
Porphyrin Cobalt(III) “Nitrene Radical” Reactivity; Hydrogen Atom Transfer from Ortho-YH Substituents to the Nitrene Moiety of Cobalt-Bound Aryl Nitrene Intermediates (Y = O, NH)


Abstract
: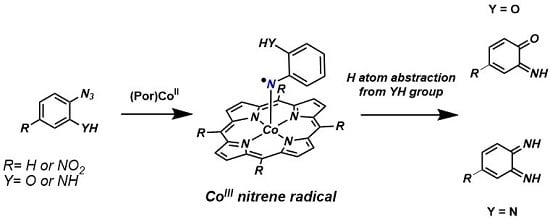
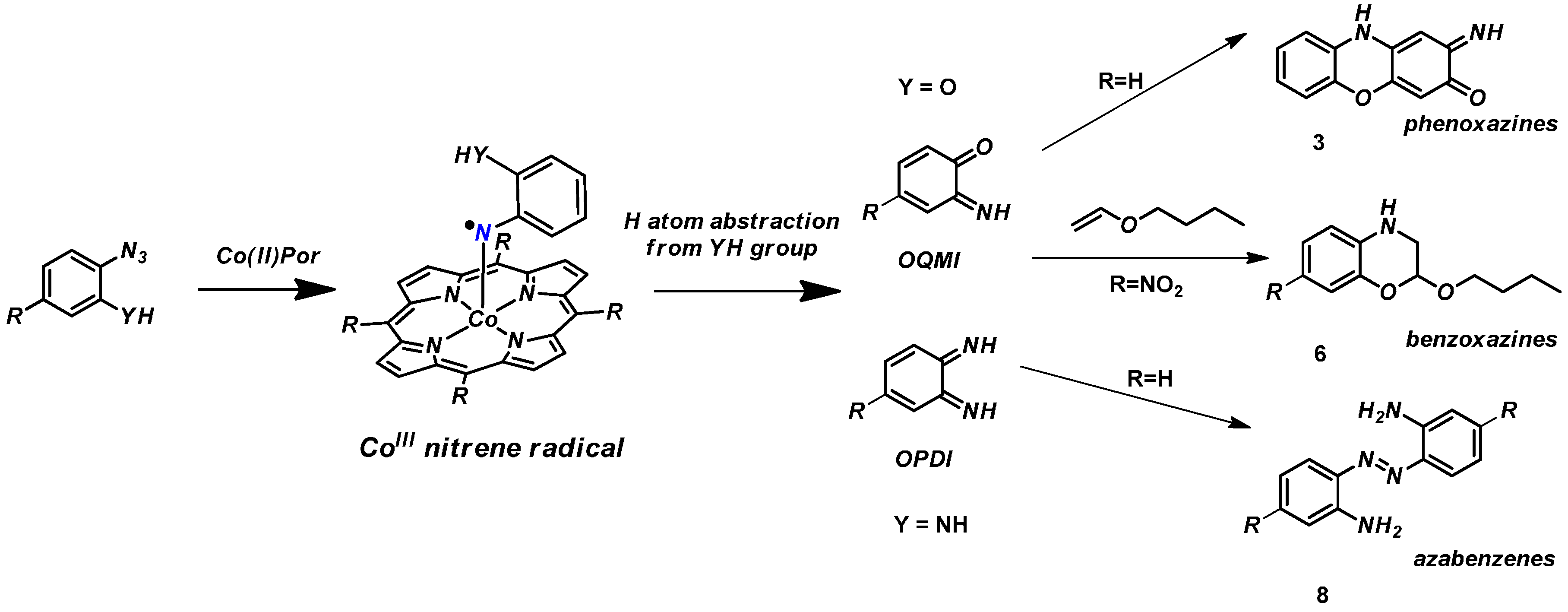
1. Introduction
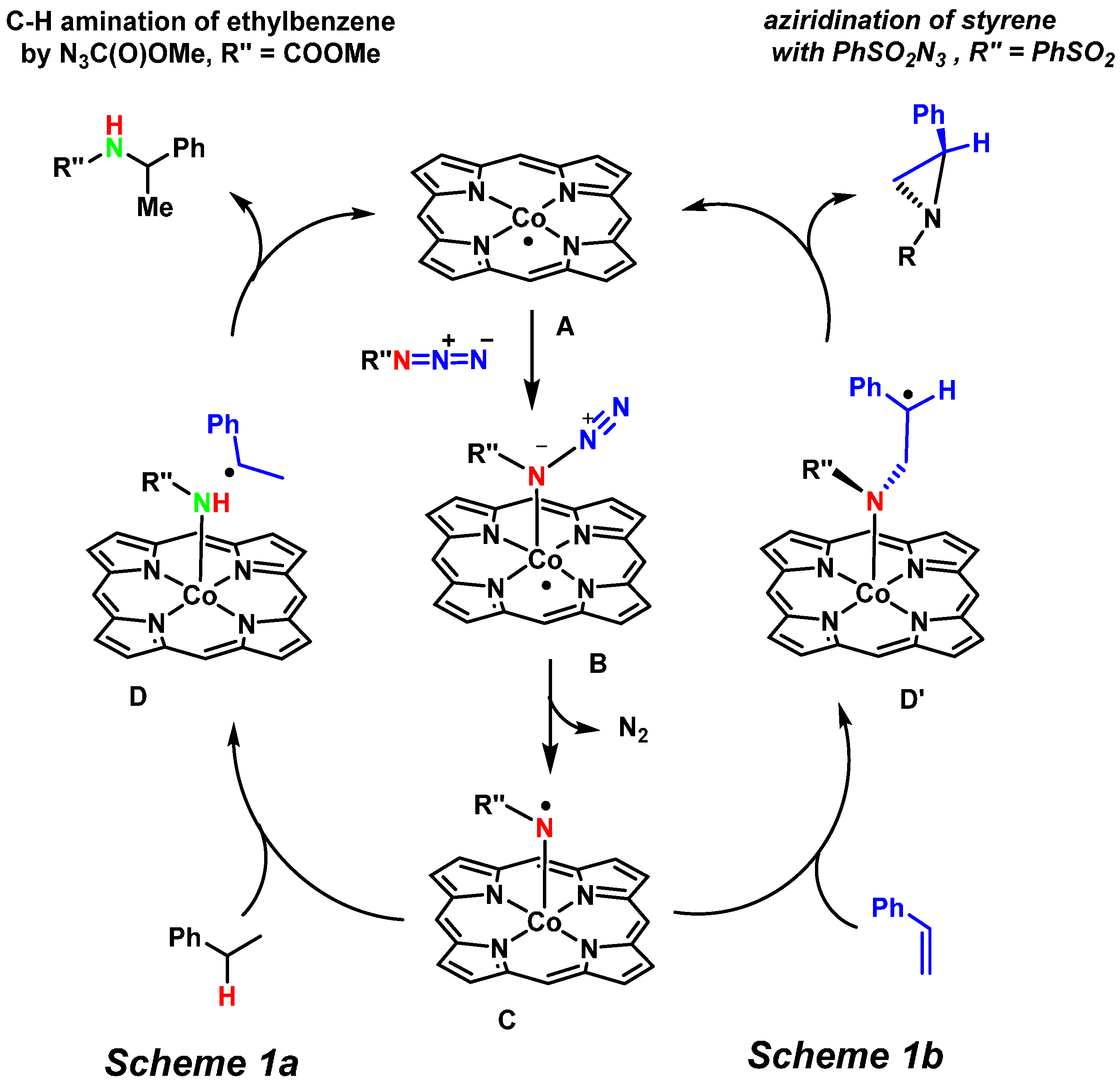
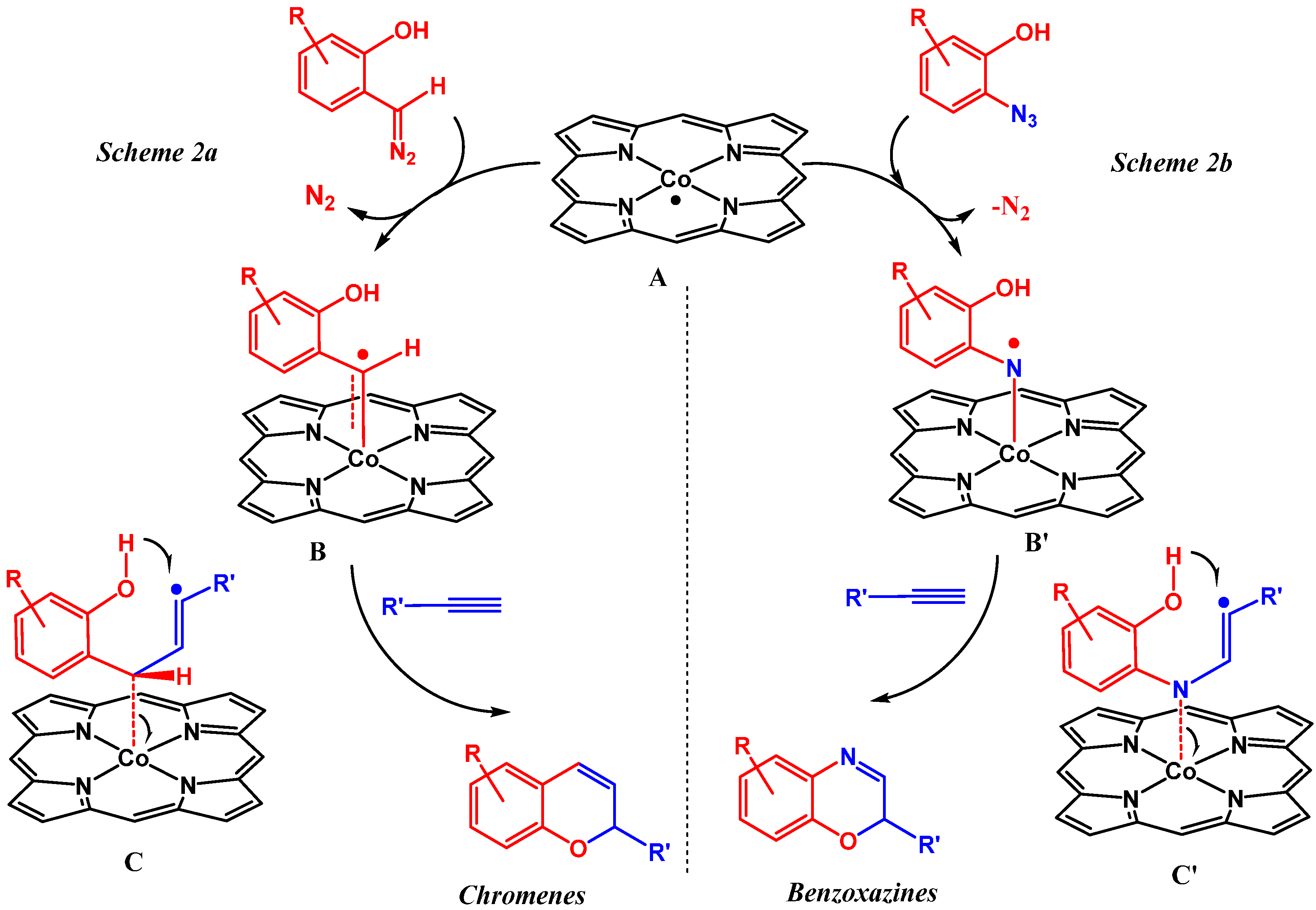
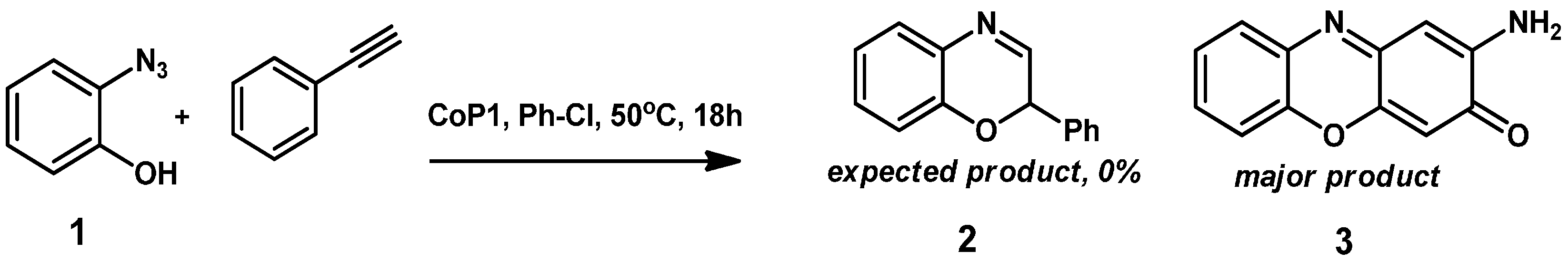
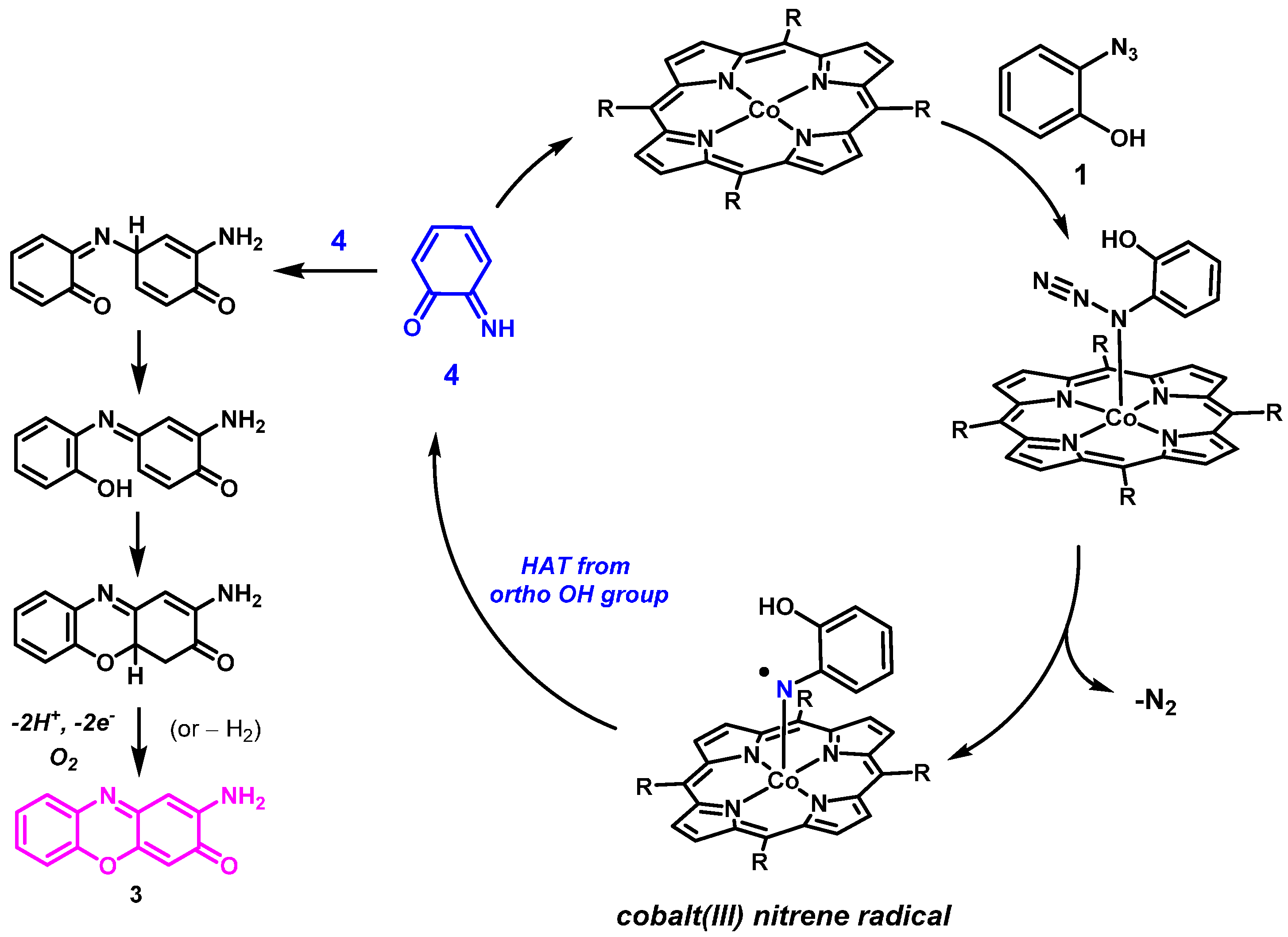
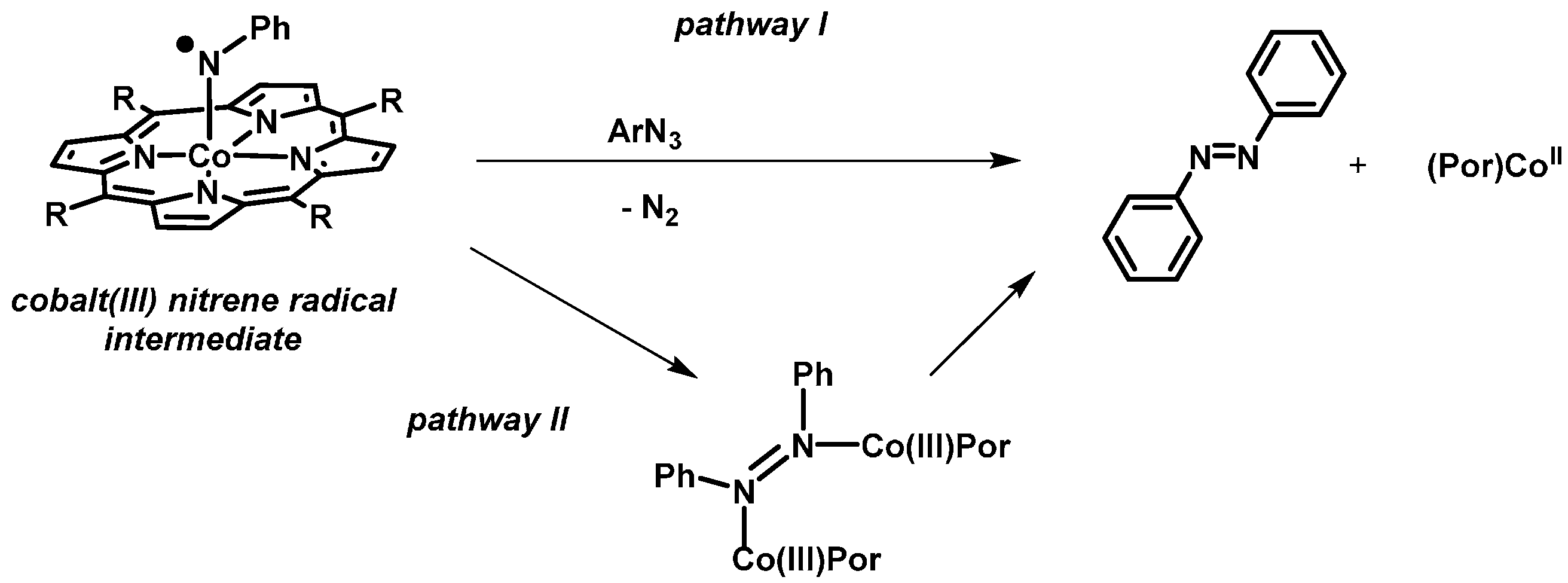
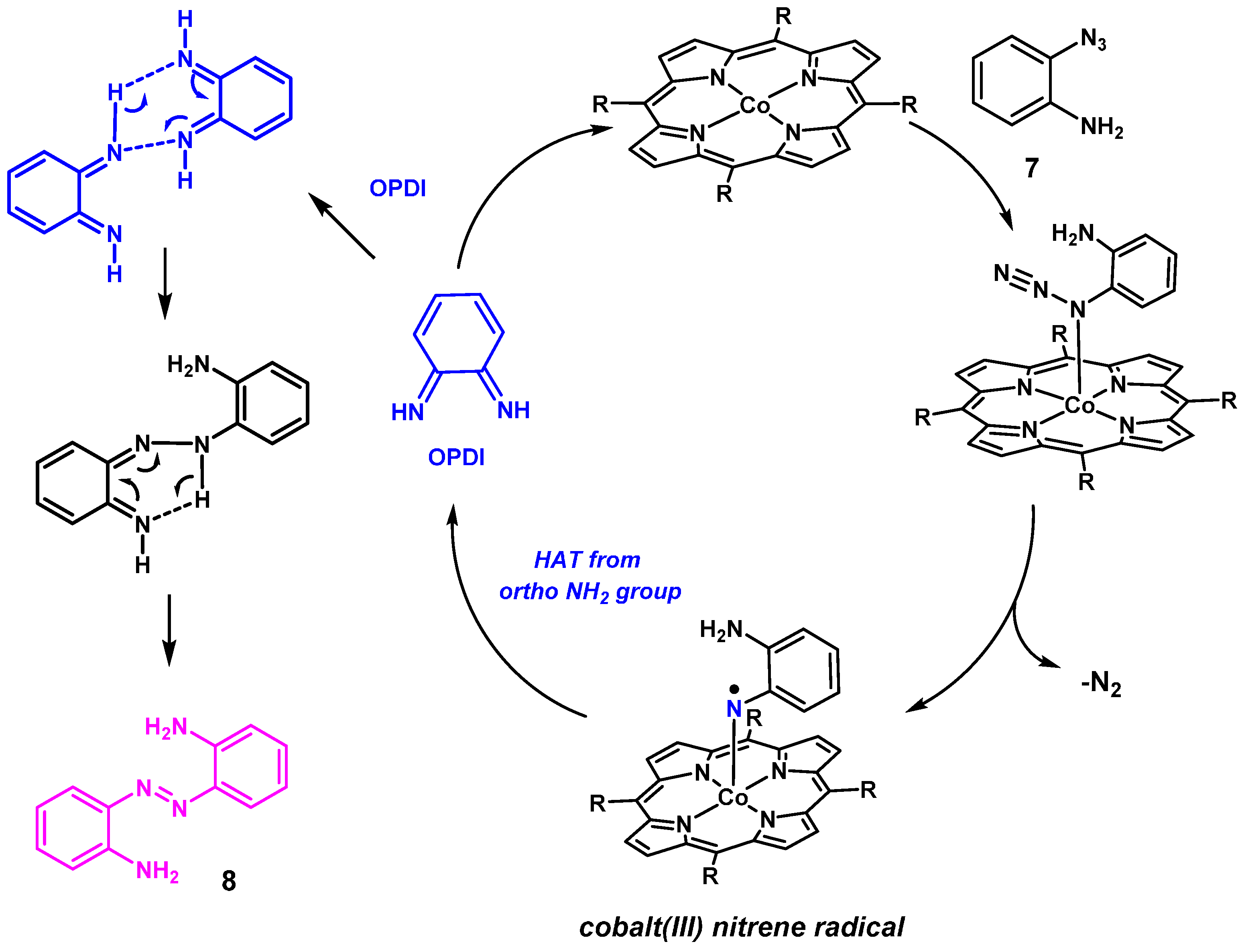
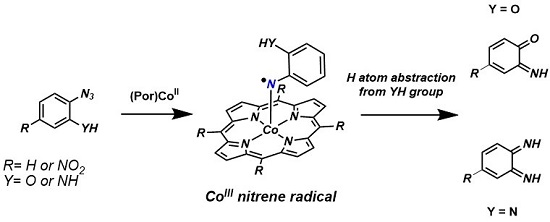
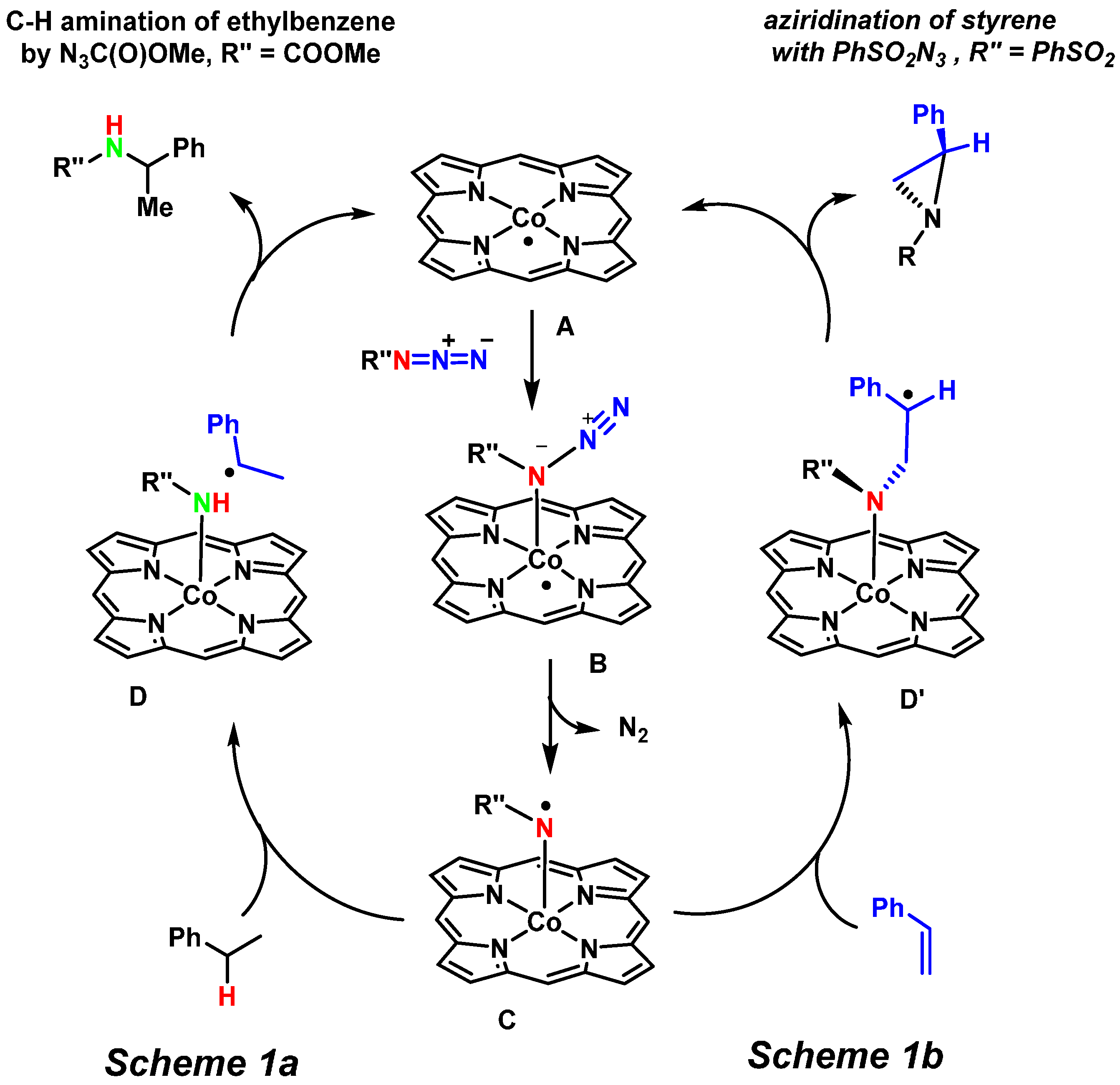
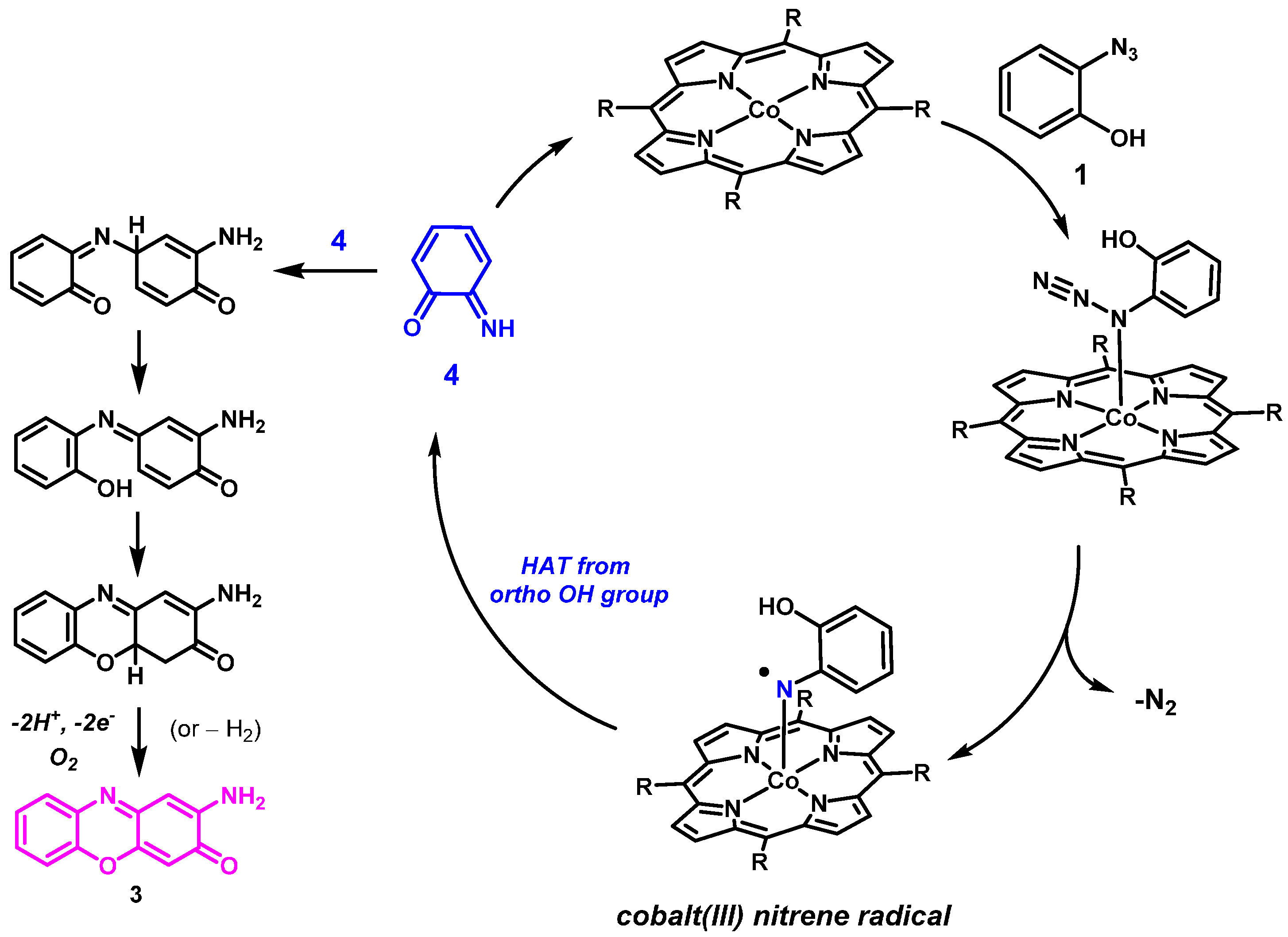
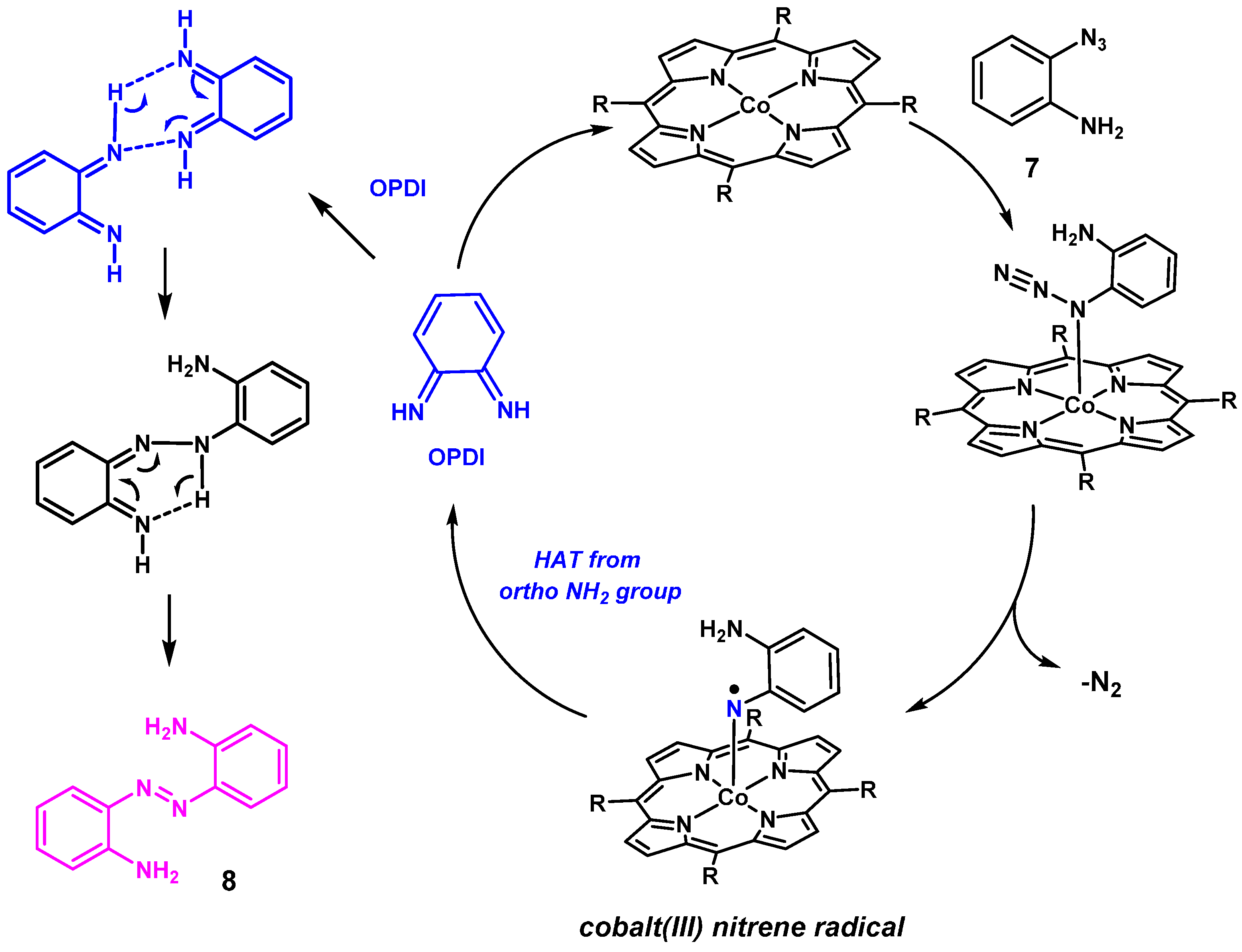
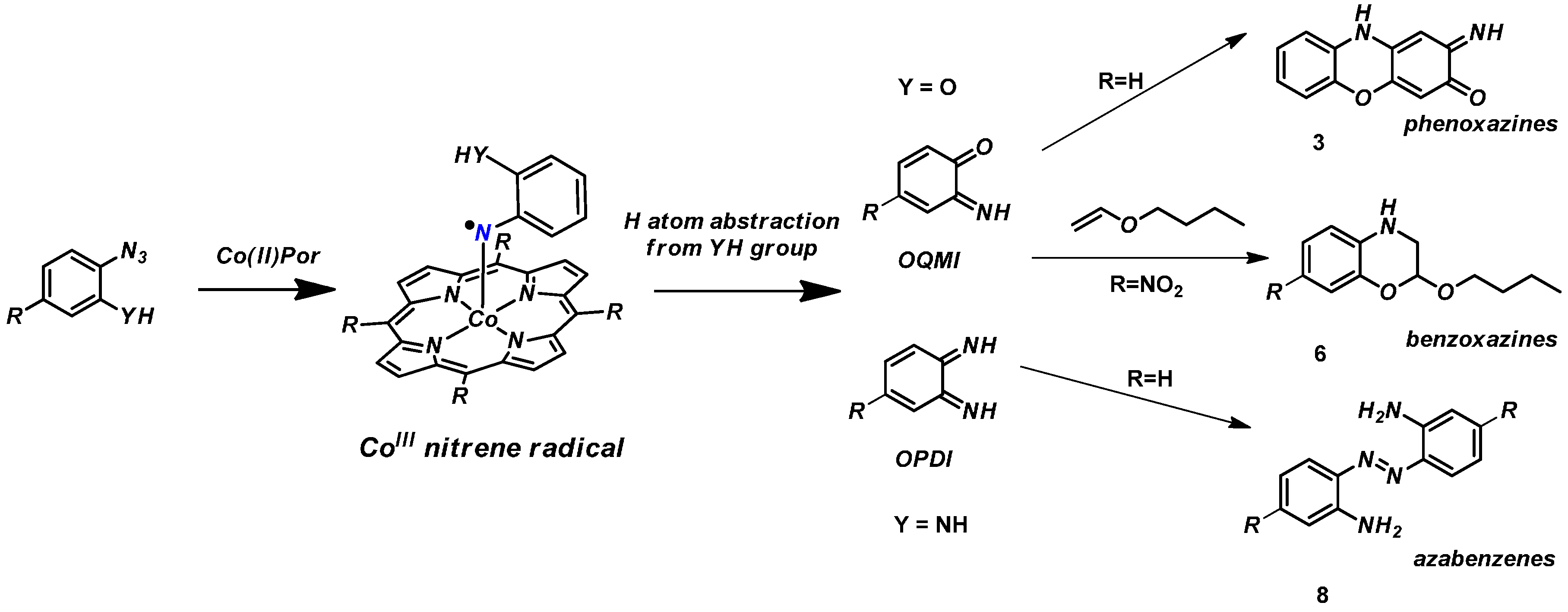
2. Results and Discussion
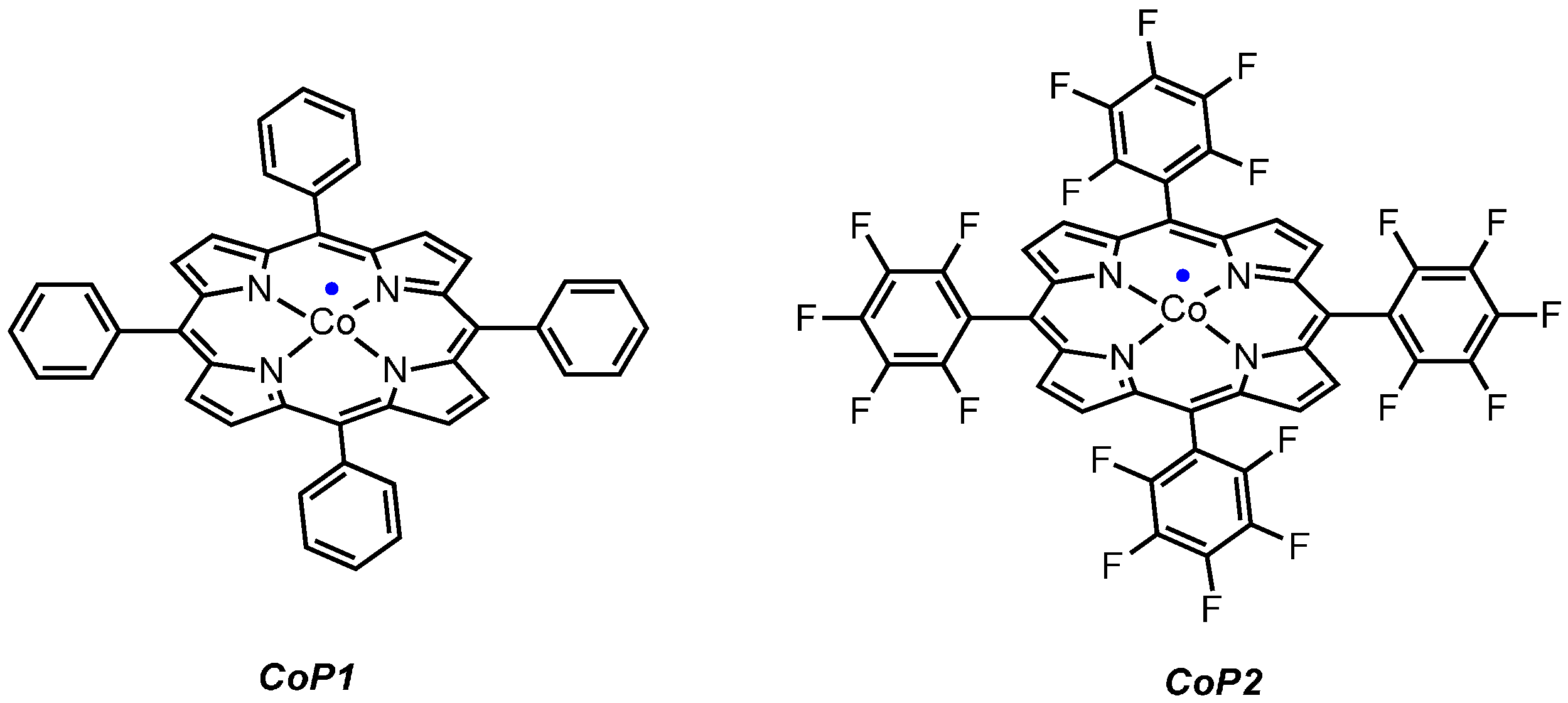
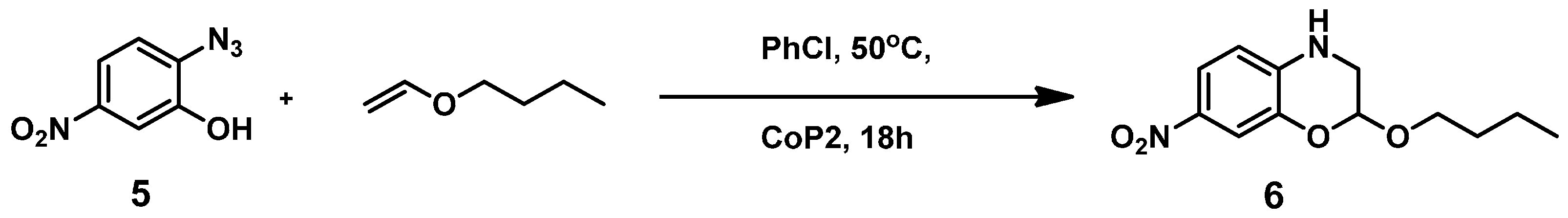
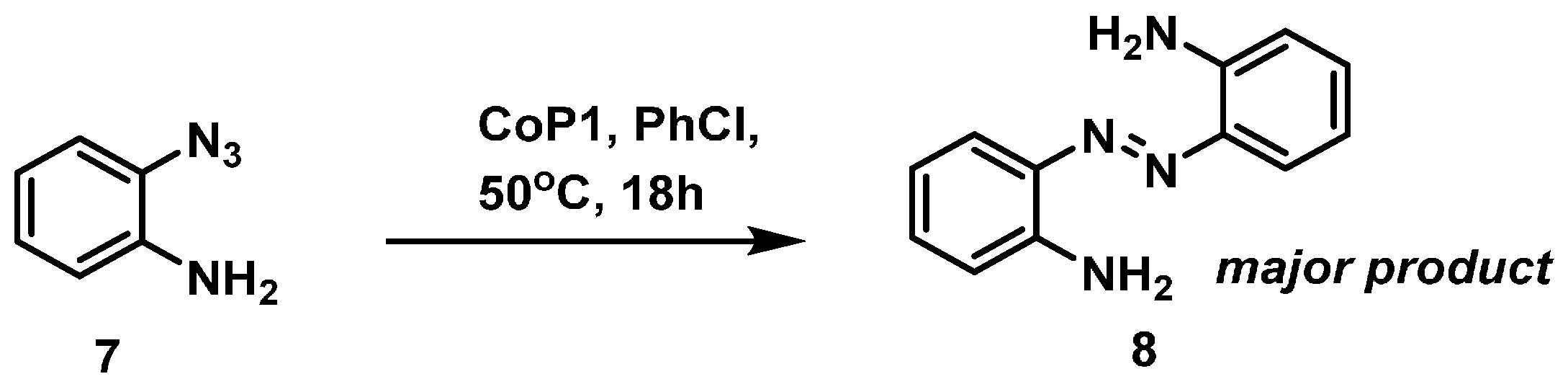
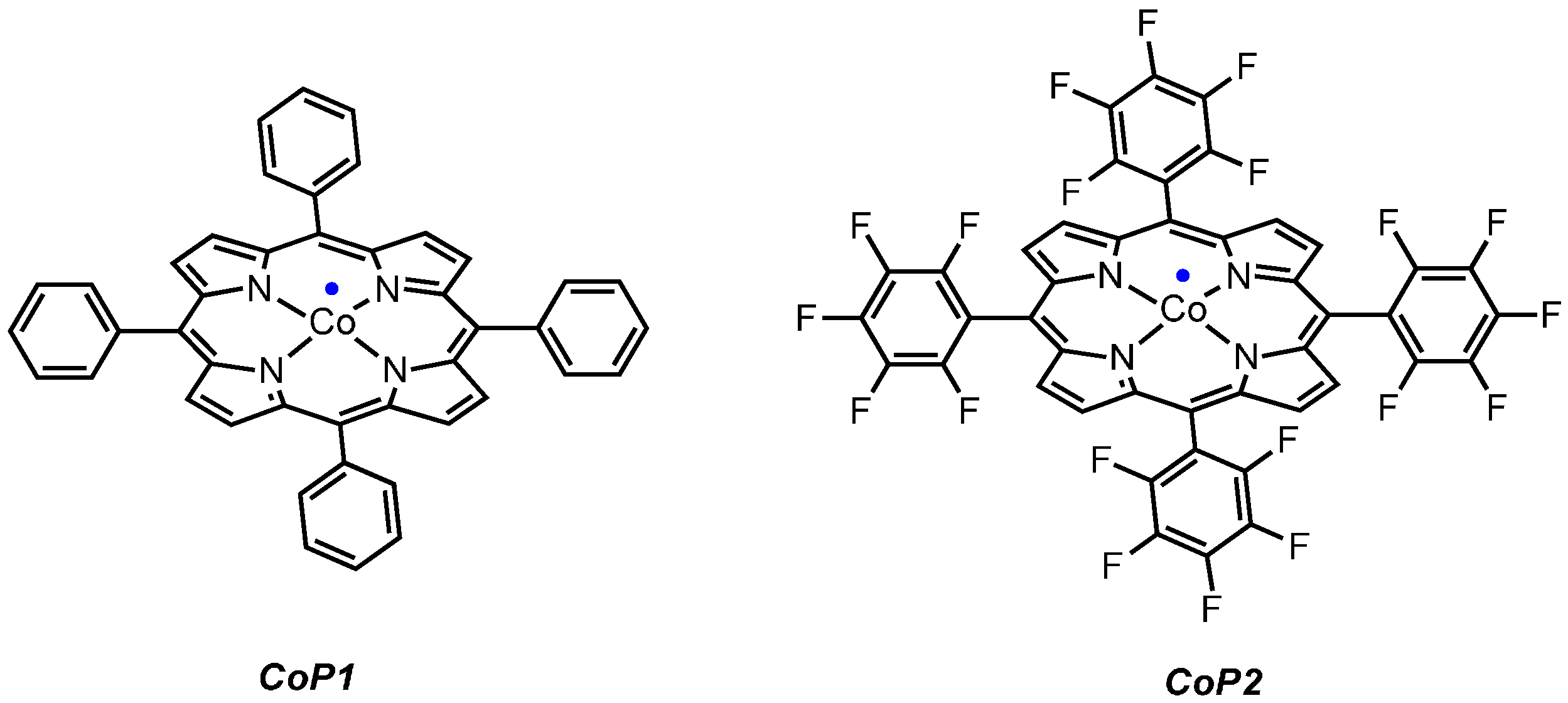
2.1. Experimental Studies
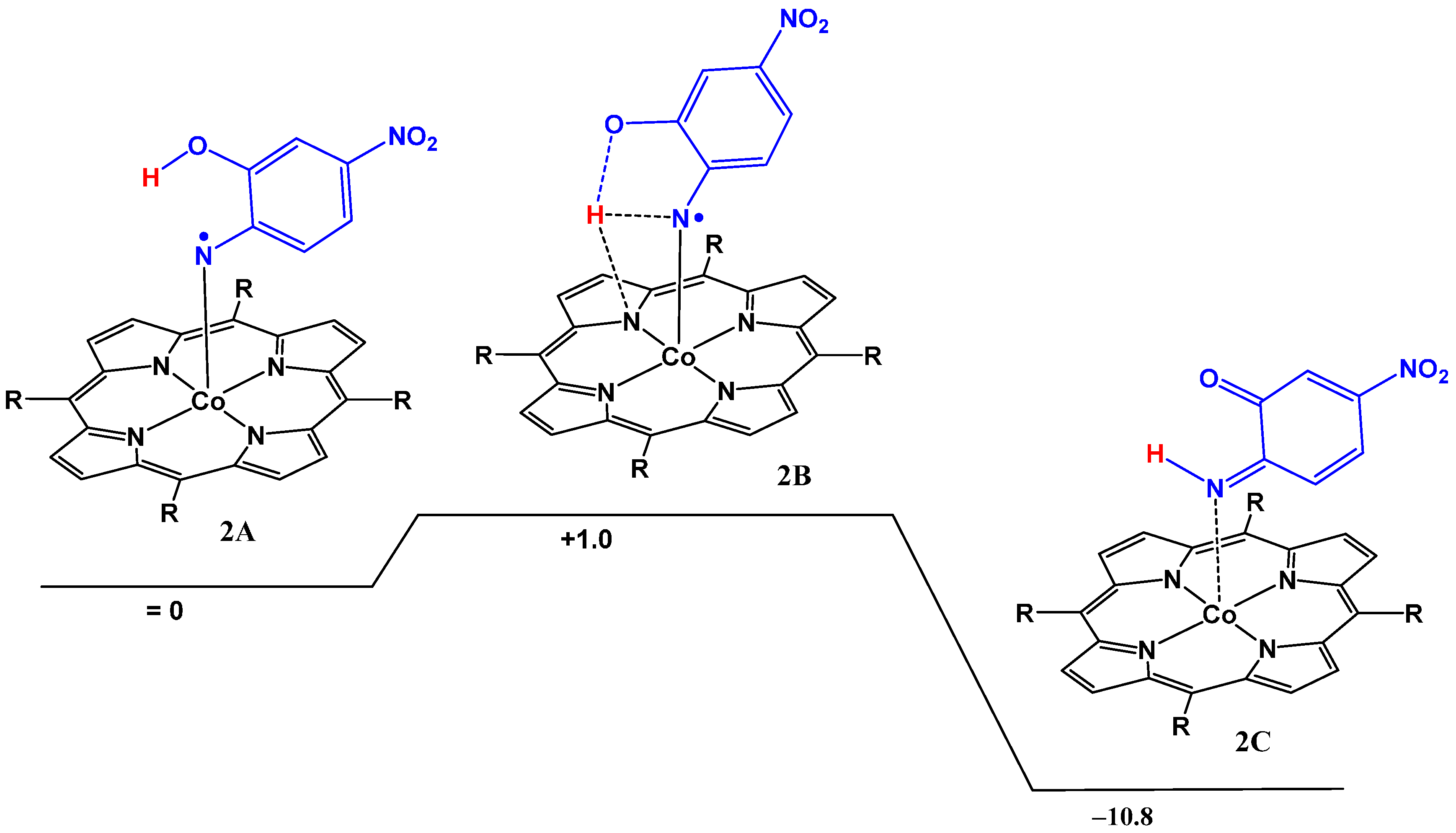
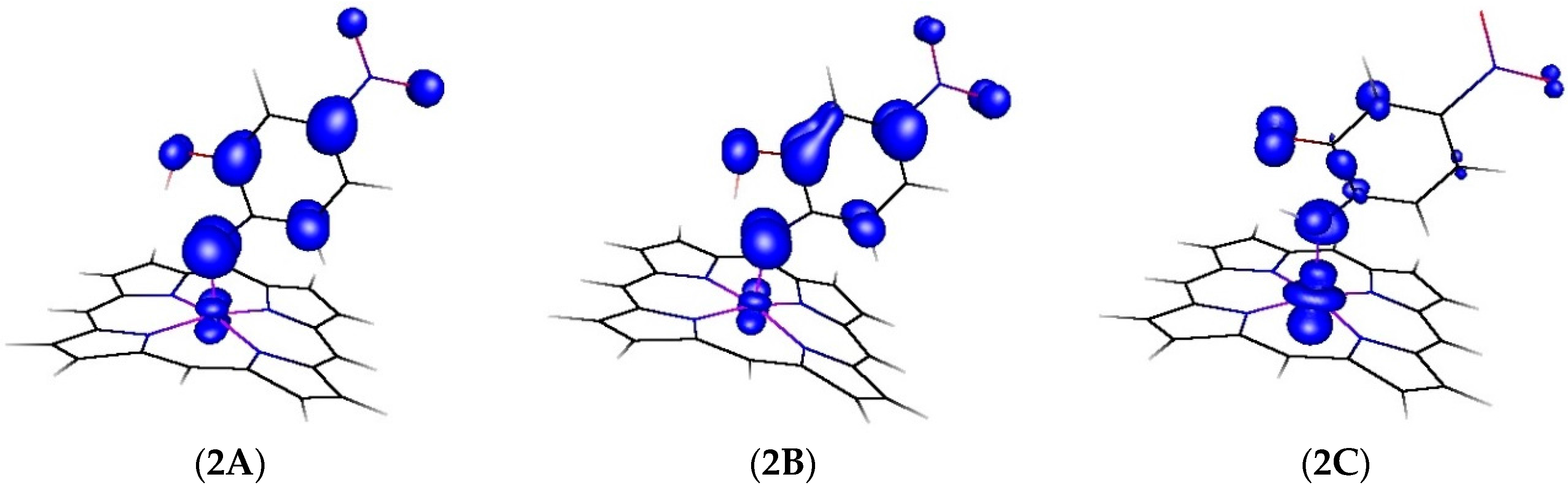
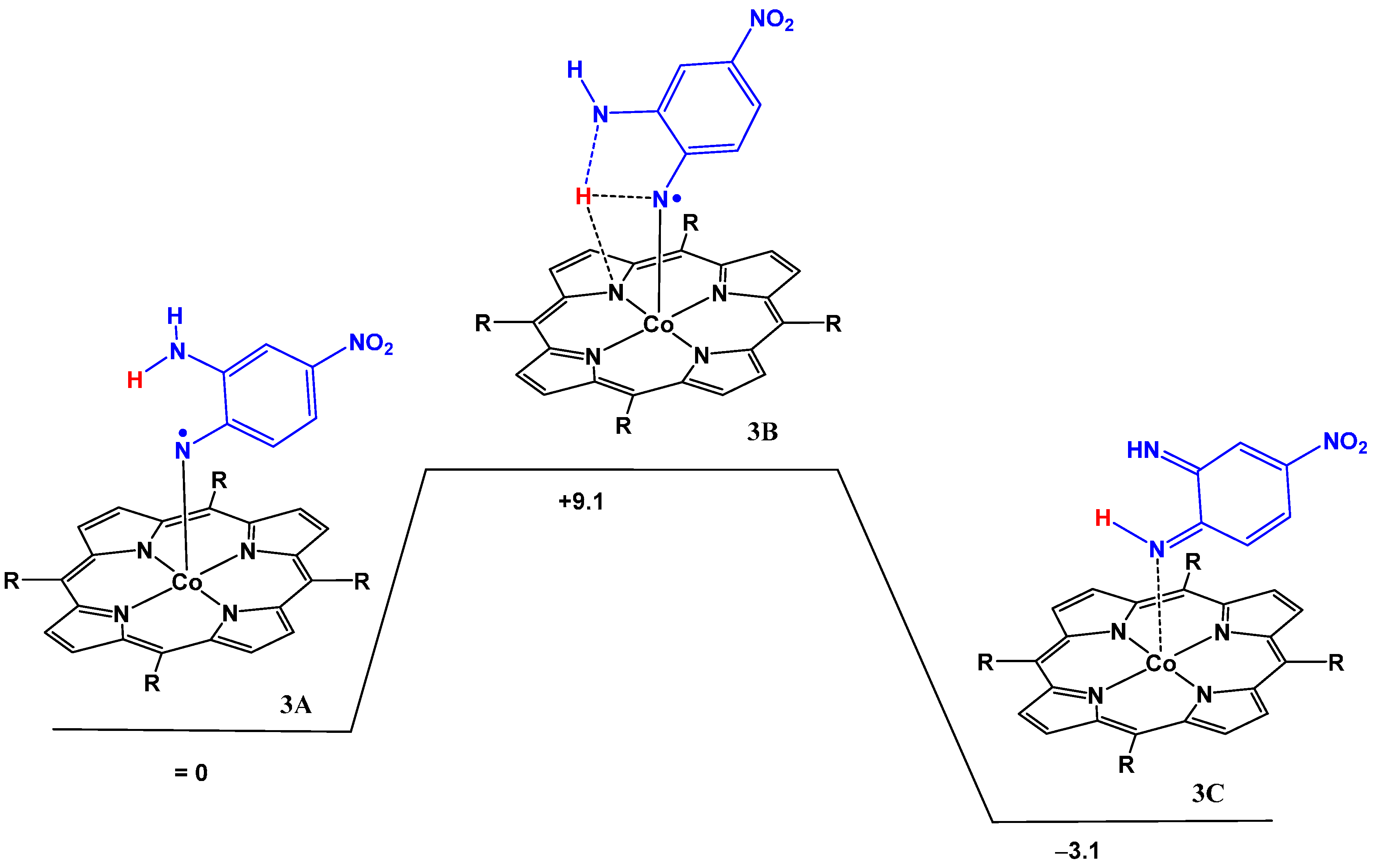
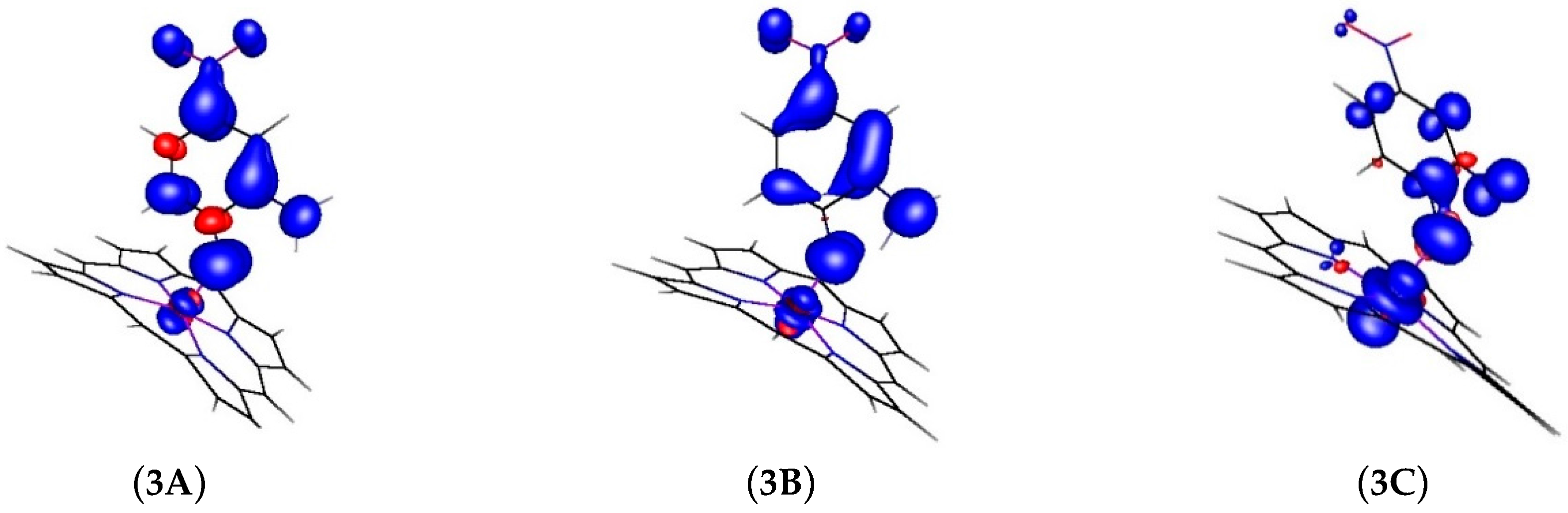
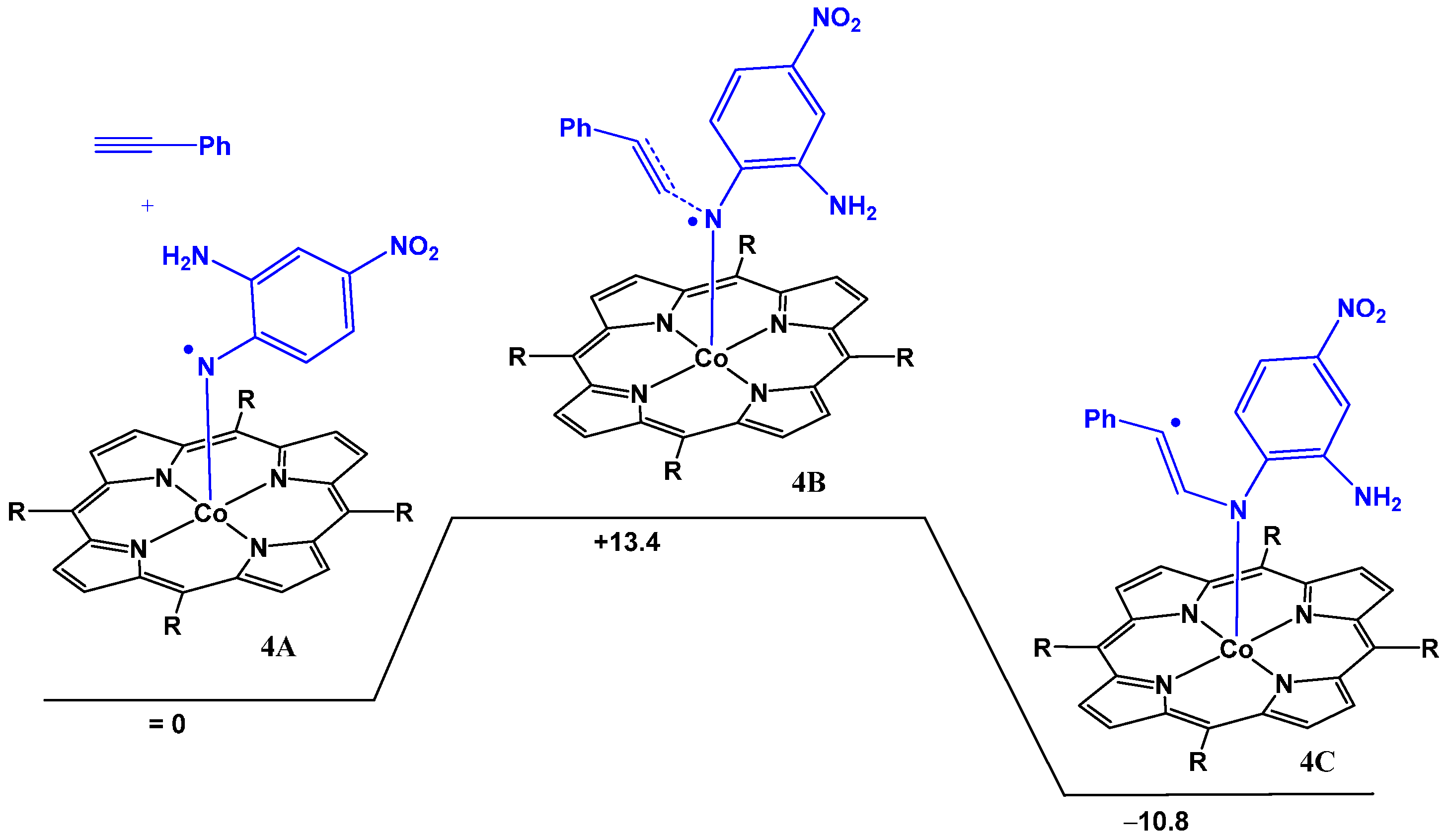
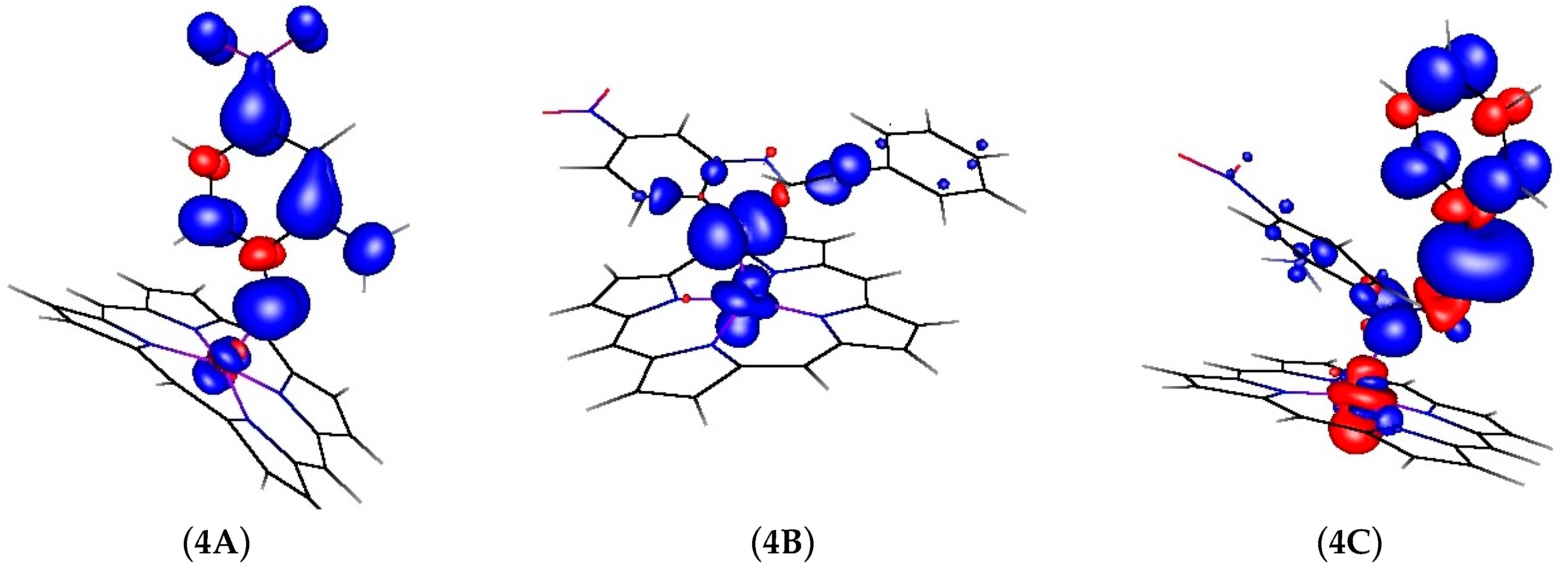
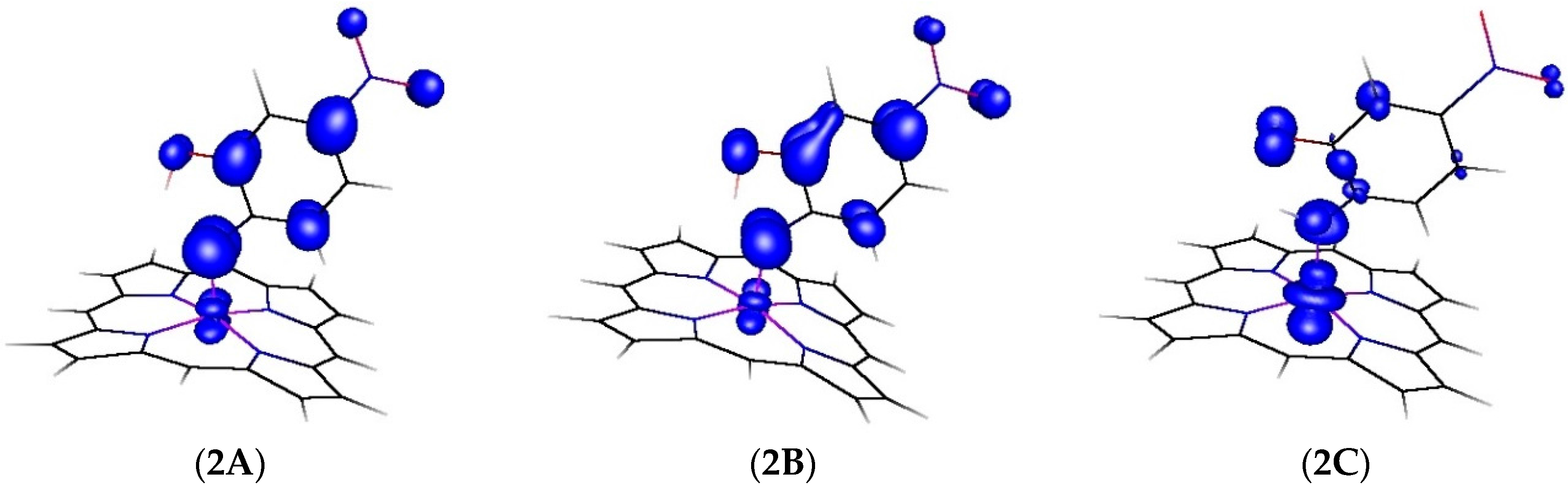
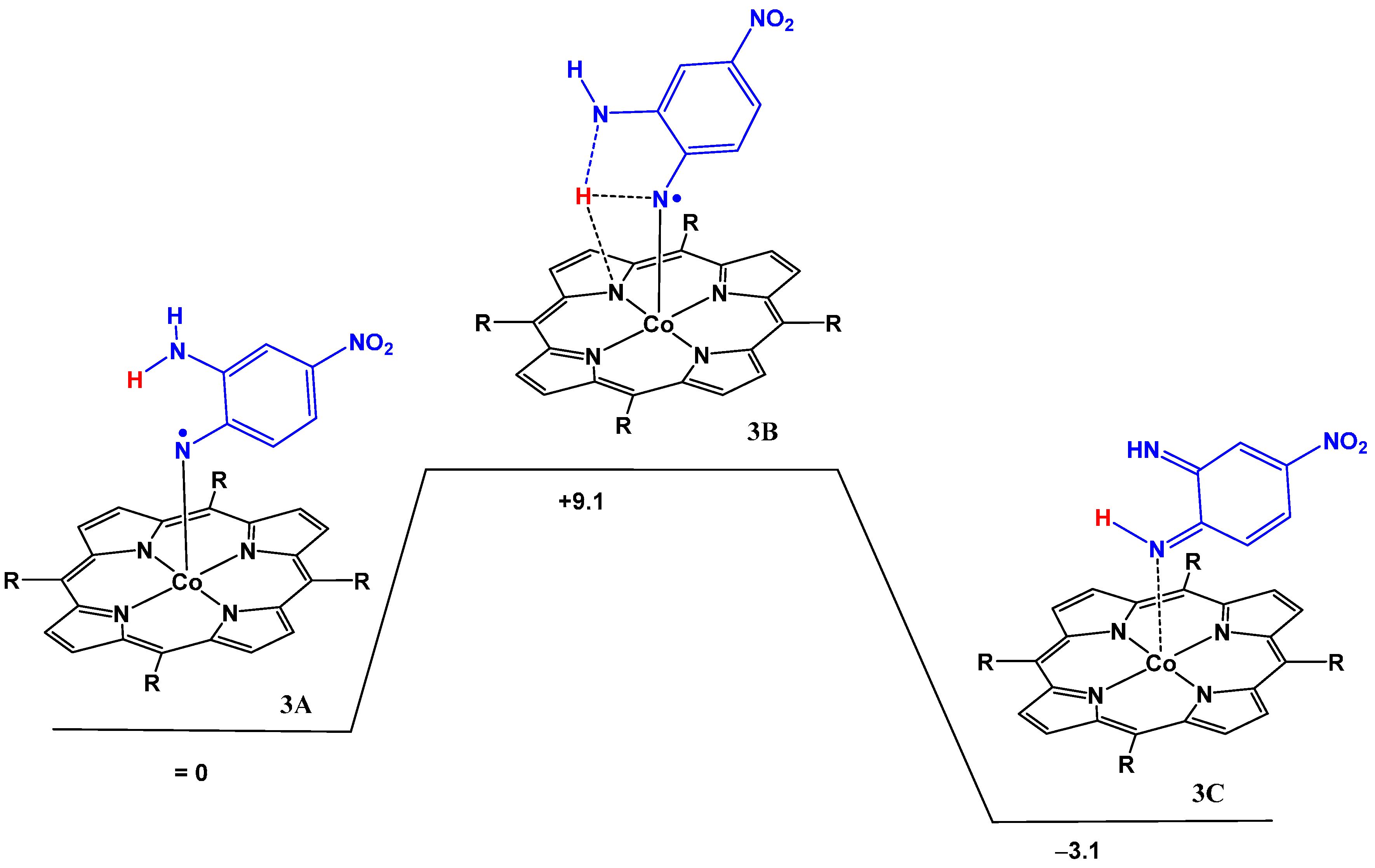
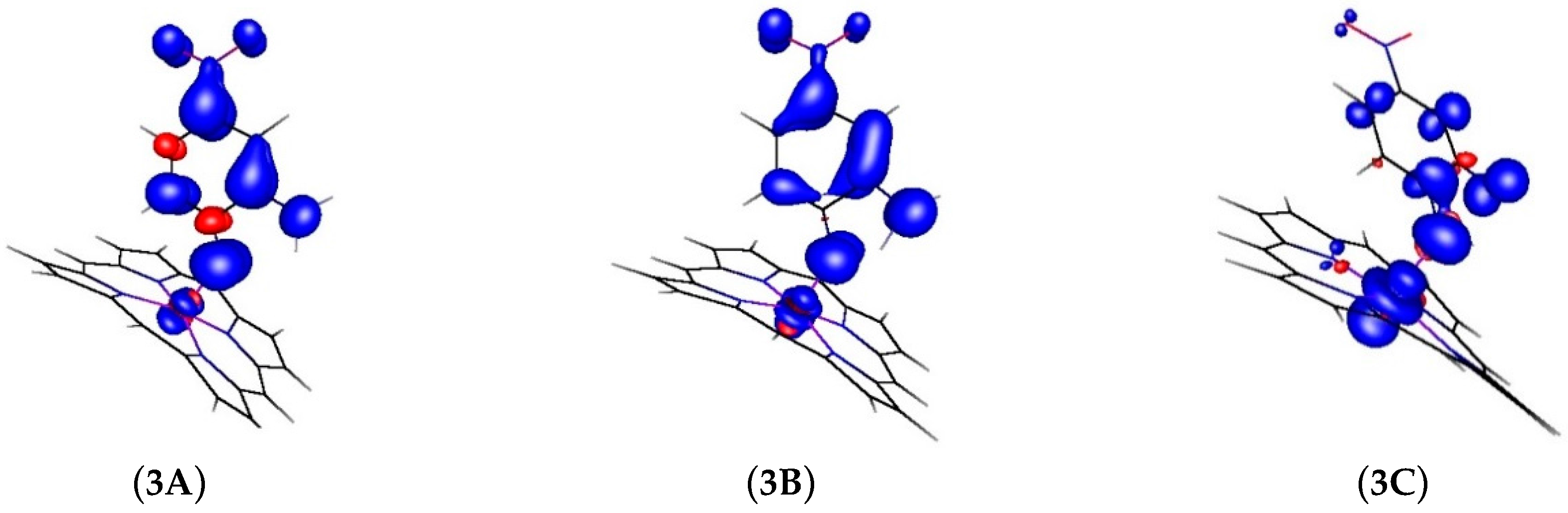
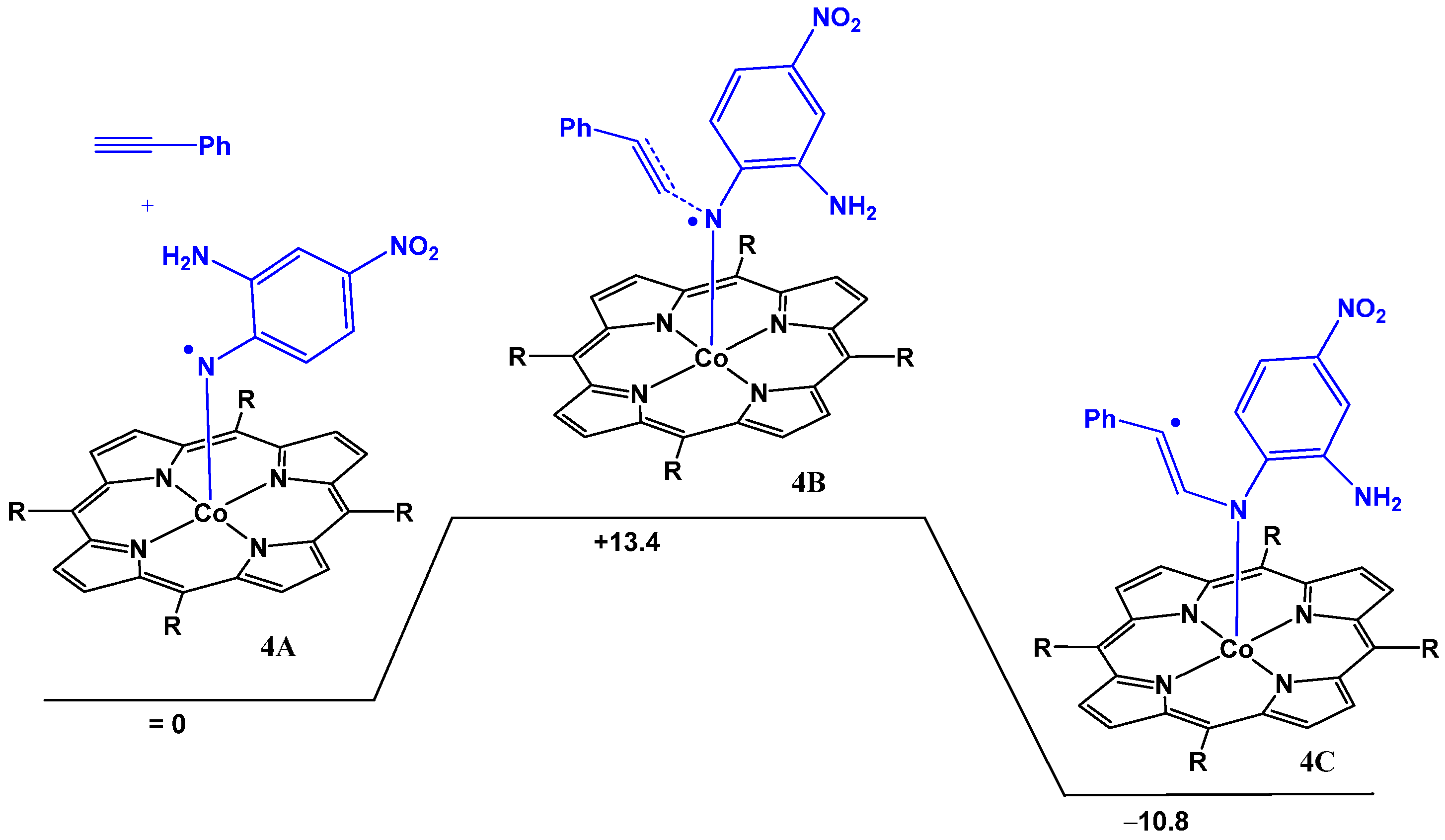
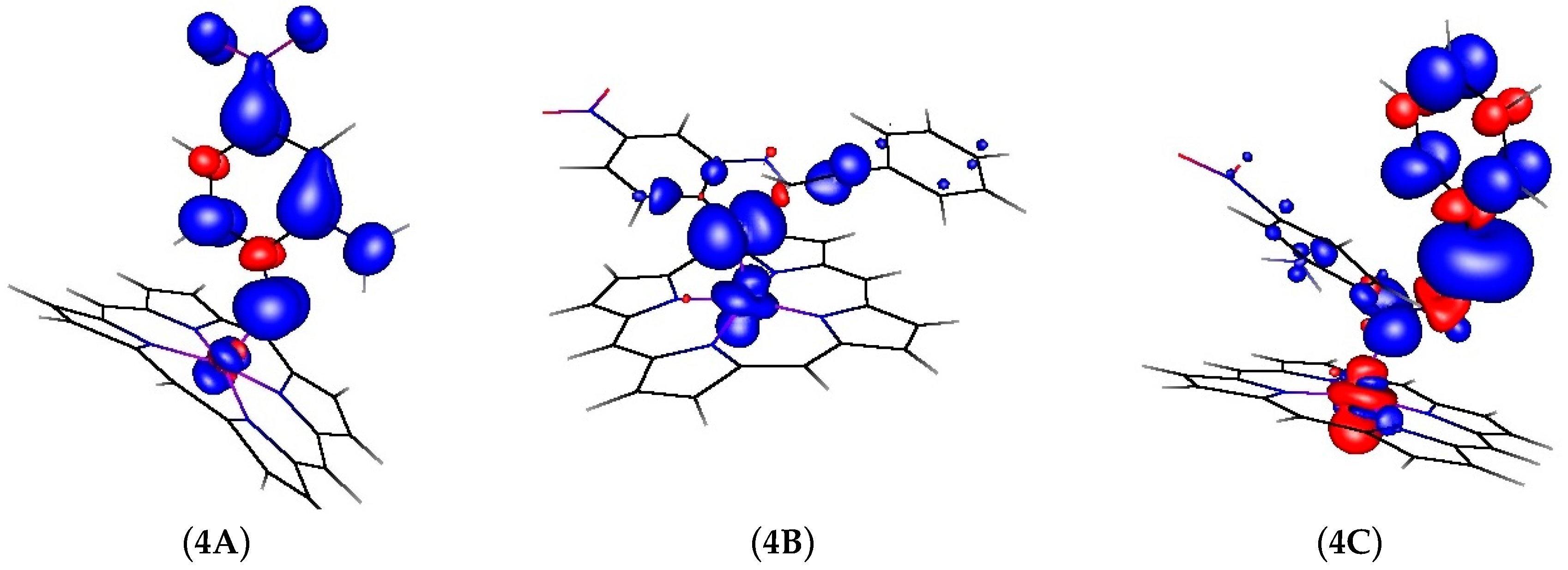
2.2. Computational Studies
3. Experimental Section
3.1. General Information
3.2. Synthetic Details
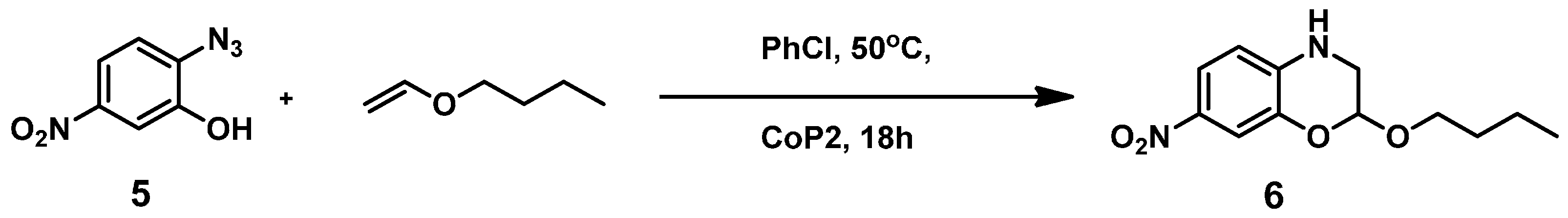
3.2.1. Synthesis of 2-Azido-5-nitrophenol (5)
3.2.2. Synthesis of the Catalyst CoP2
3.2.3. Catalytic Reaction of Compound 1 to Give Product 3
3.2.4. Catalytic Reaction of Compound 5 to Give Product 6
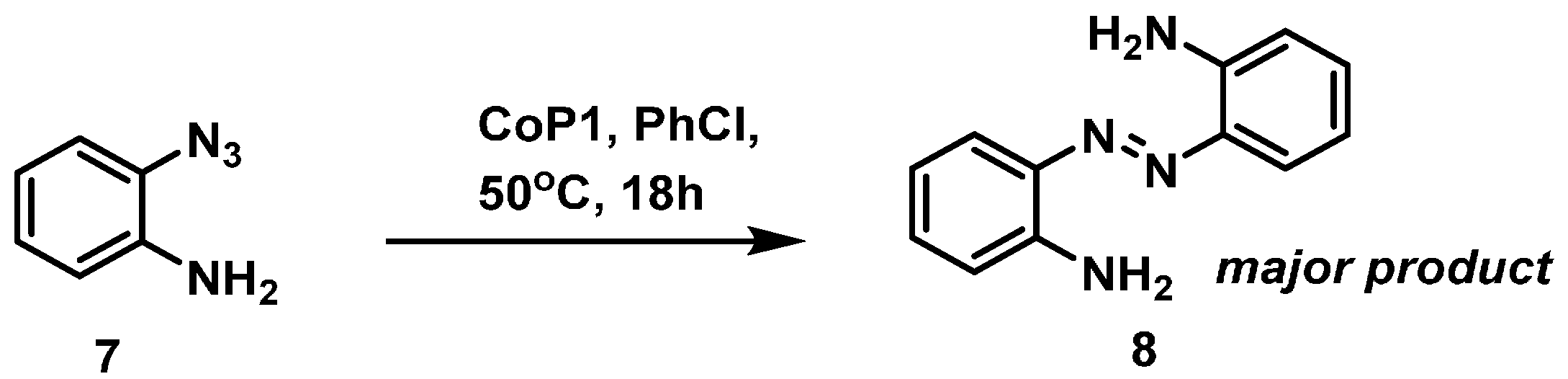
3.2.5. Catalytic Reaction of 7 to Give Compound 8
3.2.6. Catalytic Reaction of Aminophenyl Azide 7 to Give 8
3.3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davies, H.M.L.; Manning, J.R. Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Forbes, D.C. Recent advances in asymmetric catalytic metal carbene transformations. Chem. Rev. 1998, 98, 911–935. [Google Scholar] [CrossRef] [PubMed]
- Driver, T.G. Recent advances in transition metal-catalyzed N-atom transfer reactions of azides. Org. Biomol. Chem. 2010, 8, 3831–3846. [Google Scholar] [CrossRef] [PubMed]
- Godula, K.; Sames, D. C–H bond functionalization in complex organic synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Intrieri, D.; Zardi, P.; Caselli, A.; Gallo, E. Organic azides: Energetic reagents for the Inter molecular amination of C–H bonds. Chem. Commun. 2014, 50, 11440–11453. [Google Scholar] [CrossRef] [PubMed]
- Manca, G.; Gallo, E.; Intrieri, D.; Mealli, C. DFT mechanistic proposal of the ruthenium porphyrin-catalysed allylic amination by organic azides. ACS Catal. 2014, 4, 823–832. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, X.P. Catalytic C–H functionalization by metalloporphyrins: Recent developments and future directions. Chem. Soc. Rev. 2011, 40, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ruppel, J.V.; Zhang, X.P. Cobalt-catalyzed asymmetric cyclopropanation of electron-deficient olefin. J. Am. Chem. Soc. 2007, 129, 12074–12075. [Google Scholar] [CrossRef] [PubMed]
- Dzik, W.I.; Xu, X.; Zhang, X.P.; Reek, J.N.H.; de Bruin, B. Carbene radicals in CoII(por) -catalyzed olefin cyclopropanation. J. Am. Chem. Soc. 2010, 132, 10891–10902. [Google Scholar] [CrossRef] [PubMed]
- Dzik, W.I.; Zhang, X.P.; de Bruin, B. Redox noninnocence of carbene ligands: Carbene radicals in (Catalytic) C–C bond formation. Inorg. Chem. 2011, 50, 9896–9903. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Jones, J.E.; Vyas, R.; Harden, J.D.; Zhang, X.P. Cobalt-catalyzed aziridination with diphenylphosphoryl azide (DPPA): Direct synthesis of N-phosphorus-substituted aziridines from alkenes. J. Org. Chem. 2006, 71, 6655–6658. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Lyaskovskyy, V.; Domingos, S.R.; Buma, W.J.; Woutersen, S.; Troeppner, O.; Ivanović-Burmazović, I.; Lu, H.; Cui, X.; Zhang, X.P.; et al. Characterization of porphyrin-Co(III)-‘nitrene radical’ species relevant in catalytic nitrene transfer reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, Y.; Gao, G.; Zhang, X.P. Diastereoselective and enantioselective cyclopropanation of alkenes catalyzed by cobalt porphyrins. J. Org. Chem. 2003, 68, 8179–8184. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Lu, H.; Cui, Y.; Lizardi, C.L.; Arzua, T.N.; Wojtas, L.; Cui, X.; Zhang, X.P. Selective radical amination of aldehydic C(sp2)–H bonds with fluoroaryl azides via Co(II)-based metalloradical catalysis: Synthesis of N-fluoroaryl amides from aldehydes under neutral and nonoxidative conditions. Chem. Sci. 2014, 5, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.M.; Xu, X.; Lu, H.; Cui, X.; Wojtas, L.; Zhang, X.P. Effective synthesis of chiral N-fluoroaryl aziridines through enantioselective aziridination of alkenes with fluoroaryl azides. Angew. Chem. Int. Ed. 2013, 52, 5309–5313. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Hu, Y.; Jiang, H.; Wojtas, L.; Zhang, X.P. Stereoselective radical amination of electron-deficient C (sp3) H Bonds by Co(II)-based metalloradical catalysis: Direct synthesis of amino acid derivatives via C–H amination. Org. Lett. 2012, 14, 5158–5161. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Subbarayan, V.; Tao, J.; Zhang, X.P. Cobalt(II)-catalyzed Intermolecular benzylic C−H amination with 2,2,2-trichloroethoxycarbonyl azide (TrocN3). Organometallics 2010, 29, 389–393. [Google Scholar] [CrossRef]
- Lyaskovskyy, V.; Suarez, A.I.O.; Lu, H.; Jiang, H.; Zhang, X.P.; de Bruin, B. Mechanism of cobalt(II) porphyrin-catalyzed C–H amination with organic azides: Radical nature and H-atom abstraction ability of the key cobalt(III) Nitrene Intermediates. J. Am. Chem. Soc. 2011, 12264–12273. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.D.; Chirila, A.; Lu, H.; Zhang, X.P.; de Bruin, B. Carbene radicals in cobalt(II)-porphyrin-catalysed carbene carbonylation reactions; A catalytic approach to ketenes. Chem. Eur. J. 2013, 19, 12953–12958. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.D.; Mandal, S.; Otte, M.; Cui, X.; Zhang, X.P.; de Bruin, B. Metalloradical approach to 2H-Chromenes. J. Am. Chem. Soc. 2014, 136, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.I.O.; Jiang, H.; Zhang, X.P.; de Bruin, B. The radical mechanism of cobalt(II) porphyrin-catalyzed olefin aziridination and the importance of cooperative H-bonding. Dalton Trans. 2011, 40, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, V.; Ruppel, J.V.; Zhu, S.; Perman, J.A.; Zhang, X.P. Highly asymmetric cobalt-catalyzed aziridination of alkenes with trichloroethoxysulfonyl azide (TcesN3). Chem. Commun. 2009. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Perman, J.A.; Zhang, X.P. Acceptor/acceptor-substituted diazo reagents for carbene transfers: Cobalt-catalyzed asymmetric Z-cyclopropanation of alkenes with alpha-nitrodiazoacetates. Angew. Chem. Int. Ed. 2008, 47, 8460–8463. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ruppel, J.V.; Lu, H.; Wojtas, L.; Zhang, X.P. Cobalt-catalyzed asymmetric cyclopropanation with diazosulfones: Rigidification and polarization of ligand chiral environment via hydrogen bonding and cyclization. J. Am. Chem. Soc. 2008, 130, 5042–5043. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Xu, X.; Perman, J.A.; Zhang, X.P. A general and efficient cobalt(II)-based catalytic system for highly stereoselective cyclopropanation of alkenes with α-cyanodiazoacetates. J. Am. Chem. Soc. 2010, 132, 12796–12799. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E.; Ruppel, J.V.; Gao, G.; Moore, T.M.; Zhang, X.P. Cobalt-catalyzed asymmetric olefin aziridination with diphenylphosphoryl azide. J. Org. Chem. 2008, 73, 7260–7265. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Tao, J.; Jones, J.E.; Wojtas, L.; Zhang, X.P. Cobalt(II)-catalyzed intramolecular C–H amination with phosphoryl azides: Formation of 6- and 7-membered cyclophosphoramidates. Org. Lett. 2010, 12, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, J.V.; Jones, J.E.; Huff, C.A.; Kamble, R.M.; Chen, Y.; Zhang, X.P. A highly effective cobalt catalyst for olefin aziridination with azides: Hydrogen bonding guided catalyst design. Org. Lett. 2008, 10, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Capretto, D.; Rahaman, R.; He, C. Silver-catalyzed intermolecular amination of C–H groups. Angew. Chem. Int. Ed. 2007, 46, 5184–5186. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.W.; Chan, P.W.H. Highly efficient ruthenium(II) porphyrin catalyzed amidation of aldehydes. Angew. Chem. Int. Ed. 2008, 47, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Baucom, K.D.; Murry, J.A. Rh (II)-catalyzed intermolecular oxidative sulfamidation of aldehydes: A mild efficient synthesis of N-sulfonylcarboxamides. J. Am. Chem. Soc. 2007, 129, 14106–14107. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Collet, F.; Robert-Peillard, F.; Mu, P.; Dodd, R.H.; Dauban, P. Toward a synthetically useful stereoselective C–H amination of hydrocarbons. J. Am. Chem. Soc. 2008, 130, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.; Breslow, R. The hydroxylation and amidation of equilenin acetate catalyzed by chloro[5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato]manganese(III)]. Chem. Commun. 2000, 7, 531–532. [Google Scholar] [CrossRef]
- Lyaskovskyy, V.; de Bruin, B. Redox non-innocent ligands: Versatile new tools to control catalytic reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Suarez, A.I.O.; Lyaskovskyy, V.; Reek, J.N.H.; van der Vlugt, J.I.; de Bruin, B. Complexes with nitrogen-centered radical ligands: Classification, spectroscopic features, reactivity, and catalytic applications. Angew. Chem. Int. Ed. 2013, 52, 12510–12529. [Google Scholar] [CrossRef] [PubMed]
- El-Khalafy, S.H.; Hassanein, M. Oxidation of 2-aminophenol with molecular oxygen and hydrogen peroxide catalyzed by water-soluble metalloporphyrins. J. Mol. Catal. A Chem. 2012, 363–364, 148–152. [Google Scholar] [CrossRef]
- Bodipati, N.; Peddinti, R.K. Chemical generation of o-quinone monoimines for the rapid construction of 1,4-benzoxazine derivatives. Org. Biomol. Chem. 2012, 10, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Gallo, E.; Fantauzzi, S.; Morlacchi, S.; Ragaini, F.; Cenini, S. Allylic amination and aziridination of olefins by aryl azides catalyzed by CoII(tpp): A synthetic and mechanistic study. Eur. J. Inorg. Chem. 2008, 19, 3009–3019. [Google Scholar] [CrossRef]
- Ragaini, F.; Penoni, A.; Gallo, E.; Tollari, S.; Li Gotti, C.; Lapadula, M.; Mangioni, E.; Cenini, S. Amination of benzylic C–H bonds by arylazides catalyzed by CoII-porphyrin complexes: A synthetic and mechanistic study. Chem. Eur. J. 2003, 9, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Nogami, T.; Hishida, T.; Yamada, M.; Mikawa, H.; Shirota, Y. Formation and reactions of o-benzoquinone mono and di-imines. I. Bull. Chem. Soc. Jpn. 1975, 48, 3709–3714. [Google Scholar] [CrossRef]
- Bellow, J.A.; Yousif, M.; Cabelof, A.C.; Lord, R.L.; Groysman, S. Reactivity modes of an iron bis(alkoxide) complex with aryl azides: Catalytic nitrene coupling vs formation of Iron(III) imido dimers. Organometallics 2015, 34, 2917–2923. [Google Scholar] [CrossRef]
- Harrold, N.D.; Waterman, R.; Hillhouse, G.L.; Cundari, T.R. Group-transfer reactions of nickel-carbene and -nitrene complexes with organoazides and nitrous oxide that form new C=N, C=O, and N=N bonds. J. Am. Chem. Soc. 2009, 131, 12872–12873. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Moret, M.; Peters, J.C. A Ru(I) metalloradical that catalyzes nitrene coupling to azoarenes from arylazides. J. Am. Chem. Soc. 2012, 134, 6695–6706. [Google Scholar] [CrossRef] [PubMed]
- Turbomole, Version 6.5, Theoretical Chemistry Group, University of Karlsruhe: Karlsruhe, Germany, 2002.
- PQS, Version 2.4, Parallel Quantum Solutions: Fayetteville, AR, USA, 2001.
- Baker, J. An algorithm for the location of transition states. J. Comput. Chem. 1986, 7, 385–395. [Google Scholar] [CrossRef]
- Budzelaar, P.H.M. Geometry optimization using generalized, chemically meaningful constraints. J. Comput. Chem. 2007, 28, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Expected Product(s) | Obtained Product |
|---|---|---|---|
| 1 |  |  |  |
| 2 | 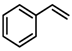 | 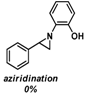 | |
| 3 | 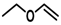 | 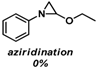 | |
| Entry | Substrate | Expected Product(s) | Obtained Product |
|---|---|---|---|
| 1 |  | 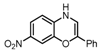 | 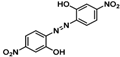 (~10%) (~10%) |
| 2 | |  | --- |
| 3 |  | 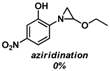 |  (80%) (80%) |
| 4 | | 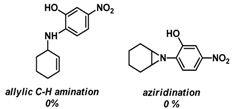 | --- |
| Atom | 2A | 2B | 2C |
|---|---|---|---|
| Co | 11.1% | 8.5% | 60.6% |
| N | 39.6% | 33.5% | 16.1% |
| O | 5.3% | 10.0% | 9.4% |
| Structure | Co–N | N–C | C–C | C–O | N–H |
|---|---|---|---|---|---|
| 2A | 1.81769 | 1.32018 | 1.47138 | 1.34208 | 1.86203 |
| 2B | 1.81731 | 1.31667 | 1.48868 | 1.30627 | 1.33591 |
| 2C | 1.92289 | 1.32157 | 1.50424 | 1.24441 | 1.03461 |
| Atom | 3A | 3B | 3C |
|---|---|---|---|
| Co | 8% | 11% | 63% |
| N | 36% | 24% | 10% |
| O | 10% | 20% | 9% |
| Structure | Co–N | N–C | C–C | C–N | N–H |
|---|---|---|---|---|---|
| 3A | 1.8411 | 1.3161 | 1.4835 | 1.3576 | 2.1848 |
| 3B | 1.8521 | 1.3126 | 1.5060 | 1.3262 | 1.3213 |
| 3C | 1.9604 | 1.3152 | 1.5022 | 1.4378 | 1.0340 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goswami, M.; Rebreyend, C.; De Bruin, B. Porphyrin Cobalt(III) “Nitrene Radical” Reactivity; Hydrogen Atom Transfer from Ortho-YH Substituents to the Nitrene Moiety of Cobalt-Bound Aryl Nitrene Intermediates (Y = O, NH). Molecules 2016, 21, 242. https://doi.org/10.3390/molecules21020242
Goswami M, Rebreyend C, De Bruin B. Porphyrin Cobalt(III) “Nitrene Radical” Reactivity; Hydrogen Atom Transfer from Ortho-YH Substituents to the Nitrene Moiety of Cobalt-Bound Aryl Nitrene Intermediates (Y = O, NH). Molecules. 2016; 21(2):242. https://doi.org/10.3390/molecules21020242
Chicago/Turabian StyleGoswami, Monalisa, Christophe Rebreyend, and Bas De Bruin. 2016. "Porphyrin Cobalt(III) “Nitrene Radical” Reactivity; Hydrogen Atom Transfer from Ortho-YH Substituents to the Nitrene Moiety of Cobalt-Bound Aryl Nitrene Intermediates (Y = O, NH)" Molecules 21, no. 2: 242. https://doi.org/10.3390/molecules21020242
APA StyleGoswami, M., Rebreyend, C., & De Bruin, B. (2016). Porphyrin Cobalt(III) “Nitrene Radical” Reactivity; Hydrogen Atom Transfer from Ortho-YH Substituents to the Nitrene Moiety of Cobalt-Bound Aryl Nitrene Intermediates (Y = O, NH). Molecules, 21(2), 242. https://doi.org/10.3390/molecules21020242