
New Insights into Molecular Mechanisms Mediating Adaptation to Exercise; A Review Focusing on Mitochondrial Biogenesis, Mitochondrial Function, Mitophagy and Autophagy


Abstract
:1. Introduction
Emerging Important Molecular Mechanisms in the Regulation of Exercise Adaptation:
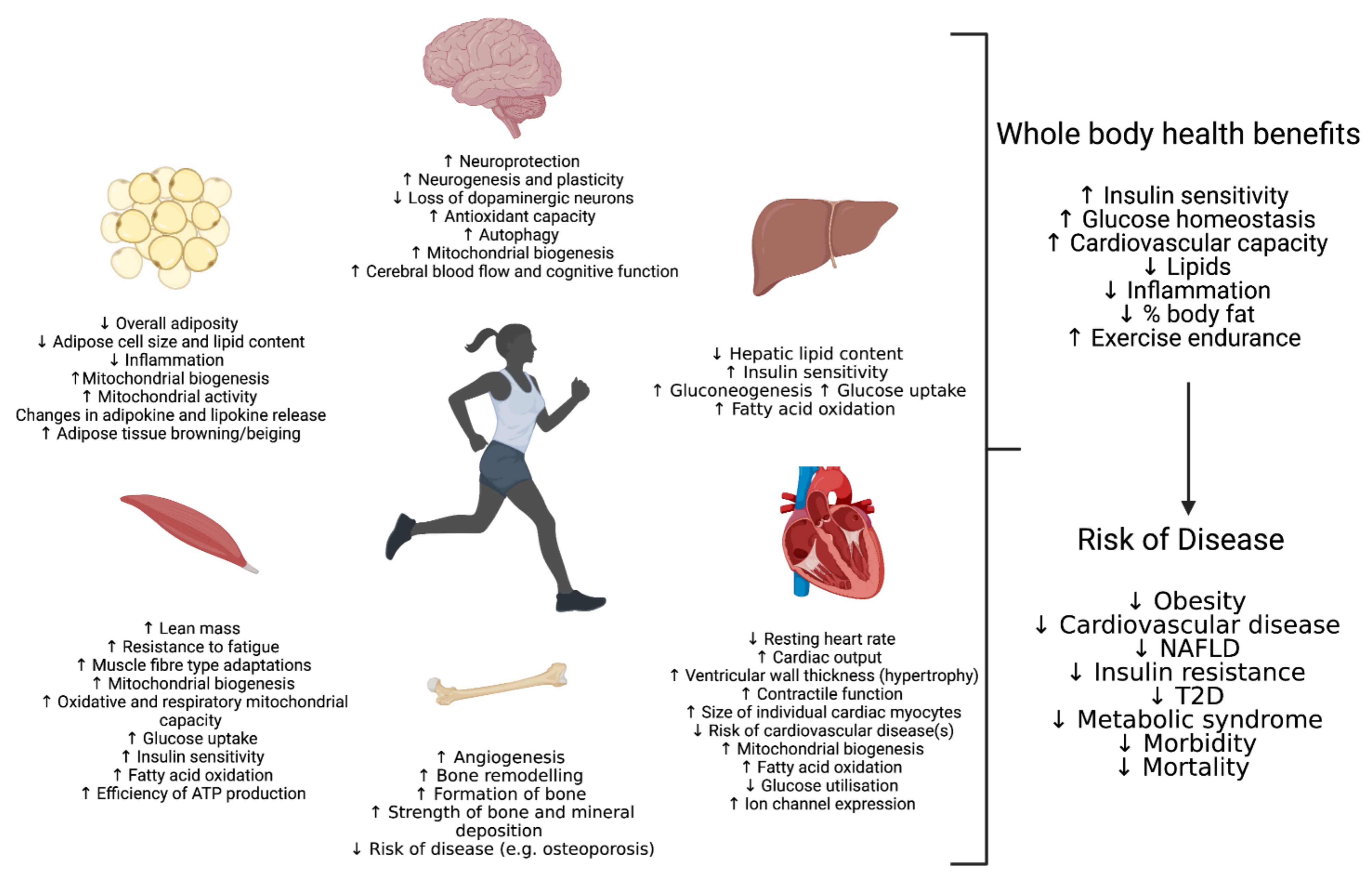
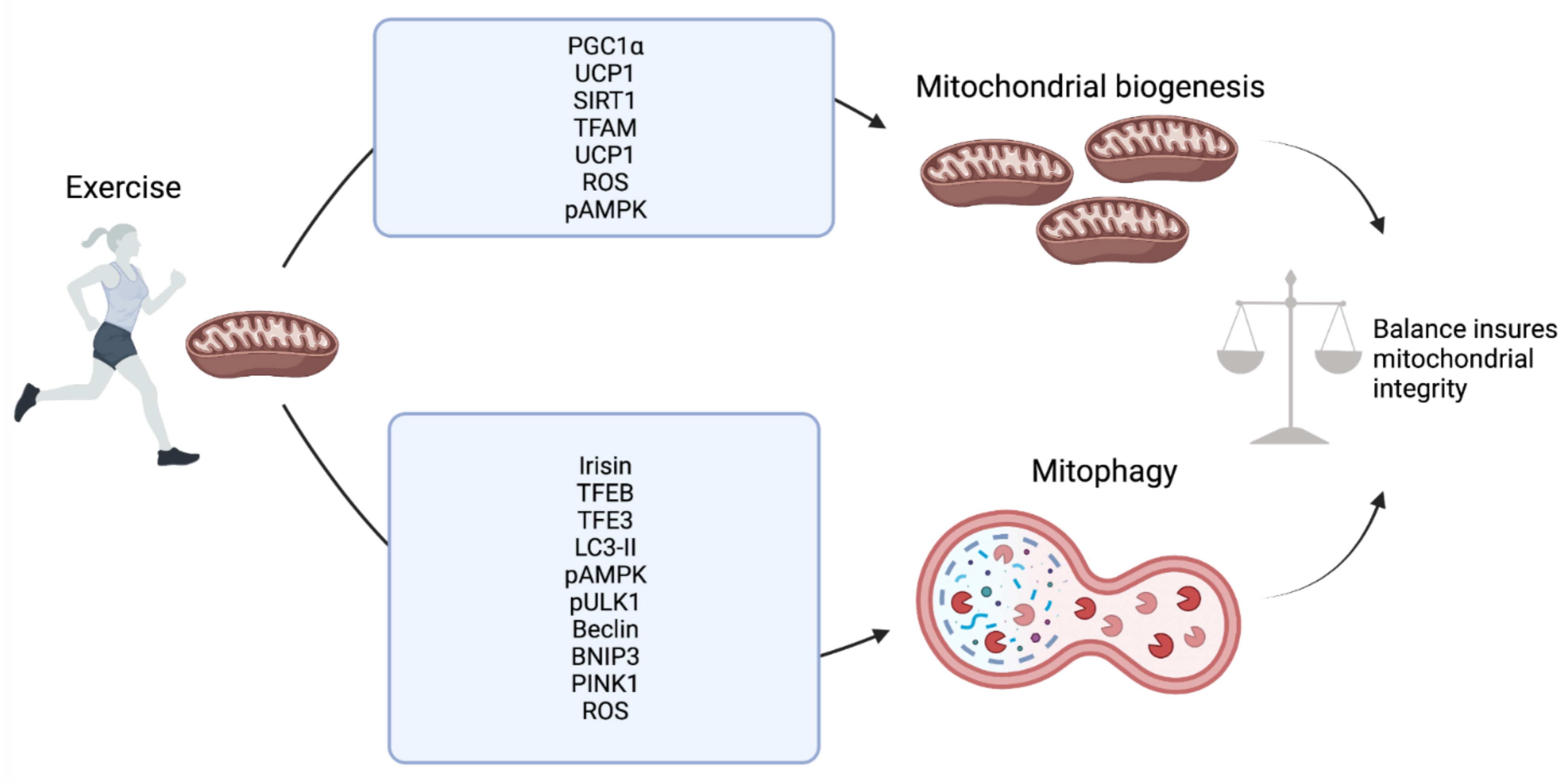
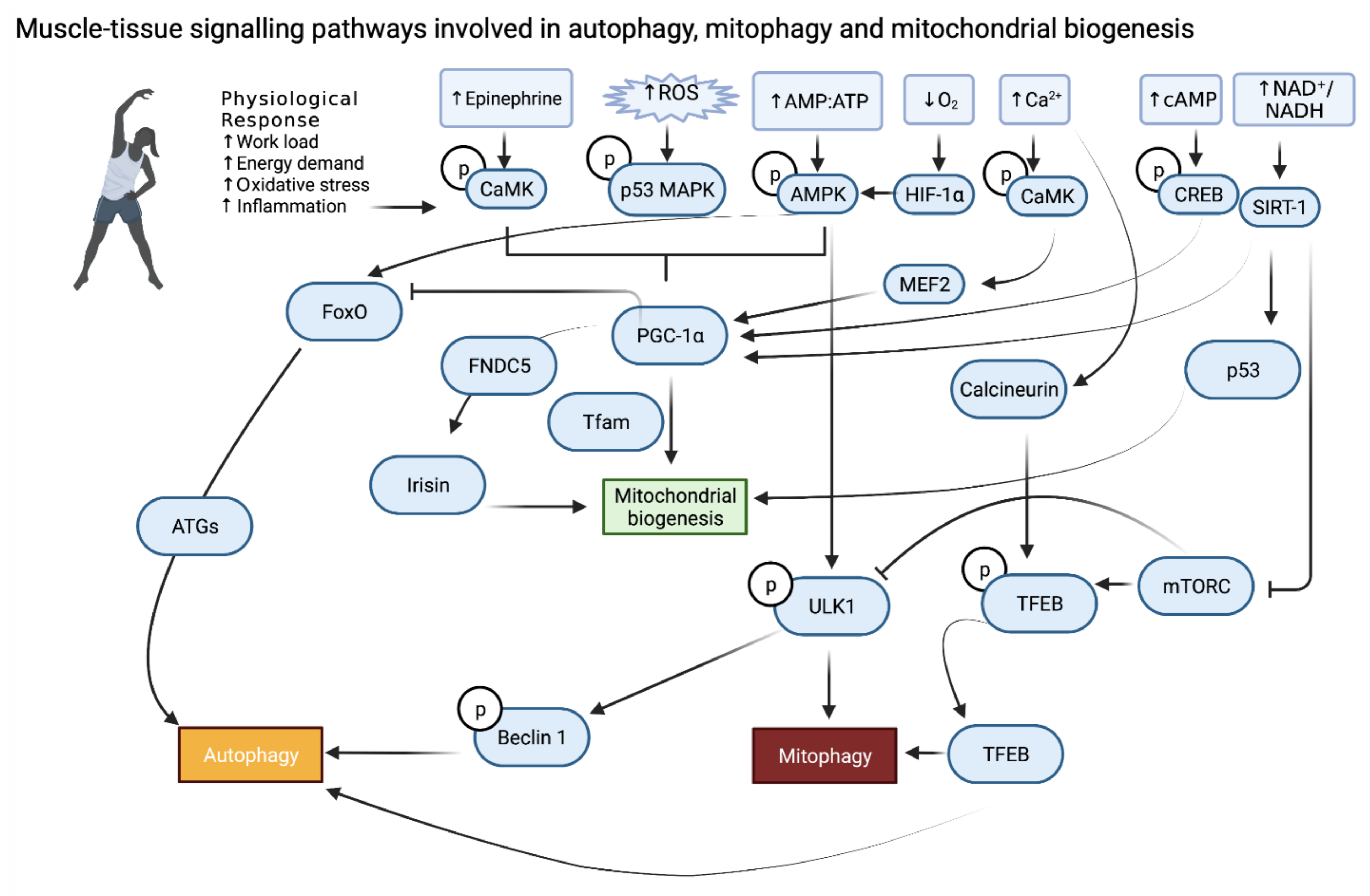
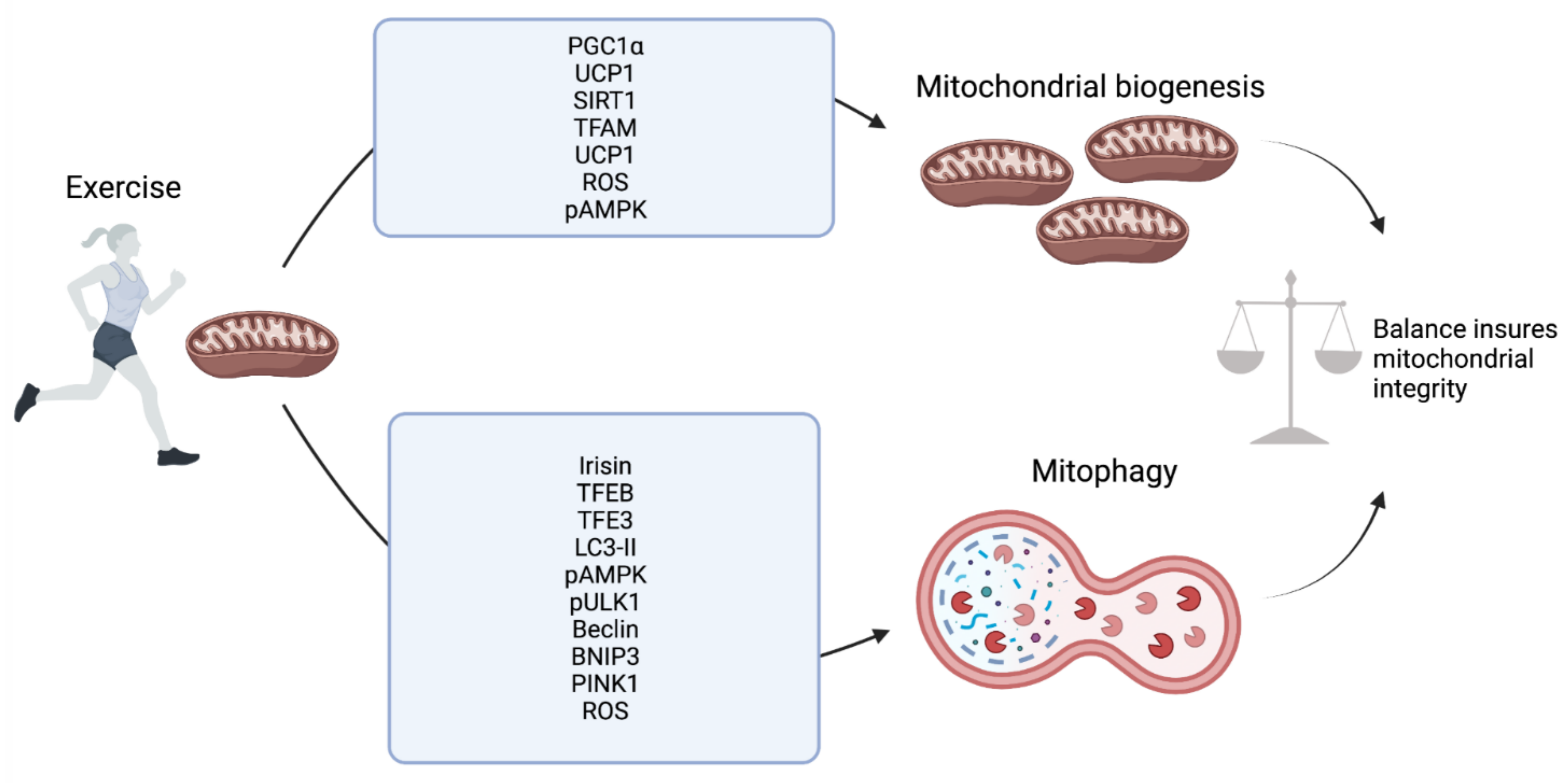
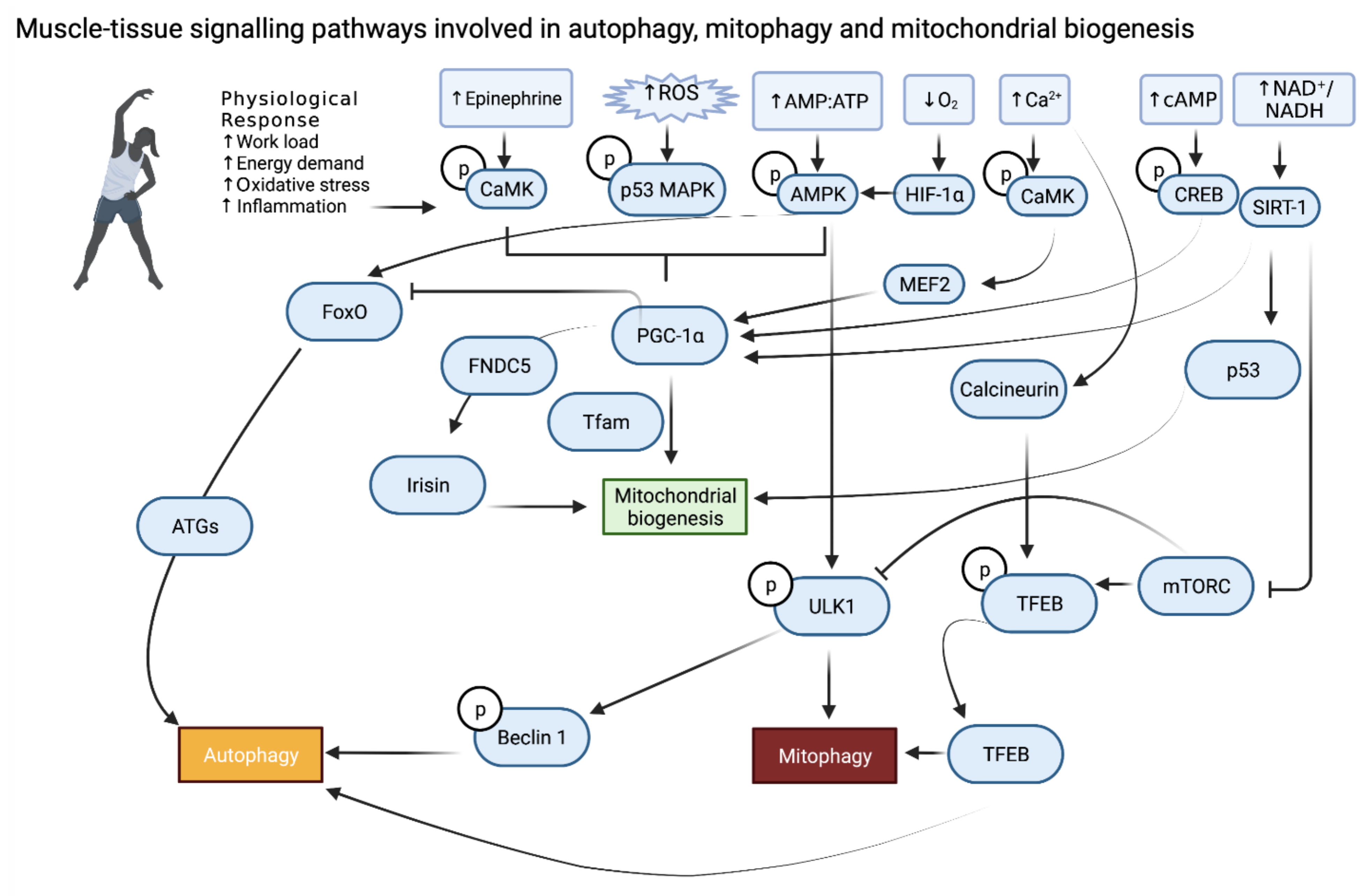
2. Skeletal Muscle
3. Liver
4. Adipose
5. Cardiovascular
6. Conclusions and Future Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manson, J.E.; Hu, F.B.; Rich-Edwards, J.W.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Speizer, F.E.; Hennekens, C.H. A Prospective Study of Walking as Compared with Vigorous Exercise in the Prevention of Coronary Heart Disease in Women. N. Engl. J. Med. 1999, 341, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.; Liu, S.; Solomon, C.G.; Willett, W.C. Diet, Lifestyle, and the Risk of Type 2 Diabetes Mellitus in Women. N. Engl. J. Med. 2001, 345, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, A.S.; Boyle, P.A.; Yu, L.; Shah, R.C.; Wilson, R.S.; Bennett, D.A. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 2012, 78, 1323–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colditz, G.A.; Cannuscio, C.C.; Frazier, A.L. Physical activity and reduced risk of colon cancer: Implications for prevention. Cancer Causes Control 1997, 8, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, S.; Saito, K.; Tanaka, S.; Maki, M.; Yachi, Y.; Asumi, M.; Sugawara, A.; Totsuka, K.; Shimano, H.; Ohashi, Y.; et al. Cardiorespiratory Fitness as a Quantitative Predictor of All-Cause Mortality and Cardiovascular Events in Healthy Men and Women. JAMA 2009, 301, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Gonzalez, M.A.; Martínez, J.A.; Hu, F.B.; Gibney, M.J.; Kearney, J. Physical inactivity, sedentary lifestyle and obesity in the European Union. Int. J. Obes. 1999, 23, 1192–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. PharmacoEconomics 2014, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.; McNamara, E.; Tainio, M.; de Sá, T.H.; Smith, A.D.; Sharp, S.J.; Edwards, P.; Woodcock, J.; Brage, S.; Wijndaele, K. Sedentary behaviour and risk of all-cause, cardiovascular and cancer mortality, and incident type 2 diabetes: A systematic review and dose response meta-analysis. Eur. J. Epidemiol. 2018, 33, 811–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of Exercise Is a Major Cause of Chronic Diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, C.; An, P.; Rice, T.; Skinner, J.S.; Wilmore, J.H.; Gagnon, J.; Pérusse, L.; Leon, A.S.; Rao, D.C. Familial aggregation ofVo 2 max response to exercise training: Results from the HERITAGE Family Study. J. Appl. Physiol. 1999, 87, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Hubal, M.J.; Gordish-Dressman, H.; Thompson, P.D.; Price, T.B.; Hoffman, E.P.; Angelopoulos, T.J.; Gordon, P.M.; Moyna, N.M.; Pescatello, L.S.; Visich, P.S.; et al. Variability in muscle size and strength gain after unilateral resistance training. Med. Sci. Sports Exerc. 2005, 37, 964–972. [Google Scholar] [PubMed]
- Bouchard, C.; Blair, S.N.; Church, T.S.; Earnest, C.P.; Hagberg, J.M.; Häkkinen, K.; Jenkins, N.T.; Karavirta, L.; E Kraus, W.; Leon, A.S.; et al. Adverse Metabolic Response to Regular Exercise: Is It a Rare or Common Occurrence? PLoS ONE 2012, 7, e37887. [Google Scholar] [CrossRef]
- Flockhart, M.; Nilsson, L.C.; Tais, S.; Ekblom, B.; Apró, W.; Larsen, F.J. Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell Metab. 2021, 33, 957–970.e6. [Google Scholar] [CrossRef] [PubMed]
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886. [Google Scholar] [CrossRef]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef]
- Hardie, D.G.; Sakamoto, K. AMPK: A Key Sensor of Fuel and Energy Status in Skeletal Muscle. Physiology 2006, 21, 48–60. [Google Scholar] [CrossRef]
- Wojtaszewski, J.F.P.; Nielsen, P.; Hansen, B.F.; Richter, E.A.; Kiens, B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J. Physiol. 2000, 528, 221–226. [Google Scholar] [CrossRef]
- Fujii, N.; Hayashi, T.; Hirshman, M.F.; Smith, J.T.; Habinowski, S.A.; Kaijser, L.; Mu, J.; Ljungqvist, O.; Birnbaum, M.; Witters, L.A.; et al. Exercise Induces Isoform-Specific Increase in 5′AMP-Activated Protein Kinase Activity in Human Skeletal Muscle. Biochem. Biophys. Res. Commun. 2000, 273, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Watson, K.; Baar, K. mTOR and the health benefits of exercise. Semin. Cell Dev. Biol. 2014, 36, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Little, J.P.; Safdar, A.; Wilkin, G.P.; Tarnopolsky, M.A.; Gibala, M.J. A practical model of low-volume high-intensity interval training induces mitochondrial biogenesis in human skeletal muscle: Potential mechanisms. J. Physiol. 2010, 588, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Energy sensing by the AMP-activated protein kinase and its effects on muscle metabolism. Proc. Nutr. Soc. 2010, 70, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Huang, J.; Geng, J.; Nair, U.; Klionsky, D.J. Atg22 Recycles Amino Acids to Link the Degradative and Recycling Functions of Autophagy. Mol. Biol. Cell 2006, 17, 5094–5104. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Kim, J.; Shaw, R.J.; Guan, K.-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of Macroautophagy by Calcium, Calmodulin-Dependent Kinase Kinase-β, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B Mutant Hampers the Lipidation of LC3 Paralogues and Causes Defects in Autophagosome Closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature 2008, 10, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Fader, C.M.; Sánchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta Bioenerg. 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Pastore, N.; Vainshtein, A.; Klisch, T.J.; Armani, A.; Huynh, T.; Herz, N.J.; Polishchuk, E.V.; Sandri, M.; Ballabio, A. TFE 3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 2017, 9, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 Coordinately Activates Protein Degradation by the Autophagic/Lysosomal and Proteasomal Pathways in Atrophying Muscle Cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; DePinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [Green Version]
- Van Der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; Van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the PI(3)K–PKB–FOXO network regulates autophagy. Nature 2012, 14, 829–837. [Google Scholar] [CrossRef]
- Zhou, J.; Liao, W.; Yang, J.; Ma, K.; Li, X.; Wang, Y.; Wang, D.; Wang, L.; Zhang, Y.; Yin, Y.; et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012, 8, 1712–1723. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Hood, D.A. The regulation of autophagy during exercise in skeletal muscle. J. Appl. Physiol. 2016, 120, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Markby, G.R.; Sakamoto, K. Transcription factor EB and TFE3: New metabolic coordinators mediating adaptive responses to exercise in skeletal muscle? Am. J. Physiol. Metab. 2020, 319, E763–E768. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Zorzano, A. Self-Eating for Muscle Fitness: Autophagy in the Control of Energy Metabolism. Dev. Cell 2020, 54, 268–281. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2010, 12, 9–14. [Google Scholar] [CrossRef]
- Cairns, G.; Thumiah-Mootoo, M.; Burelle, Y.; Khacho, M. Mitophagy: A New Player in Stem Cell Biology. Biology 2020, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Memme, J.M.; Hood, D.A. Molecular Basis for the Therapeutic Effects of Exercise on Mitochondrial Defects. Front. Physiol. 2020, 11, 615038. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.-K.; Lee, S.-H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2020, 17, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. Erratum: AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.; Kapralov, O.; Tyurin, V.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2016, 168, 224–238.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjottem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Okamoto, K.; Omiya, S.; Taneike, M.; Yamaguchi, O.; Otsu, K. A Mammalian Mitophagy Receptor, Bcl2-L-13, Recruits the ULK1 Complex to Induce Mitophagy. Cell Rep. 2019, 26, 338–345.e6. [Google Scholar] [CrossRef]
- Paul, M.H.; Sperling, E. Cyclophorase System XXIII. Correlation of Cyclophorase Activity and Mitochondrial Density in Striated Muscle. Exp. Biol. Med. 1952, 79, 352–354. [Google Scholar] [CrossRef]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Rossi, E.; Ruiz-Carrillo, A.; Rossi, G.; Rocchi, M.; Didonato, S.; Zuffardi, O.; Zeviani, M. Chromosomal localization of mitochondrial transcription factor A (TCF6), single-stranded DNA-binding protein (SSBP), and Endonuclease G (ENDOG), three human housekeeping genes involved in mitochondrial biogenesis. Genomics 1995, 25, 559–564. [Google Scholar] [CrossRef]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.N.; Clark, J.P.; Anderson, R.M. Mitochondrial regulator PGC-1a—Modulating the modulator. Curr. Opin. Endocr. Metab. Res. 2019, 5, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Uguccioni, G.; Vainshtein, A.; D’Souza, D. Mechanisms of Exercise-Induced Mitochondrial Biogenesis in Skeletal Muscle: Implications for Health and Disease. Compr. Physiol. 2011, 1, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise Training-Induced Regulation of Mitochondrial Quality. Exerc. Sport Sci. Rev. 2012, 40, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of Mitochondrial Biogenesis in Skeletal Muscle by CaMK. Science 2002, 296, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Ojuka, E.O.; Jones, T.E.; Han, D.H.; Chen, M.; Holloszy, J.O. Raising Ca2+in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 2003, 17, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Tran-scriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Wallberg, A.E.; Yamamura, S.; Malik, S.; Spiegelman, B.M.; Roeder, R.G. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1alpha. Mol. Cell 2003, 12, 1137–1149. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Conde Alonso, S.; Ofori, E.; Auwerx, J.; et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Janssen, I.; Heymsfield, S.B.; Baumgartner, R.N.; Ross, R. Estimation of skeletal muscle mass by bioelectrical impedance analysis. J. Appl. Physiol. 2000, 89, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Zierath, J.R.; Hawley, J.A. Skeletal muscle fiber type: Influence on contractile and metabolic properties. PLoS Biol. 2004, 2, e348. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Vihko, V. Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1984, 45, 97–106. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Schiavinato, A.; Castagnaro, S.; Bertaggia, E.; Sandri, M.; Bonaldo, P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 2011, 7, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Mansueto, G.; Armani, A.; Viscomi, C.; D’Orsi, L.; De Cegli, R.; Polishchuk, E.V.; Lamperti, C.; Di Meo, I.; Romanello, V.; Marchet, S.; et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017, 25, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Jamart, C.; Naslain, D.; Gilson, H.; Francaux, M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E964–E974. [Google Scholar] [CrossRef]
- Jamart, C.; Benoit, N.; Raymackers, J.M.; Kim, H.J.; Kim, C.K.; Francaux, M. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur. J. Appl. Physiol. 2012, 112, 3173–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collodet, C.; Foretz, M.; Deak, M.; Bultot, L.; Metairon, S.; Viollet, B.; Lefebvre, G.; Raymond, F.; Parisi, A.; Civiletto, G.; et al. AMPK promotes induction of the tumor suppressor FLCN through activation of TFEB independently of mTOR. FASEB J. 2019, 33, 12374–12391. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Rosato, A.S.; Prezioso, C.; For-rester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nature 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Desjardins, E.M.; Armani, A.; Sandri, M.; Hood, D.A. PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skelet. Muscle 2015, 5, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lettieri Barbato, D.; Tatulli, G.; Aquilano, K.; Ciriolo, M.R. FoxO1 controls lysosomal acid lipase in adipocytes: Implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis 2013, 4, e861. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Carson, B.P.; Garcia-Roves, P.M.; Chibalin, A.V.; Sarsfield, F.M.; Barron, N.; McCaffrey, N.; Moyna, N.M.; Zierath, J.R.; O’Gorman, D.J. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 2010, 588, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.W.; Erlich, A.T.; Hood, D.A. Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet Muscle 2018, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordsborg, N.B.; Lundby, C.; Leick, L.; Pilegaard, H. Relative workload determines exercise-induced increases in PGC-1alpha mRNA. Med. Sci. Sports Exerc. 2010, 42, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Drake, J.C.; Yan, Z. Exercise-Induced Mitophagy in Skeletal Muscle and Heart. Exerc. Sport Sci. Rev. 2019, 47, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Granata, C.; Oliveira, R.S.; Little, J.P.; Renner, K.; Bishop, D.J. Sprint-interval but not continuous exercise increases PGC-1alpha protein content and p53 phosphorylation in nuclear fractions of human skeletal muscle. Sci. Rep. 2017, 7, 44227. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.J.; Botella, J.; Genders, A.J.; Lee, M.J.; Saner, N.J.; Kuang, J.; Yan, X.; Granata, C. High-Intensity Exercise and Mitochondrial Biogenesis: Current Controversies and Future Research Directions. Physiology 2019, 34, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Bishop, D.; Tarnopolsky, M.A.; Gibala, M.J. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1alpha and activates mitochondrial biogenesis in human skeletal muscle. Am. J. Physiol. Regul. Integr Comp. Physiol. 2011, 300, R1303–R1310. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, M.M.; Kristensen, C.M.; Tøndering, A.S.; Lassen, S.B.; Ringholm, S.; Pilegaard, H. Impact of liver PGC-1α on exercise and exercise training-induced regulation of hepatic autophagy and mitophagy in mice on HFF. Physiol. Rep. 2018, 6, e13731. [Google Scholar] [CrossRef]
- Murray, C.J.; Barber, R.M.; Foreman, K.J.; Abbasoglu Ozgoren, A.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Abraham, J.P.; Abubakar, I.; Abu-Raddad, L.J.; et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: Quantifying the epidemiological transition. Lancet 2015, 386, 2145–2191. [Google Scholar] [CrossRef] [Green Version]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Principles of Exercise Prescription, and How They Influence Exercise-Induced Changes of Transcription Factors and Other Regulators of Mitochondrial Biogenesis. Sports Med. 2018, 48, 1541–1559. [Google Scholar] [CrossRef]
- Burgess, S.C.; He, T.; Yan, Z.; Lindner, J.; Sherry, A.D.; Malloy, C.R.; Browning, J.D.; Magnuson, M.A. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007, 5, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Shen, W.; Liu, Z.; Guan, S.; Liu, J.; Ding, S. Endurance exercise causes mitochondrial and oxidative stress in rat liver: Effects of a combination of mitochondrial targeting nutrients. Life Sci. 2010, 86, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 294, G619–G626. [Google Scholar] [CrossRef] [Green Version]
- Rector, R.S.; Uptergrove, G.M.; Morris, E.M.; Borengasser, S.J.; Laughlin, M.H.; Booth, F.W.; Thyfault, J.P.; Ibdah, J.A. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 300, G874–G883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoene, M.; Lehmann, R.; Hennige, A.M.; Pohl, A.K.; Häring, H.U.; Schleicher, E.D.; Weigert, C. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. J. Physiol. 2009, 587, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Go, K.L.; Leeuwenburgh, C.; Kim, J.S. Autophagy in the liver: Cell’s cannibalism and beyond. Arch. Pharm. Res. 2016, 39, 1050–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Rodriguez, S.; Petersen, P.S.; Seldin, M.M.; Bowman, C.E.; Wolfgang, M.J.; Wong, G.W. Loss of CTRP5 improves insulin action and hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E1036–E1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Lee, J.O.; Kim, N.; Kim, J.K.; Kim, H.I.; Lee, Y.W.; Kim, S.J.; Choi, J.I.; Oh, Y.; Kim, J.H.; et al. Irisin, a Novel Myokine, Regulates Glucose Uptake in Skeletal Muscle Cells via AMPK. Mol. Endocrinol. 2015, 29, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunadi, J.W.; Tarawan, V.M.; Daniel Ray, H.R.; Wahyudianingsih, R.; Lucretia, T.; Tanuwijaya, F.; Lesmana, R.; Supratman, U.; Setiawan, I. Different training intensities induced autophagy and histopathology appearances potentially associated with lipid metabolism in wistar rat liver. Heliyon 2020, 6, e03874. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Importance of the ‘crossover’ concept in exercise metabolism. Clin. Exp. Pharm. Physiol. 1997, 24, 889–895. [Google Scholar] [CrossRef]
- Chen, Z.P.; Stephens, T.J.; Murthy, S.; Canny, B.J.; Hargreaves, M.; Witters, L.A.; Kemp, B.E.; McConell, G.K. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes 2003, 52, 2205–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romijn, J.A.; Coyle, E.F.; Sidossis, L.S.; Gastaldelli, A.; Horowitz, J.F.; Endert, E.; Wolfe, R.R. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am. J. Physiol. Endocrinol. Metab. 1993, 265, E380–E391. [Google Scholar] [CrossRef] [Green Version]
- Tarnopolsky, L.J.; MacDougall, J.D.; Atkinson, S.A.; Tarnopolsky, M.A.; Sutton, J.R. Gender differences in substrate for endurance exercise. J. Appl. Physiol. 1990, 68, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Koonen, D.P.Y.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.-L.M.; Ong, H.; Vance, D.E.; Dyck, J.R.B. Increased Hepatic CD36 Expression Contributes to Dyslipidemia Associated With Diet-Induced Obesity. Diabetes 2007, 56, 2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Tarnopolsky, M.A.; Atkinson, S.A.; Phillips, S.M.; MacDougall, J.D. Carbohydrate loading and metabolism during exercise in men and women. J. Appl. Physiol. 1995, 78, 1360–1368. [Google Scholar] [CrossRef]
- Romijn, J.A.; Coyle, E.F.; Sidossis, L.S.; Rosenblatt, J.; Wolfe, R.R. Substrate metabolism during different exercise intensities in endurance-trained women. J. Appl. Physiol. 2000, 88, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.E.; Huang, T.-Y.; Simon, J.; Batdorf, H.M.; Essajee, N.M.; Scott, M.C.; Waskom, C.M.; Brown, J.M.; Burke, S.J.; Collier, J.J.; et al. Low-intensity exercise induces acute shifts in liver and skeletal muscle substrate metabolism but not chronic adaptations in tissue oxidative capacity. J. Appl. Physiol. 2019, 127, 143–156. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Laye, M.J.; Rector, R.S.; Borengasser, S.J.; Naples, S.P.; Uptergrove, G.M.; Ibdah, J.A.; Booth, F.W.; Thyfault, J.P. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J. Appl. Physiol. 2009, 106, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Haase, T.N.; Ringholm, S.; Leick, L.; Biensø, R.S.; Kiilerich, K.; Johansen, S.; Nielsen, M.M.; Wojtaszewski, J.F.; Hidalgo, J.; Pedersen, P.A.; et al. Role of PGC-1α in exercise and fasting-induced adaptations in mouse liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1501–R1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Kotani, K.; Tanaka, K.; Egucih, Y.; Anzai, K. Therapeutic Approaches to Nonalcoholic Fatty Liver Disease: Exercise Intervention and Related Mechanisms. Front. Endocrinol. 2018, 9, 588. [Google Scholar] [CrossRef]
- Delgado, T.C.; Pinheiro, D.; Caldeira, M.; Castro, M.M.C.A.; Geraldes, C.F.G.C.; López-Larrubia, P.; Cerdán, S.; Jones, J.G. Sources of hepatic triglyceride accumulation during high-fat feeding in the healthy rat. NMR Biomed. 2009, 22, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [Green Version]
- González-Rodríguez, Á.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death. Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.C.; Liu, C.H.; Tsai, Y.C.; Li, S.J.; Chen, C.Y.; Chu, C.H.; Chen, M.F. Time-dependent cellular response in the liver and heart in a dietary-induced obese mouse model: The potential role of ER stress and autophagy. Eur. J. Nutr. 2016, 55, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zeng, J.; Gu, Q. Exercise restores bioavailability of hydrogen sulfide and promotes autophagy influx in livers of mice fed with high-fat diet. Can. J. Physiol. Pharmacol. 2017, 95, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Barroso, E.; Rodríguez-Calvo, R.; Serrano-Marco, L.; Astudillo, A.M.; Balsinde, J.; Palomer, X.; Vázquez-Carrera, M. The PPARβ/δ Activator GW501516 Prevents the Down-Regulation of AMPK Caused by a High-Fat Diet in Liver and Amplifies the PGC-1α-Lipin 1-PPARα Pathway Leading to Increased Fatty Acid Oxidation. Endocrinology 2011, 152, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Ghareghani, P.; Shanaki, M.; Ahmadi, S.; Khoshdel, A.R.; Rezvan, N.; Meshkani, R.; Delfan, M.; Gorgani-Firuzjaee, S. Aerobic endurance training improves nonalcoholic fatty liver disease (NAFLD) features via miR-33 dependent autophagy induction in high fat diet fed mice. Obes. Res. Clin. Pract. 2018, 12, 80–89. [Google Scholar] [CrossRef]
- Liu, X.; Niu, Y.; Yuan, H.; Huang, J.; Fu, L. AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism 2015, 64, 658–665. [Google Scholar] [CrossRef]
- Rosa-Caldwell, M.E.; Lee, D.E.; Brown, J.L.; Brown, L.A.; Perry, R.A.; Greene, E.S.; Carvallo Chaigneau, F.R.; Washington, T.A.; Greene, N.P. Moderate physical activity promotes basal hepatic autophagy in diet-induced obese mice. Appl. Physiol. Nutr. Metab. 2016, 42, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhu, Y.-Y.; Wang, L.; Teng, T.; Zhou, M.; Wang, S.-G.; Tian, Y.-Z.; Du, L.; Yin, X.-X.; Sun, Y. Mangiferin ameliorates fatty liver via modulation of autophagy and inflammation in high-fat-diet induced mice. Biomed. Pharmacother. 2017, 96, 328–335. [Google Scholar] [CrossRef]
- Greene, N.P.; Lee, D.E.; Brown, J.L.; Rosa, M.E.; Brown, L.A.; Perry, R.A.; Henry, J.N.; Washington, T.A. Mitochondrial quality control, promoted by PGC-1α, is dysregulated by Western diet-induced obesity and partially restored by moderate physical activity in mice. Physiol. Rep. 2015, 3, e12470. [Google Scholar] [CrossRef]
- Keuper, M.; Jastroch, M.; Yi, C.X.; Fischer-Posovszky, P.; Wabitsch, M.; Tschöp, M.H.; Hofmann, S.M. Spare mitochondrial respiratory capacity permits human adipocytes to maintain ATP homeostasis under hypoglycemic conditions. Faseb J. 2014, 28, 761–770. [Google Scholar] [CrossRef]
- Martin, S.D.; McGee, S.L. The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochim. Et Biophys. Acta (BBA) - Gen. Subj. 2014, 1840, 1303–1312. [Google Scholar] [CrossRef]
- Talior, I.; Yarkoni, M.; Bashan, N.; Eldar-Finkelman, H. Increased glucose uptake promotes oxidative stress and PKC-delta activation in adipocytes of obese, insulin-resistant mice. Am. J. Physiol Endocrinol. Metab. 2003, 285, E295–E302. [Google Scholar] [CrossRef] [Green Version]
- Granneman, J.G.; Li, P.; Zhu, Z.; Lu, Y. Metabolic and cellular plasticity in white adipose tissue I: Effects of beta3-adrenergic receptor activation. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E608–E616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, J.M.; Grimsrud, P.A.; Wright, W.S.; Xu, X.; Foncea, R.E.; Graham, D.W.; Brestoff, J.R.; Wiczer, B.M.; Ilkayeva, O.; Cianflone, K.; et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 2010, 59, 1132–1142. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Masschelin, P.M.; Cox, A.R.; Chernis, N.; Hartig, S.M. The Impact of Oxidative Stress on Adipose Tissue Energy Balance. Front. Physiol. 2020, 10. [Google Scholar] [CrossRef]
- Jastroch, M.; Withers, K.W.; Taudien, S.; Frappell, P.B.; Helwig, M.; Fromme, T.; Hirschberg, V.; Heldmaier, G.; McAllan, B.M.; Firth, B.T.; et al. Marsupial uncoupling protein 1 sheds light on the evolution of mammalian nonshivering thermogenesis. Physiol. Genom. 2008, 32, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijers, S.L.; Saris, W.H.; van Marken Lichtenbelt, W.D. Recent advances in adaptive thermogenesis: Potential implications for the treatment of obesity. Obes. Rev. 2009, 10, 218–226. [Google Scholar] [CrossRef]
- Young, P.; Arch, J.R.; Ashwell, M. Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett. 1984, 167, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Cousin, B.; Cinti, S.; Morroni, M.; Raimbault, S.; Ricquier, D.; Pénicaud, L.; Casteilla, L. Occurrence of brown adipocytes in rat white adipose tissue: Molecular and morphological characterization. J. Cell Sci. 1992, 103 Pt 4, 931–942. [Google Scholar] [CrossRef]
- Rong, J.X.; Qiu, Y.; Hansen, M.K.; Zhu, L.; Zhang, V.; Xie, M.; Okamoto, Y.; Mattie, M.D.; Higashiyama, H.; Asano, S.; et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 2007, 56, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Lanza, I.R.; Swain, J.M.; Sarr, M.G.; Nair, K.S.; Jensen, M.D. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J. Clin. Endocrinol. Metab. 2014, 99, E209–E216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burguera, B.; Proctor, D.; Dietz, N.; Guo, Z.; Joyner, M.; Jensen, M.D. Leg free fatty acid kinetics during exercise in men and women. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E113–E117. [Google Scholar] [CrossRef]
- Wolfe, R.R.; Klein, S.; Carraro, F.; Weber, J.M. Role of triglyceride-fatty acid cycle in controlling fat metabolism in humans during and after exercise. Am. J. Physiol. Endocrinol. Metab. 1990, 258, E382–E389. [Google Scholar] [CrossRef]
- Sutherland, L.N.; Bomhof, M.R.; Capozzi, L.C.; Basaraba, S.A.U.; Wright, D.C. Exercise and adrenaline increase PGC-1α mRNA expression in rat adipose tissue. J. Physiol. 2009, 587, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Tiraby, C.; Tavernier, G.; Lefort, C.; Larrouy, D.; Bouillaud, F.; Ricquier, D.; Langin, D. Acquirement of Brown Fat Cell Features by Human White Adipocytes*. J. Biol. Chem. 2003, 278, 33370–33376. [Google Scholar] [CrossRef] [Green Version]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; Lee, M.Y.; Takahashi, H.; So, K.; Hitchcox, K.M.; Markan, K.R.; Hellbach, K.; Hirshman, M.F.; et al. A novel role for subcutaneous adipose tissue in exercise-induced improvements in glucose homeostasis. Diabetes 2015, 64, 2002–2014. [Google Scholar] [CrossRef] [Green Version]
- Trevellin, E.; Scorzeto, M.; Olivieri, M.; Granzotto, M.; Valerio, A.; Tedesco, L.; Fabris, R.; Serra, R.; Quarta, M.; Reggiani, C.; et al. Exercise training induces mitochondrial biogenesis and glucose uptake in subcutaneous adipose tissue through eNOS-dependent mechanisms. Diabetes 2014, 63, 2800–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.V.; Bikopoulos, G.; Hung, S.; Ceddia, R.B. Thermogenic capacity is antagonistically regulated in classical brown and white subcutaneous fat depots by high fat diet and endurance training in rats: Impact on whole-body energy expenditure. J. Biol. Chem. 2014, 289, 34129–34140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, J.G.; Murholm, M.; Carey, A.L.; Biensø, R.S.; Basse, A.L.; Allen, T.L.; Hidalgo, J.; Kingwell, B.A.; Febbraio, M.A.; Hansen, J.B.; et al. Role of IL-6 in exercise training- and cold-induced UCP1 expression in subcutaneous white adipose tissue. PLoS ONE 2014, 9, e84910. [Google Scholar] [CrossRef] [Green Version]
- De Matteis, R.; Lucertini, F.; Guescini, M.; Polidori, E.; Zeppa, S.; Stocchi, V.; Cinti, S.; Cuppini, R. Exercise as a new physiological stimulus for brown adipose tissue activity. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 582–590. [Google Scholar] [CrossRef]
- Sidossis, L.S.; Porter, C.; Saraf, M.K.; Børsheim, E.; Radhakrishnan, R.S.; Chao, T.; Ali, A.; Chondronikola, M.; Mlcak, R.; Finnerty, C.C.; et al. Browning of Subcutaneous White Adipose Tissue in Humans after Severe Adrenergic Stress. Cell Metab. 2015, 22, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallknecht, B.; Vinten, J.; Ploug, T.; Galbo, H. Increased activities of mitochondrial enzymes in white adipose tissue in trained rats. Am. J. Physiol. Endocrinol. Metab. 1991, 261, E410–E414. [Google Scholar] [CrossRef]
- Xu, X.; Ying, Z.; Cai, M.; Xu, Z.; Li, Y.; Jiang, S.Y.; Tzan, K.; Wang, A.; Parthasarathy, S.; He, G.; et al. Exercise ameliorates high-fat diet-induced metabolic and vascular dysfunction, and increases adipocyte progenitor cell population in brown adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1115–R1125. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.; Sifuentes, A.; Holland, W.L. Regulation of glucose and lipid homeostasis by adiponectin: Effects on hepatocytes, pancreatic β cells and adipocytes. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Baar, K.; Davidsen, P.K.; Atherton, P.J. Is irisin a human exercise gene? Nature 2012, 488, E9–E10. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Knaub, L.A.; Olivera-Fragoso, L.F.; Keller, A.C.; Balasubramaniam, V.; Watson, P.A.; Reusch, J.E.B. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1624–H1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Young, R.S.; Ayala, J.E.; Hunley, C.F.; James, F.D.; Bracy, D.P.; Kang, L.; Wasserman, D.H. Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1399–R1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: Role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E519–E528. [Google Scholar] [CrossRef] [Green Version]
- Salma, N.; Song, J.S.; Kawakami, A.; Devi, S.P.; Khaled, M.; Cacicedo, J.M.; Fisher, D.E. Tfe3 and Tfeb Transcriptionally Regulate Peroxisome Proliferator-Activated Receptor γ2 Expression in Adipocytes and Mediate Adiponectin and Glucose Levels in Mice. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Tao, Z.; Zheng, L.D.; Brooke, J.P.; Smith, C.M.; Liu, D.; Long, Y.C.; Cheng, Z. FoxO1 interacts with transcription factor EB and differentially regulates mitochondrial uncoupling proteins via autophagy in adipocytes. Cell Death. Discov. 2016, 2, 16066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, T.D.; Zhang, X.; Jeong, S.J.; He, A.; Song, E.; Bhattacharya, S.; Holloway, K.B.; Lodhi, I.J.; Razani, B. TFEB drives PGC-1α expression in adipocytes to protect against diet-induced metabolic dysfunction. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Negoita, F.; Säll, J.; Morén, B.; Stenkula, K.; Göransson, O. Salt-inducible kinase 2 regulates TFEB and is required for autophagic flux in adipocytes. Biochem. Biophys. Res. Commun. 2019, 508, 775–779. [Google Scholar] [CrossRef]
- Mendham, A.E.; Larsen, S.; George, C.; Adams, K.; Hauksson, J.; Olsson, T.; Fortuin-de Smidt, M.C.; Nono Nankam, P.A.; Hakim, O.; Goff, L.M.; et al. Exercise training results in depot-specific adaptations to adipose tissue mitochondrial function. Sci. Rep. 2020, 10, 3785. [Google Scholar] [CrossRef]
- Jackson, A.S.; Stanforth, P.R.; Gagnon, J.; Rankinen, T.; Leon, A.S.; Rao, D.C.; Skinner, J.S.; Bouchard, C.; Wilmore, J.H. The effect of sex, age and race on estimating percentage body fat from body mass index: The Heritage Family Study. Int. J. Obes. 2002, 26, 789–796. [Google Scholar] [CrossRef] [Green Version]
- White, U.A.; Tchoukalova, Y.D. Sex dimorphism and depot differences in adipose tissue function. Biochim. Et Biophys. Acta 2014, 1842, 377–392. [Google Scholar] [CrossRef] [Green Version]
- Enguix, N.; Pardo, R.; González, A.; López, V.M.; Simó, R.; Kralli, A.; Villena, J.A. Mice lacking PGC-1β in adipose tissues reveal a dissociation between mitochondrial dysfunction and insulin resistance. Mol. Metab. 2013, 2, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Boss, O.; Samec, S.; Desplanches, D.; Mayet, M.-H.; Seydoux, J.; Muzzin, P.; Giacobino, J.-P. Effect of endurance training on mRNA expression of uncoupling proteins 1, 2, and 3 in the rat. FASEB J. 1998, 12, 335–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuboyama-Kasaoka, N.; Tsunoda, N.; Maruyama, K.; Takahashi, M.; Kim, H.; Ikemoto, S.; Ezaki, O. Up-Regulation of Uncoupling Protein 3 (UCP3) mRNA by Exercise Training and Down-Regulation of UCP3 by Denervation in Skeletal Muscles. Biochem. Biophys. Res. Commun. 1998, 247, 498–503. [Google Scholar] [CrossRef]
- Oh, K.S.; Kim, E.Y.; Yoon, M.; Lee, C.M. Swim training improves leptin receptor deficiency-induced obesity and lipid disorder by activating uncoupling proteins. Exp. Mol. Med. 2007, 39, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, F.; Glanville, E.J. Low Levels of Physical Activity Increase Metabolic Responsiveness to Cold in a Rat (Rattus fuscipes). PLoS ONE 2010, 5, e13022. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.; Rioux, B.V.; Goulet, E.D.B.; Johanssen, N.M.; Swift, D.L.; Bouchard, D.R.; Loewen, H.; Sénéchal, M. Effect of an acute exercise bout on immediate post-exercise irisin concentration in adults: A meta-analysis. Scand. J. Med. Sci. Sports 2018, 28, 16–28. [Google Scholar] [CrossRef]
- Ioannilli, L.; Ciccarone, F.; Ciriolo, M.R. Adipose Tissue and FoxO1: Bridging Physiology and Mechanisms. Cells 2020, 9, 849. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Rinaldi, B.; Corbi, G.; Conti, V.; Stiuso, P.; Boccuti, S.; Rengo, G.; Rossi, F.; Filippelli, A. Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res. 2008, 11, 139–150. [Google Scholar] [CrossRef]
- Kurylowicz, A. Role of Sirtuins in Adipose Tissue Development and Metabolism. In Adipose Tissue-An Update; IntechOpen: London, UK, 2019. [Google Scholar]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177. [Google Scholar] [CrossRef]
- Kim, S.H.; Asaka, M.; Higashida, K.; Takahashi, Y.; Holloszy, J.O.; Han, D.-H. β-Adrenergic stimulation does not activate p38 MAP kinase or induce PGC-1α in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E844–E852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, L.G.; Lopes, M.A.; Batista, M.L., Jr. Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Åkerström, T.C.A.; Nielsen, A.R.; Fischer, C.P. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007, 103, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, S.H.; Min, Y.-K.; Yang, H.-M.; Lee, J.-B.; Lee, M.-S. Acute Exercise Induces FGF21 Expression in Mice and in Healthy Humans. PLoS ONE 2013, 8, e63517. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Vidal, P.; Stanford, K.I. Exercise-Induced Adaptations to Adipose Tissue Thermogenesis. Front. Endocrinol. 2020, 11, 270. [Google Scholar] [CrossRef]
- Ortega, S.P.; Chouchani, E.T.; Boudina, S. Stress turns on the heat: Regulation of mitochondrial biogenesis and UCP1 by ROS in adipocytes. Adipocyte 2017, 6, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, M.H.; Davis, M.J.; Secher, N.H.; van Lieshout, J.J.; Arce-Esquivel, A.A.; Simmons, G.H.; Bender, S.B.; Padilla, J.; Bache, R.J.; Merkus, D.; et al. Peripheral Circulation. Compr. Physiol. 2012, 2, 321–447. [Google Scholar]
- Hiura, M.; Nariai, T.; Ishii, K.; Sakata, M.; Oda, K.; Toyohara, J.; Ishiwata, K. Changes in cerebral blood flow during steady-state cycling exercise: A study using oxygen-15-labeled water with PET. J. Cereb. Blood Flow Metab. 2014, 34, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandrino, F.; Molinari, G.; Smeraldi, A.; Odaglia, G.; Masperone, M.A.; Sardanelli, F. Magnetic resonance imaging of athlete’s heart: Myocardial mass, left ventricular function, and cross-sectional area of the coronary arteries. Eur. Radiol. 2000, 10, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Kozàkovà, M.; Galetta, F.; Gregorini, L.; Bigalli, G.; Franzoni, F.; Giusti, C.; Palombo, C. Coronary Vasodilator Capacity and Epicardial Vessel Remodeling in Physiological and Hypertensive Hypertrophy. Hypertension 2000, 36, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Konhilas, J.P.; Kelly, D.P.; Leinwand, L.A. Molecular Mechanisms Underlying Cardiac Adaptation to Exercise. Cell Metab. 2017, 25, 1012–1026. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; Patterson, N.L.; McMullen, J.R. Understanding Key Mechanisms of Exercise-Induced Cardiac Protection to Mitigate Disease: Current Knowledge and Emerging Concepts. Physiol. Rev. 2018, 98, 419–475. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Santos-Lozano, A.; Joyner, M.; Carrera-Bastos, P.; Picazo, O.; Zugaza, J.L.; Izquierdo, M.; Ruilope, L.M.; Lucia, A. Exercise benefits in cardiovascular disease: Beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 2018, 15, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Miao, W.; Ma, J.; Xv, Z.; Bo, H.; Li, J.; Zhang, Y.; Ji, L.L. Acute Exercise-Induced Mitochondrial Stress Triggers an Inflammatory Response in the Myocardium via NLRP3 Inflammasome Activation with Mitophagy. Oxid Med. Cell Longev. 2016, 2016, 1987149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, E.; Schwalm, C.; Naslain, D.; Nielens, H.; Francaux, M.; Deldicque, L. Regular Endurance Exercise Promotes Fission, Mitophagy, and Oxidative Phosphorylation in Human Skeletal Muscle Independently of Age. Front. Physiol. 2019, 10, 1088. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.V.; Klionsky, D.J. Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr. Opin. Cell Biol. 1998, 10, 523–529. [Google Scholar] [CrossRef]
- Ju, J.S.; Jeon, S.I.; Park, J.Y.; Lee, J.Y.; Lee, S.C.; Cho, K.J.; Jeong, J.M. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J. Physiol. Sci. 2016, 66, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Brandt, N.; Gunnarsson, T.P.; Bangsbo, J.; Pilegaard, H. Exercise and exercise training-induced increase in autophagy markers in human skeletal muscle. Physiol. Rep. 2018, 6, e13651. [Google Scholar] [CrossRef]
- Lo Verso, F.; Carnio, S.; Vainshtein, A.; Sandri, M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 2014, 10, 1883–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yue, W.; Chen, H. The correlation between autophagy and tamoxifen resistance in breast cancer. Int J. Clin. Exp. Pathol 2019, 12, 2066–2074. [Google Scholar]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pan, S.-S.; Wan, D.-F.; Lu, J.; Huang, Y. H2O2 Signaling-Triggered PI3K Mediates Mitochondrial Protection to Participate in Early Cardioprotection by Exercise Preconditioning. Oxidative Med. Cell. Longev. 2018, 2018, 1916841. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, J.; Cretoiu, D.; Li, G.; Xiao, J. Exercise-mediated regulation of autophagy in the cardiovascular system. J. Sport Health Sci. 2020, 9, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I. Autophagic Vacuolar Myopathy. Semin. Pediatric Neurol. 2006, 13, 90–95. [Google Scholar] [CrossRef]
- Lee, Y.; Kang, E.-B.; Kwon, I.; Cosio-Lima, L.; Cavnar, P.; Javan, G. Cardiac Kinetophagy Coincides with Activation of Anabolic Signaling. Med. Sci. Sports Exerc. 2016, 48 2, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Iemitsu, M.; Naito, H.; Kakigi, R.; Kakehashi, C.; Maeda, S.; Akema, T. Single bout of running exercise changes LC3-II expression in rat cardiac muscle. Biochem. Biophys. Res. Commun. 2011, 414, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Hsu, H.-C.; Lee, B.-C.; Lin, H.-J.; Chen, Y.-H.; Huang, H.-C.; Ho, Y.-L.; Chen, M.-F. Exercise training improves cardiac function in infarcted rabbits: Involvement of autophagic function and fatty acid utilization. Eur. J. Heart Fail. 2010, 12, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Bei, Y.; Lin, S.; Zhang, H.; Zhou, Y.; Jiang, J.; Chen, P.; Shen, S.; Xiao, J.; Li, X. Exercise Training Protects Against Acute Myocardial Infarction via Improving Myocardial Energy Metabolism and Mitochondrial Biogenesis. Cell. Physiol. Biochem. 2015, 37, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; Urabe, Y.; Kent, R.L.; Vinciguerra, S.; Cooper, G. Cellular versus myocardial basis for the contractile dysfunction of hypertrophied myocardium. Circ. Res. 1991, 68, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Lehman, J.J.; Barger, P.M.; Kelly, D.P. Regulatory networks controlling mitochondrial energy production in the developing, hypertrophied, and diabetic heart. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 371–382. [Google Scholar] [CrossRef]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Sharov, V.G.; Goussev, A.; Lesch, M.; Goldstein, S.; Sabbah, H.N. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. J. Mol. Cell Cardiol. 1998, 30, 1757–1762. [Google Scholar] [CrossRef]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.D.; Lucena, J.R.; et al. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayod, S.; Del Valle, J.; Canudas, A.M.; Lalanza, J.F.; Sanchez-Roige, S.; Camins, A.; Escorihuela, R.M.; Pallàs, M. Long-term treadmill exercise induces neuroprotective molecular changes in rat brain. J. Appl. Physiol. 2011, 111, 1380–1390. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Caldwell, M.E.; Brown, J.L.; Lee, D.E.; Blackwell, T.A.; Turner, K.W.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Washington, T.A.; Greene, N.P. Autophagy activation, not peroxisome proliferator-activated receptor γ coactivator 1α, may mediate exercise-induced improvements in glucose handling during diet-induced obesity. Exp. Physiol. 2017, 102, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Santos-Alves, E.; Marques-Aleixo, I.; Rizo-Roca, D.; Torrella, J.R.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci. 2015, 135, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Lenhare, L.; Crisol, B.M.; Silva, V.R.R.; Katashima, C.K.; Cordeiro, A.V.; Pereira, K.D.; Luchessi, A.D.; da Silva, A.S.R.; Cintra, D.E.; Moura, L.P.; et al. Physical exercise increases Sestrin 2 protein levels and induces autophagy in the skeletal muscle of old mice. Exp. Gerontol. 2017, 97, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: "One-Size-Fits-All" and "Precision Medicine" Strategies. Pharmaceutics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C. Caloric restriction and exercise “mimetics”: Ready for prime time? Pharm. Res. 2016, 103, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, D.; Bottani, E.; Segala, A.; Marchet, S.; Rossi, F.; Orlando, F.; Malavolta, M.; Carruba, M.O.; Lamperti, C.; Provinciali, M.; et al. Targeting Multiple Mitochondrial Processes by a Metabolic Modulator Prevents Sarcopenia and Cognitive Decline in SAMP8 Mice. Front. Pharm. 2020, 11, 1171. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Tissue | Metabolic Mechanism | Effect of Exercise on Metabolic Mechanism | Effect on Physiology | Reference |
|---|---|---|---|---|
| Adipose | PGC-1α | Increases expression | Enhances mitochondrial biogenesis | [167] |
| PGC1-B | Not exercise-induced | Contributes to WAT browning Induces UCP1 expression. | [190] | |
| Adrenaline | Increases expression | Enhances PGC-1α mRNA expression post-exercise Enhances mitochondrial biogenesis | [167] | |
| UCP1 | Increases expression | Exercise-dependent increase in UCP1 drives WAT browning, regulated by a balance of mitophagy and mitochondrial biogenesis | [191,192,193,194] | |
| Irisin | Increased release from muscle | Promotes WAT UCP1-mediated thermogenesis Regulates muscle-adipose tissue cross-talk enhancing WAT browning, mediated by PGC-1α. Facilitates autophagy | [178,195] | |
| TFEB | Increased TFEB expression and nuclear translocation | TFEB induction by FOXO1 increases autophagy decreases UCP1 expression | [184,196] | |
| SIK2 | Not exercise-induced | Induces autophagic flux and TFEB expression | [186] | |
| SIRT1 | Increased activity | Induces deacetylation of PPARγ and PGC-1α and recruits adipose browning coactivators including PRDm16 | [197,198,199] | |
| Norepinepherine | Increased activity | Induces PGC-1α via p38 MAPK activation and subsequent ATF2 activation. | [200] | |
| Myokine response (IL-6, IL-10, IL1ra) | Increased | Important in anti-inflammatory response which also mediated by mitophagy to regulate inflammatory tone in response to exercise. | [201,202] | |
| eNOS | Increases response to exercise | Increases mitochondrial biogenesis | [170] | |
| FGF21 | Increases in response to exercise | Increases mitochondrial biogenesis | [203,204] | |
| Prdm16 | Increases in response to exercise | Induces upregulation of thermogenic genes and WAT adipocyte browning | [205] | |
| ROS | Increases in response to exercise | Induces mitochondrial biogenesis and induces WAT adipocyte browning | [206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, F.L.; Markby, G.R. New Insights into Molecular Mechanisms Mediating Adaptation to Exercise; A Review Focusing on Mitochondrial Biogenesis, Mitochondrial Function, Mitophagy and Autophagy. Cells 2021, 10, 2639. https://doi.org/10.3390/cells10102639
Roberts FL, Markby GR. New Insights into Molecular Mechanisms Mediating Adaptation to Exercise; A Review Focusing on Mitochondrial Biogenesis, Mitochondrial Function, Mitophagy and Autophagy. Cells. 2021; 10(10):2639. https://doi.org/10.3390/cells10102639
Chicago/Turabian StyleRoberts, Fiona Louise, and Greg Robert Markby. 2021. "New Insights into Molecular Mechanisms Mediating Adaptation to Exercise; A Review Focusing on Mitochondrial Biogenesis, Mitochondrial Function, Mitophagy and Autophagy" Cells 10, no. 10: 2639. https://doi.org/10.3390/cells10102639
APA StyleRoberts, F. L., & Markby, G. R. (2021). New Insights into Molecular Mechanisms Mediating Adaptation to Exercise; A Review Focusing on Mitochondrial Biogenesis, Mitochondrial Function, Mitophagy and Autophagy. Cells, 10(10), 2639. https://doi.org/10.3390/cells10102639