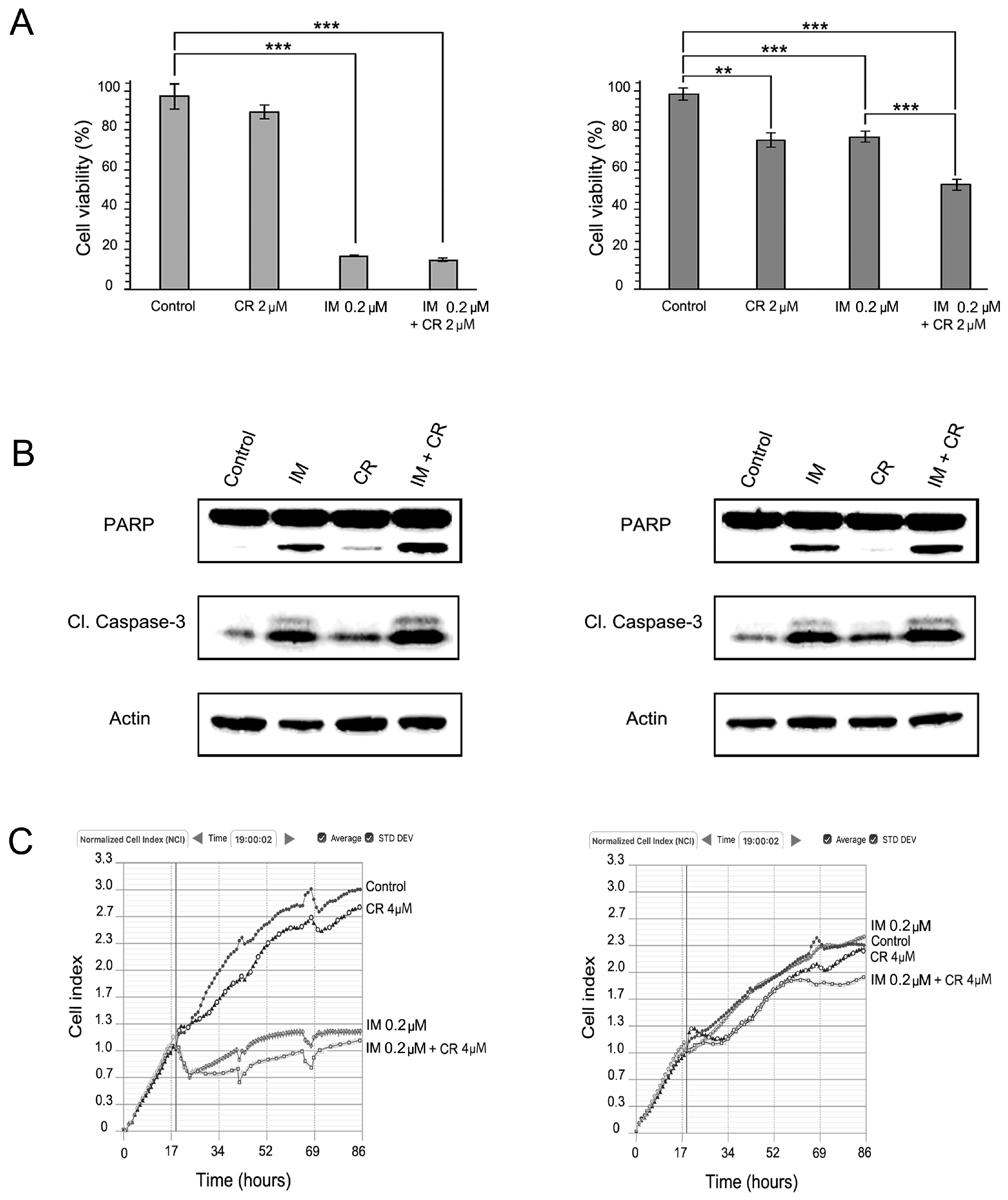
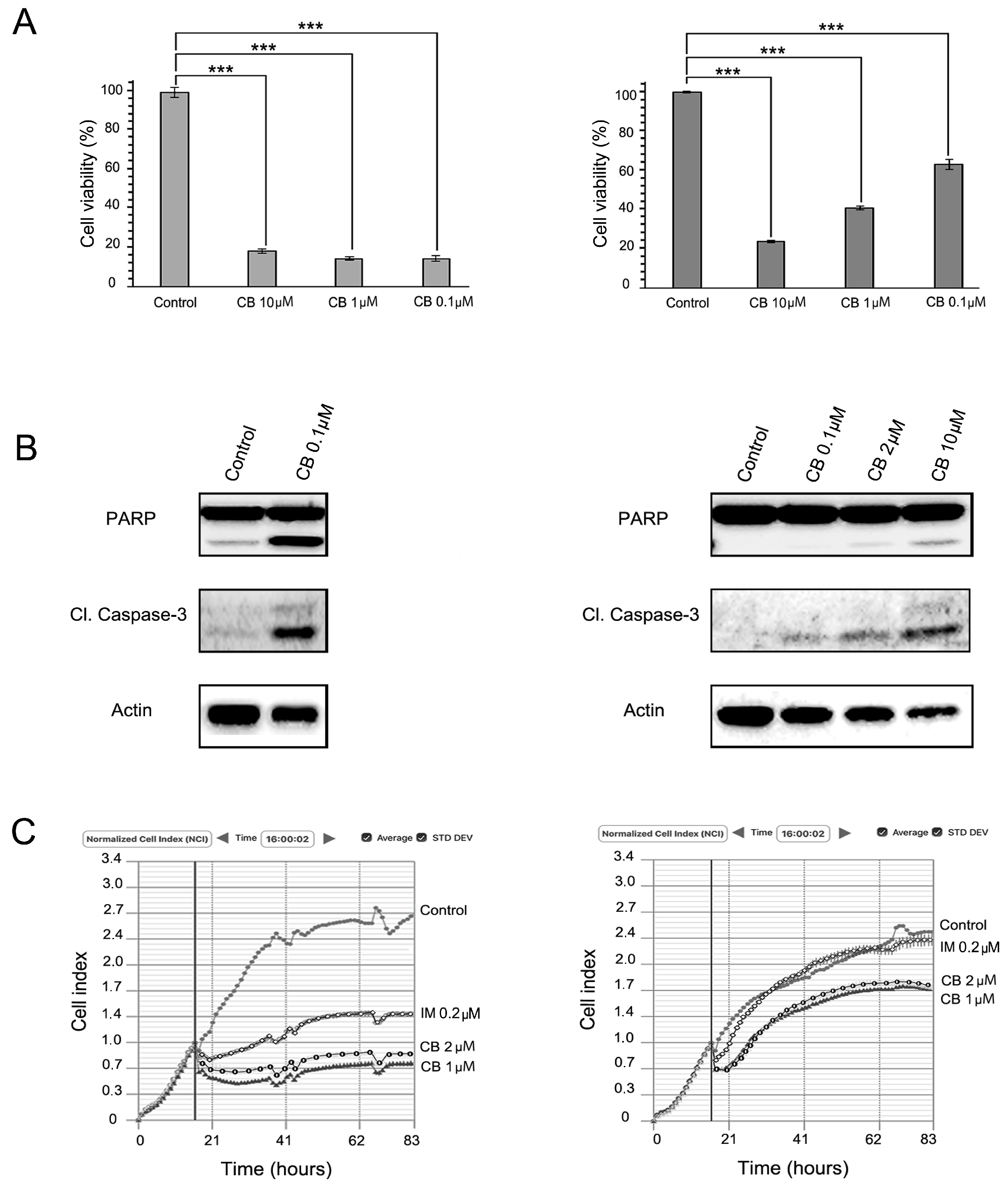
2.2. MET Inhibition Enhances the Cytotoxic Effects of IM in GIST T-1R Cells
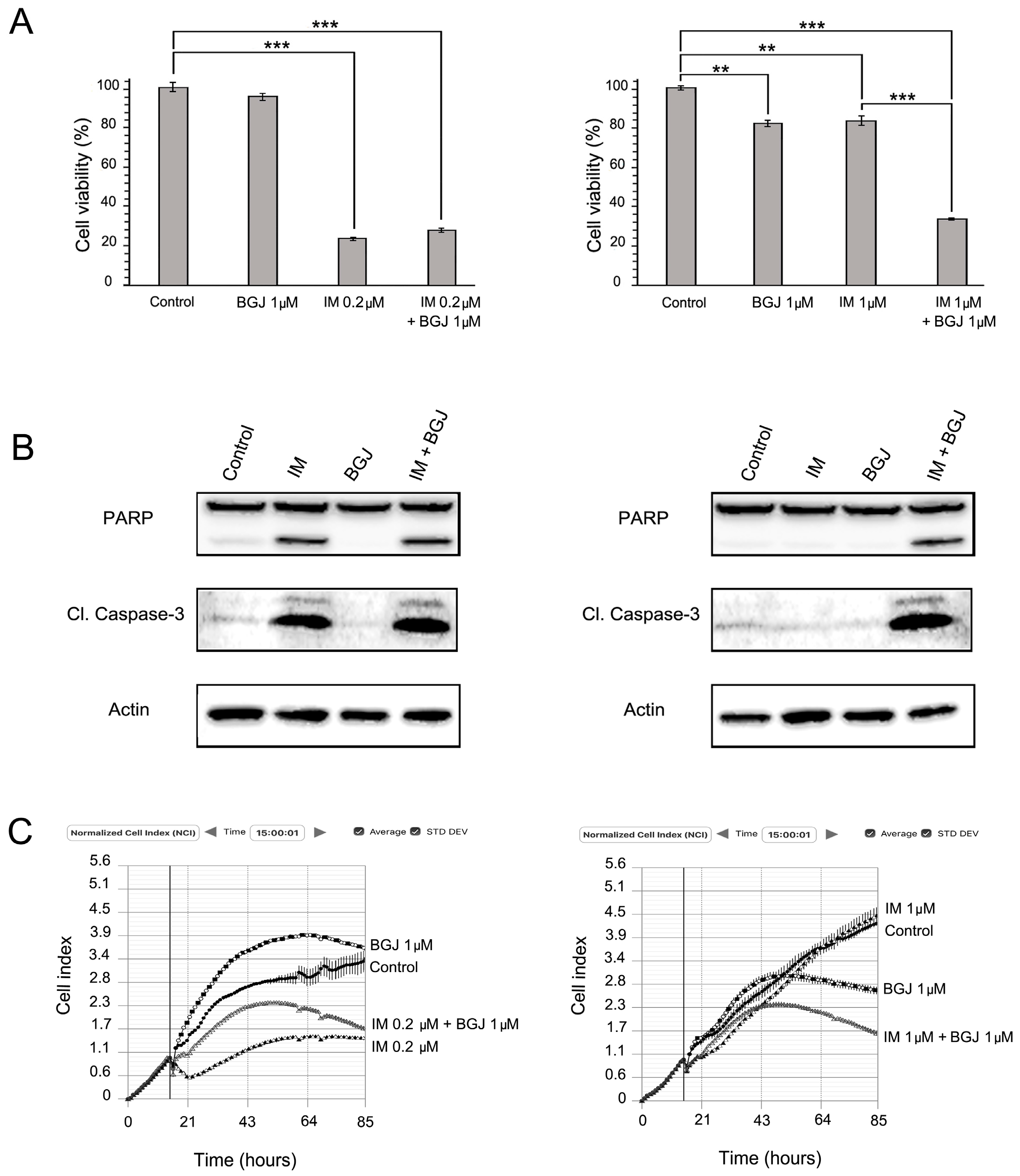
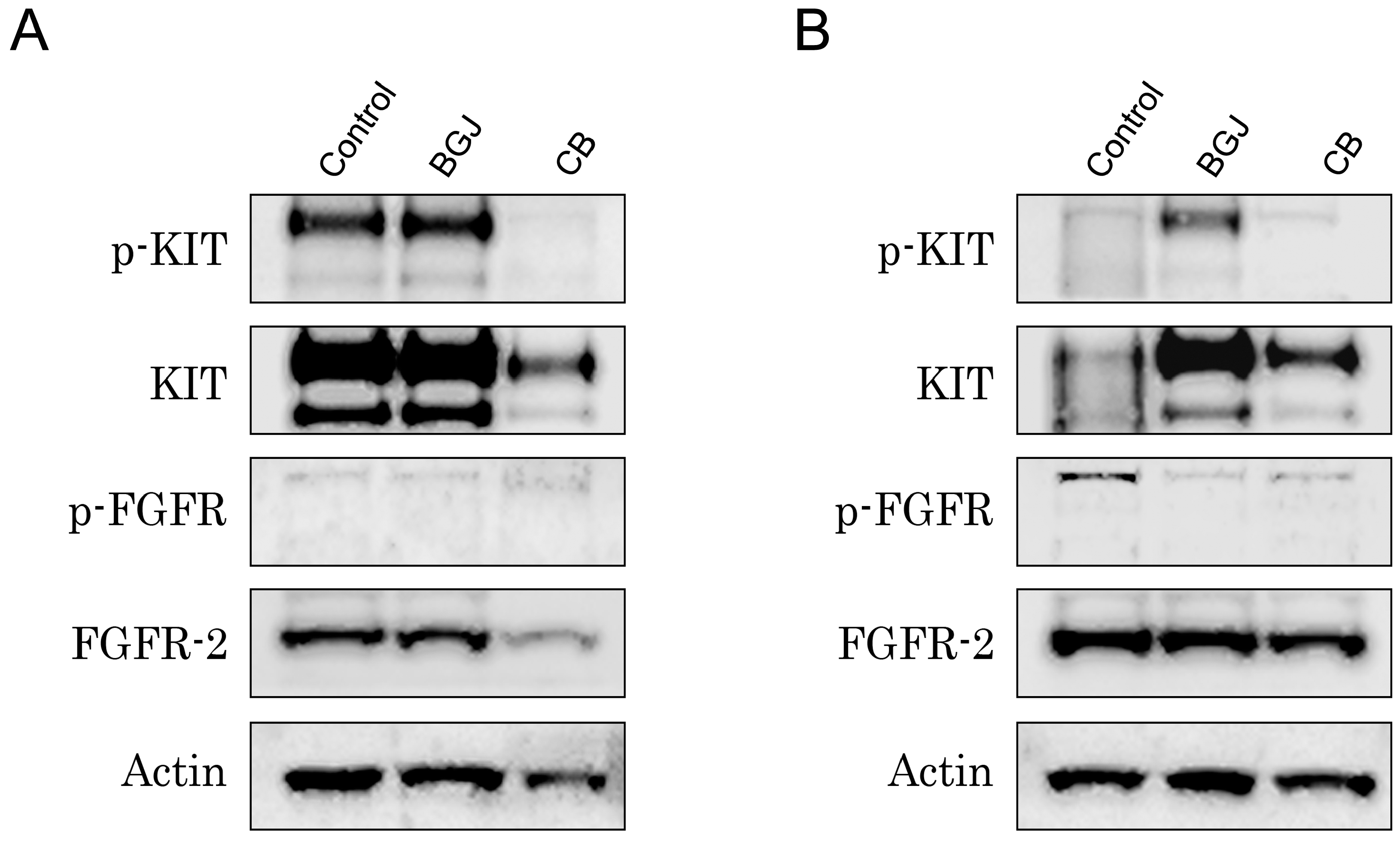
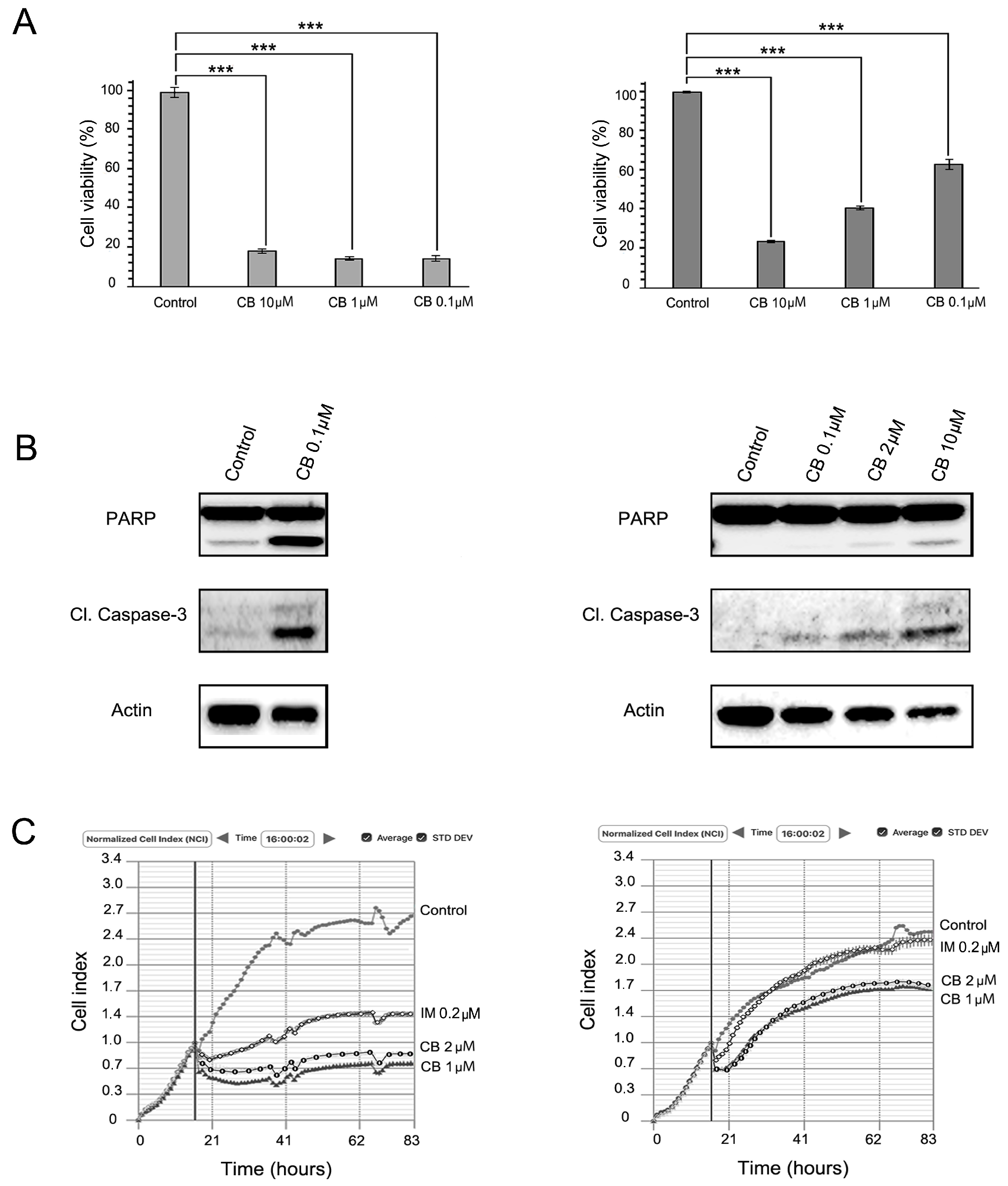
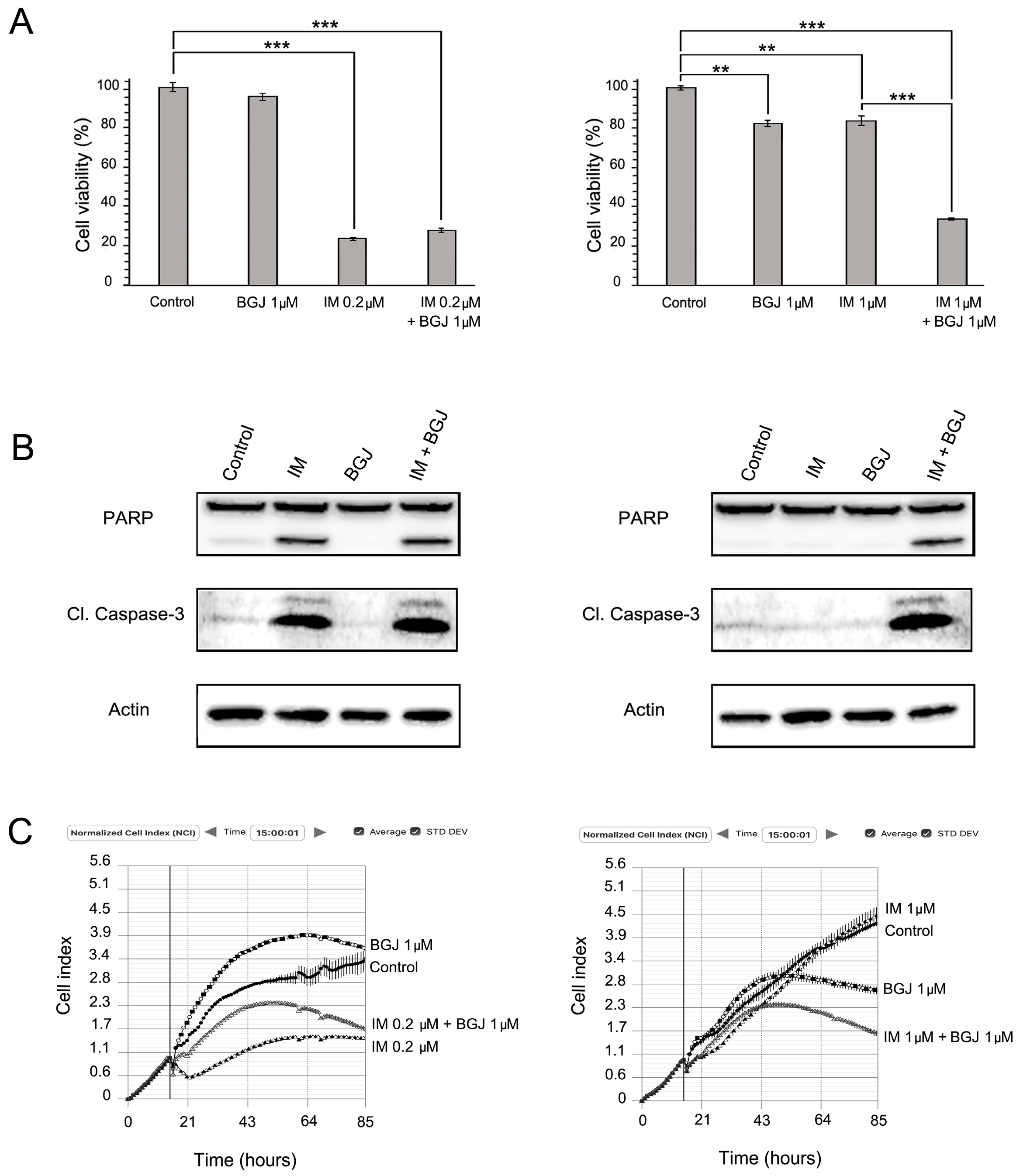
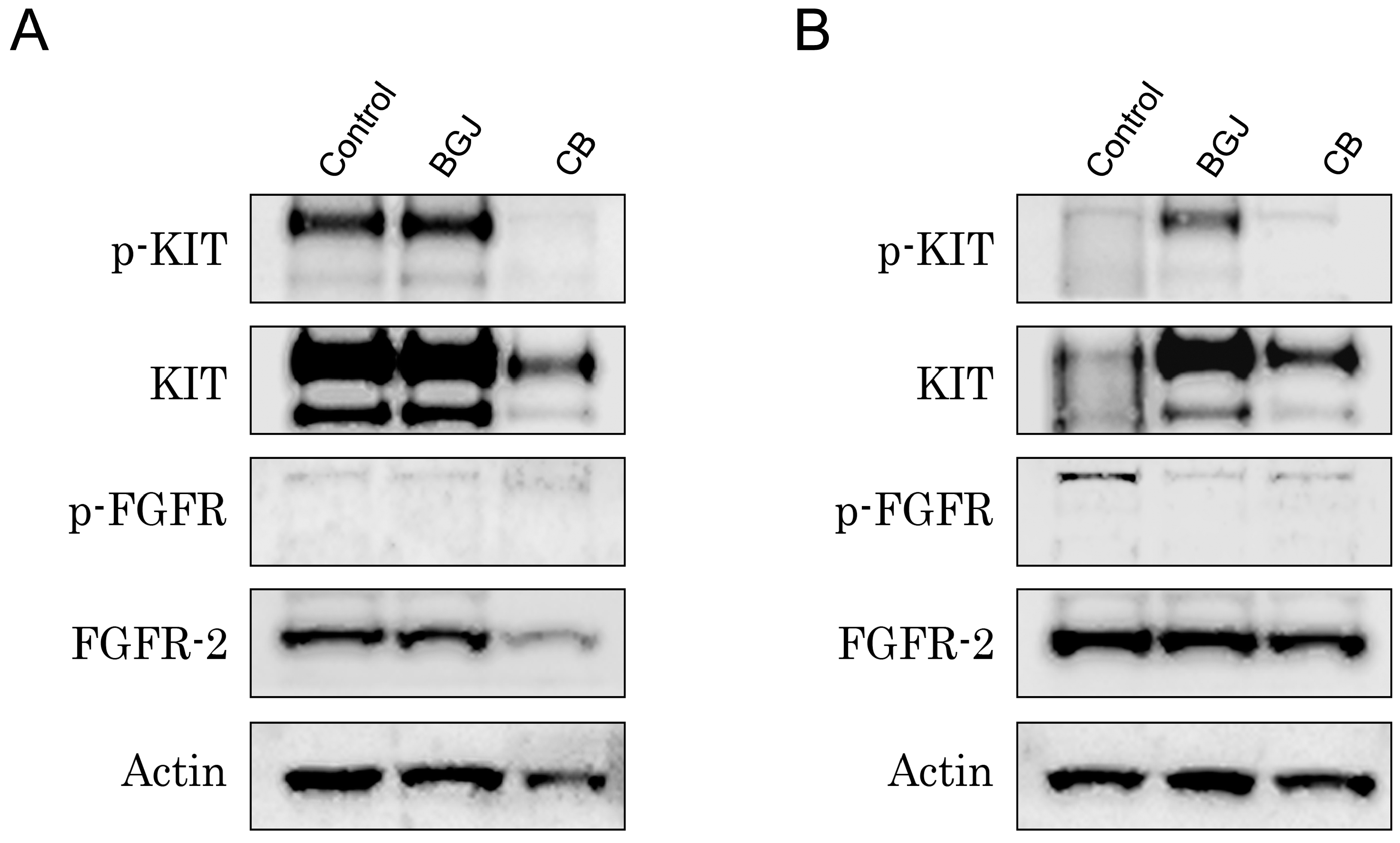
2.3. FGFR Inhibition Substantially Enhances the Cytotoxic Effects of IM in IM-Resistant GISTs
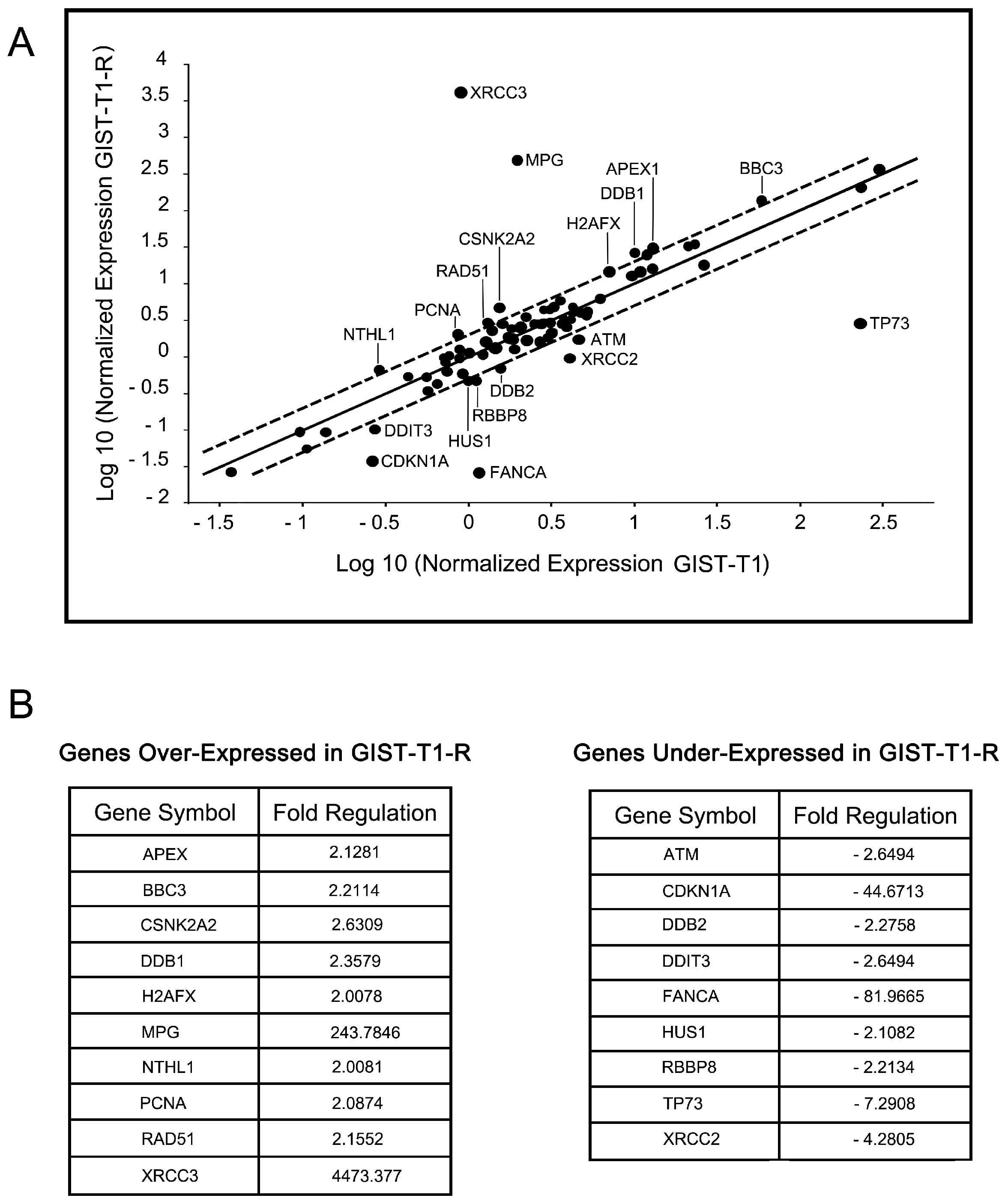
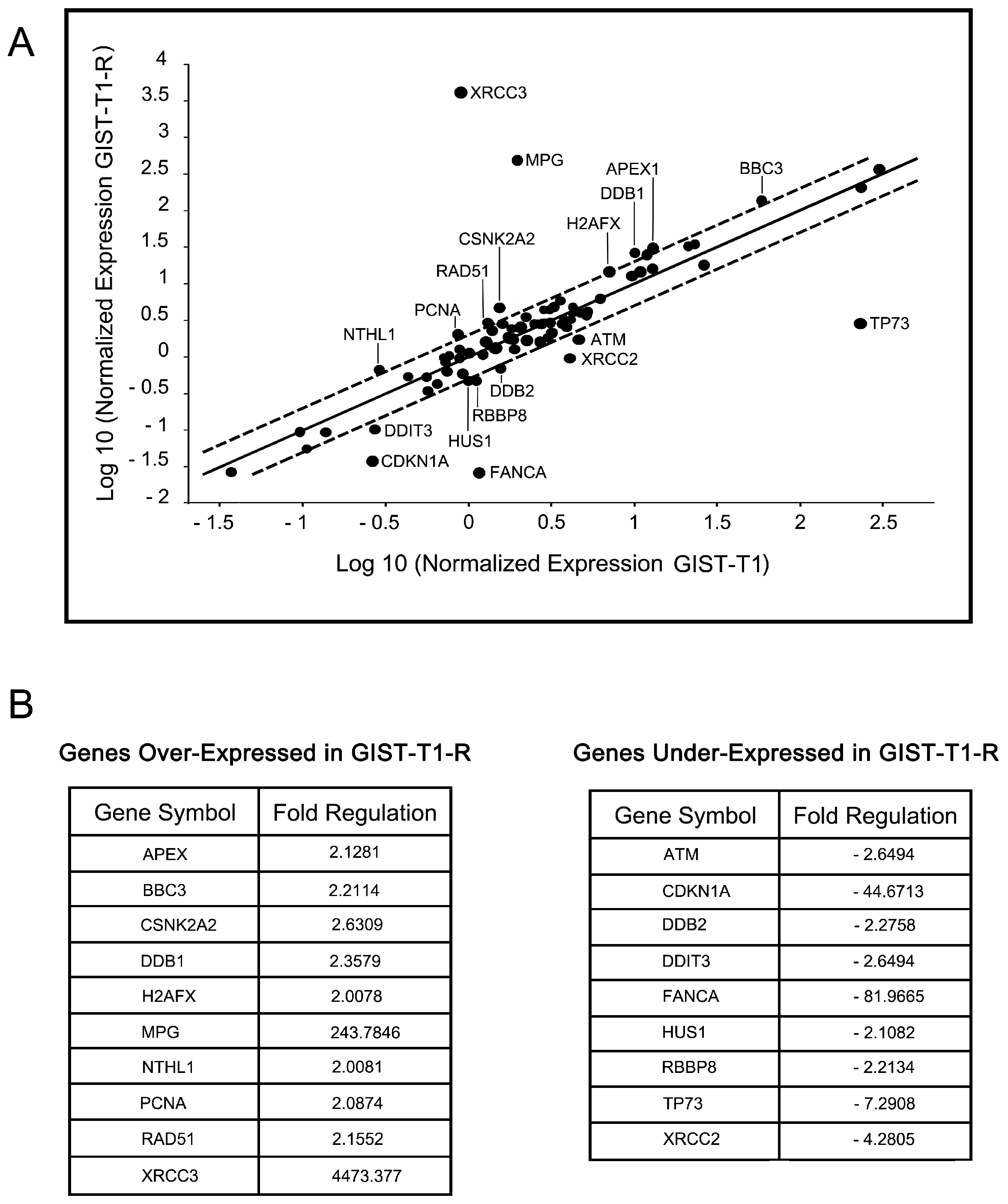
2.4. IM-Resistant GISTs Are Less Sensitive to Certain DNA-Damaging Chemotherapeutic Agents
3. Discussion
4. Materials and Methods
4.1. Chemical Compounds
4.2. Antibodies
4.3. Cell Lines and Culture Conditions
4.4. Cellular Survival MTS-Based Assay
4.5. Real-Time Monitoring of Cell Proliferation
4.6. Western Blotting Analysis
4.7. Phosphokinase Arrays
4.8. DNA Damage Signaling RT Profiler PCR Array
4.9. Genetic Analysis
4.10. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Willis, N.A.; Jacks, T.; Griffin, J.D.; Singer, S.; Fletcher, C.D.; Fletcher, J.A.; Demetri, G.D. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: Biological and clinical implications. Oncogene 2001, 20, 5054–5058. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Verweij, J.; Casali, P.G.; Zalcberg, J.; LeCesne, A.; Reichardt, P.; Blay, J.Y.; Issels, R.; van Oosterom, A.; Hogendoorn, P.C.; Van Glabbeke, M.; et al. Progression-free survival in gastrointestinal stromal tumors with high-dose imatinib: Randomized trial. Lancet 2004, 364, 1127–1132. [Google Scholar] [CrossRef]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [PubMed]
- Miselli, F.C.; Casieri, P.; Negri, T.; Orsenigo, M.; Lagonigro, M.S.; Gronchi, A.; Fiore, M.; Casali, P.G.; Bertulli, R.; Carbone, A.; et al. c-Kit/PDGFRA gene status alterations possibly related to primary imatinib resistance in gastrointestinal stromal tumors. Clin. Cancer Res. 2007, 13, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Sakurama, K.; Noma, K.; Takaoka, M.; Tomono, Y.; Watanabe, N.; Hatakeyama, S.; Ohmori, O.; Hirota, S.; Motoki, T.; Shirakawa, Y.; et al. Inhibition of focal adhesion kinase as a potential therapeutic strategy for imatinib-resistant gastrointestinal stromal tumor. Mol. Cancer Ther. 2009, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Tarn, C.; Rink, L.; Merkel, E.; Flieder, D.; Pathak, H.; Koumbi, D.; Testa, J.R.; Eisenberg, B.; von Mehren, M.; Godwin, A.K. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 8387–8392. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-mediated reactivation of MAPK signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.P.; Goodman, V.; Jiang, J.X.; Mahjoob, K.; Verbois, S.L.; Morse, D.; Dagher, R.; Justice, R.; Pazdur, R. Food and Drug Administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist 2007, 12, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. GRID study investigators. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Mahadevan, D.; Theiss, N.; Morales, C.; Stejskal, A.E.; Cooke, L.S.; Zhu, M.; Kurtzman, D.; Swart, R.; Ong, E.; Qi, W. Novel receptor tyrosine kinase targeted combination therapies for imatinib-resistant gastrointestinal stromal tumors (GIST). Oncotarget 2015, 6, 1954–1966. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.A.; Zeng, S.; Seifert, A.M.; Kim, T.S.; Sorenson, E.C.; Greer, J.B.; Beckman, M.J.; Santamaria-Barria, J.A.; Crawley, M.H.; Green, B.L.; et al. Pharmacological inhibition of KIT activates MET signaling in gastrointestinal stromal tumors. Cancer Res. 2015, 75, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Yeap, B.Y.; Solomon, B.J.; Riely, G.J.; Gainor, J.; Engelman, J.A.; Shapiro, G.I.; Costa, D.B.; Ou, S.H.; Butaney, M.; et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol. 2011, 12, 1004–1012. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; et al. METEOR Investigators. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef]
- Boichuk, S.; Lee, D.J.; Mehalek, K.R.; Makielski, K.R.; Wozniak, A.; Seneviratne, D.S.; Korzeniewski, N.; Cuevas, R.; Parry, J.A.; Brown, M.F.; et al. Unbiased compound screening identifies unexpected drug sensitivities and novel treatment options for gastrointestinal stromal tumors. Cancer Res. 2014, 74, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Ramazanov, B.; Duesing, A. Imatinib enchances the sensitivity of gastrointestinal stromal tumors to the topoisomerase II inhibitors. Adv. Mol. Oncol. 2015, 1, 76–81. [Google Scholar] [CrossRef]
- Pessetto, Z.Y.; Ma, Y.; Hirst, J.J.; von Mehren, M.; Weir, S.J.; Godwin, A.K. Drug repurposing identifies a synergistic combination therapy with imatinib mesylate for gastrointestinal stromal tumor. Mol. Cancer Ther. 2014, 13, 2276–2287. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar] [PubMed]
- Takahashi, T.; Elzawahry, A.; Mimaki, S.; Furukawa, E.; Nakatsuka, R.; Nakamura, H.; Nishigaki, T.; Serada, S.; Naka, T.; Hirota, S.; et al. Genomic and transcriptomic analysis of imatinib resistance in gastrointestinal stromal tumors. Genes Chromosomes Cancer 2017, 56, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 15, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Semrad, T.J.; Mack, P.C. Fibroblast Growth Factor Signaling in Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2012, 13, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; O’Connell, M.J.; Pilcher, W.; Reeder, J.E. Fibroblast growth factor receptor-3 expression in meningiomas with stimulation of proliferation by the phosphoinositide 3 kinase-Akt pathway. J. Neurosurg. 2010, 112, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.; Knowles, M.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lamerdin, J.E.; Tebbs, R.S.; Schild, D.; Tucker, J.D.; Shen, M.R.; Brookman, K.W.; Siciliano, M.J.; Walter, C.A.; Fan, W.; et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol. Cell 1998, 1, 783–793. [Google Scholar] [CrossRef]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Sonobe, H.; Toyonaga, S.; Yamasaki, I.; Shuin, T.; Takano, A.; Araki, K.; Akimaru, K.; Yuri, K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab. Investig. 2002, 82, 663–665. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds used in current study are available from the authors. |


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules 2017, 22, 2152. https://doi.org/10.3390/molecules22122152
Boichuk S, Galembikova A, Dunaev P, Valeeva E, Shagimardanova E, Gusev O, Khaiboullina S. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules. 2017; 22(12):2152. https://doi.org/10.3390/molecules22122152
Chicago/Turabian StyleBoichuk, Sergei, Aigul Galembikova, Pavel Dunaev, Elena Valeeva, Elena Shagimardanova, Oleg Gusev, and Svetlana Khaiboullina. 2017. "A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance" Molecules 22, no. 12: 2152. https://doi.org/10.3390/molecules22122152
APA StyleBoichuk, S., Galembikova, A., Dunaev, P., Valeeva, E., Shagimardanova, E., Gusev, O., & Khaiboullina, S. (2017). A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules, 22(12), 2152. https://doi.org/10.3390/molecules22122152