
Transcriptional Regulatory Systems in Pseudomonas: A Comparative Analysis of Helix-Turn-Helix Domains and Two-Component Signal Transduction Networks

 and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Understanding the Genomic Sequences of P. aeruginosa PAO1 and P. putida KT2440: An Overview
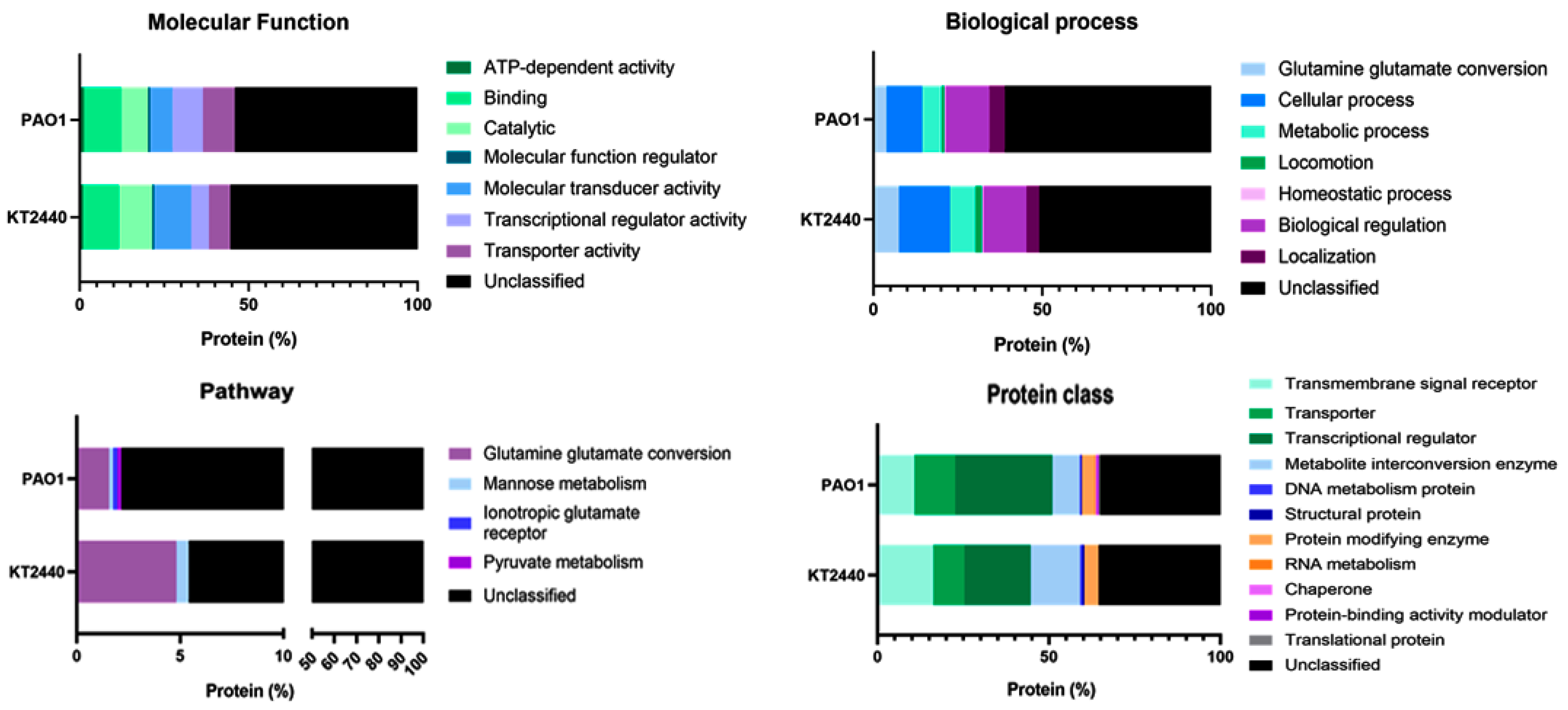
2.2. The Set of DNA-Binding Transcriptional Regulators in the Non-Pathogenic P. putida KT2440 and Pathogenic P. aeruginosa PAO1 Strains
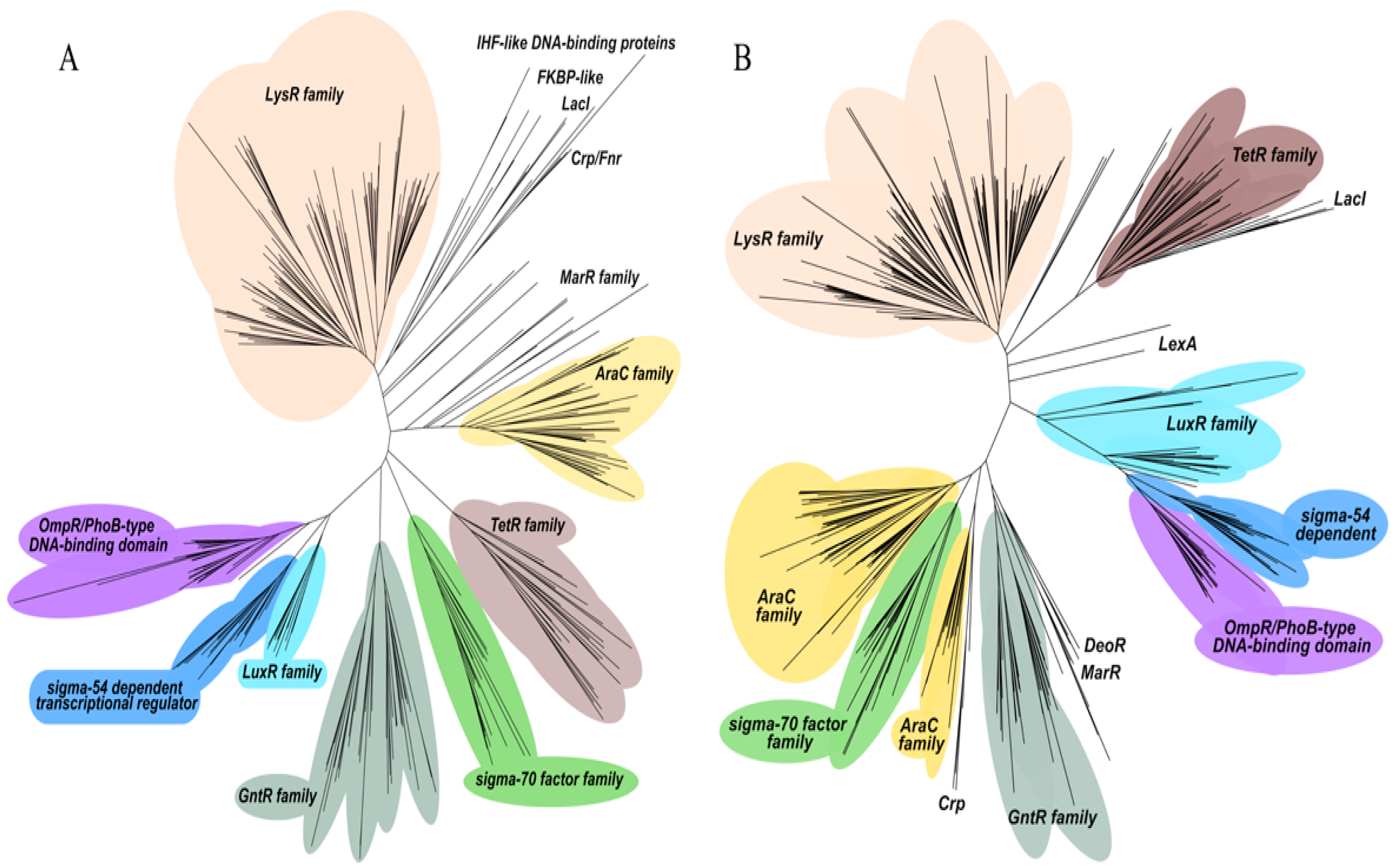
2.3. Categorization of Transcriptional Regulators in P. putida KT2440 and P. aeruginosa PAO1 into Regulatory Protein Families
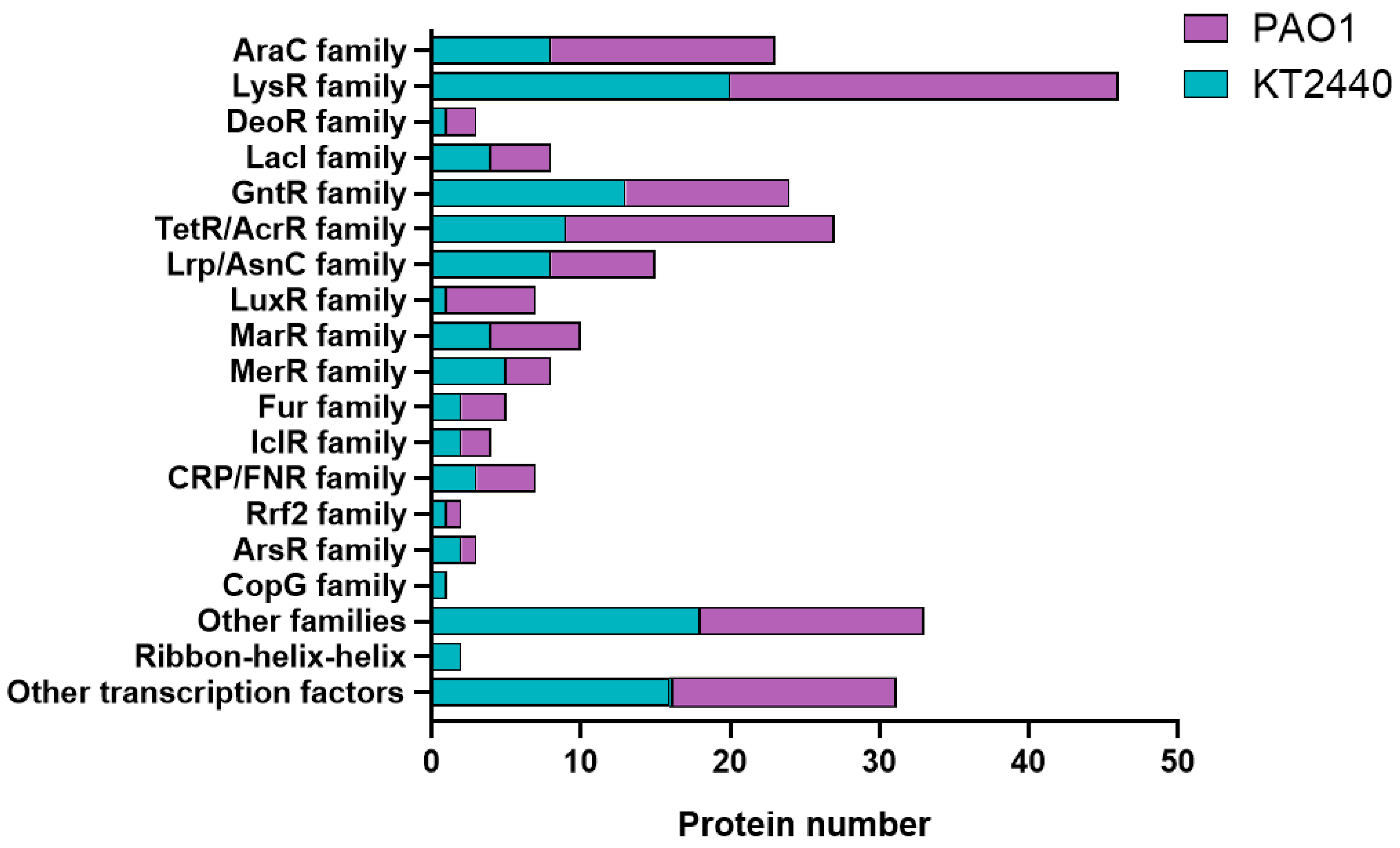
2.4. A Differential Repertoire Transcriptional Regulator Protein Is Evident in Pathogenic and Non-Pathogenic Pseudomonas Strains
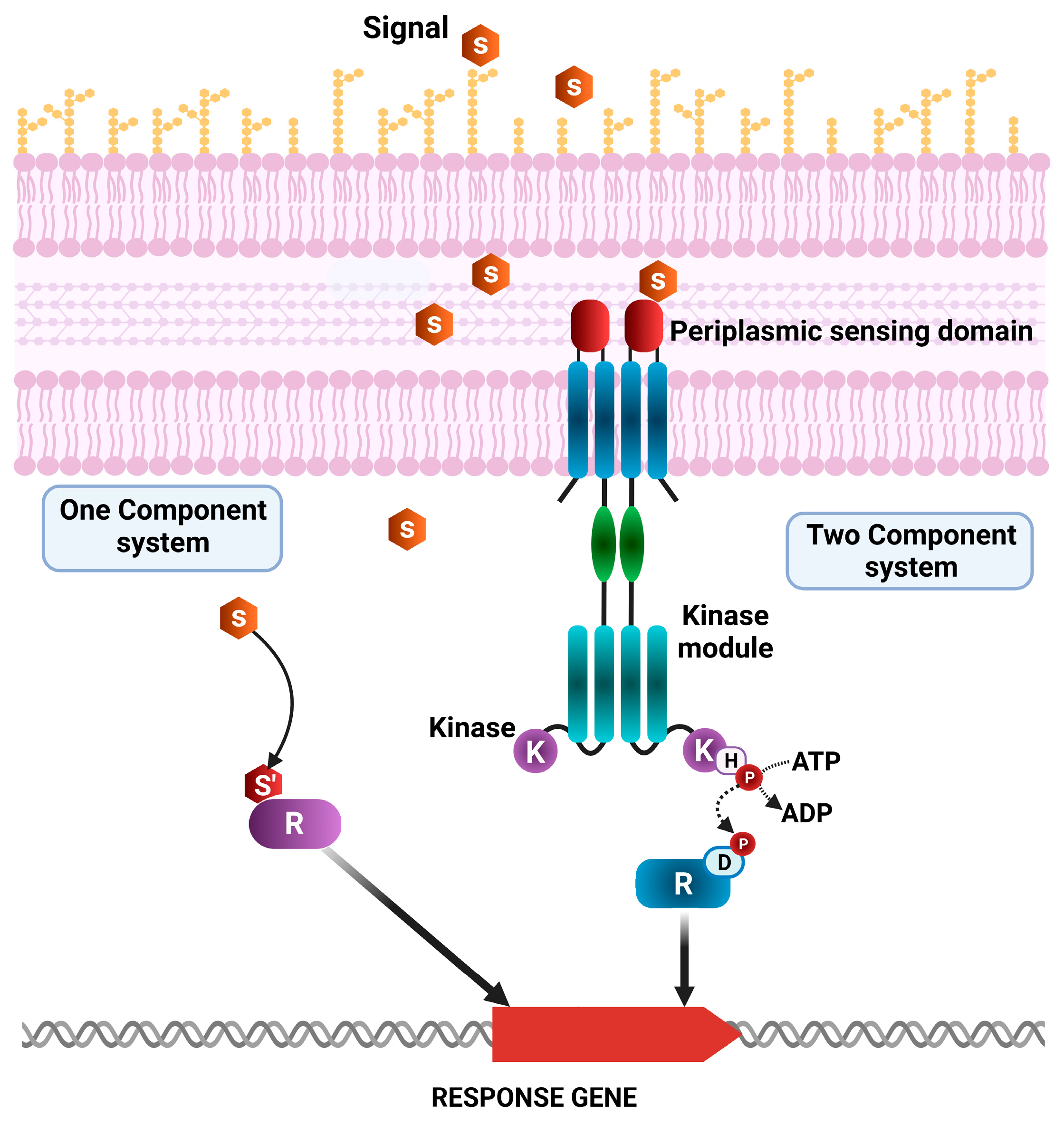
2.5. Two-Component System Function in Pathogenic and Non-Pathogenic Pseudomonas Strains
2.6. Forecasting and Choice of Transcriptional Regulators Associated with Pathogenicity
3. Material and Methods
3.1. Genomic Analysis
3.2. Distribution of DNA-Binding Proteins
3.3. Phylogenetic Analysis
3.4. Classification Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wani, A.K.; Akhtar, N.; Sher, F.; Navarrete, A.A.; Américo-Pinheiro, J.H.P. Microbial adaptation to different environmental conditions: Molecular perspective of evolved genetic and cellular systems. Arch. Microbiol. 2022, 204, 144. [Google Scholar] [CrossRef]
- Narciso-da-Rocha, C.; Manaia, C.M. Multidrug resistance phenotypes are widespread over different bacterial taxonomic groups thriving in surface water. Sci. Total Environ. 2016, 563–564, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abram, K.Z.; Jun, S.-R.; Udaondo, Z. Pseudomonas aeruginosa Pangenome: Core and Accessory Genes of a Highly Resourceful Opportunistic Pathogen. Adv. Exp. Med. Biol. 2022, 1386, 3–28. [Google Scholar] [CrossRef]
- Goldberg, J.B. Why is Pseudomonas aeruginosa a pathogen? F1000 Biol. Rep. 2010, 2, 29. [Google Scholar] [CrossRef]
- Daddaoua, A.; Fillet, S.; Fernández, M.; Udaondo, Z.; Krell, T.; Ramos, J.L. Genes for carbon metabolism and the ToxA virulence factor in Pseudomonas aeruginosa are regulated through molecular interactions of PtxR and PtxS. PLoS ONE 2012, 7, e39390. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gómez, J.M.; Santiago, C.M.; Udaondo, Z.; Garitaonaindia, M.T.; Krell, T.; Ramos, J.-L.; Daddaoua, A. Full Transcriptomic Response of Pseudomonas aeruginosa to an Inulin-Derived Fructooligosaccharide. Front. Microbiol. 2020, 11, 202. [Google Scholar] [CrossRef]
- Udaondo, Z.; Molina, L.; Daniels, C.; Gómez, M.J.; Molina-Henares, M.A.; Matilla, M.A.; Roca, A.; Fernández, M.; Duque, E.; Segura, A.; et al. Metabolic potential of the organic-solvent tolerant Pseudomonas putida DOT-T1E deduced from its annotated genome. Microb. Biotechnol. 2013, 6, 598–611. [Google Scholar] [CrossRef]
- Molina-Santiago, C.; Udaondo, Z.; Gómez-Lozano, M.; Molin, S.; Ramos, J.-L. Global transcriptional response of solvent-sensitive and solvent-tolerant Pseudomonas putida strains exposed to toluene. Environ. Microbiol. 2017, 19, 645–658. [Google Scholar] [CrossRef]
- Abram, K.Z.; Udaondo, Z. Towards a better metabolic engineering reference: The microbial chassis. Microb. Biotechnol. 2020, 13, 17–18. [Google Scholar] [CrossRef]
- Duque, E.; Udaondo, Z.; Molina, L.; de la Torre, J.; Godoy, P.; Ramos, J.L. Providing octane degradation capability to Pseudomonas putida KT2440 through the horizontal acquisition of oct genes located on an integrative and conjugative element. Environ. Microbiol. Rep. 2022, 14, 934–946. [Google Scholar] [CrossRef]
- Pizarro-Tobías, P.; Fernández, M.; Niqui, J.L.; Solano, J.; Duque, E.; Ramos, J.; Roca, A. Restoration of a Mediterranean forest after a fire: Bioremediation and rhizoremediation field-scale trial. Microb. Biotechnol. 2014, 8, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Molina, L.; Segura, A.; Duque, E.; Ramos, J.-L. The versatility of Pseudomonas putida in the rhizosphere environment. Adv. Appl. Microbiol. 2020, 110, 149–180. [Google Scholar] [CrossRef]
- Mikkelsen, H.; McMullan, R.; Filloux, A. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in ladS. PLoS ONE 2011, 6, e29113. [Google Scholar] [CrossRef]
- Chastre, J.; Fagon, J.-Y. Ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 2002, 165, 867–903. [Google Scholar] [CrossRef]
- Hassett, D.J.; Korfhagen, T.R.; Irvin, R.T.; Schurr, M.J.; Sauer, K.; Lau, G.W.; Sutton, M.D.; Yu, H.; Hoiby, N. Pseudomonas aeruginosa biofilm infections in cystic fibrosis: Insights into pathogenic processes and treatment strategies. Expert Opin. Ther. Targets 2010, 14, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Montie, T.C.; Doyle-Huntzinger, D.; Craven, R.C.; Holder, I.A. Loss of virulence associated with absence of flagellum in an isogenic mutant of Pseudomonas aeruginosa in the burned-mouse model. Infect. Immun. 1982, 38, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Montie, T.C.; Craven, R.C.; Holder, I.A. Flagellar preparations from Pseudomonas aeruginosa: Isolation and characterization. Infect. Immun. 1982, 35, 281–288. [Google Scholar] [CrossRef]
- Cano, P.G.; Santacruz, A.; Trejo, F.M.; Sanz, Y. BifidobacteriumCECT 7765 improves metabolic and immunological alterations associated with obesity in high-fat diet-fed mice. Obesity 2013, 21, 2310–2321. [Google Scholar] [CrossRef]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Ohara, T.; Itoh, K. Significance of Pseudomonas aeruginosa colonization of the gastrointestinal tract. Intern. Med. 2003, 42, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- von Klitzing, E.; Bereswill, S.; Heimesaat, M.M. Multidrug-Resistant Pseudomonas aeruginosa Induce Systemic Pro-Inflammatory Immune Responses in Colonized Mice. Eur. J. Microbiol. Immunol. 2017, 7, 200–209. [Google Scholar] [CrossRef]
- Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Hancock, R.E.W.; Martínez, J.L. The intrinsic resistome of Pseudomonas aeruginosa to β-lactams. Virulence 2011, 2, 144–146. [Google Scholar] [CrossRef]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Genes Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef]
- Hall, J.P.J.; Brockhurst, M.A.; Harrison, E. Sampling the mobile gene pool: Innovation via horizontal gene transfer in bacteria. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2017, 372, 20160424. [Google Scholar] [CrossRef]
- Hogardt, M.; Heesemann, J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 2010, 300, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Mah, T.-F.; Pitts, B.; Pellock, B.; Walker, G.C.; Stewart, P.S.; O’Toole, G.A. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 2003, 426, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.K.; Yeung, A.T.Y.; Hancock, R.E.W. Antibiotic resistance in Pseudomonas aeruginosa biofilms: Towards the development of novel anti-biofilm therapies. J. Biotechnol. 2014, 191, 121–130. [Google Scholar] [CrossRef]
- Iglewski, B.H.; Kabat, D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc. Natl. Acad. Sci. USA 1975, 72, 2284–2288. [Google Scholar] [CrossRef]
- Ortiz-Castro, R.; Pelagio-Flores, R.; Méndez-Bravo, A.; Ruiz-Herrera, L.F.; Campos-García, J.; López-Bucio, J. Pyocyanin, a virulence factor produced by Pseudomonas aeruginosa, alters root development through reactive oxygen species and ethylene signaling in Arabidopsis. Mol. Plant. Microbe. Interact. 2014, 27, 364–378. [Google Scholar] [CrossRef]
- Matar, G.M.; Ramlawi, F.; Hijazi, N.; Khneisser, I.; Abdelnoor, A.M. Transcription levels of Pseudomonas aeruginosa exotoxin a gene and severity of symptoms in patients with otitis externa. Curr. Microbiol. 2002, 45, 350–354. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.L.; Kirienko, N.V.; Ausubel, F.M. Host translational inhibition by Pseudomonas aeruginosa Exotoxin A Triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 2012, 11, 364–374. [Google Scholar] [CrossRef]
- Yates, S.P.; Jørgensen, R.; Andersen, G.R.; Merrill, A.R. Stealth and mimicry by deadly bacterial toxins. Trends Biochem. Sci. 2006, 31, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hamood, A.N.; Ohman, D.E.; West, S.E.; Iglewski, B.H. Isolation and characterization of toxin A excretion-deficient mutants of Pseudomonas aeruginosa PAO1. Infect. Immun. 1992, 60, 510–517. [Google Scholar] [CrossRef]
- Durand, E.; Verger, D.; Rêgo, A.T.; Chandran, V.; Meng, G.; Fronzes, R.; Waksman, G. Structural biology of bacterial secretion systems in gram-negative pathogens--potential for new drug targets. Infect. Disord. Drug Targets 2009, 9, 518–547. [Google Scholar] [CrossRef]
- Bleves, S.; Viarre, V.; Salacha, R.; Michel, G.P.F.; Filloux, A.; Voulhoux, R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int. J. Med. Microbiol. 2010, 300, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, L.P.; Wood, T.E.; Howard, S.A.; Maggiorelli, F.; Nolan, L.M.; Wettstadt, S.; Filloux, A. RsmA and AmrZ orchestrate the assembly of all three type VI secretion systems in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2017, 114, 7707–7712. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef]
- Ulrich, L.E.; Koonin, E.V.; Zhulin, I.B. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 2005, 13, 52–56. [Google Scholar] [CrossRef]
- Mascher, T. Intramembrane-sensing histidine kinases: A new family of cell envelope stress sensors in Firmicutes bacteria. FEMS Microbiol. Lett. 2006, 264, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Hazelbauer, G.L.; Falke, J.J.; Parkinson, J.S. Bacterial chemoreceptors: High-performance signaling in networked arrays. Trends Biochem. Sci. 2008, 33, 9–19. [Google Scholar] [CrossRef]
- Galperin, M.Y. A census of membrane-bound and intracellular signal transduction proteins in bacteria: Bacterial IQ, extroverts and introverts. BMC Microbiol. 2005, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Nixon, B.T.; Ronson, C.W.; Ausubel, F.M. Two-component regulatory systems responsive to environmental stimuli share strongly conserved domains with the nitrogen assimilation regulatory genes ntrB and ntrC. Proc. Natl. Acad. Sci. USA 1986, 83, 7850–7854. [Google Scholar] [CrossRef]
- Bijlsma, J.J.E.; Groisman, E.A. Making informed decisions: Regulatory interactions between two-component systems. Trends Microbiol. 2003, 11, 359–366. [Google Scholar] [CrossRef]
- Bourret, R.B.; Silversmith, R.E. Two-component signal transduction. Curr. Opin. Microbiol. 2010, 13, 113–115. [Google Scholar] [CrossRef]
- Cheung, J.; Hendrickson, W.A. Sensor domains of two-component regulatory systems. Curr. Opin. Microbiol. 2010, 13, 116–123. [Google Scholar] [CrossRef]
- Groisman, E.A. Feedback Control of Two-Component Regulatory Systems. Annu. Rev. Microbiol. 2016, 70, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Gislason, A.S.; Choy, M.; Bloodworth, R.A.M.; Qu, W.; Stietz, M.S.; Li, X.; Zhang, C.; Cardona, S.T. Competitive Growth Enhances Conditional Growth Mutant Sensitivity to Antibiotics and Exposes a Two-Component System as an Emerging Antibacterial Target in Burkholderia cenocepacia. Antimicrob. Agents Chemother. 2017, 61, e00790-16. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Groisman, E.A. Signal integration in bacterial two-component regulatory systems. Genes Dev. 2008, 22, 2601–2611. [Google Scholar] [CrossRef]
- Nelson, K.E.; Weinel, C.; Paulsen, I.T.; Dodson, R.J.; Hilbert, H.; Martins dos Santos, V.A.P.; Fouts, D.E.; Gill, S.R.; Pop, M.; Holmes, M.; et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002, 4, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Belda, E.; van Heck, R.G.A.; José Lopez-Sanchez, M.; Cruveiller, S.; Barbe, V.; Fraser, C.; Klenk, H.-P.; Petersen, J.; Morgat, A.; Nikel, P.I.; et al. The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ. Microbiol. 2016, 18, 3403–3424. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Tettmann, B.; Dötsch, A.; Armant, O.; Fjell, C.D.; Overhage, J. Knockout of extracytoplasmic function sigma factor ECF-10 affects stress resistance and biofilm formation in Pseudomonas putida KT2440. Appl. Environ. Microbiol. 2014, 80, 4911–4919. [Google Scholar] [CrossRef] [PubMed]
- Brune, I.; Brinkrolf, K.; Kalinowski, J.; Pühler, A.; Tauch, A. The individual and common repertoire of DNA-binding transcriptional regulators of Corynebacterium glutamicum, Corynebacterium efficiens, Corynebacterium diphtheriae and Corynebacterium jeikeium deduced from the complete genome sequences. BMC Genomics 2005, 6, 86. [Google Scholar] [CrossRef]
- Lindquist, S.; Lindberg, F.; Normark, S. Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC beta-lactamase gene. J. Bacteriol. 1989, 171, 3746–3753. [Google Scholar] [CrossRef]
- Schell, M.A. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 1993, 47, 597–626. [Google Scholar] [CrossRef]
- Deng, W.; Li, C.; Xie, J. The underling mechanism of bacterial TetR/AcrR family transcriptional repressors. Cell. Signal. 2013, 25, 1608–1613. [Google Scholar] [CrossRef]
- Colclough, A.L.; Scadden, J.; Blair, J.M.A. TetR-family transcription factors in Gram-negative bacteria: Conservation, variation and implications for efflux-mediated antimicrobial resistance. BMC Genomics 2019, 20, 731. [Google Scholar] [CrossRef]
- Routh, M.D.; Su, C.-C.; Zhang, Q.; Yu, E.W. Structures of AcrR and CmeR: Insight into the mechanisms of transcriptional repression and multi-drug recognition in the TetR family of regulators. Biochim. Biophys. Acta 2009, 1794, 844–851. [Google Scholar] [CrossRef]
- Scharf, B.E. Summary of useful methods for two-component system research. Curr. Opin. Microbiol. 2010, 13, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Utsumi, R. Introduction to bacterial signal transduction networks. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2008; Volume 631, pp. 1–6. [Google Scholar] [CrossRef]
- Gotoh, Y.; Eguchi, Y.; Watanabe, T.; Okamoto, S.; Doi, A.; Utsumi, R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 2010, 13, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Schaefers, M.M.; Liao, T.L.; Boisvert, N.M.; Roux, D.; Yoder-Himes, D.; Priebe, G.P. An Oxygen-Sensing Two-Component System in the Burkholderia cepacia Complex Regulates Biofilm, Intracellular Invasion, and Pathogenicity. PLoS Pathog. 2017, 13, e1006116. [Google Scholar] [CrossRef]
- Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef]
- Barrett, J.F.; Isaacson, R.E. Chapter 12. Bacterial Virulence as a Potential Target for Therapeutic Intervention. In Annual Reports in Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 1995; Volume 30, pp. 111–118. [Google Scholar]
- Rodrigue, A.; Quentin, Y.; Lazdunski, A.; Méjean, V.; Foglino, M. Two-component regulatory systems in Pseudomonas aeruginosa: Why so many, and what are they doing? Microbiology 2000, 8, 498–504. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCS Family | P. putida KT2440 | P. aeruginosa PAO1 | Key Functions |
|---|---|---|---|
| OmpR family | |||
| PhoR-PhoB | PP_5321 (phoR)/PP_5320 (phoB) | PA5361 (phoR)/PA5360 (phoB) | Phosphate starvation response |
| PhoQ-PhoP | PP_1187 (phoQ)/PP_1186 (phoP) | PA1180 (phoQ)/PA1179 (phoP) | Magnesium transport |
| EnvZ-OmpR | PP_0247 (envZ)/PP_0246 (ompR) | PA5199 (envZ)/PA5200 (ompR) | Osmotic stress response |
| RstB-RstA | PP_1182 (rstB)/PP_1181 (rstA) | PA1158 (rstB)/PA1157 (rstA) | Envelope stress response |
| CpxA-CpxR | nd/PP_3372 (cpxR) | nd/PA3204 (cpxR) | Envelope stress response |
| CusS-CusR | PP_1437 (cusS)/PP_1438 (czcR-III), PP_2157 (cusS)/PP_2158 (copR-I), PP_5384 (copS)/PP_5383, (copR-II) Others: PP_0030 (czrSA)/nd | PA1438 (cusS)/PA1437 (cusR), PA4886 (cusS)/PA4885 (cusR), PA2524 (cusS)/PA2523 (cusR), PA2810 (cusS)/PA2809 (cusR) | Copper resistance and heavy metal tolerance |
| QseC-QseB | PP_2714 (qseC)/PP_2713 (qseB) | PA4777 (qseC)/PA4776 (qseB), PA2480 (qseC)/PA2479 (qseB) | Quorum-sensing |
| KdpD-KdpE | PP_4158 (kdpD)/PP_4157 (kdpE) | PA1636 (kdpD)/PA1637 (kdpE) | Potassium transport |
| TctE-TctD | PP_1421 (tctE)/PP_1420 (tctD) | PA0757 (tctE)/PA0756 (tctD) | Tricarboxylic acid transport |
| PfeS-PfeR | PP_0533 (pfeS-I)/PP_0534(pfeR), PP_1652 (pfeSII)/PP_1651 (pfeR) | PA2687 (pfeS)/PA2686 (pfeR), PA0930 (pirS)/PA0929 (pirR) | Iron acquisition |
| Unclassified | PP_3453 (nd)/PP_3454 (nd), PP_2403 (nd), PP_2907 (nd), PP_4224 (nd)/nd | PA3206 (cpxS)/nd, nd/PA4101 (bfmR), nd/PA2657 (bqsR), PA3191 (gltS)/PA3192 (gltR) | Miscellaneous roles in signal transduction and response regulation |
| NarL family | |||
| UhpB-UhpA | PP_2671 (uhpB)/PP_0410 (uhpA) | PA1980 (eraR)/PA0410 (uhpA) | Hexose phosphate uptake |
| BarA-UvrY | PP_1650 (gacS)/nd, nd/PP_4099 (uvrY) | PA0928 (gacS)/PA2586 (gacA) | Central carbon metabolism regulation |
| EvgS-EvgA | PP_2100 (evgS)/PP_2101 (evgA), PP_3413 (evgS)/nd, nd/PP_1090 (bvgA) | PA3946 (rocS1)/PA3948(rocA1), PA2583 (evgS)/nd, nd/PA3045 (evgA) | Acid tolerance, drug resistance, virulence regulation |
| LytTR family | |||
| AlgZ-AlgR | nd/PP_0185 (algR) | PA5262 (algZ)/PA5261 (algR) | Alginate biosynthesis regulation |
| NtrC family | |||
| GlnL-GlnG | PP_5047 (glnL)/PP_5048 (glnG) | PA5124 (ntrB)/PA5125 (ntrC) | Nitrogen regulation |
| DctB-DctD | PP_0264 (nd)/PP_0263 (dctD-I), PP_1402 (dctB)/PP_1401 (dctD-III) | PA5165 (dctB)/PA5166 (dctD), PA5512 (mifS)/PA5511 (mifR) | C4-dicarboxylate transport regulation |
| KinB-AlgB | PP_0132 (kinB)/PP_0133 (algB) | PA5484 (kinB)/PA5483 (algB) | Alginate biosynthesis regulation |
| Unclassified | PP_2945 (flgS)/nd | PA2571 (flgS)/nd | Miscellaneous roles in signal transduction and response regulation |
| CheA family | |||
| CheA-CheYBV | PP_4338 (cheA)/PP_4337 (cheBA), nd/PP_4340 (cheY), nd/PP_3759 (cheB), nd/PP_0802 (cheV), nd/PP_2128 (cheV), nd/PP_4393 (nd) | PA0178 (cheA)/nd, PA1458 (cheA)/PA1459 (cheB), nd/PA0179 (nd), nd/PA1456 (cheY), nd/PA0173 (nd), nd/PA3349 (cheV) | Chemotaxis |
| ChpA-ChpB/PilGH | nd/PP_4988 (nd), PP_4992 (pilG)/PP_4991 (pilH) | PA0413(chpA)/PA0414 (chpB), PA0408 (pilG)/PA0409 (pilH) | Chemosensory signal transduction and motility regulation |
| WspE-WspRF | PP_1492 (wspE)/PP_1494 (wspR) | PA3703 (wspF)/PA3702 (wspR), PA3704 (wspE)/PA3702 (wspR) | Chemosensory regulation; biofilm formation |
| Cph1-Rcp1 | PP_2356 (cph1)/nd | nd/nd | Light-response regulation |
| Other families | |||
| FlrB-FlrC | PP_4371 (atoC)/PP_4372 (fleS) | PA1098 (fleS)/PA1099 (fleR) | Flagellar synthesis regulation |
| AauS-AauR | PP_1066 (dctD-II)/PP_1067 (nd) | PA1335 (auuR)/PA1336 (auuS) | Acidic amino acid utilization regulation |
| RegB-RegA | PP_0887 (nd)/PP_0888 (regA) | PA4494 (roxS)/PA4493 (roxR) | Redox and oxidative stress response regulation |
| SagS-HptB-HsbR | PP_4362 (nd)/PP_4363 (nd), nd/PP_2664 (nd), nd/PP_1875 (nd), nd/PP_4173 (nd) | PA2824 (sagS)/nd, nd/PA1611 (nd), PA1976 (ercS)/nd, PA3345 (hptB)/PA3346 (hsbR) | Swarming, biofilm formation, and signaling |
| NasS-NasT | PP_2093 (nasS)/PP_2094 (nasT) | PA1786 (nasS)/PA1785 (nasT), nd/PA3363 (amiR) | Nitrate response regulation |
| Unclassified | PP_4781 (nd)/PP_4824 (nd) | PA3974 (ladS)/PA4856 (retS) | |
| Virulence Factors | Gene Name | P. aeruginosa PAO1 | P. putida KT2440 |
|---|---|---|---|
| Adherence | |||
| Type IV pili | pilA | PA4525 | PP_0634 |
| pilB | PA4526 | nd | |
| pilC | PA4527 | PP_0633 | |
| pilD | PA4528 | PP_0632 | |
| pilE | PA4556 | PP_0611 | |
| pilF | PA3805 | PP_0851 | |
| pilM | PA5044 | PP_5083 | |
| pilN | PA5043 | PP_5082 | |
| pilO | PA5042 | nd | |
| pilP | PA5041 | PP_5081 | |
| pilQ | PA5040 | PP_5080 | |
| pilT | PA0395 | PP_5093 | |
| pilU | PA0396 | nd | |
| pilV | PA4551 | nd | |
| pilW | PA4552 | nd | |
| pilX | PA4553 | nd | |
| pilY1 | PA4554 | nd | |
| pilY2 | PA4555 | nd | |
| pilZ | PA2960 | nd | |
| fimT | PA4549 | nd | |
| fimU | PA4550 | nd | |
| fimV | PA3115 | PP_1993 | |
| pilR | PA4547 | nd | |
| pilS | PA4546 | nd | |
| pilG | PA0408 | PP_4992 | |
| pilH | PA0409 | PP_4991 | |
| pilI | PA0410 | PP_4990 | |
| pilJ | PA0411 | PP_4989 | |
| pilK | PA0412 | nd | |
| chpA | PA0413 | PP_4988 | |
| chpB | PA0414 | nd | |
| chpC | PA0415 | PP_4987 | |
| chpD | PA0416 | nd | |
| chpE | PA0417 | nd | |
| Effector delivery system | |||
| Exolysin | exlA | nd | PP_1449 |
| exlB | nd | PP_1450 | |
| HSI-1 | tagR | PA0071 | nd |
| tagS | PA0072 | nd | |
| tagT | PA0073 | nd | |
| ppkA | PA0074 | nd | |
| pppA | PA0075 | nd | |
| tagF | PA0076 | nd | |
| icmF1 | PA0077 | nd | |
| dotU1 | PA0078 | nd | |
| hsiJ1 | PA0079 | nd | |
| lip1 | PA0080 | nd | |
| fha1 | PA0081 | nd | |
| hsiA1 | PA0082 | nd | |
| hsiB1 | PA0083 | nd | |
| hsiC1 | PA0084 | nd | |
| hcp1 | PA0085 | nd | |
| hsiE1 | PA0086 | nd | |
| hsiF1 | PA0087 | nd | |
| hsiG1 | PA0088 | nd | |
| hsiH1 | PA0089 | nd | |
| clpV1 | PA0090 | nd | |
| vgrG1a | PA0091 | nd | |
| HSI-1 T6SS | tse1 | PA1844 | nd |
| tse2 | PA2702 | nd | |
| tse3 | PA3484 | nd | |
| tse7 | PA0099 | nd | |
| tse4 | PA2774 | nd | |
| tse5 | PA2684 | nd | |
| tse6 | PA0093 | nd | |
| HSI-2 | vgrG | PA1511 | nd |
| hcpA | PA1512 | nd | |
| tssA | PA1656 | nd | |
| tssB | PA1657 | nd | |
| tssC | PA1658 | nd | |
| tssE | PA1659 | nd | |
| tssF | PA1660 | nd | |
| tssG | PA1661 | nd | |
| tssH | PA1662 | nd | |
| tssJ | PA1666 | nd | |
| tssK | PA1667 | nd | |
| icmH | PA1668 | nd | |
| tssM | PA1669 | nd | |
| HSI-2 T6SS | pldA | PA3487 | nd |
| vgrG2b | PA0262 | nd | |
| HSI-3 | tssA | PA2360 | nd |
| tssM | PA2361 | nd | |
| icmH | PA2362 | nd | |
| tssK | PA2363 | nd | |
| tssB | PA2365 | nd | |
| tssC | PA2366 | nd | |
| hcp | PA2367 | nd | |
| tssE | PA2368 | nd | |
| tssF | PA2369 | nd | |
| tssG | PA2370 | nd | |
| tssH | PA2371 | nd | |
| tssI | PA2373 | nd | |
| HSI-3 T6SS | pldB | PA5089 | nd |
| LasA | lasA | PA1871 | nd |
| LasB | lasB | PA3724 | nd |
| Putida K1-T6SS | tssA1 | nd | PP_3088 |
| hcp1 | nd | PP_3089 | |
| tssM1 | nd | PP_3090 | |
| tagF1 | nd | PP_5561 | |
| tssL1 | nd | PP_3092 | |
| tssK1 | nd | PP_3093 | |
| tssJ1 | nd | PP_3094 | |
| tssH | nd | PP_3095 | |
| tssG1 | nd | PP_3096 | |
| tssF1 | nd | PP_3097 | |
| tssE1 | nd | PP_3098 | |
| tssC1 | nd | PP_3099 | |
| tssB1 | nd | PP_3100 | |
| vgrG1 | nd | PP_3106 | |
| Putida K2-T6SS | tssM2 | nd | PP_4071 |
| tssA2 | nd | PP_4072 | |
| vasl2 | nd | PP_4073 | |
| tssB2 | nd | PP_4074 | |
| tssE2 | nd | PP_4076 | |
| tssF2 | nd | PP_4077 | |
| tssG2 | nd | PP_4078 | |
| tssJ2 | nd | PP_4079 | |
| tssK2 | nd | PP_4080 | |
| tssL2 | nd | PP_4081 | |
| hcp2 | nd | PP_4082 | |
| Putida K3-T6SS | vgrG3 | nd | PP_2614 |
| hcp3 | nd | PP_2615 | |
| tssL3 | nd | PP_2616 | |
| tssK3 | nd | PP_2617 | |
| tssJ3 | nd | PP_2618 | |
| fha3 | nd | PP_2619 | |
| tssG3 | nd | PP_2620 | |
| tssF3 | nd | PP_2621 | |
| tssE3 | nd | PP_2622 | |
| tssC3 | nd | PP_2623 | |
| tssB3 | nd | PP_2624 | |
| vasl3 | nd | PP_2625 | |
| tssA3 | nd | PP_2626 | |
| tssM3 | nd | PP_2627 | |
| Putida-T6SS | tke1 | nd | PP_3103 |
| tke2 | nd | PP_3108 | |
| tke4 | nd | PP_4085 | |
| tke5 | nd | PP_2612 | |
| tke6 | nd | PP_0646 | |
| tke7 | nd | PP_4885 | |
| tke9 | nd | PP_3388 | |
| tke10 | nd | PP_4048 | |
| TTSS | pscU | PA1690 | nd |
| pscT | PA1691 | nd | |
| pscS | PA1692 | nd | |
| pscR | PA1693 | nd | |
| pscQ | PA1694 | nd | |
| pscP | PA1695 | nd | |
| pscO | PA1696 | nd | |
| pscN | PA1697 | nd | |
| popN | PA1698 | nd | |
| pcr1 | PA1699 | nd | |
| pcr2 | PA1700 | nd | |
| pcr3 | PA1701 | nd | |
| pcr4 | PA1702 | nd | |
| pcrD | PA1703 | nd | |
| pcrR | PA1704 | nd | |
| pcrG | PA1705 | nd | |
| pcrV | PA1706 | nd | |
| pcrH | PA1707 | nd | |
| popB | PA1708 | nd | |
| popD | PA1709 | nd | |
| exsC | PA1710 | nd | |
| exsE | PA1711 | nd | |
| exsB | PA1712 | nd | |
| exsA | PA1713 | nd | |
| exsD | PA1714 | nd | |
| pscB | PA1715 | nd | |
| pscC | PA1716 | nd | |
| pscD | PA1717 | nd | |
| pscE | PA1718 | nd | |
| pscF | PA1719 | nd | |
| pscG | PA1720 | nd | |
| pscH | PA1721 | nd | |
| pscI | PA1722 | nd | |
| pscJ | PA1723 | nd | |
| pscK | PA1724 | nd | |
| pscL | PA1725 | nd | |
| TTSS effector proteins | exoS | PA3841 | nd |
| exoT | PA0044 | nd | |
| exoY | PA2191 | nd | |
| Motility | |||
| Flagella | flgB | PA1077 | PP_4391 |
| flgC | PA1078 | PP_4390 | |
| flgD | PA1079 | PP_4389 | |
| flgE | PA1080 | PP_4388 | |
| flgF | PA1081 | PP_4386 | |
| flgG | PA1082 | PP_4385 | |
| flgH | PA1083 | PP_4384 | |
| flgI | PA1084 | PP_4383 | |
| flgJ | PA1085 | PP_4382 | |
| flgK | PA1086 | PP_4381 | |
| flgL | PA1087 | PP_4380 | |
| fliC | PA1092 | PP_4378 | |
| fleI | PA1093 | PP_4377 | |
| fliD | PA1094 | PP_4376 | |
| fliS | PA1095 | PP_4375 | |
| fleP | PA1096 | PP_4374 | |
| fleQ | PA1097 | PP_4373 | |
| fleS | PA1098 | PP_4372 | |
| fleR | PA1099 | PP_4371 | |
| fliE | PA1100 | PP_4370 | |
| fliF | PA1101 | PP_4369 | |
| fliG | PA1102 | PP_4368 | |
| fliH | PA1103 | PP_4367 | |
| fliI | PA1104 | PP_4366 | |
| fliJ | PA1105 | PP_4365 | |
| fliK | PA1441 | PP_4361 | |
| fliL | PA1442 | PP_4359 | |
| fliM | PA1443 | PP_4358 | |
| fliN | PA1444 | PP_4357 | |
| fliO | PA1445 | PP_4356 | |
| fliP | PA1446 | PP_4355 | |
| fliQ | PA1447 | PP_4354 | |
| fliR | PA1448 | PP_4353 | |
| flhB | PA1449 | PP_4352 | |
| flhA | PA1452 | PP_4344 | |
| flhF | PA1453 | PP_4343 | |
| fleN | PA1454 | PP_4342 | |
| fliA | PA1455 | PP_4341 | |
| flgA | PA3350 | PP_4394 | |
| flgM | PA3351 | PP_4395 | |
| flgN | PA3352 | PP_4396 | |
| motB | PA4953 | PP_4904 | |
| motA | PA4954 | PP_4905 | |
| motC | PA1460 | PP_4336 | |
| motD | PA1461 | PP_4335 | |
| motY | PA3526 | PP_1087 | |
| Exotoxin | |||
| ExoA | toxA | PA1148 | nd |
| Non-hemolytic phospholipase C | plcN | PA3319 | nd |
| Phospholipase C | plcB | PA0026 | nd |
| PLC | plcH | PA0844 | nd |
| Exoenzyme | |||
| Alkaline protease | aprA | PA1249 | nd |
| Protease IV | prpL | PA4175 | nd |
| Immune modulation | |||
| Lipopolysaccharide (LPS) | nd | PA3141 | nd |
| PA3142 | nd | ||
| PA3143 | nd | ||
| PA3145 | nd | ||
| PA3146 | nd | ||
| PA3147 | nd | ||
| PA3148 | nd | ||
| PA3149 | nd | ||
| PA3150 | nd | ||
| PA3151 | nd | ||
| PA3152 | nd | ||
| PA3153 | nd | ||
| PA3154 | nd | ||
| PA3155 | nd | ||
| PA3156 | nd | ||
| PA3157 | nd | ||
| PA3158 | nd | ||
| PA3160 | nd | ||
| Rhamnolipid | rhlA | PA3479 | nd |
| rhlB | PA3478 | nd | |
| rhlC | PA1130 | nd | |
| Biofilm | |||
| Acylhomoserine lactone synthase | hdtS | PA0005 | PP_0058 |
| Alginate | algD | PA3540 | PP_1288 |
| alg8 | PA3541 | PP_1287 | |
| alg44 | PA3542 | PP_1286 | |
| algK | PA3543 | PP_1285 | |
| algE | PA3544 | PP_1284 | |
| algG | PA3545 | PP_1283 | |
| algX | PA3546 | PP_1282 | |
| algL | PA3547 | PP_1281 | |
| algI | PA3548 | PP_1280 | |
| algJ | PA3549 | PP_1279 | |
| algF | PA3550 | PP_1278 | |
| algA | PA3551 | PP_1277 | |
| algC | PA5322 | PP_5288 | |
| algU | PA0762 | PP_1427 | |
| mucA | PA0763 | PP_1428 | |
| mucB | PA0764 | PP_1429 | |
| mucC | PA0765 | nd | |
| mucD | PA0766 | PP_1430 | |
| algR | PA5261 | PP_0185 | |
| algZ | PA5262 | nd | |
| algW | PA4446 | PP_1301 | |
| mucE | PA4033 | nd | |
| mucP | PA3649 | PP_1598 | |
| algP/algR3 | PA5253 | PP_0194 | |
| algQ | PA5255 | PP_0191 | |
| Quorum-sensing | |||
| rhlR | PA3477 | nd | |
| rhlI | PA3476 | nd | |
| lasR | PA1430 | nd | |
| lasI | PA1432 | nd | |
| Nutritional/Metabolic factor | |||
| Pyochelin | pchI | PA4222 | nd |
| pchH | PA4223 | nd | |
| pchG | PA4224 | nd | |
| pchF | PA4225 | nd | |
| pchE | PA4226 | nd | |
| pchR | PA4227 | nd | |
| pchD | PA4228 | nd | |
| pchC | PA4229 | nd | |
| pchB | PA4230 | nd | |
| pchA | PA4231 | nd | |
| fptA | PA4221 | nd | |
| Pyocyanin | phzA1 | PA4210 | nd |
| phzB1 | PA4211 | nd | |
| phzC1 | PA4212 | nd | |
| phzD1 | PA4213 | nd | |
| phzE1 | PA4214 | nd | |
| phzF1 | PA4215 | nd | |
| phzG1 | PA4216 | nd | |
| phzA2 | PA1899 | nd | |
| phzB2 | PA1900 | nd | |
| phzC2 | PA1901 | nd | |
| phzD2 | PA1902 | nd | |
| phzE2 | PA1903 | nd | |
| phzF2 | PA1904 | nd | |
| phzG2 | PA1905 | nd | |
| phzM | PA4209 | nd | |
| phzS | PA4217 | nd | |
| phzH | PA0051 | nd | |
| Pyoverdine | pvdQ | PA2385 | PP_2901 |
| pvdA | PA2386 | PP_3796 | |
| pvdP | PA2392 | PP_4212 | |
| pvdM | PA2393 | PP_4213 | |
| pvdN | PA2394 | PP_4214 | |
| pvdO | PA2395 | PP_4215 | |
| pvdF | PA2396 | nd | |
| pvdE | PA2397 | PP_4216 | |
| pvdD | PA2399 | PP_4219 | |
| pvdJ | PA2400 | nd | |
| pvdI | PA2402 | nd | |
| pvdH | PA2413 | PP_4223 | |
| pvdL | PA2424 | PP_4243 | |
| pvdG | PA2425 | nd | |
| pvdS | PA2426 | PP_4244 | |
| pvdY | PA2427 | PP_4245 | |
| fpvA | PA2398 | PP_4217 | |
| Antimicrobial activity/Competitive advantage | |||
| Hydrogen cyanide production | hcnA | PA2193 | nd |
| hcnB | PA2194 | nd | |
| hcnC | PA2195 | nd | |
| Regulation | |||
| GacS/GacA | gacS | PA0928 | PP_1650 |
| gacA | PA2586 | PP_4099 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udaondo, Z.; Schilder, K.A.; Blesa, A.R.M.; Tena-Garitaonaindia, M.; Mangana, J.C.; Daddaoua, A. Transcriptional Regulatory Systems in Pseudomonas: A Comparative Analysis of Helix-Turn-Helix Domains and Two-Component Signal Transduction Networks. Int. J. Mol. Sci. 2025, 26, 4677. https://doi.org/10.3390/ijms26104677
Udaondo Z, Schilder KA, Blesa ARM, Tena-Garitaonaindia M, Mangana JC, Daddaoua A. Transcriptional Regulatory Systems in Pseudomonas: A Comparative Analysis of Helix-Turn-Helix Domains and Two-Component Signal Transduction Networks. International Journal of Molecular Sciences. 2025; 26(10):4677. https://doi.org/10.3390/ijms26104677
Chicago/Turabian StyleUdaondo, Zulema, Kelsey Aguirre Schilder, Ana Rosa Márquez Blesa, Mireia Tena-Garitaonaindia, José Canto Mangana, and Abdelali Daddaoua. 2025. "Transcriptional Regulatory Systems in Pseudomonas: A Comparative Analysis of Helix-Turn-Helix Domains and Two-Component Signal Transduction Networks" International Journal of Molecular Sciences 26, no. 10: 4677. https://doi.org/10.3390/ijms26104677
APA StyleUdaondo, Z., Schilder, K. A., Blesa, A. R. M., Tena-Garitaonaindia, M., Mangana, J. C., & Daddaoua, A. (2025). Transcriptional Regulatory Systems in Pseudomonas: A Comparative Analysis of Helix-Turn-Helix Domains and Two-Component Signal Transduction Networks. International Journal of Molecular Sciences, 26(10), 4677. https://doi.org/10.3390/ijms26104677