Ring Expansion of Vinylaziridines through the Strain-Release Pericyclic Reaction: Recent Developments and Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
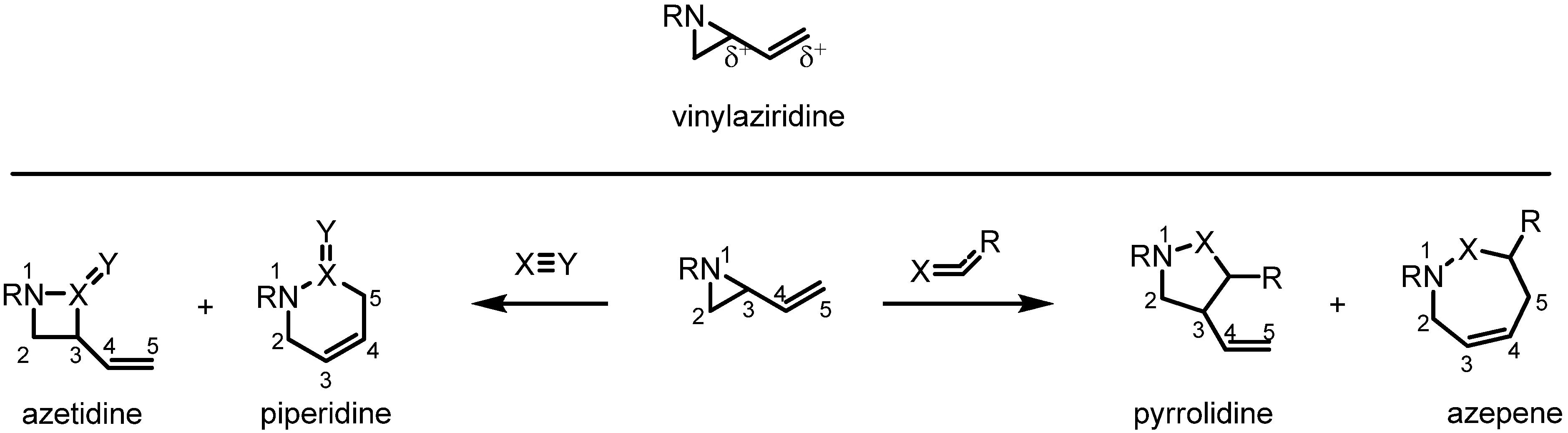
:1. Introduction

2. Results and Discussion
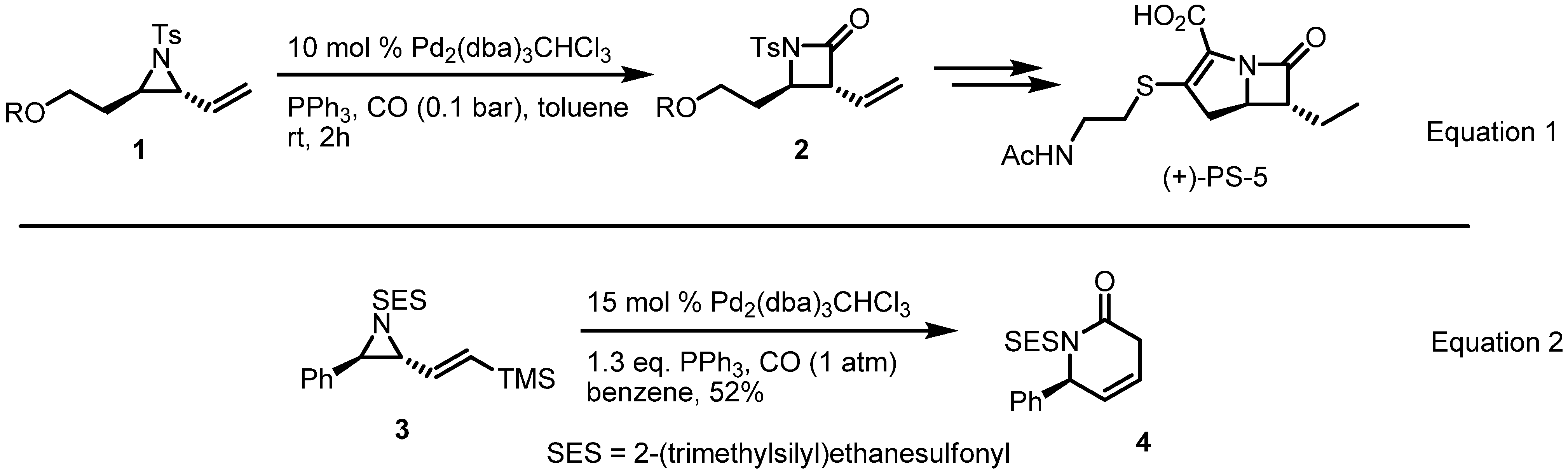
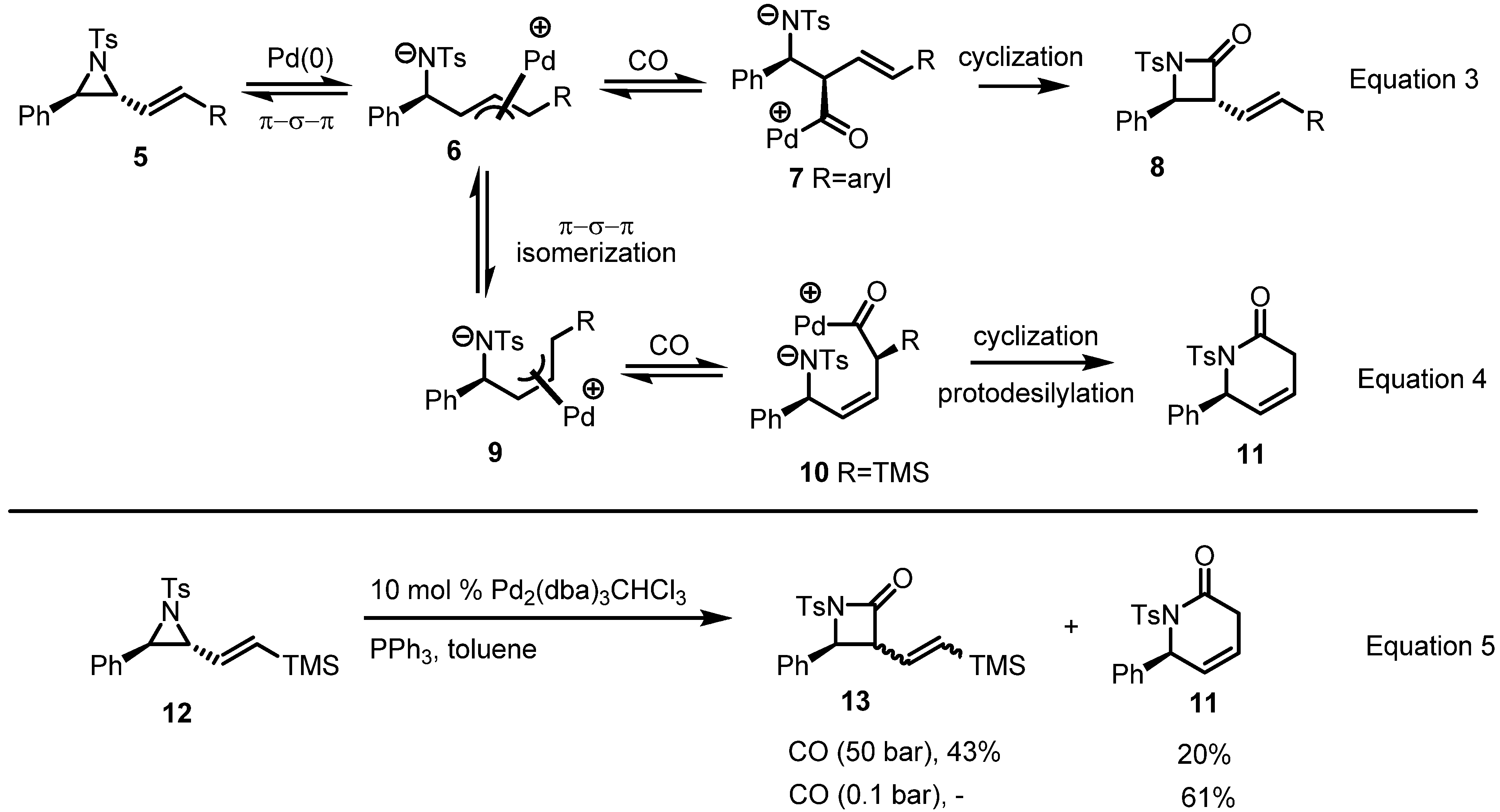
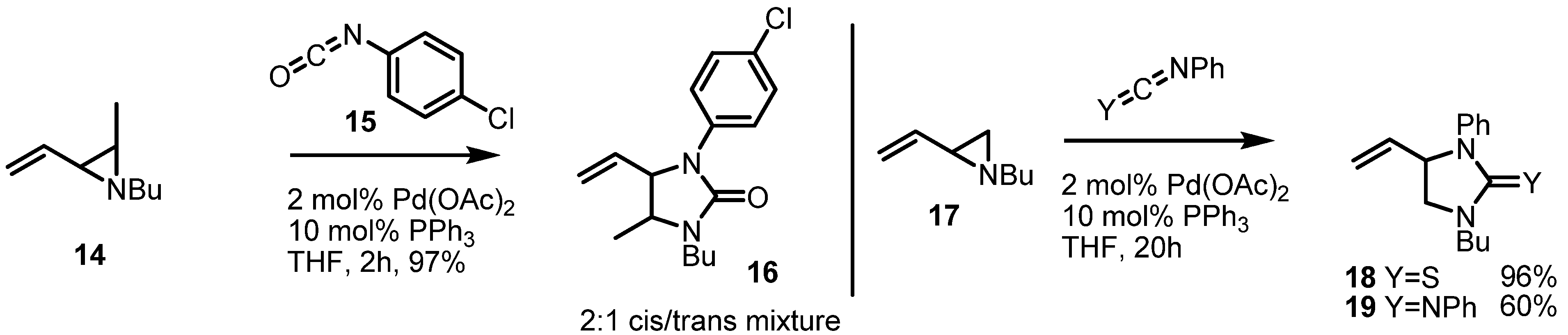
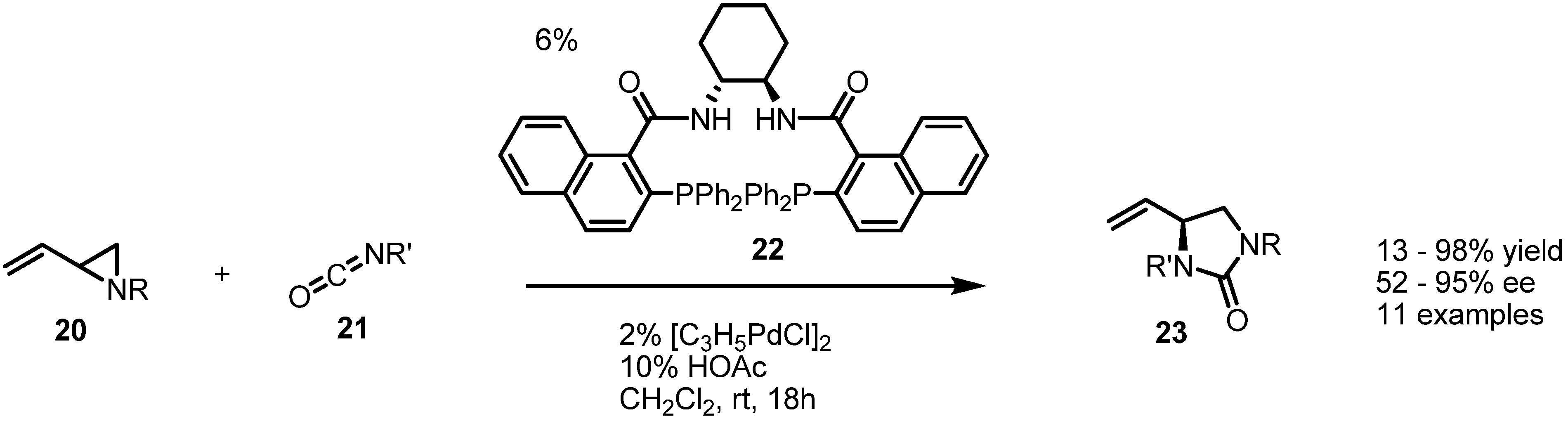
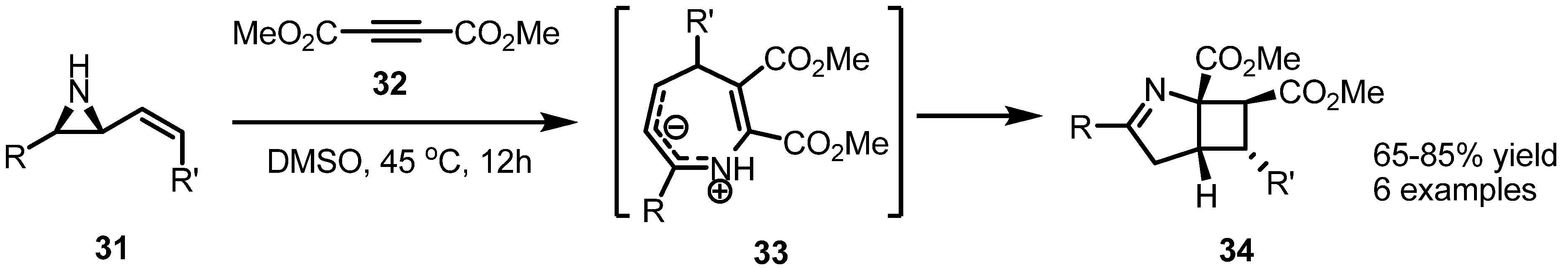
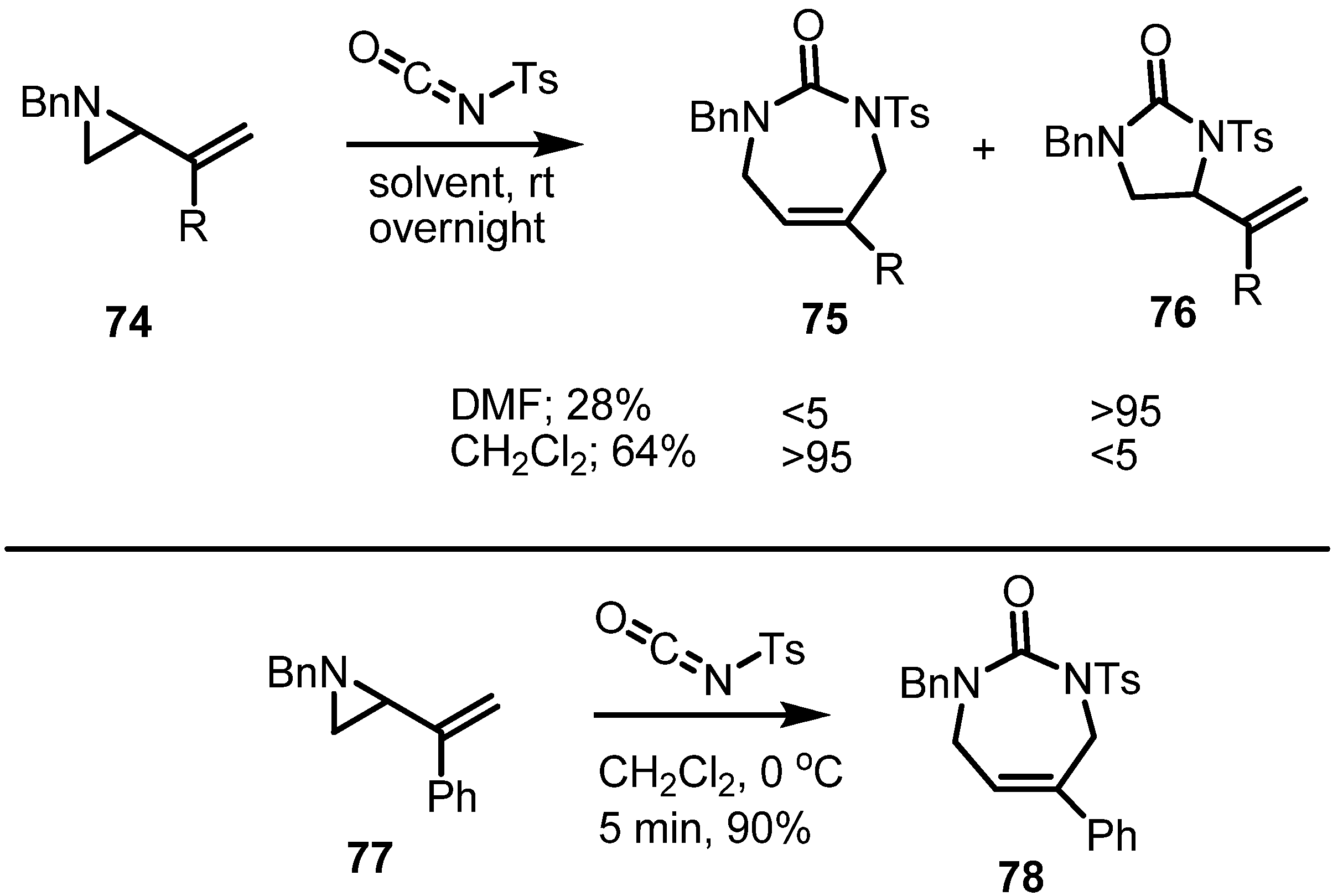
2.1. Azetidines from Vinylaziridines
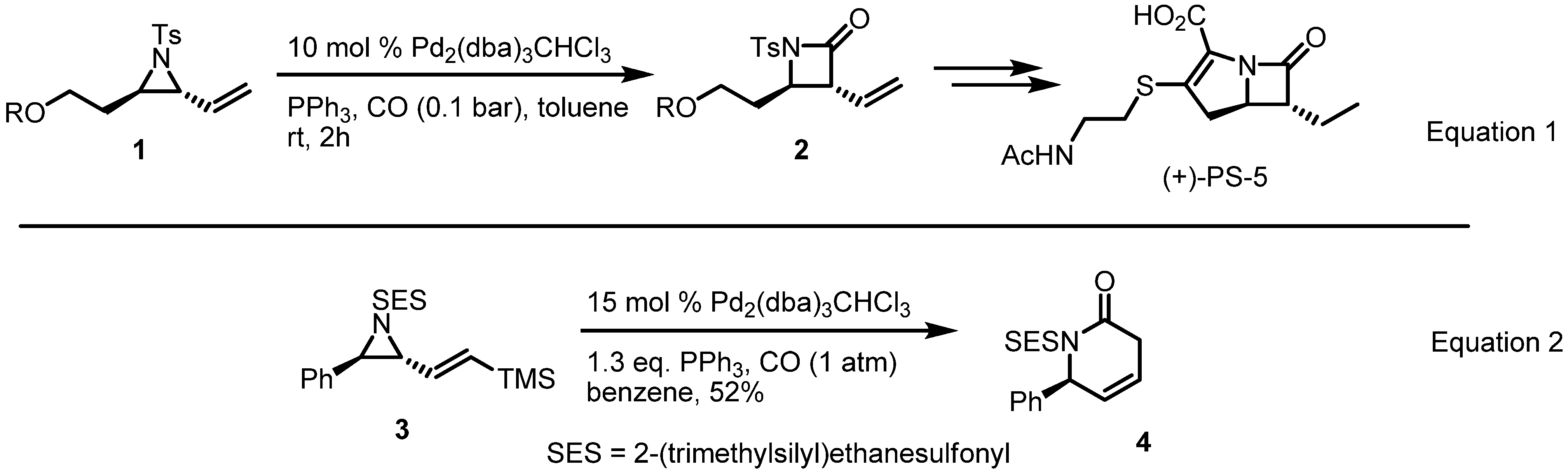
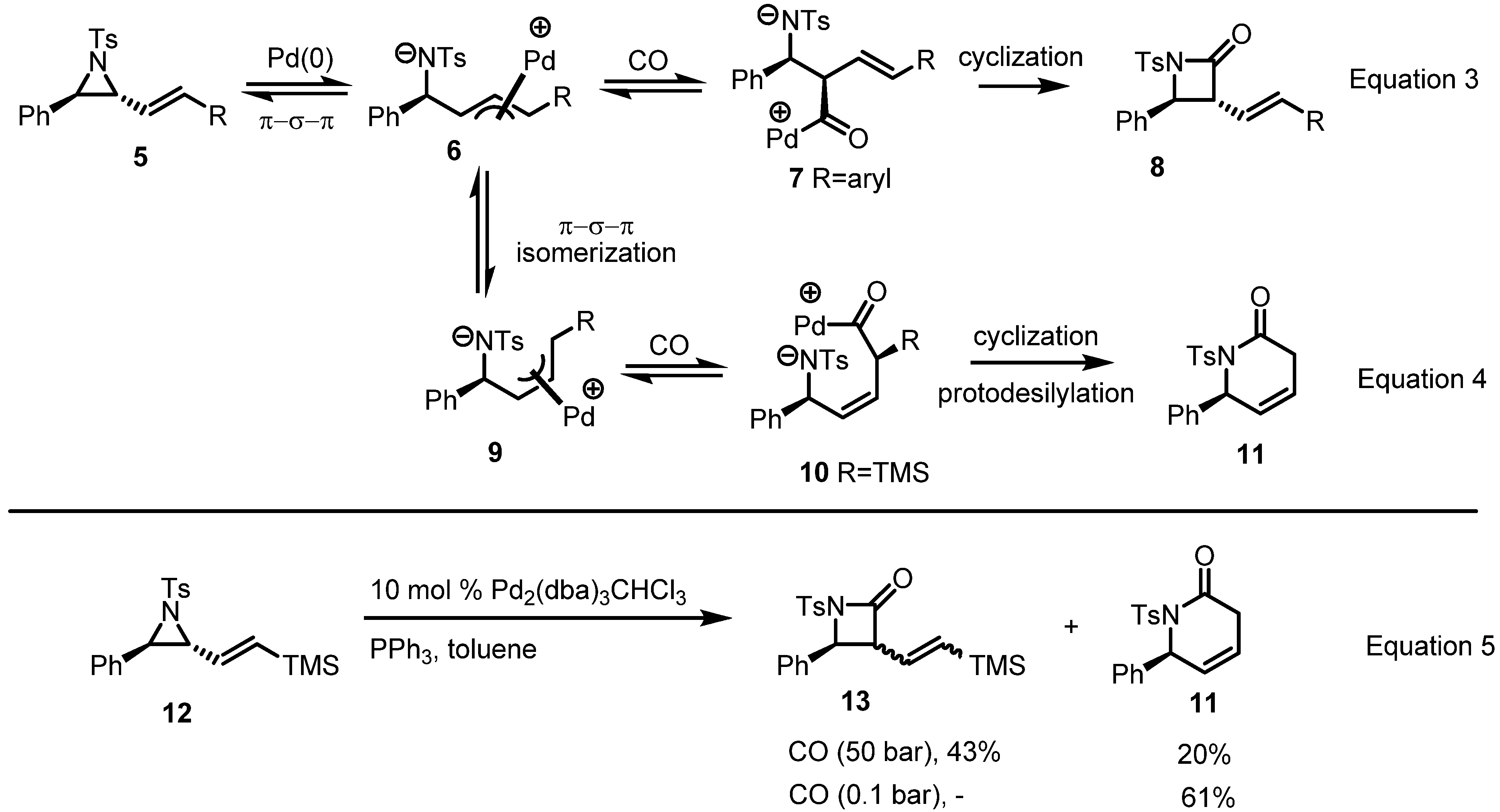
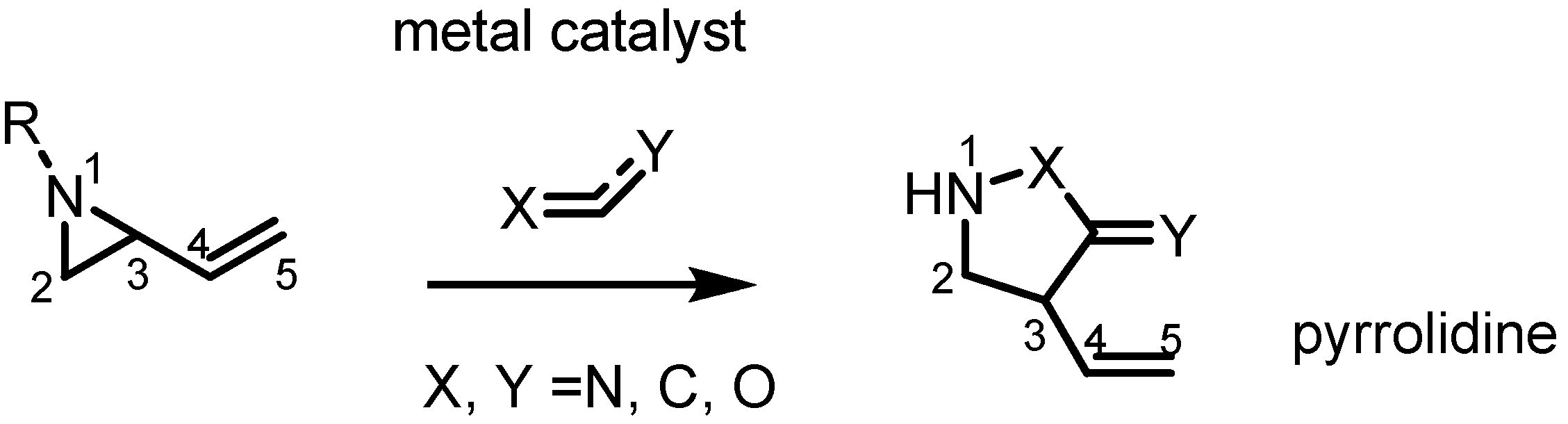
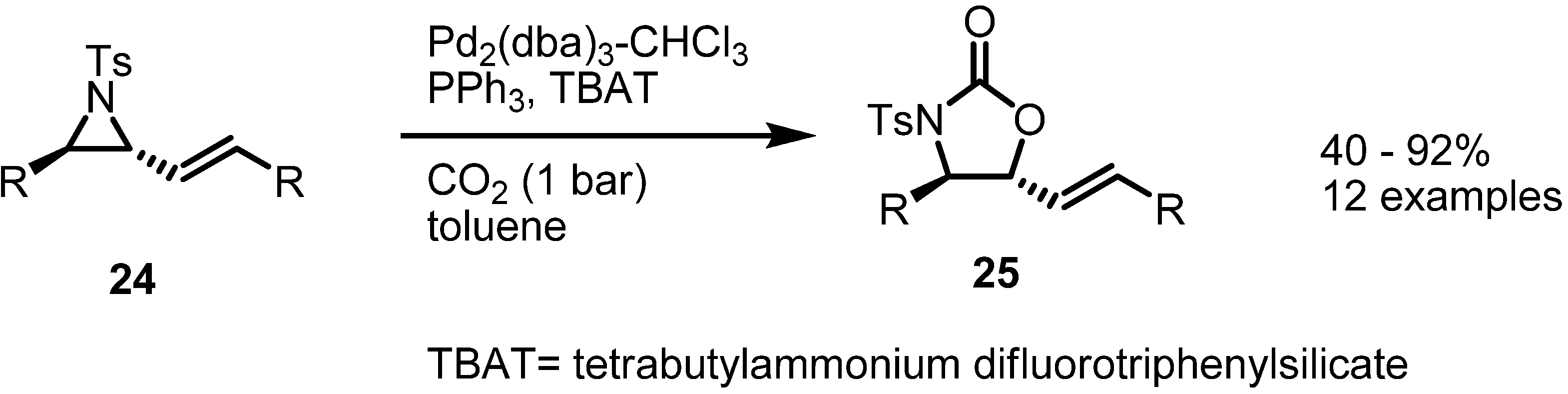
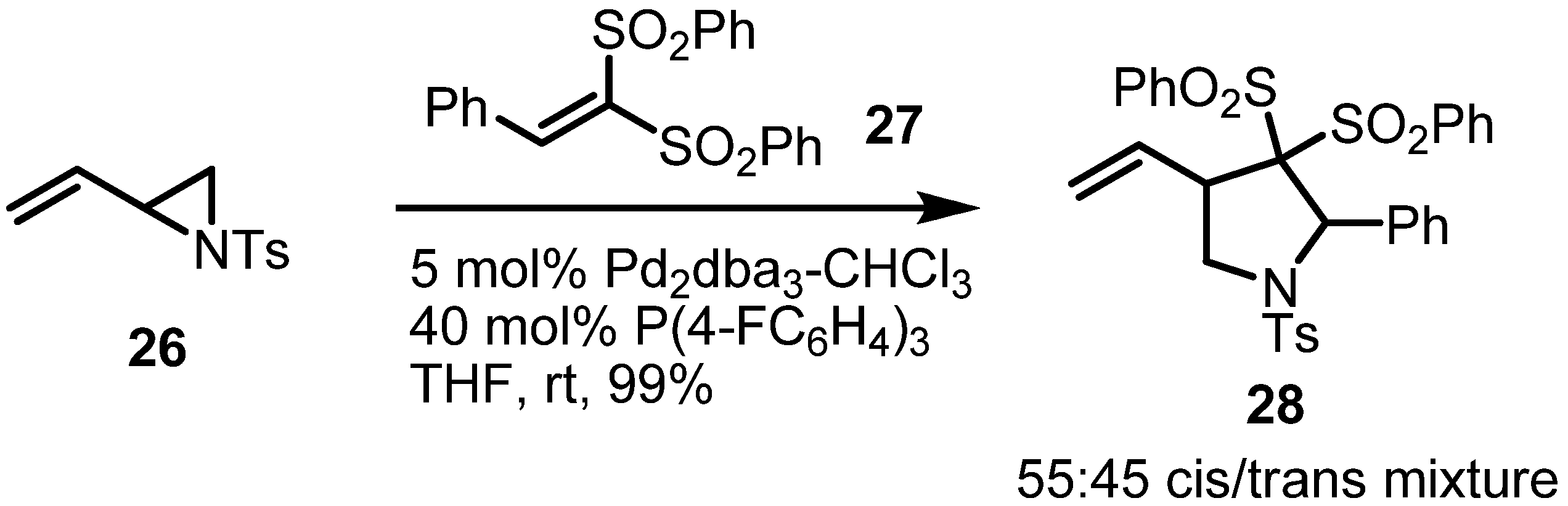
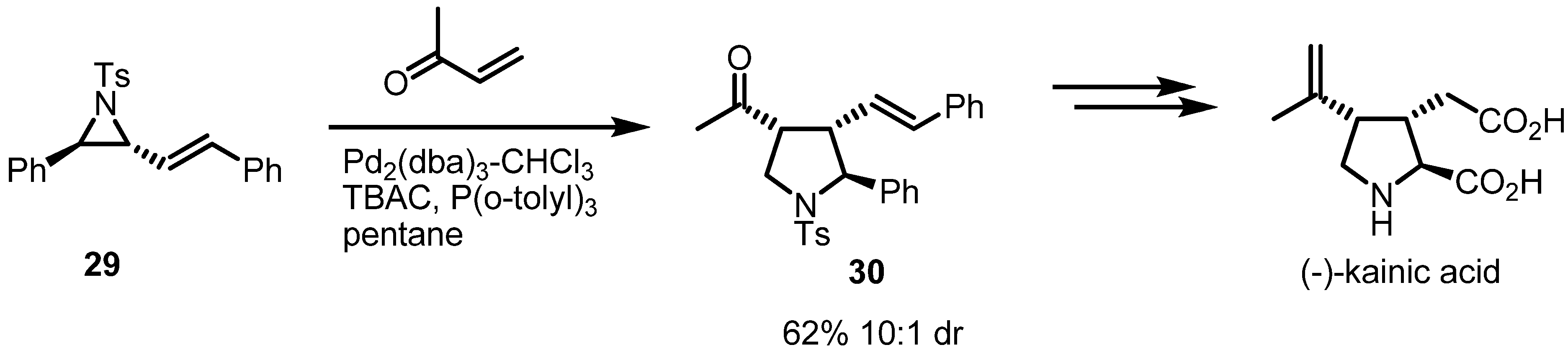
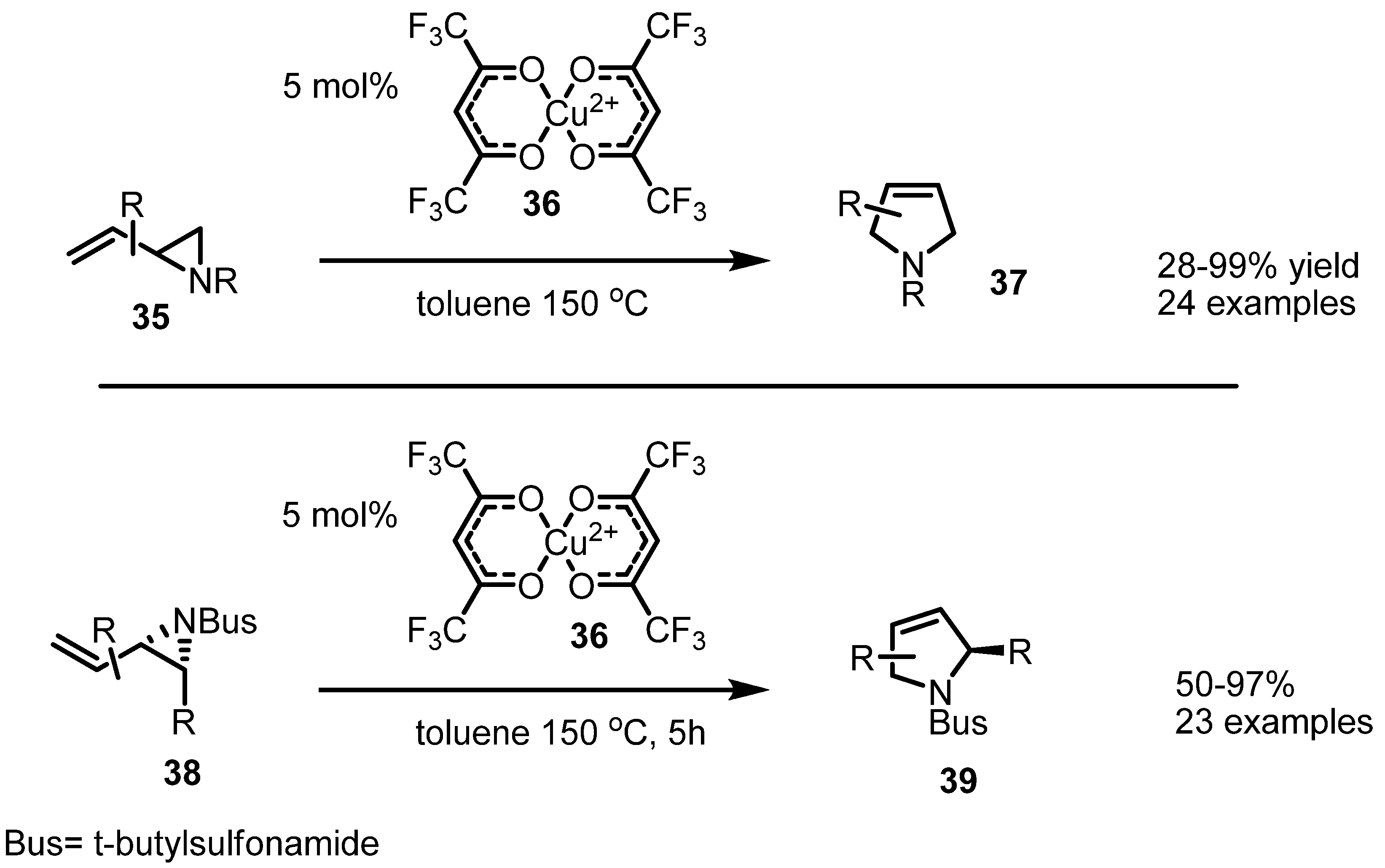
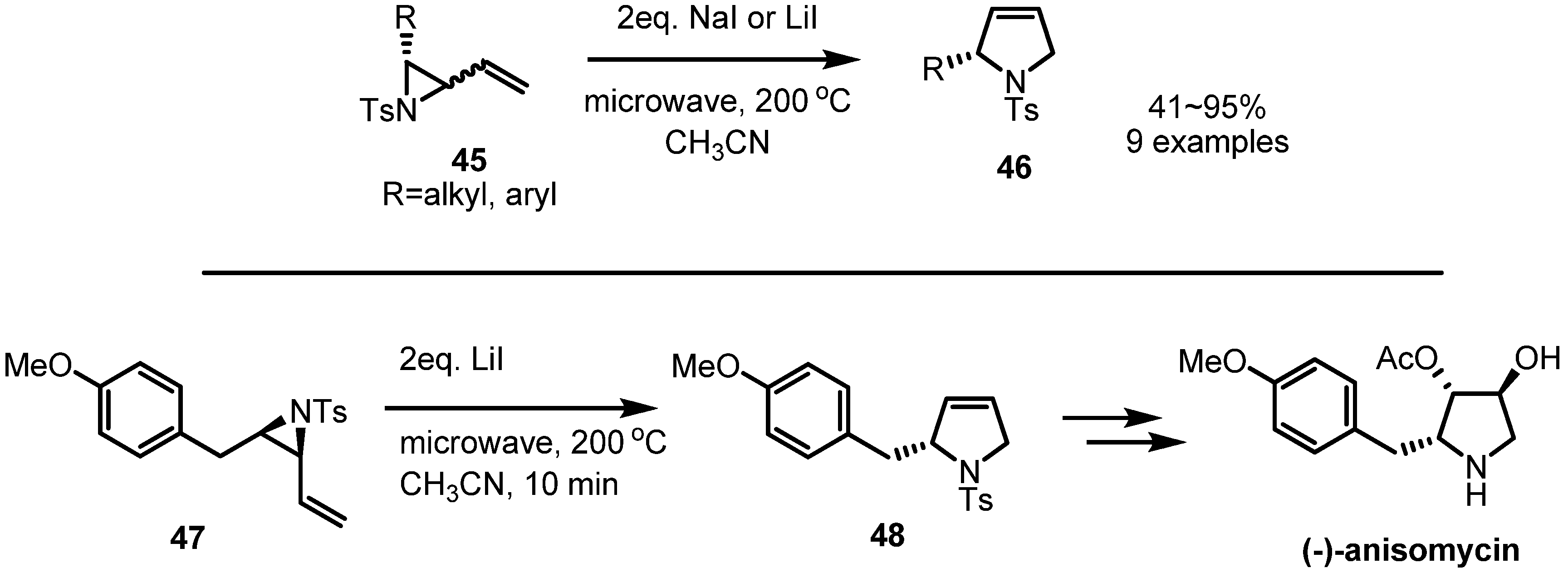
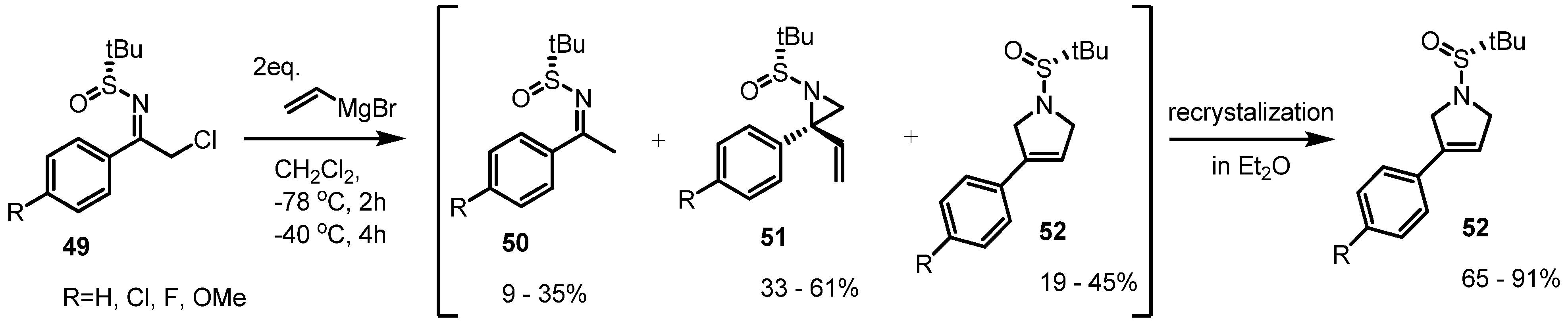
2.2. Pyrrolidines from Vinylaziridines
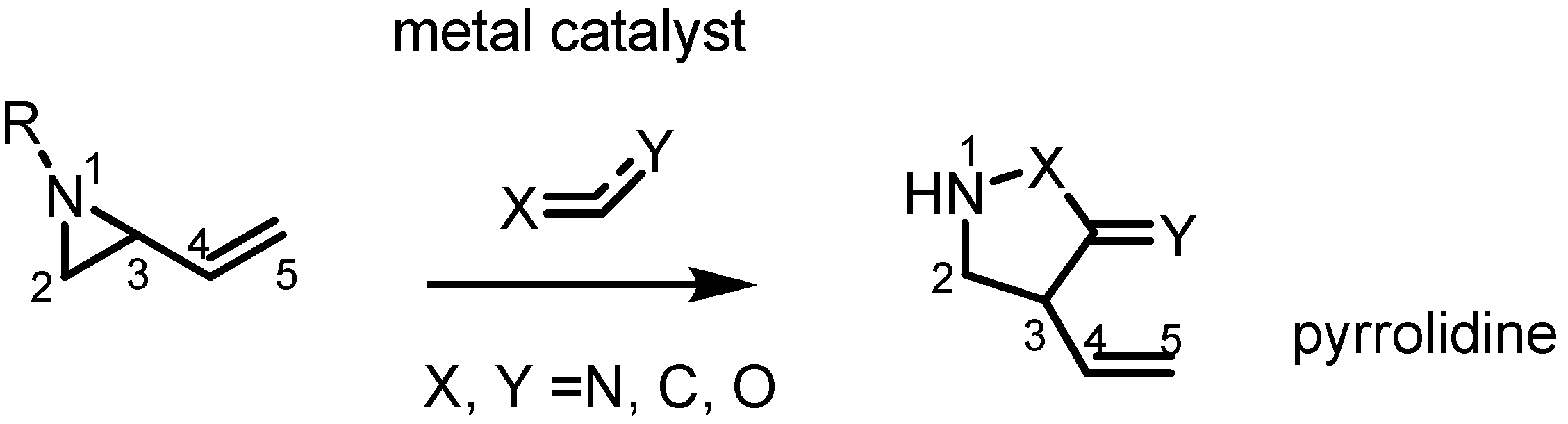
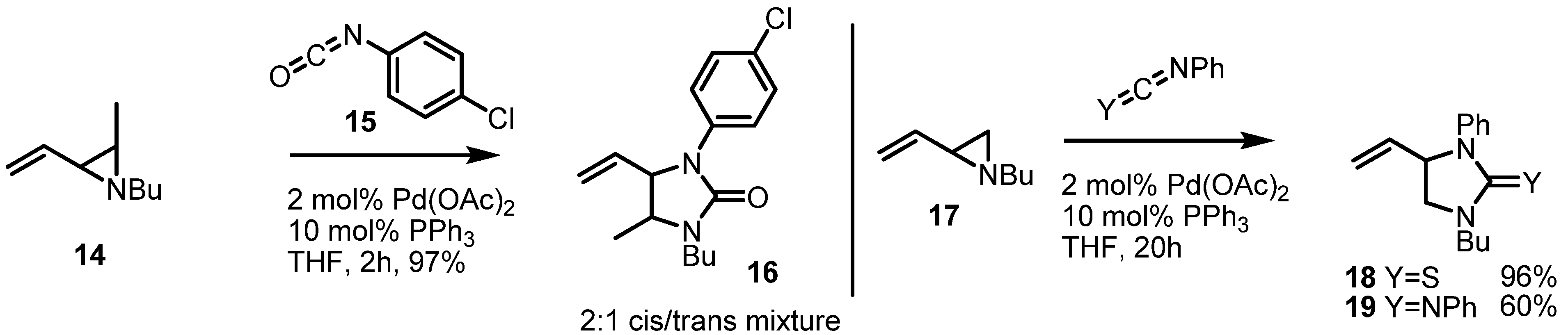
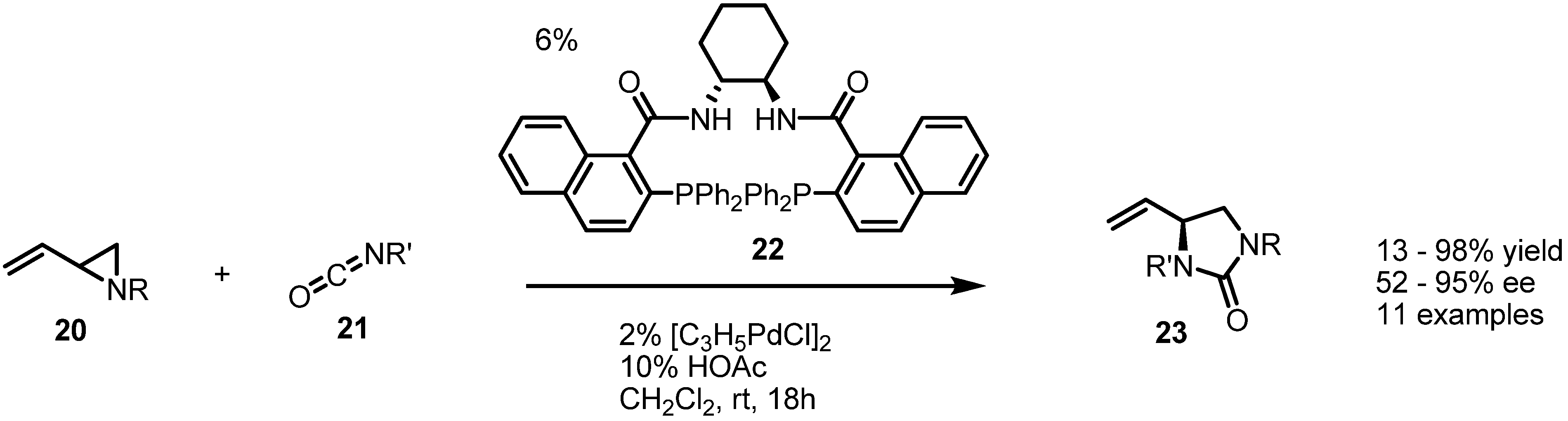
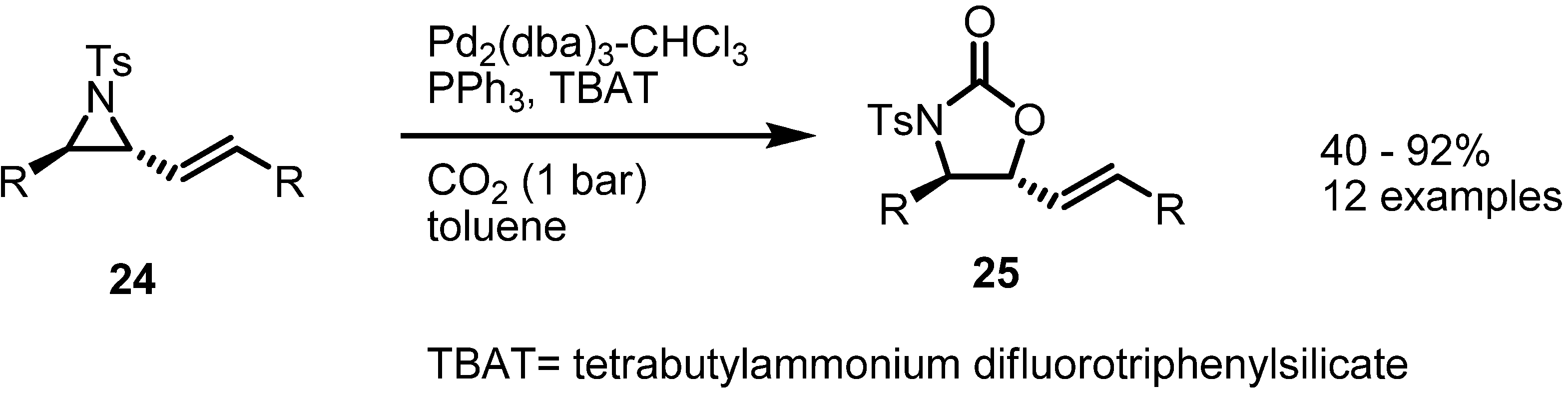

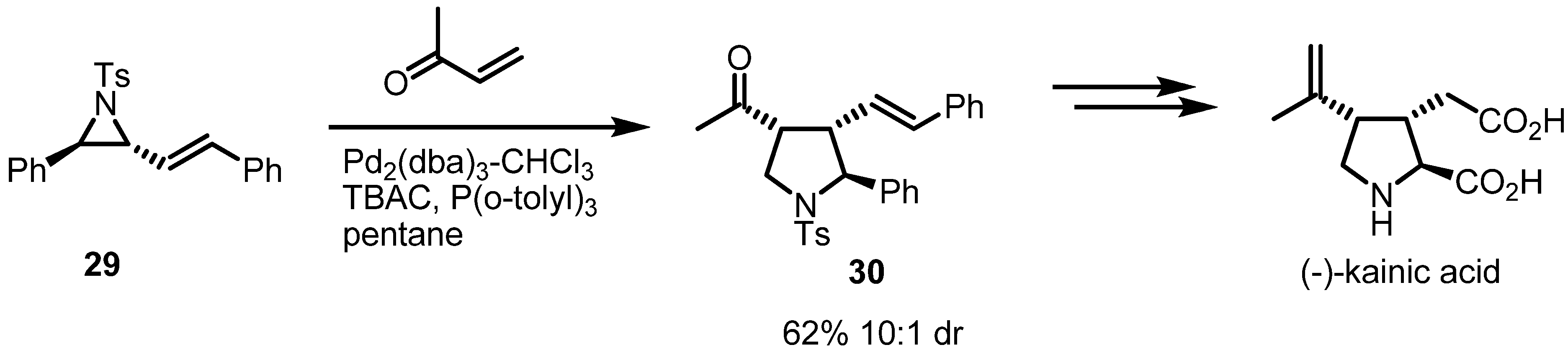
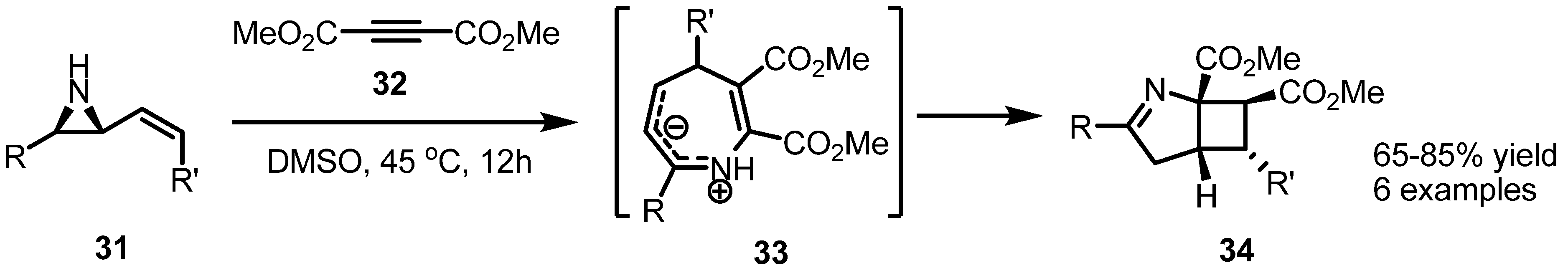
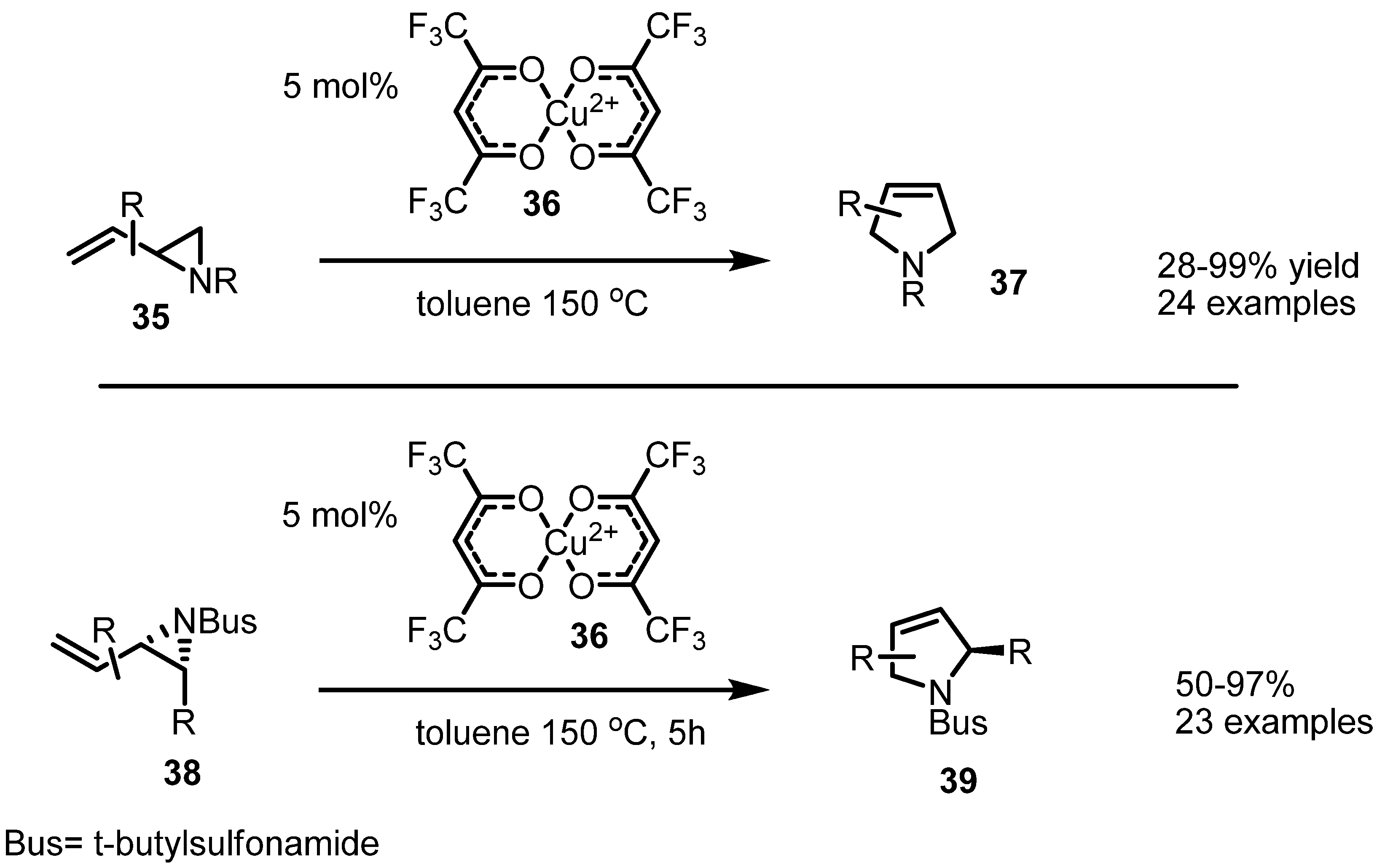
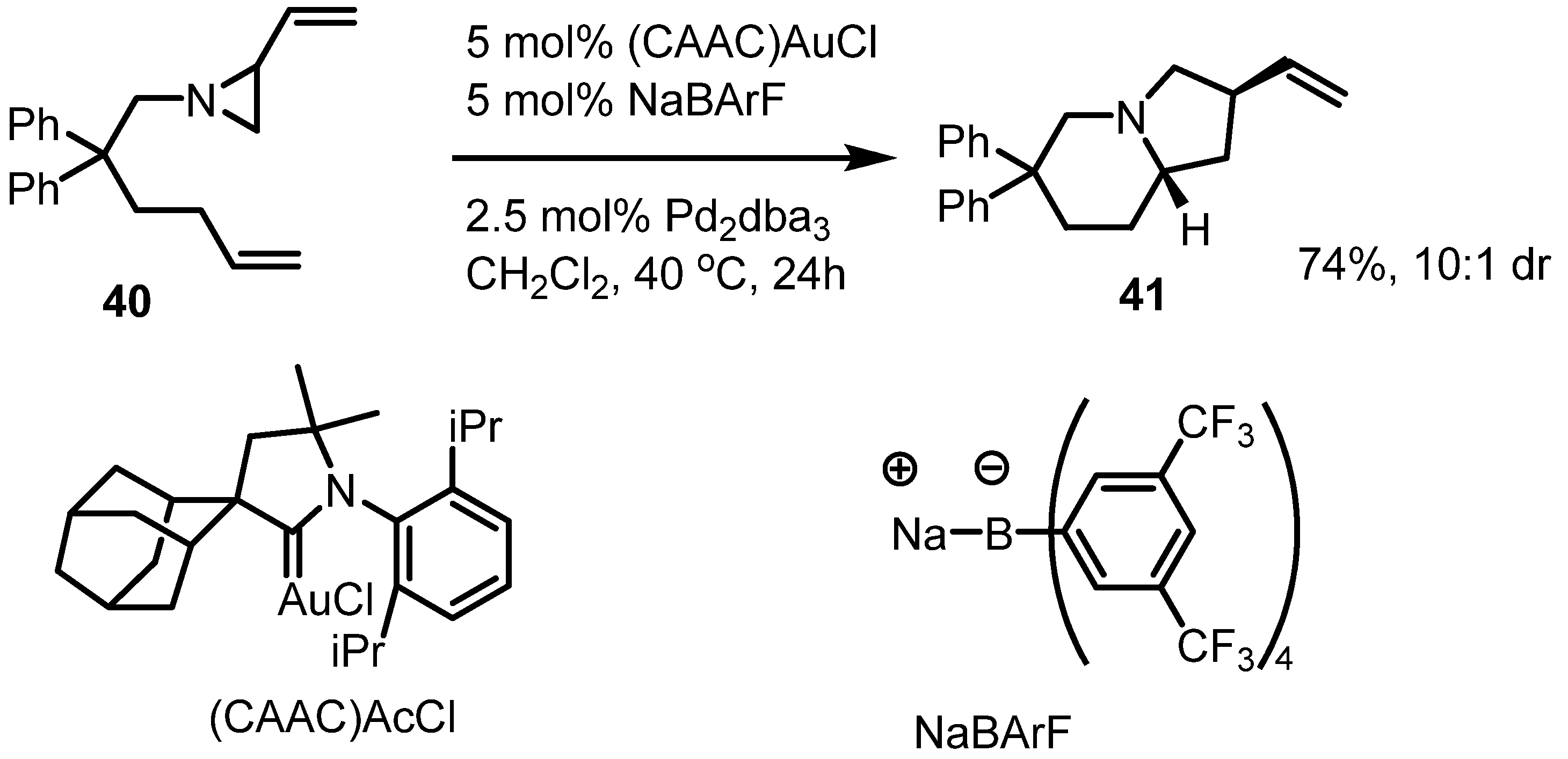
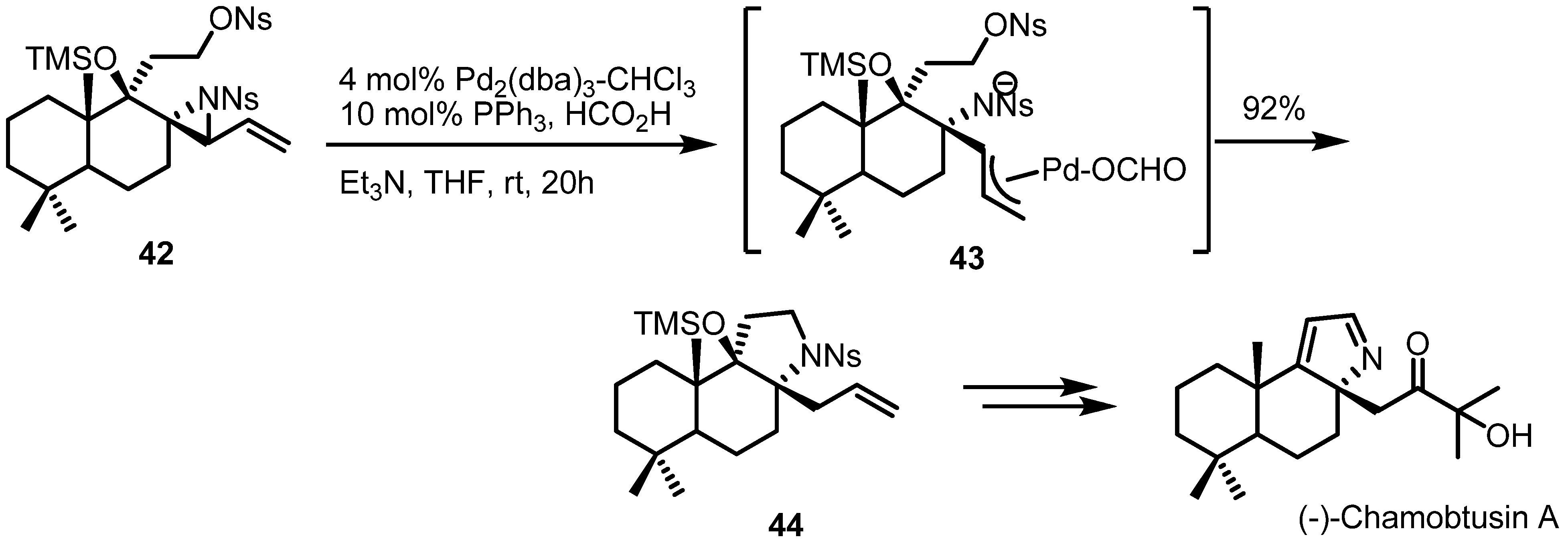
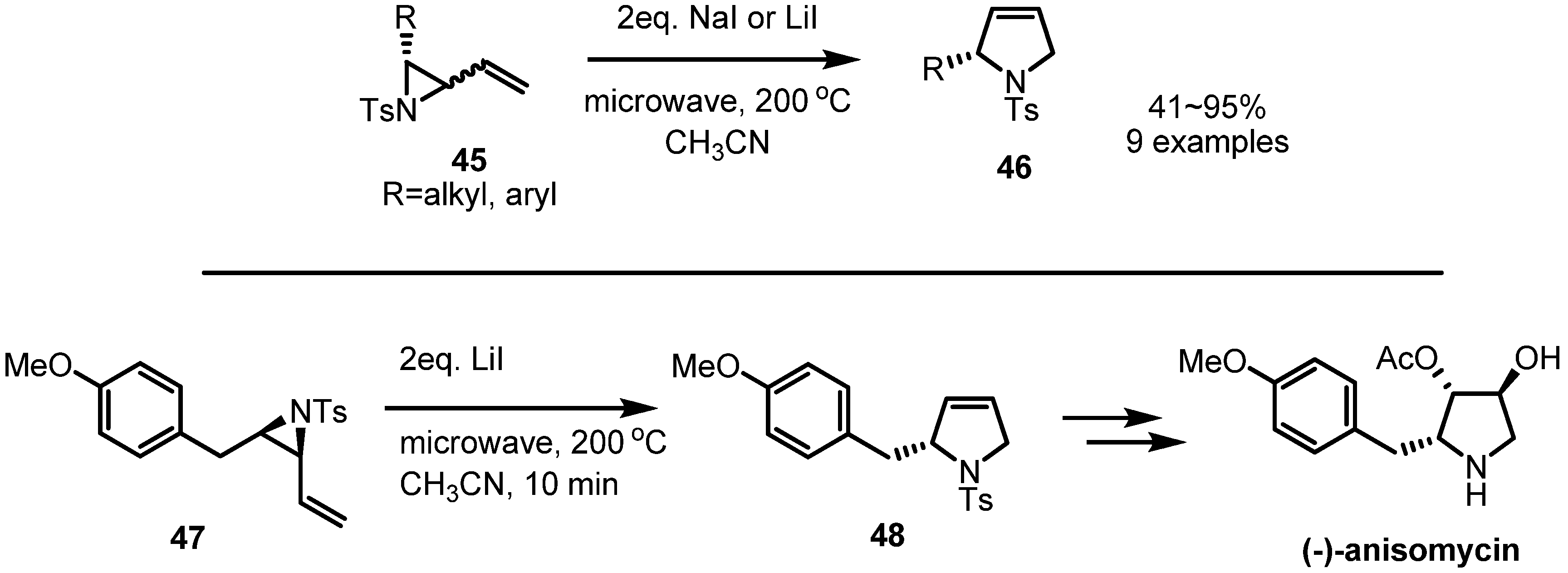
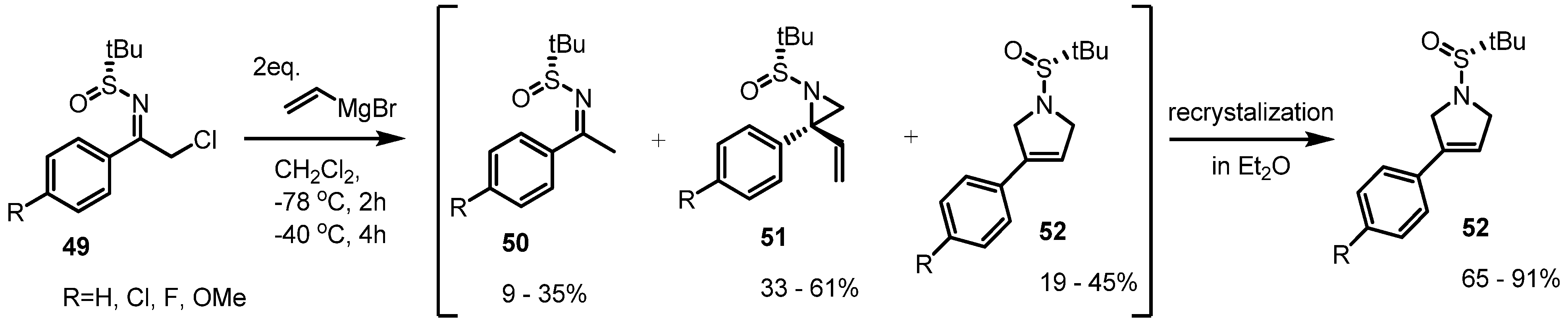
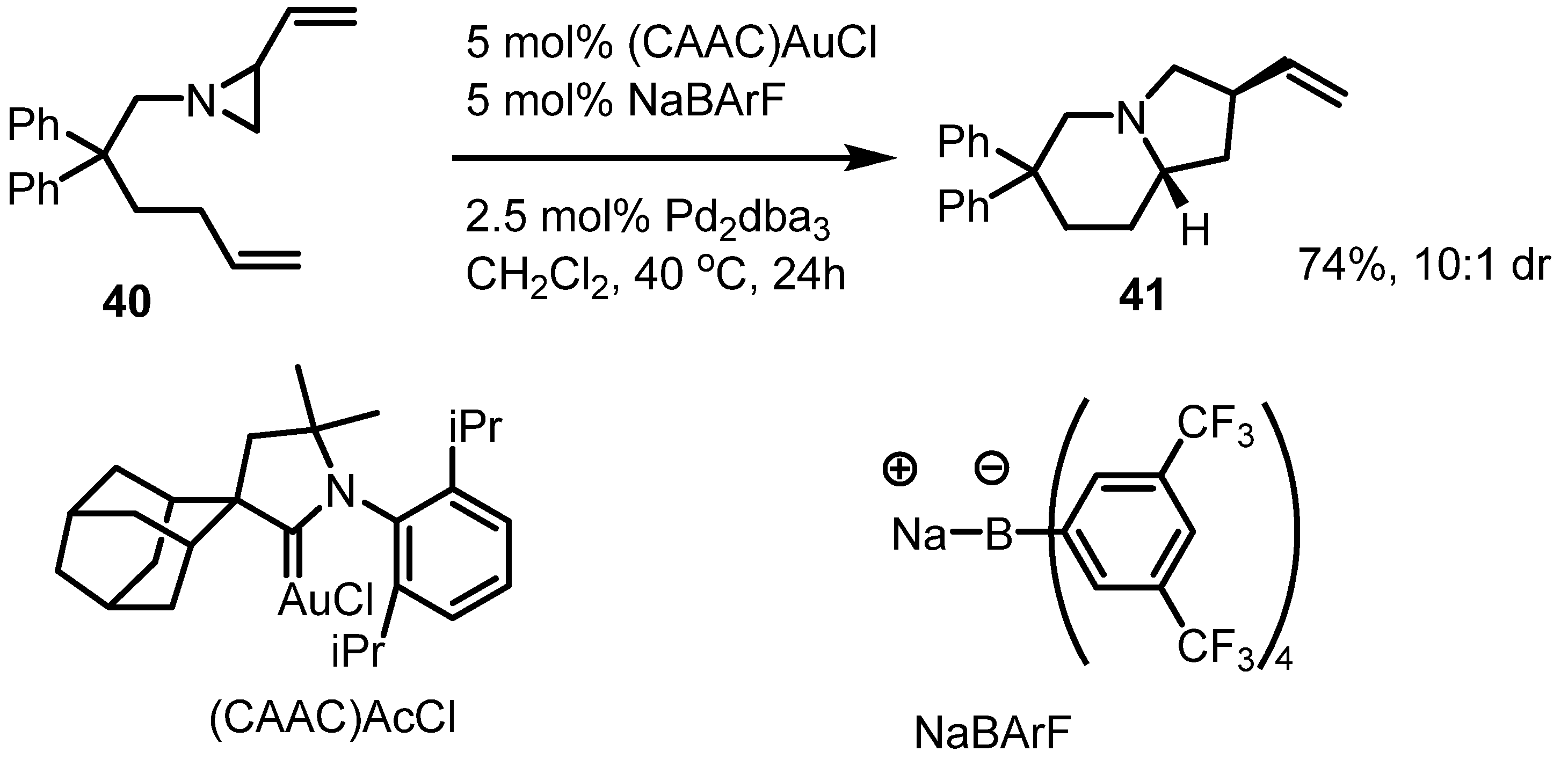
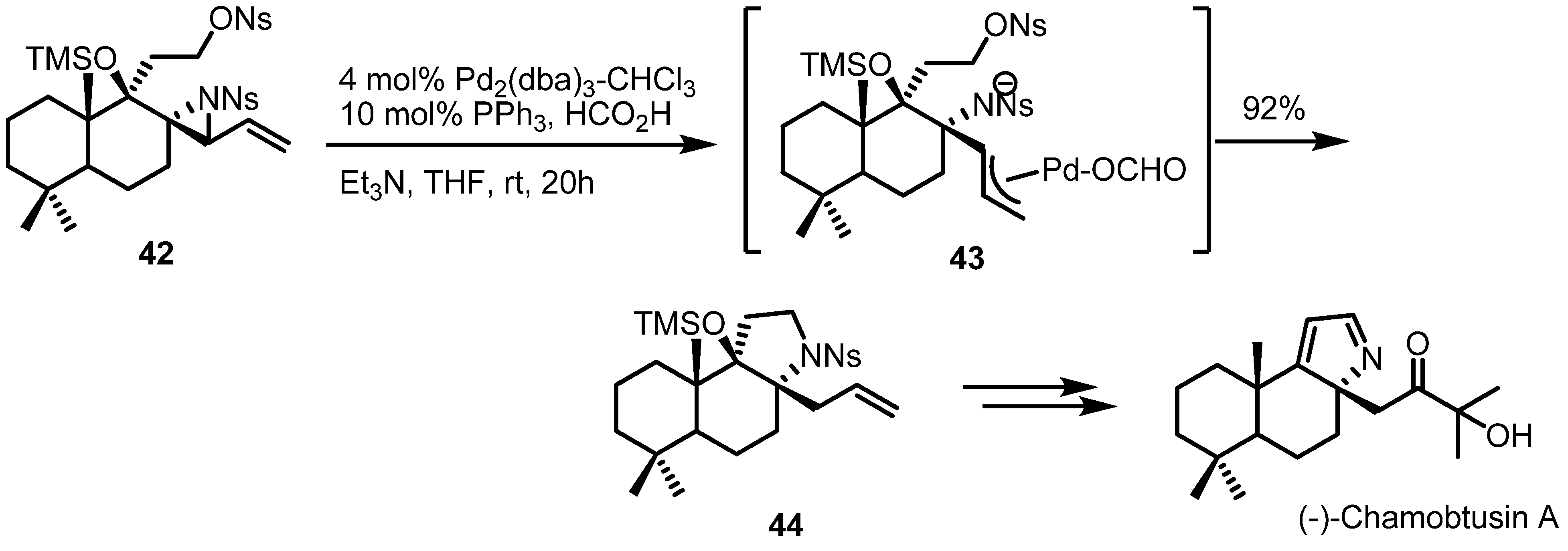
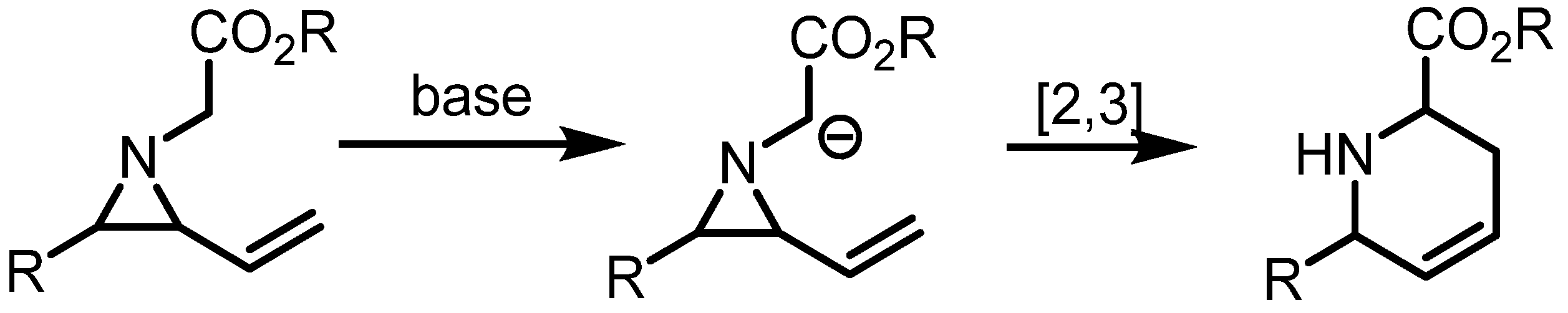
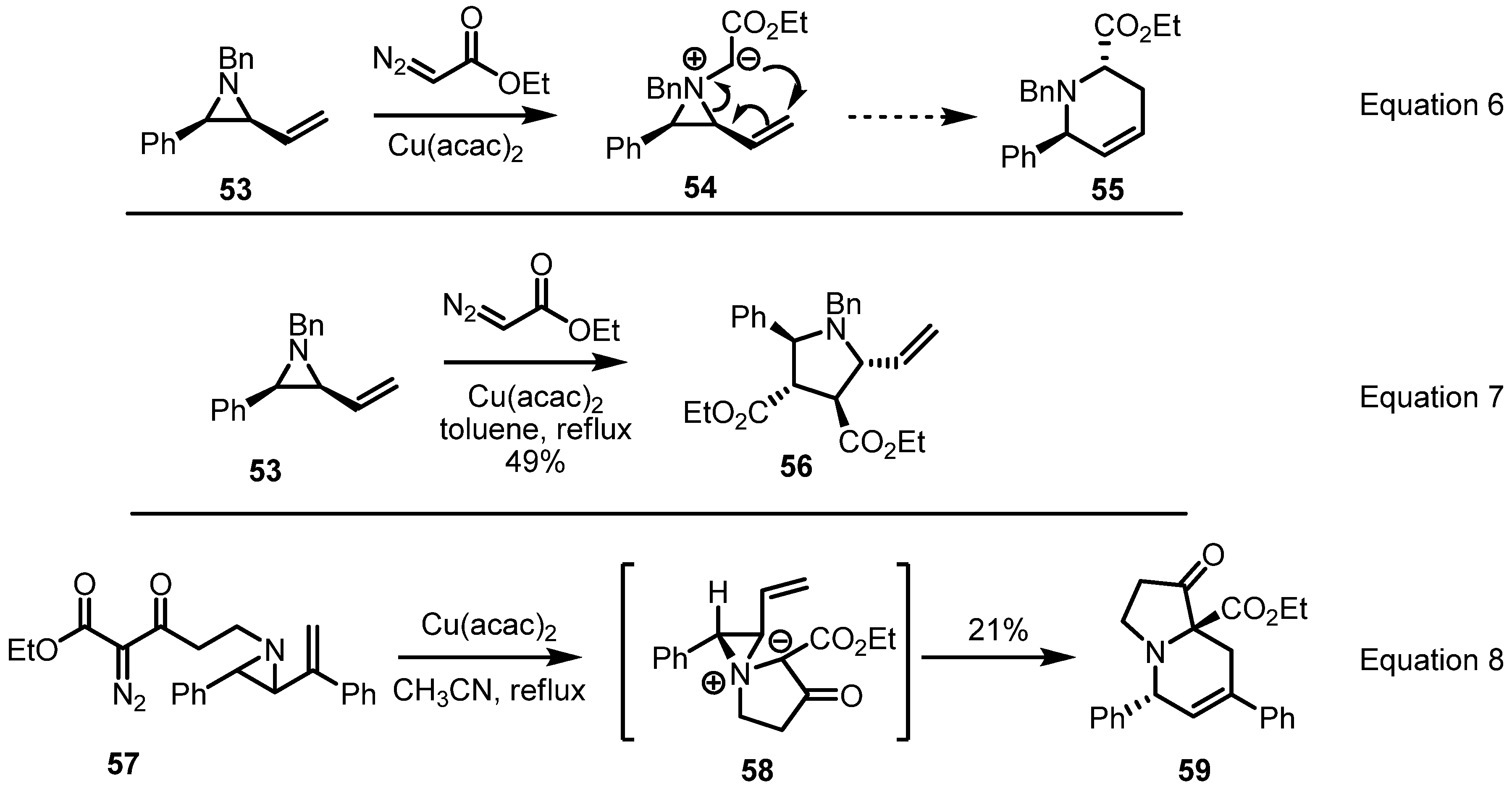
2.3. Piperidines from Vinylaziridines

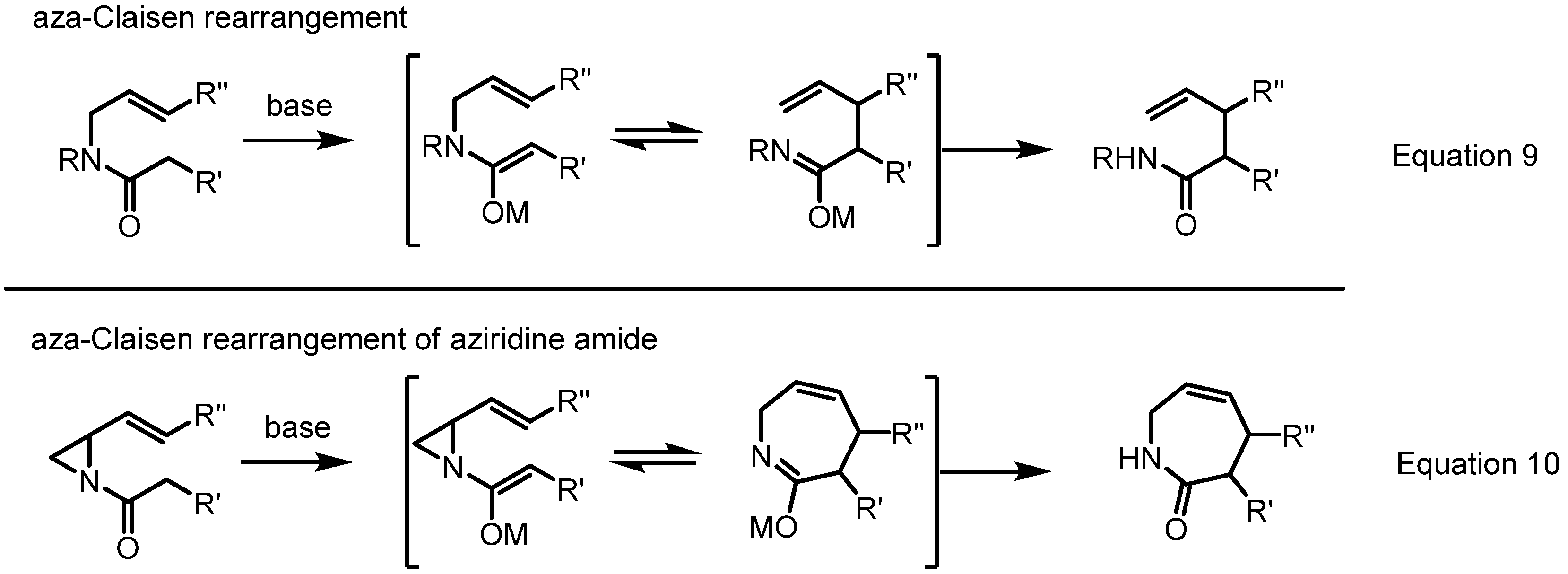
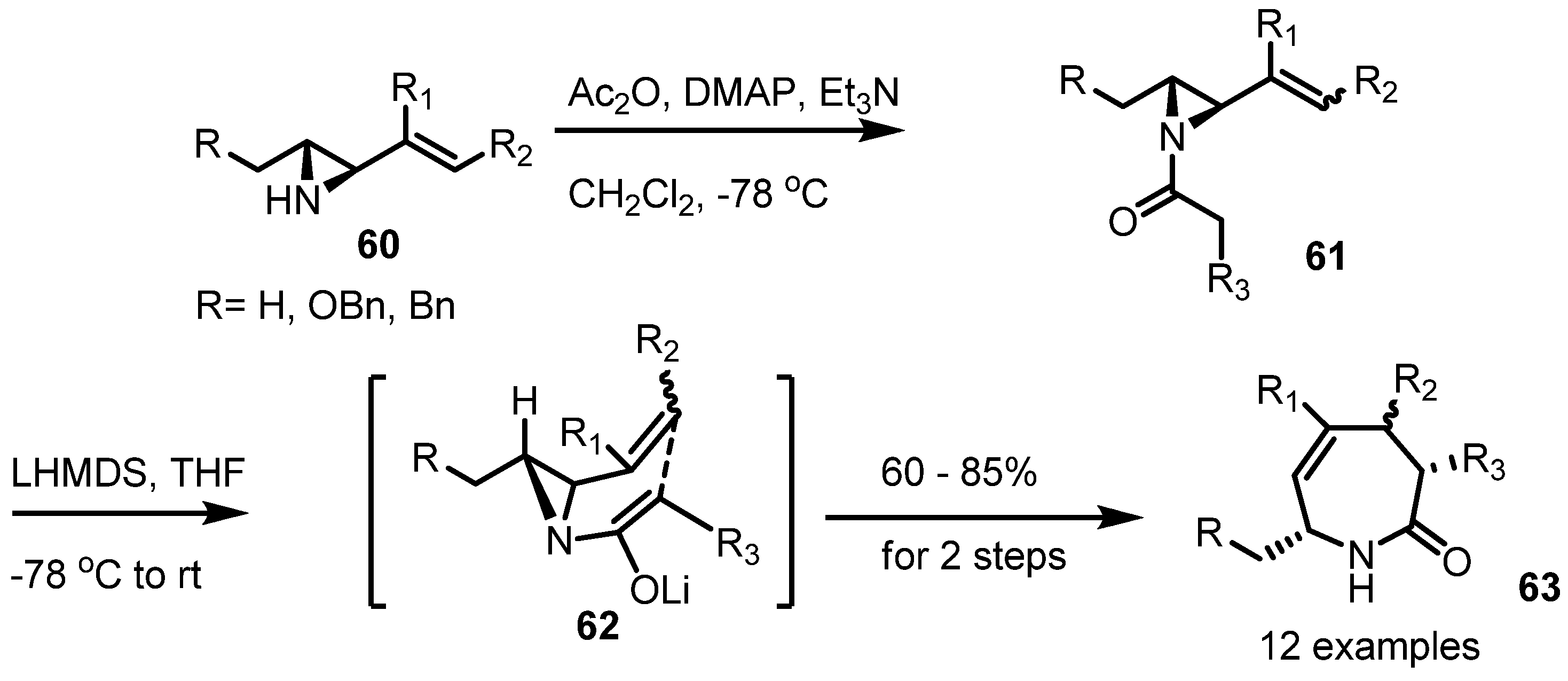
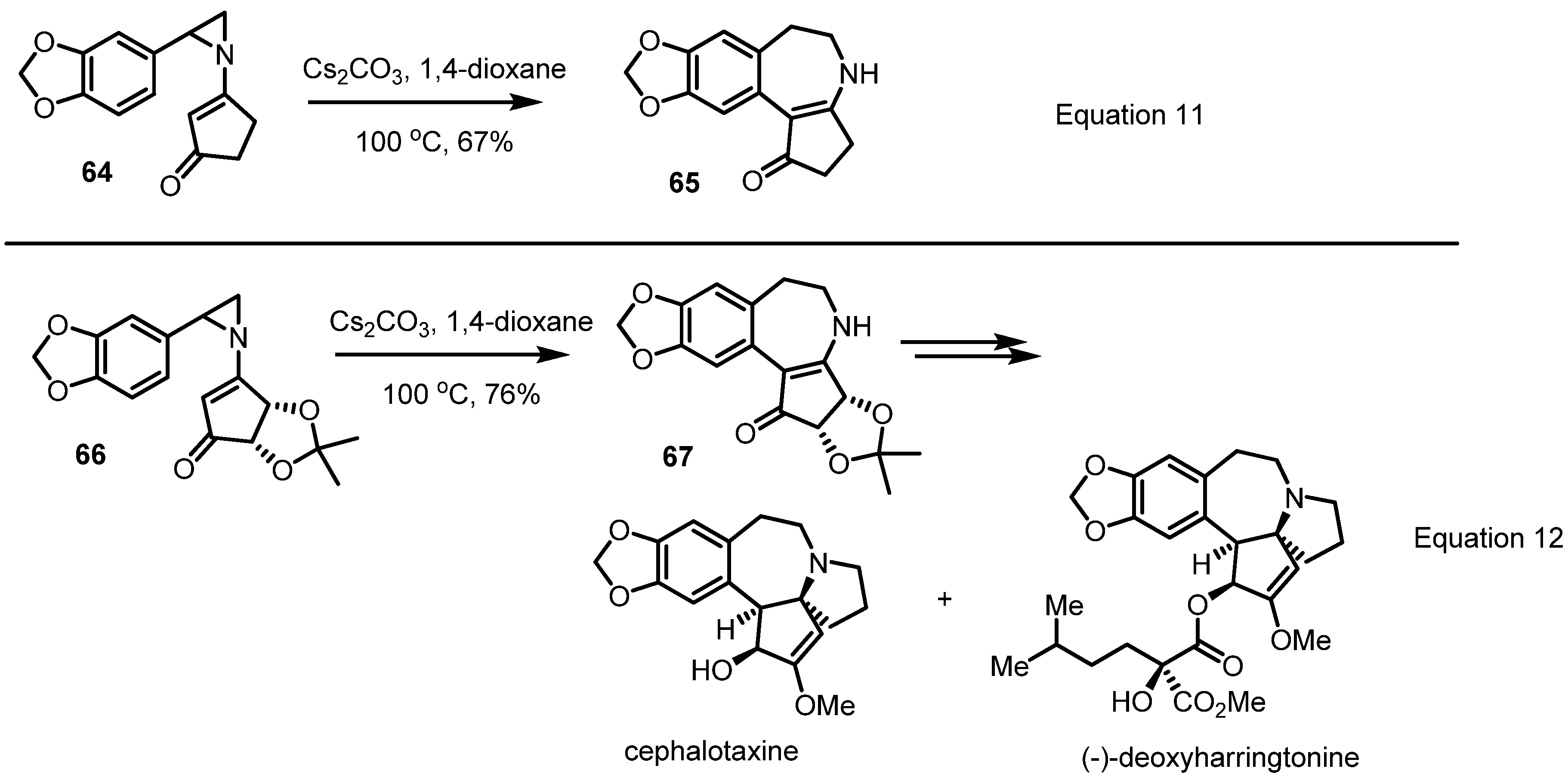
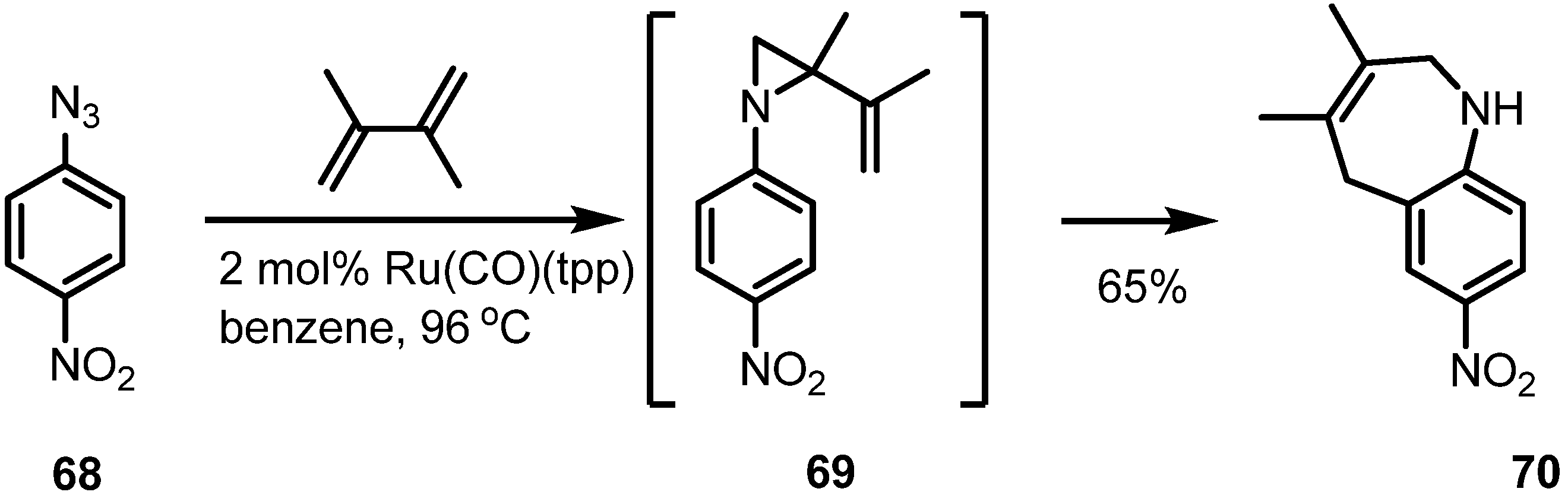
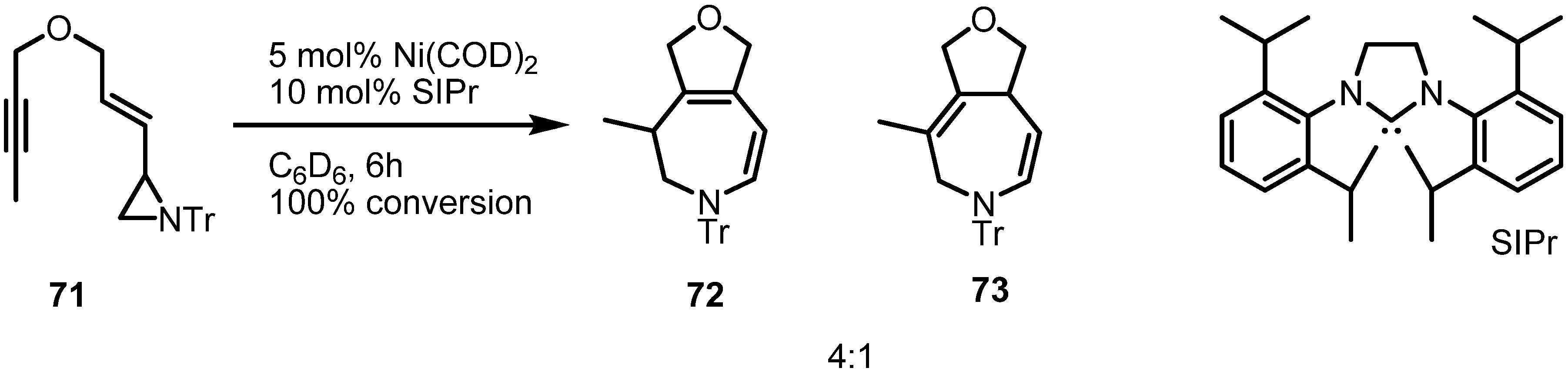
2.4. Azepines from Vinylaziridines
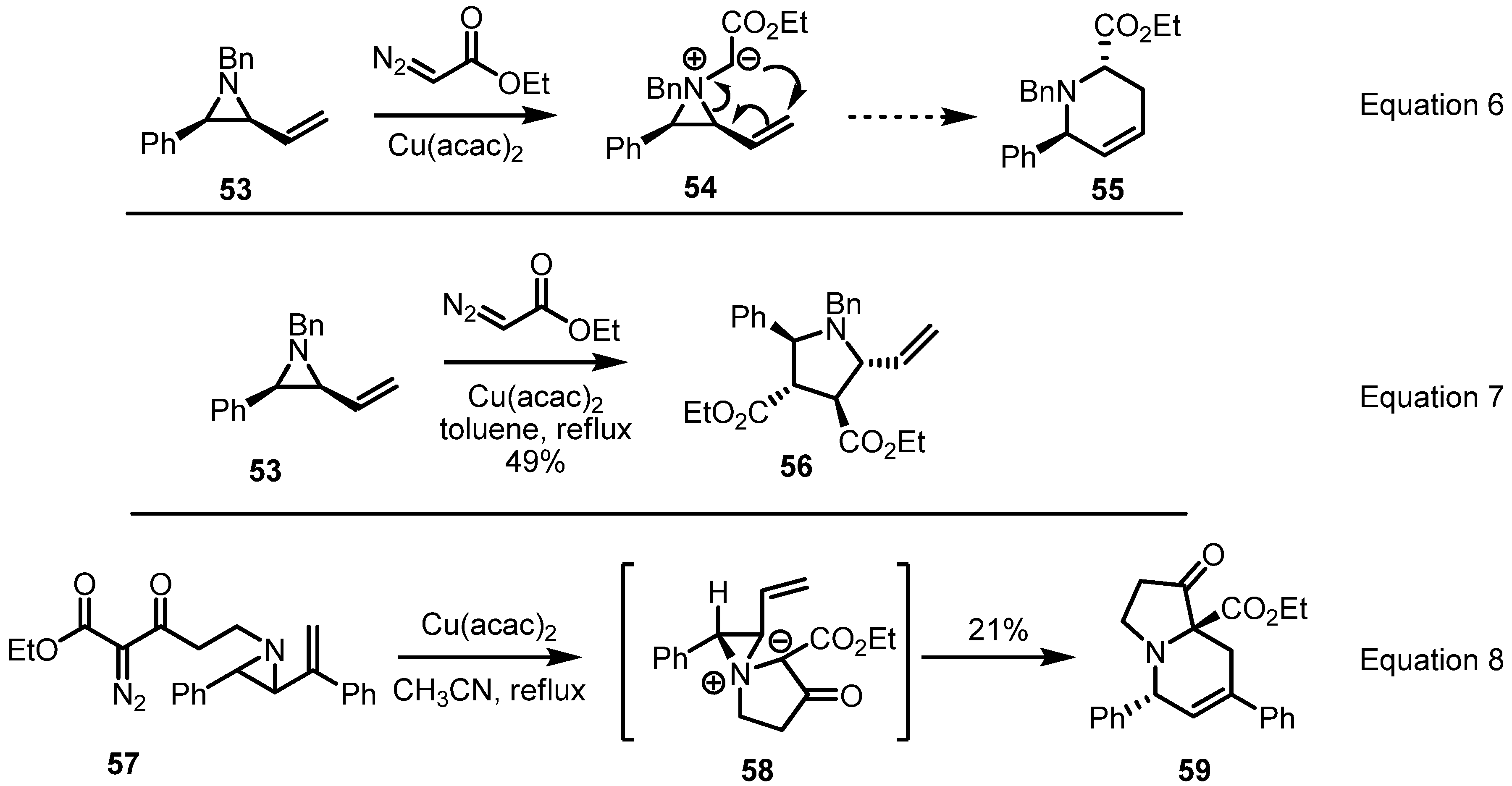
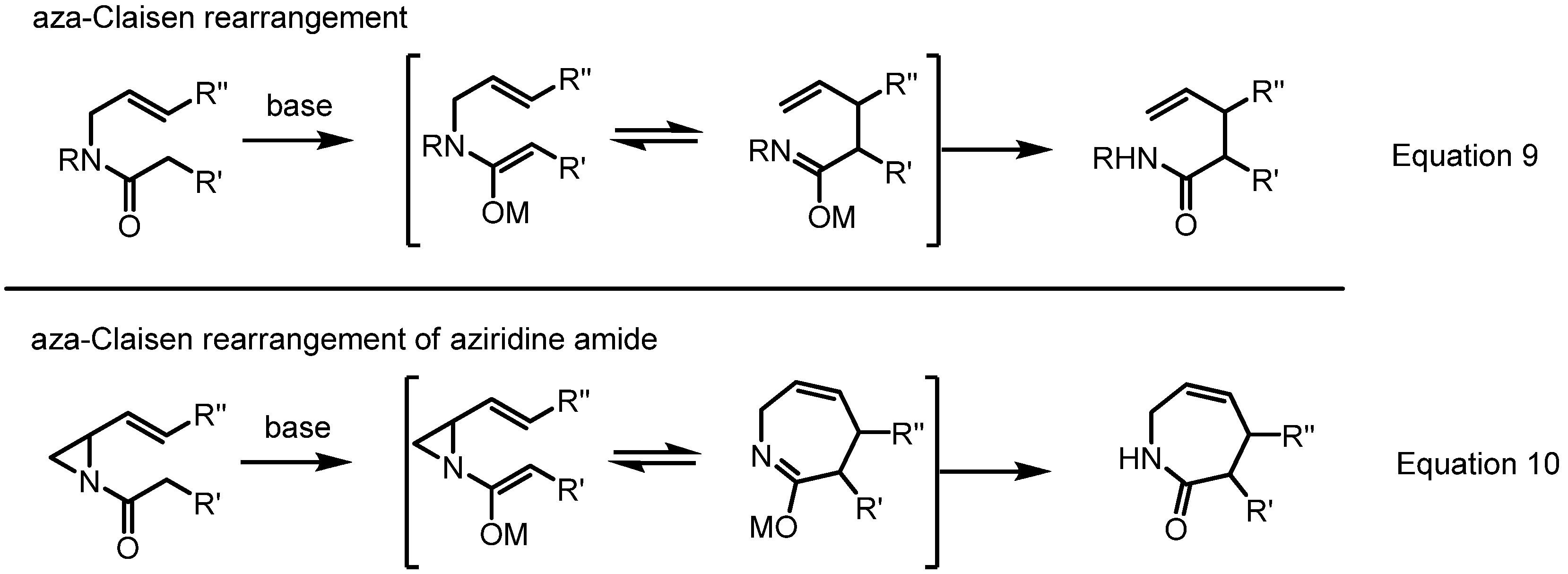
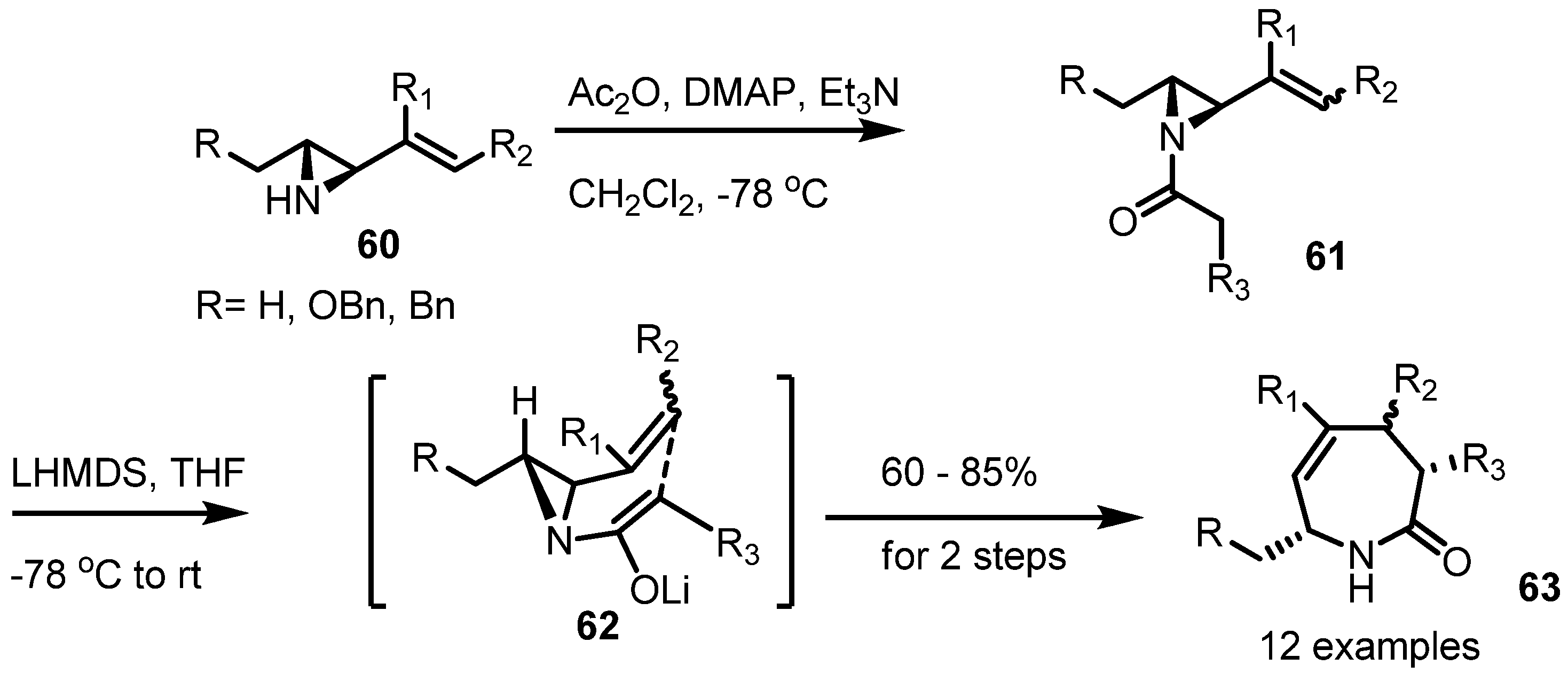
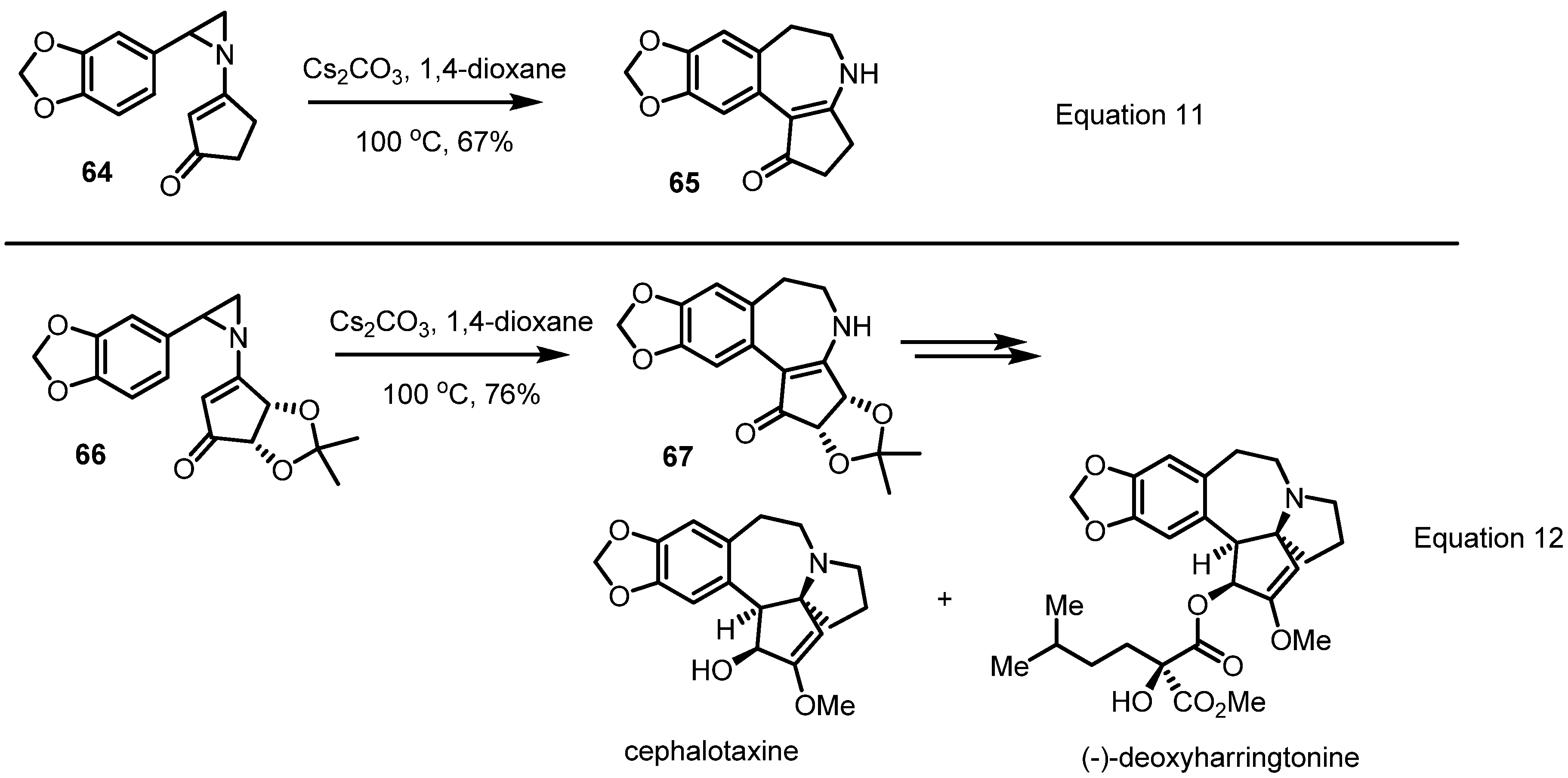
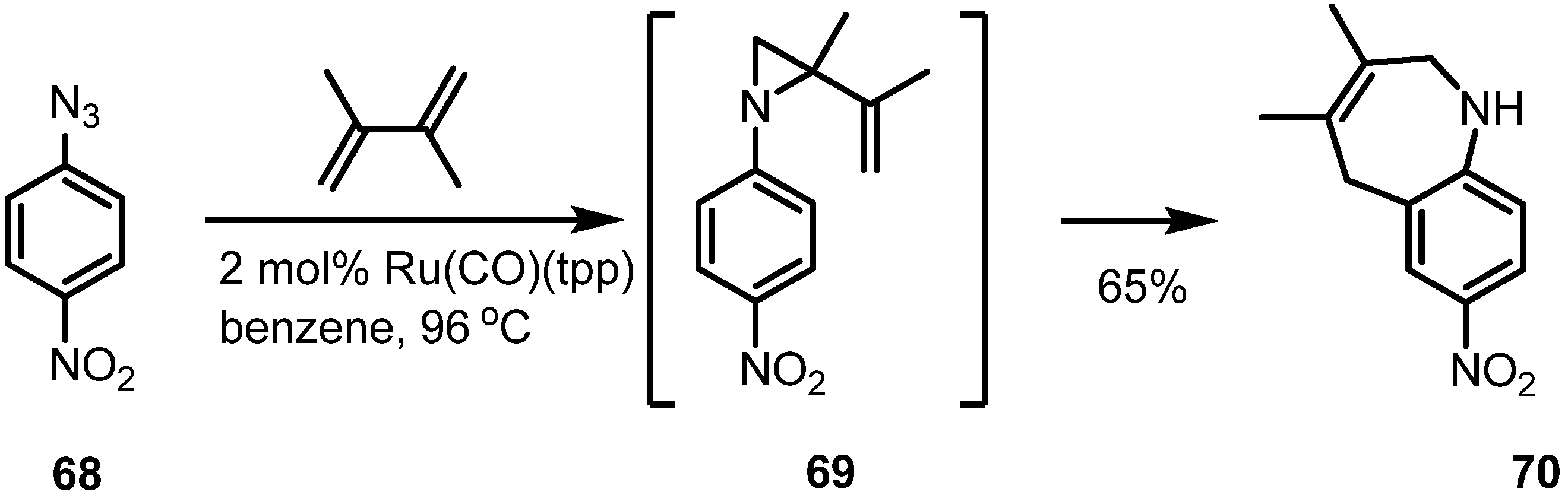
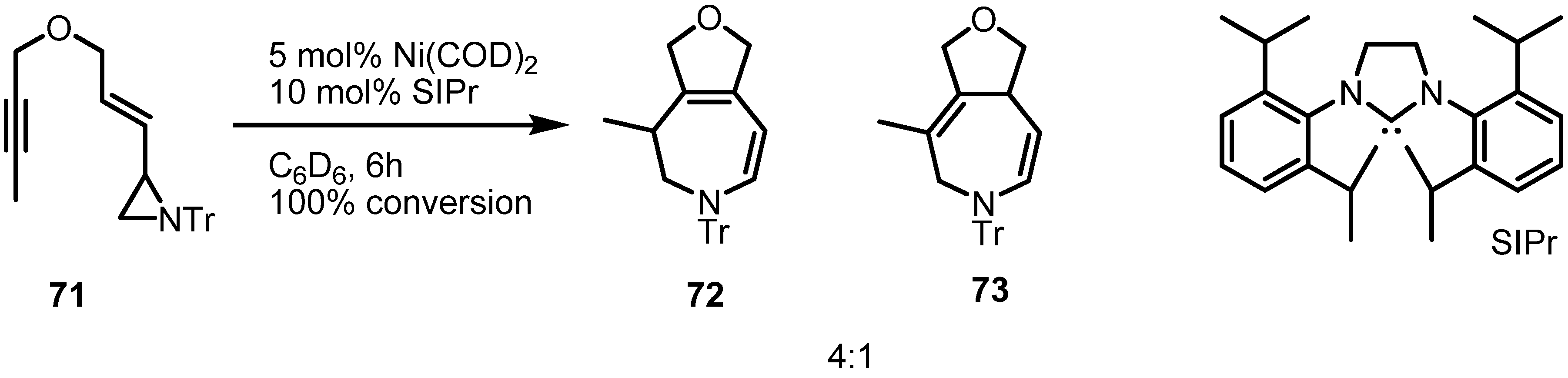
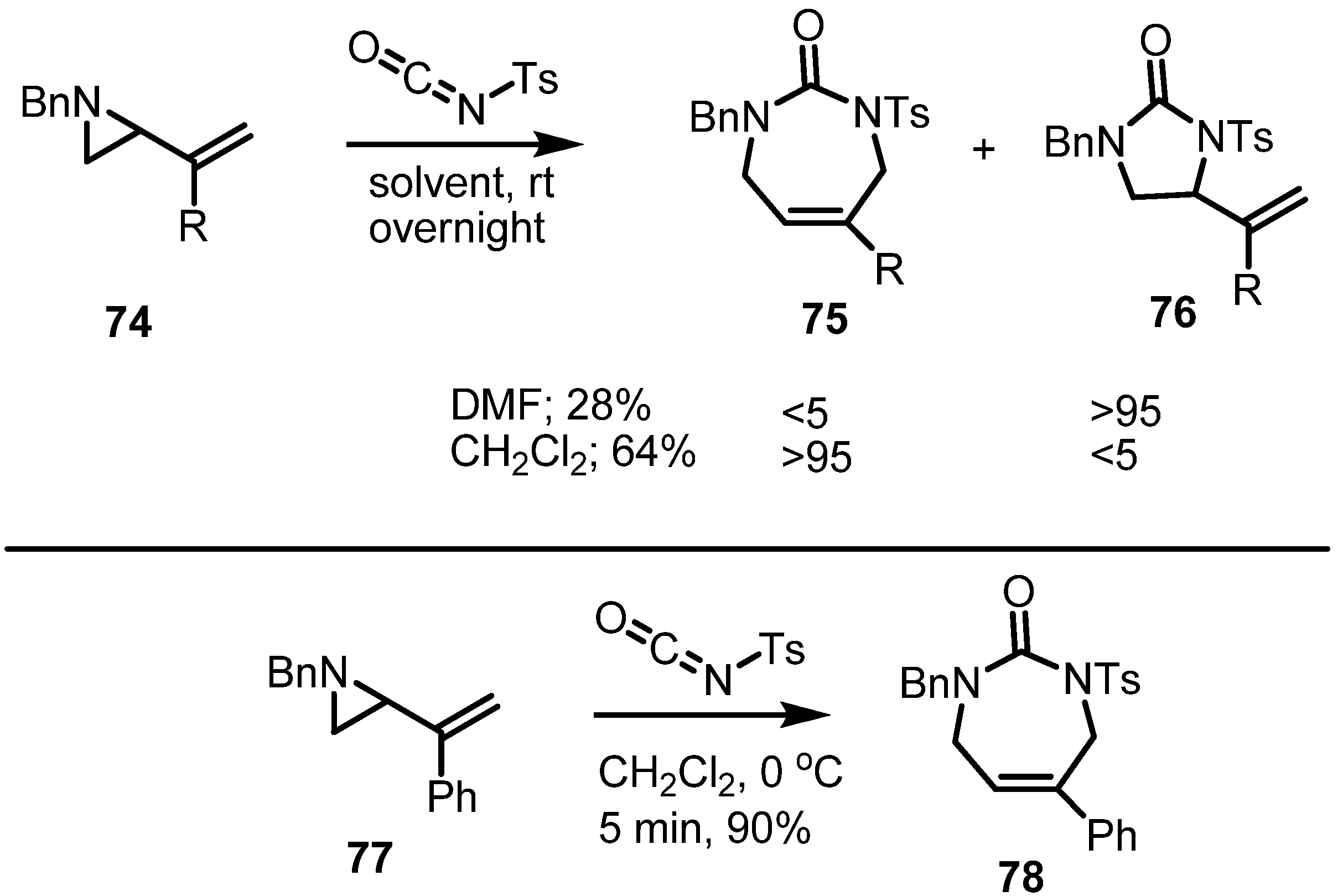
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cardoso, A.L.; Pinho e Melo, T.M.V.D. Aziridines in formal [3+2] cycloadditions: Synthesis of five-membered heterocycles. Eur. J. Org. Chem. 2012, 33, 6479–6501. [Google Scholar]
- Scheiner, P. Rearrangements of a 2-vinylaziridine. J. Org. Chem. 1967, 32, 2628–2630. [Google Scholar] [CrossRef]
- Stogryn, E.L.; Brois, S.J. The valence isomerization of 1,2-divinylaziridines. Synthetic and kinetic studies. J. Am. Chem. Soc. 1967, 89, 605–609. [Google Scholar] [CrossRef]
- Tanner, D.; Somfai, P. Palladium-catalyzed transformation of a chiral vinylaziridine to a β-lactam. An enantioselective route to the carbapenem (+)-PS-5. Bioorg. Med. Chem. Lett. 1993, 3, 2415–2418. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Alonso, E.; Fang, G.; Ferrara, M.; Hynd, G.; Porcelloni, M. Application of chiral sulfides to catalytic asymmetric aziridination and cyclopropanation with in situ generation of the diazo compound. Angew. Chem. Int. Ed. Engl. 2001, 40, 1433–1436. [Google Scholar] [CrossRef]
- Fontana, F.; Tron, G.C.; Barbero, N.; Ferrini, S.; Thomas, S.P.; Aggarwal, V.K. Stereoselective synthesis of trans-beta-lactams by palladium-catalysed carbonylation of vinylaziridines. Chem. Commun. 2010, 46, 267–269. [Google Scholar]
- Branchadell, V.; Moreno-Mañas, M.; Pleixats, R. Theoretical study on the regioselectivity of nucleophilic attack in silyl-substituted (diphosphino)(η3-allyl) palladium cations. Organometallics 2002, 21, 2407–2412. [Google Scholar] [CrossRef]
- Butler, D.C.; Inman, G.A.; Alper, H. Room temperature ring-opening cyclization reactions of 2-vinylaziridines with isocyanates, carbodiimides, and isothiocyanates catalyzed by [Pd(OAc)2]/PPh3. J. Org. Chem. 2000, 65, 5887–5890. [Google Scholar] [CrossRef]
- Trost, B.M.; Fandrick, D.R. Dynamic kinetic asymmetric cycloadditions of isocyanates to vinylaziridines. J. Am. Chem. Soc. 2003, 125, 11836–11837. [Google Scholar] [CrossRef]
- Fontana, F.; Chen, C.C.; Aggarwal, V.K. Palladium-catalyzed insertion of CO2 into vinylaziridines: New route to 5-vinyloxazolidinones. Org. Lett. 2011, 13, 3454–3457. [Google Scholar] [CrossRef]
- Aoyagi, K.; Nakamura, H.; Yamamoto, Y. Palladium-catalyzed aminoallylation of activated olefins with allylic halides and phthalimide. J. Org. Chem. 2002, 67, 5977–5980. [Google Scholar] [CrossRef]
- Lowe, M.A.; Ostovar, M.; Ferrini, S.; Chen, C.C.; Lawrence, P.G.; Fontana, F.; Calabrese, A.A.; Aggarwal, V.K. Palladium-mediated annulation of vinylaziridines with michael acceptors: Stereocontrolled synthesis of substituted pyrrolidines and its application in a formal synthesis of (−)-α-kainic acid. Angew. Chem. Int. Ed. Engl. 2011, 123, 6494–6498. [Google Scholar] [CrossRef]
- Baktharaman, S.; Afagh, N.; Vandersteen, A.; Yudin, A.K. Unprotected vinylaziridines: Facile synthesis and cascade transformations. Org. Lett. 2010, 12, 240–243. [Google Scholar] [CrossRef]
- Brichacek, M.; Lee, D.; Njardarson, J.T. Lewis Acid catalyzed [1,3]-sigmatropic rearrangement of vinylaziridines. Org. Lett. 2008, 10, 5023–5026. [Google Scholar] [CrossRef]
- Mack, D.J.; Njardarson, J.T. New Mechanistic insights into the copper catalyzed ring expansion of vinylaziridines: Evidence in support of a copper(I) mediated pathway. Chem. Sci. 2012, 3, 3321–3325. [Google Scholar] [CrossRef]
- Brichacek, M.; Navarro Villalobos, M.; Plichta, A.; Njardarson, J.T. Stereospecific ring expansion of chiral vinylaziridines. Org. Lett. 2011, 13, 1110–1113. [Google Scholar] [CrossRef]
- Hirner, J.J.; Roth, K.E.; Shi, Y.; Blum, S.A. Mechanistic Studies of azaphilic versus carbophilic activation by gold (I) in the gold/palladium dual-catalyzed rearrangement of alkenyl vinylaziridines. Organometallics 2012, 31, 6843–6850. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoyagi, S. Total Synthesis of (−)-Chamobtusin A. Org. Lett. 2012, 14, 6374–6376. [Google Scholar] [CrossRef]
- Hirner, S.; Somfai, P. Microwave-assisted rearrangement of vinylaziridines to 3-pyrrolines: Formal synthesis of (−)-anisomycin. Synlett 2005, 20, 3099–3102. [Google Scholar]
- Buchanan, J.G.; MacLean, K.A.; Wightman, R.H.; Paulsen, H. A new synthesis of (–)-anisomycin and its demethoxy analogue from D-ribose. J. Chem. Soc. Perkin Trans. 1 1985, 1463–1470. [Google Scholar]
- Leemans, E.; Colpaert, F.; Mangelinckx, S.; de Brabandere, S.; Denolf, B.; de Kimpe, N. Synthesis of 3-aryl-3-pyrrolines and 3-arylpyrroles via spontaneous rearrangement of N-sulfinyl 2-aryl-2-vinylaziridines. Synlett 2011, 5, 674–678. [Google Scholar]
- Coldham, I.; Collis, A.J.; Mould, R.J.; Rathmell, R.E. Ring expansion of aziridines to piperidines using the aza-wittig rearrangement. Tetrahedron Lett. 1995, 36, 3557–3560. [Google Scholar]
- Ahman, J.; Somfai, P. Enantioselective total synthesis of (−)-indolizidines 209B and 209D via a highly efficient aza-[2,3]-wittig rearrangement of vinylaziridines. Tetrahedron 1995, 51, 9747–9756. [Google Scholar] [CrossRef]
- Rowlands, G.J.; Barnes, W.K. Studies on the [2,3]-Stevens rearrangement of aziridinium ions. Tetrahedron Lett. 2004, 45, 5347–5350. [Google Scholar] [CrossRef]
- Lindström, U.M.; Somfai, P. Aza-[3,3]-claisen enolate rearrangement in vinylaziridines: Stereoselective synthesis of mono-, di-, and trisubstituted seven-membered lactams. Chem. Eur. J. 2001, 7, 94–98. [Google Scholar] [CrossRef]
- Eckelbarger, J.D.; Wilmot, J.T.; Gin, D.Y. Strain-release rearrangement of N-vinyl-2-arylaziridines. Total synthesis of the anti-leukemia alkaloid (−)-deoxyharringtonine. J. Am. Chem. Soc. 2006, 128, 10370–10371. [Google Scholar] [CrossRef]
- Fantauzzi, S.; Gallo, E.; Caselli, A.; Piangiolino, C.; Ragaini, F.; Re, N.; Cenini, S. Rearrangement of N-aryl-2-vinylaziridines to benzoazepines and dihydropyrroles: A synthetic and theoretical study. Chem. Eur. J. 2009, 15, 1241–1251. [Google Scholar] [CrossRef]
- Zuo, G.; Zhang, K.; Louie, J. Nickel-catalyzed reactions of vinylaziridines and aziridinylen-ynes. Tetrahedron Lett. 2008, 49, 6797–6799. [Google Scholar] [CrossRef]
- Kanno, E.; Yamanoi, K.; Koya, S.; Azumaya, I.; Masu, H.; Yamasaki, R.; Saito, S. [5+2] Cycloaddition reaction of 2-vinylaziridines and sulfonyl isocyanates. Synthesis of seven-membered cyclic ureas. J. Org. Chem. 2012, 77, 2142–2148. [Google Scholar] [CrossRef]
- Sample Availability: Not availability.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heo, Y.M.; Paek, S.-M. Ring Expansion of Vinylaziridines through the Strain-Release Pericyclic Reaction: Recent Developments and Applications. Molecules 2013, 18, 9650-9662. https://doi.org/10.3390/molecules18089650
Heo YM, Paek S-M. Ring Expansion of Vinylaziridines through the Strain-Release Pericyclic Reaction: Recent Developments and Applications. Molecules. 2013; 18(8):9650-9662. https://doi.org/10.3390/molecules18089650
Chicago/Turabian StyleHeo, Yu Mi, and Seung-Mann Paek. 2013. "Ring Expansion of Vinylaziridines through the Strain-Release Pericyclic Reaction: Recent Developments and Applications" Molecules 18, no. 8: 9650-9662. https://doi.org/10.3390/molecules18089650
APA StyleHeo, Y. M., & Paek, S.-M. (2013). Ring Expansion of Vinylaziridines through the Strain-Release Pericyclic Reaction: Recent Developments and Applications. Molecules, 18(8), 9650-9662. https://doi.org/10.3390/molecules18089650