Abstract
In Austria, newborns have been screened for cystic fibrosis (CF) by analyzing immunoreactive trypsinogen (IRT) from dried blood spots (DBS)s for nearly 20 years. Recently, pancreatitis-associated protein (PAP) analysis was introduced as a second-tier test with the aim of reducing recalls for second DBS cards while keeping sensitivity high. For 28 months, when IRT was elevated (65–130 ng/mL), PAP was measured from the first DBS (n = 198,927) with a two-step cut-off applied. For the last 12 months of the observation period (n = 85,421), an additional IRT×PAP cut-off was introduced. If PAP or IRT×PAP were above cut-off, a second card was analyzed for IRT and in case of elevated values identified as screen-positive. Above 130 ng/mL IRT in the first DBS, newborns were classified as screen-positive. IRT analysis of first DBS resulted in 1961 (1%) tests for PAP. In the first 16 months, 26 of 93 screen-positive were confirmed to have CF. Two false-negatives have been reported (sensitivity = 92.8%). Importantly, less than 30% of families compared to the previous IRT-IRT screening scheme had to be contacted causing distress. Adding IRT×PAP caused a marginally increased number of second cards and sweat tests to be requested during this period (15 and 3, respectively) compared to the initial IRT-PAP scheme. One case of confirmed CF was found due to IRT×PAP, demonstrating an increase in sensitivity. Thus, the relatively simple and economical algorithm presented here performs effectively and may be a useful model for inclusion of CF into NBS panels or modification of existing schemes.
1. Introduction
Cystic fibrosis (CF) is an autosomal recessive inherited disease characterized by accumulation of viscous secretion in the pancreas, lungs, and other organs, severely impairing their function [1]. It is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, encoding an important regulator for the transport of chloride and bicarbonate ions across epithelial cell membranes. Although there is no cure for CF, early diagnosis through newborn screening (NBS) combined with appropriate prompt treatment results in significant benefits for children born with CF [2,3]. These benefits outweigh risks of diagnostic uncertainty such as false-positive screening results. Therefore, NBS for CF is established in many countries or regions around the world and continues to be added to screening programmes [4].
The primary parameter is always immunoreactive trypsinogen (IRT) concentration in blood sampled as dried blood spots (DBS)s during the first days of life [5]. If positive (above cut-off), measurement of pancreatitis-associated protein (PAP) and/or DNA mutation analysis of the CFTR gene from the first card and a second card requested for a further IRT measurement are possible with different algorithms and procedures [6]. Thus, several strategies with numerous variances to screen newborns for CF exist. Importantly, a sweat test is used to confirm a diagnosis of CF for all screen-positive children [7].
In Austria, newborns have been screened for CF within the national NBS program since 1997 [8]. The Austrian NBS is centrally organized with a single laboratory at the Medical University of Vienna responsible for the analysis of about 85,000 newborns in Austria per year. Treatment and follow-up of affected patients are well organized routines in Austria, particularly for children and adolescents [9]. Until May 2017, IRT was the only parameter determined from DBSs. In the case of a positive result from the first card, a second card was obtained (IRT-IRT strategy) and a positive IRT value from this second card was referred to as “screen-positive”, resulting in a call for a sweat test for final diagnosis. Despite the high sensitivity (>95% on average), the positive predictive value (PPV) of this IRT-IRT algorithm was less than 15% and therefore markedly below an acceptable value [10]. Even more problematic were the high numbers of second cards (0.9%) requested using this algorithm, causing anxiety and stress in about 800 families in Austria per year.
The discovery of pancreatitis-associated protein (PAP) [11] as a suitable biomarker for second-tier analysis from the first DBSs as well as different genetic approaches for second or third-tier testing [12,13] opened new possibilities prompting us to review the CF screening algorithm in Austria. Due to ethical, legal, practical, and economic concerns [6,14,15], inclusion of genetic tests into the screening algorithm had a priori been excluded for further consideration. Therefore, an IRT-PAP-IRT algorithm including a fail-safe strategy and a two-step cut-off for PAP was adjusted from published variants [16] and implemented in the Austrian NBS in May 2017. Although widely published [16,17,18,19,20], a two-step IRT-dependent PAP cut-off is somewhat arbitrary and a more dynamic cut-off determination was sought. Weidler et al. showed that the IRT×PAP product showed better discrimination for classical CF than PAP alone as a second-tier screening parameter [21]. Therefore, after a 16-month evaluation period of the new algorithm, we combined these variants by including an IRT×PAP product cut-off into the scheme for the last 12 months of the observation period presented here. We compared performance parameters to other published algorithms, thereby showing that this relatively simple and economically inexpensive algorithm performs effectively.
2. Materials and Methods
2.1. Patients
In Austria, newborns are screened for CF as part of the regular NBS program. DBSs are recommended to be sampled between the 36th and 72nd hour of life and sent to the NBS laboratory located at the Department of Pediatrics and Adolescent Medicine of the Medical University of Vienna. If samples are taken before the 36th hour of life or before completed 32nd gestational week, a second card is requested and handled as a first card concerning CF screening irrespective of the initial result (Figure 1). The same accounts for cards of insufficient sampling quality or not enough blood. Within the observation period of May 2017 to August 2019, 198,927 DBS cards of newborns were received, which were suitable as first cards of the CF screening. This number corresponds to >99.5% of newborns born in Austria within this period.
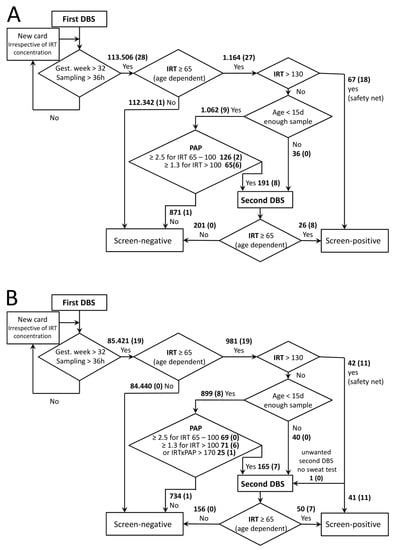
Figure 1.
CF Screening schemes in Austria. Algorithms for the first period (A) and the second period (B) are depicted. Bold numbers give numbers of cards within indicated categories. The number in brackets is the number of confirmed positives within these categories. IRT and PAP cut-off concentrations are given in ng/mL, IRT × PAP in ng2/mL2.
2.2. DBS Tests
IRT blood concentrations were measured using the AutoDELFIA Neonatal IRT kit (Perkin-Elmer, Turku, Finland) according to manufacturer’s instructions. When IRT results were between 65 and 130 ng/mL and the newborn was less than 15 days old, PAP was measured as described below [22]. For children older than 14 days, measurement of samples with elevated IRT (age dependent cut-off values: up to 4 weeks of age, 65 ng/mL and; 5th to 6th week of life, 50 ng/mL; 7th to 9th week of life, 30 ng/mL) was repeated in duplicate. When the repeat analysis confirmed the raised IRT measurement, a second card was requested. If the initial result was above 130 ng/mL, measurement was also repeated in duplicate and if the mean exceeded the cut-off, patients were regarded as screen-positive. CF screening is repeated for all second cards requested for suspected biotinidase deficiency as low biotinidase activities can indicate sample deterioration due to heat and humidity [23].
MucoPAPII (Dynabio, Marseille, France) was used to determine PAP blood concentrations in duplicate according to the manufacturer’s instructions. Cut-offs for PAP were ≥2.5 and ≥1.3 ng/mL for IRT 65–100 and 100–130 ng/mL, respectively. For comparison with earlier publications, these values are obtained with the MucoPAP II Kit, which was altered by the manufacturer in January 2017, resulting in a shift of values to 83% compared to the previous kit as determined by a series of comparisons in our laboratory. The PAP cut-offs were adjusted accordingly from previously published values [19]. In addition to these dual cut-offs, in the last 12 months of the observation period, a PAP×IRT cut-off [21] was introduced. To avoid a loss of sensitivity compared to the previous screening algorithm, values exceeding either of these cut-offs led to request of a second card. The PAP×IRT cut-off was set to 170 ng2/mL2, resulting in a smoothing of the step between the two PAP cut-off values, as depicted by the solid line in Figure 2B. PAP values rise with age, and age-dependent cut-offs are not available yet [22]. Therefore, for second cards, PAP was not measured and age-dependent cut-offs were applied for IRT as given above, and in the case of exceeding these values, patients were regarded as screen-positive.
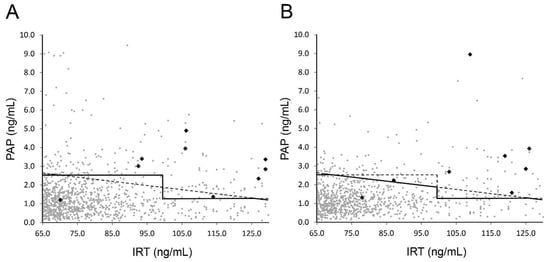
Figure 2.
PAP vs. IRT values from first DBSs. Values of samples with IRT between 65 and 130 ng/mL eligible for PAP analysis according to the screening scheme for (A) first period and (B) second period are plotted. Solid lines indicate used cut-off values for request of second card; dashed lines give comparison to alternative strategies (see main text). Black data points indicate confirmed CF cases.
2.3. CF Diagnosis
Parents of screen-positive newborns are referred to a pediatric CF center for a sweat test to be performed. A sweat chloride concentration above 60 mmol/L is considered CF-positive, leading to genetic testing and diagnostic, as well as therapeutic work up. Borderline sweat test results (30 to 60 mmol/L chloride) are followed by the diagnostic procedure recommended by the European CF Society [24]: Patients undergo a repeat sweat test and further evaluation in a specialist CF center, including detailed clinical assessment and extensive CFTR gene mutation analysis. In a first step, a targeted detection approach is applied using kits for multiplex allele-specific PCR amplification, which generates fluorescently labelled fragments that are analyzed by capillary electrophoresis. Used kits are, for instance, the CF-EU2 kit (Elucigene, Manchester, UK), identifying 50 mutations in total covering about 95% of mutations in Austria and the CFTR 68 kit (Devyse, Hagersten, Sweden) detecting 68 mutations. In case of inconclusive results, which means no or just one detected mutation, sequencing of the CFTR exon, and, if still inconclusive, the whole gene locus by next generation sequencing is performed. Additionally, structural variants are identified by multiplex ligation-dependent probe amplification.
All results of sweat tests and most of the follow-up diagnostic results are reported to the NBS laboratory by the 10 CF centers in Austria. Since clinically diagnosed cases of CF are also routinely reported to the national NBS laboratory whatever their screening test result, the calculated sensitivity reported here has a high probability of accuracy. A small possibility exists that not all clinically diagnosed cases are notified to the national laboratory. Negative screen results for babies found clinically with meconium ileus are not classified as false-negatives.
3. Results
In the first period (16 months), 113,506 DBSs of newborns were screened for CF, 112,342 of which were initially screen-negative and not further considered.
Interestingly, a large proportion of screen-positives (67 out of 93) and the majority of confirmed positives (18 out of 28, for details of confirmed CF cases see Table 1) were found via the safety net (IRT above 130 ng/mL). Eight of the 9 other positive cases were detected via PAP analysis, which included 1062 (0.9%) samples with one false-negative (Figure 1A). This false-negative case with an IRT of 71 ng/mL and a PAP of 1.3 ng/mL (Figure 2A) was identified due to family history, but was a foreign citizen who left Austria after birth and therefore we do not have any further information. Sample quality of all false-negatives described here appeared sufficient not only in the receiving inspection but also in retrospective examination.

Table 1.
Newborn screening and genetic testing results of CF cases detected by the Austrian newborn screening within the overall observation period, sorted by initial IRT.
The majority of confirmed CF cases had an initial IRT value above 100 ng/mL. Only three confirmed positives, including the false-negative case, fell into the 65–100 ng/mL value category that accounted for 126 of the requested 191 second cards. Altogether, as the main performance indicators, a PPV of 29.2% and a sensitivity of 92.8% were determined (Table 2). A single patient was diagnosed with CF out of the screen-negative cases, resulting in a false-negative for IRT screening (Figure 1A). This false-negative case with an IRT of 56 was identified due to clinical symptoms of a period of a hypochloraemic alkalosis and a wheezy bronchitis; the sweat test was borderline and the genetic analysis revealed the N1303K and D110H mutations with pancreatic sufficiency. When patients were sorted according to their initial IRT values (Table 1), DF508 homozygotes were widely distributed and no conclusions on associations between distinct mutations and the quantities of IRT and PAP could be made. This would require a much higher number of cases.
Table 2.
Comparison of different CF screening algorithms in Austrian newborn screening.
In the second period, the determination of screen-positives and confirmed cases via the safety net occurred in similar proportions as in the first period (Figure 1 B, Table 2). In these 12 months, due to the additional IRT × PAP criterion, 25 (0.03%) more second cards and 3 more sweat tests (screen- positive) had to be requested. Notably, one out of the confirmed positive cases was detected due to the IRT×PAP criterion (Figure 1B) and would not have been detected via the previous algorithm (Figure 2B; ID1, Table 1). Again, one false-negative case occurred in the PAP-dependent branch of the screening algorithm: A patient with IRT of 78 ng/mL and PAP of 1.31 ng/mL (Figure 2B) was reported to us as CF-positive. The patient was detected due to family history including known CF cases and consanguinity. The sweat test was borderline, the genetic analysis revealed a homozygous 2789 + 5G > A mutation with pancreatic sufficiency.
A comparison of the two periods and the previous screening based solely on IRT, as well as a hypothetical screening based only on an IRT×PAP cut-off value of 170 ng2/mL2, is given in Table 2. Data show that introducing PAP into screening markedly reduced numbers of second cards requested as well false-positive screening results, as sensitivity remained high. Differences between the strategies appear small, but in the first period one confirmed case would have remained undetected by the IRT×PAP only strategy, while in the second period one case would have been missed by IRT-PAP (Figure 2).
4. Discussion
This study shows how introducing a PAP measurement into an existing NBS algorithm using the conventional IRT-IRT protocol reduces the need to request second cards by more than 70%. The PPV raised by more than 50% without a substantial decrease in sensitivity due to the reduction of false-positives. Therefore, our CF screening now compared very satisfactorily to other CF screening strategies [10,25]. We also show that the inclusion of an IRT×PAP cut-off has the potential to further improve the screening yield. In the first period, one confirmed case would have remained undetected by the IRT×PAP only strategy. Therefore, we decided to combine the previous dual cut-off with the IRT×PAP criterion to reduce the probability of false-negatives. In our case, this strategy caused only minimally more second cards, and one additional confirmed case was found screen-positive due to the IRT×PAP(>170 ng2/mL2) cut-off.
Previous studies showed that concerning cost effectiveness, IRT-PAP protocols are superior to IRT-DNA protocols [6]. Another advantage of strategies like the Austrian one to determine the genetic background only after diagnosis by sweat chloride testing is that there is no ethically problematic carrier detection and less detection of CF screen-positive, inconclusive diagnosis (CFSPID) cases [26]. CFSPID is a common problem occurring with mutation analysis-based CF screening strategies. This has led to the widespread view that the individual’s phenotype rather than an equivocal genotype should be treated [27]. On the other hand, taking into account the successful reduction of CF rates by effective carrier screening strategies [28], a lack of carrier detection could also be regarded as disadvantageous depending on ethics varying from country to country. Patients with CFSPID may benefit from detection by NBS [29], although uncertainty remains challenging for families and caregivers and strategies for the follow-up of these patients are not commonly established yet [27]. Several European countries do not use DNA testing for CF screening [30], and neonatal genetic screening is often not adequately addressed in European laws [31]. In conditions with permissive laws and readily available financing, genetic analysis including F508del CFTR mutation analysis and next generation sequencing is definitely a reasonable choice to reduce second cards and screening (false-)positive, as exemplified by the Danish 3-Tier system [32], or the German and Dutch algorithms that also include analysis of PAP [19,33].
In conclusion, we here present a relatively simple and economical algorithm that performs effectively. This may be a useful model for NBS programs considering inclusion of CF into their screening panels or modification of existing schemes. However, our data on only a low number of confirmed positive cases also underline that there is still room for optimization of screening algorithms, ideally by multinational initiatives.
Author Contributions
Conceptualization, M.Z.; formal analysis, M.Z., A.S., S.R.; investigation, A.S., S.R.; resources, P.B., V.B., E.E., H.E., M.K., J.R., C.T., F.W., A.Z., S.R.; data curation, A.S.; writing—original draft preparation, M.Z.; writing—review and editing, P.B., V.B., E.E., H.E., M.K., J.R., C.T., F.W., A.Z., S.R., M.Z.; visualization, A.S., M.Z.; supervision, M.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Ethics Committee of the Medical University of Vienna (No. 2034/2020, approved 15 October 2020).
Informed Consent Statement
Patient consent was waived due to the retrospective character of the study by the Ethics Committee of the Medical University of Vienna.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brown, S.D.; White, R.; Tobin, P. Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. J. Am. Acad. PAs 2017, 30, 23–27. [Google Scholar] [CrossRef]
- Grosse, S.D.; Rosenfeld, M.; Devine, O.J.; Lai, H.J.; Farrell, P.M. Potential impact of newborn screening for cystic fibrosis on child survival: A systematic review and analysis. J. Pediatr. 2006, 149, 362–366. [Google Scholar] [CrossRef]
- Dijk, F.N.; McKay, K.; Barzi, F.; Gaskin, K.J.; Fitzgerald, D.A. Improved survival in cystic fibrosis patients diagnosed by newborn screening compared to a historical cohort from the same centre. Arch. Dis. Child. 2011, 96, 1118–1123. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef]
- Crossley, J.R.; Elliott, R.B.; Smith, P.A. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet 1979, 1, 472–474. [Google Scholar] [CrossRef]
- Schmidt, M.; Werbrouck, A.; Verhaeghe, N.; De Wachter, E.; Simoens, S.; Annemans, L.; Putman, K. Strategies for newborn screening for cystic fibrosis: A systematic review of health economic evaluations. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2018. [Google Scholar] [CrossRef]
- De Boeck, K.; Vermeulen, F.; Dupont, L. The diagnosis of cystic fibrosis. Presse Med. (Paris France 1983) 2017, 46, e97–e108. [Google Scholar] [CrossRef]
- Pollak, A.; Kasper, D.C. Austrian Newborn Screening Program: A perspective of five decades. J. Perinat. Med. 2014, 42, 151–158. [Google Scholar] [CrossRef]
- Frischer, T.; Eber, E.; Ellemunter, H.; Zacharasiewicz, A.; Kaluza, I.; Riedler, J.; Renner, S. Cystic fibrosis in Austria. Wien. Klin. Wochenschr. 2017, 129, 527–532. [Google Scholar] [CrossRef]
- Barben, J.; Castellani, C.; Dankert-Roelse, J.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.; Munck, A.; Sands, D.; Sommerburg, O.; et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J. Cyst. Fibros. 2017, 16, 207–213. [Google Scholar] [CrossRef]
- Barthellemy, S.; Maurin, N.; Roussey, M.; Ferec, C.; Murolo, S.; Berthezene, P.; Iovanna, J.L.; Dagorn, J.C.; Sarles, J. Evaluation of 47,213 infants in neonatal screening for cystic fibrosis, using pancreatitis-associated protein and immunoreactive trypsinogen assays. Arch. Pediatrie Organe Off. La Soc. Fr. Pediatrie 2001, 8, 275–281. [Google Scholar] [CrossRef]
- Baker, M.W.; Groose, M.; Hoffman, G.; Rock, M.; Levy, H.; Farrell, P.M. Optimal DNA tier for the IRT/DNA algorithm determined by CFTR mutation results over 14 years of newborn screening. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2011, 10, 278–281. [Google Scholar] [CrossRef][Green Version]
- Cordovado, S.K.; Hendrix, M.; Greene, C.N.; Mochal, S.; Earley, M.C.; Farrell, P.M.; Kharrazi, M.; Hannon, W.H.; Mueller, P.W. CFTR mutation analysis and haplotype associations in CF patients. Mol. Genet. Metab. 2012, 105, 249–254. [Google Scholar] [CrossRef]
- Seror, V.; Cao, C.; Roussey, M.; Giorgi, R. PAP assays in newborn screening for cystic fibrosis: A population-based cost-effectiveness study. J. Med. Screen 2016, 23, 62–69. [Google Scholar] [CrossRef]
- Raimond, V.; Sambuc, C.; Pibouleau, L. Ethics Evaluation Revealing Decision-Maker Motives: A Case of Neonatal Screening. Int. J. Technol. Assess. Health Care 2018, 34, 189–195. [Google Scholar] [CrossRef]
- Sommerburg, O.; Krulisova, V.; Hammermann, J.; Lindner, M.; Stahl, M.; Muckenthaler, M.; Kohlmueller, D.; Happich, M.; Kulozik, A.E.; Votava, F.; et al. Comparison of different IRT-PAP protocols to screen newborns for cystic fibrosis in three central European populations. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2014, 13, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Krulisova, V.; Balascakova, M.; Skalicka, V.; Piskackova, T.; Holubova, A.; Paderova, J.; Krenkova, P.; Dvorakova, L.; Zemkova, D.; Kracmar, P.; et al. Prospective and parallel assessments of cystic fibrosis newborn screening protocols in the Czech Republic: IRT/DNA/IRT versus IRT/PAP and IRT/PAP/DNA. Eur. J. Pediatr. 2012, 171, 1223–1229. [Google Scholar] [CrossRef]
- Sarles, J.; Giorgi, R.; Berthezene, P.; Munck, A.; Cheillan, D.; Dagorn, J.C.; Roussey, M. Neonatal screening for cystic fibrosis: Comparing the performances of IRT/DNA and IRT/PAP. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2014, 13, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Hammermann, J.; Lindner, M.; Stahl, M.; Muckenthaler, M.; Kohlmueller, D.; Happich, M.; Kulozik, A.E.; Stopsack, M.; Gahr, M.; et al. Five years of experience with biochemical cystic fibrosis newborn screening based on IRT/PAP in Germany. Pediatric Pulmonol. 2015, 50, 655–664. [Google Scholar] [CrossRef]
- Vernooij-van Langen, A.M.; Loeber, J.G.; Elvers, B.; Triepels, R.H.; Gille, J.J.; Van der Ploeg, C.P.; Reijntjens, S.; Dompeling, E.; Dankert-Roelse, J.E. Novel strategies in newborn screening for cystic fibrosis: A prospective controlled study. Thorax 2012, 67, 289–295. [Google Scholar] [CrossRef]
- Weidler, S.; Stopsack, K.H.; Hammermann, J.; Sommerburg, O.; Mall, M.A.; Hoffmann, G.F.; Kohlmuller, D.; Okun, J.G.; Macek, M., Jr.; Votava, F.; et al. A product of immunoreactive trypsinogen and pancreatitis-associated protein as second-tier strategy in cystic fibrosis newborn screening. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2016, 15, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Vernooij-van Langen, A.M.; Loeber, J.G.; Elvers, B.; Triepels, R.H.; Roefs, J.; Gille, J.J.; Reijntjens, S.; Dompeling, E.; Dankert-Roelse, J.E. The influence of sex, gestational age, birth weight, blood transfusion, and timing of the heel prick on the pancreatitis-associated protein concentration in newborn screening for cystic fibrosis. J. Inherit. Metab. Dis. 2013, 36, 147–154. [Google Scholar] [CrossRef]
- Adam, B.W.; Hall, E.M.; Sternberg, M.; Lim, T.H.; Flores, S.R.; O’Brien, S.; Simms, D.; Li, L.X.; De Jesus, V.R.; Hannon, W.H. The stability of markers in dried-blood spots for recommended newborn screening disorders in the United States. Clin. Biochem. 2011, 44, 1445–1450. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Castellani, C.; Southern, K.W.; Brownlee, K.; Dankert Roelse, J.; Duff, A.; Farrell, M.; Mehta, A.; Munck, A.; Pollitt, R.; Sermet-Gaudelus, I.; et al. European best practice guidelines for cystic fibrosis neonatal screening. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2009, 8, 153–173. [Google Scholar] [CrossRef]
- Munck, A.; Mayell, S.J.; Winters, V.; Shawcross, A.; Derichs, N.; Parad, R.; Barben, J.; Southern, K.W. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015, 14, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Doull, I. Devil in the detail of newborn screening for cystic fibrosis. Arch. Dis. Child. 2018. [Google Scholar] [CrossRef]
- Castellani, C.; Picci, L.; Tridello, G.; Casati, E.; Tamanini, A.; Bartoloni, L.; Scarpa, M.; Assael, B.M. Cystic fibrosis carrier screening effects on birth prevalence and newborn screening. Genet. Med. 2016, 18, 145–151. [Google Scholar] [CrossRef]
- Munck, A. Inconclusive Diagnosis after Newborn Screening for Cystic Fibrosis. Int. J. Neonatal Screen. 2020, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Bergougnoux, A.; Lopez, M.; Girodon, E. The Role of Extended CFTR Gene Sequencing in Newborn Screening for Cystic Fibrosis. Int. J. Neonatal Screen. 2020, 6, 23. [Google Scholar] [CrossRef]
- Esquerda, M.; Palau, F.; Lorenzo, D.; Cambra, F.J.; Bofarull, M.; Cusi, V.; Interdisciplinar en Bioetica, G. Ethical questions concerning newborn genetic screening. Clin. Genet. 2021, 99, 93–98. [Google Scholar] [CrossRef]
- Skov, M.; Bækvad-Hansen, M.; Hougaard, D.M.; Skogstrand, K.; Lund, A.M.; Pressler, T.; Olesen, H.V.; Duno, M. Cystic fibrosis newborn screening in Denmark: Experience from the first 2 years. Pediatric Pulmonol. 2020, 55, 549–555. [Google Scholar] [CrossRef]
- Dankert-Roelse, J.E.; Bouva, M.J.; Jakobs, B.S.; Janssens, H.M.; de Winter-de Groot, K.M.; Schönbeck, Y.; Gille, J.J.P.; Gulmans, V.A.M.; Verschoof-Puite, R.K.; Schielen, P.C.J.I.; et al. Newborn blood spot screening for cystic fibrosis with a four-step screening strategy in the Netherlands. J. Cyst. Fibros. 2019, 18, 54–63. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).