
Undescribed Phyllocladane-Type Diterpenoids from Callicarpa giraldii Hesse ex Rehd. and Their Anti-Neuroinflammatory Activity

 , ,
, ,
Abstract

1. Introduction
2. Results
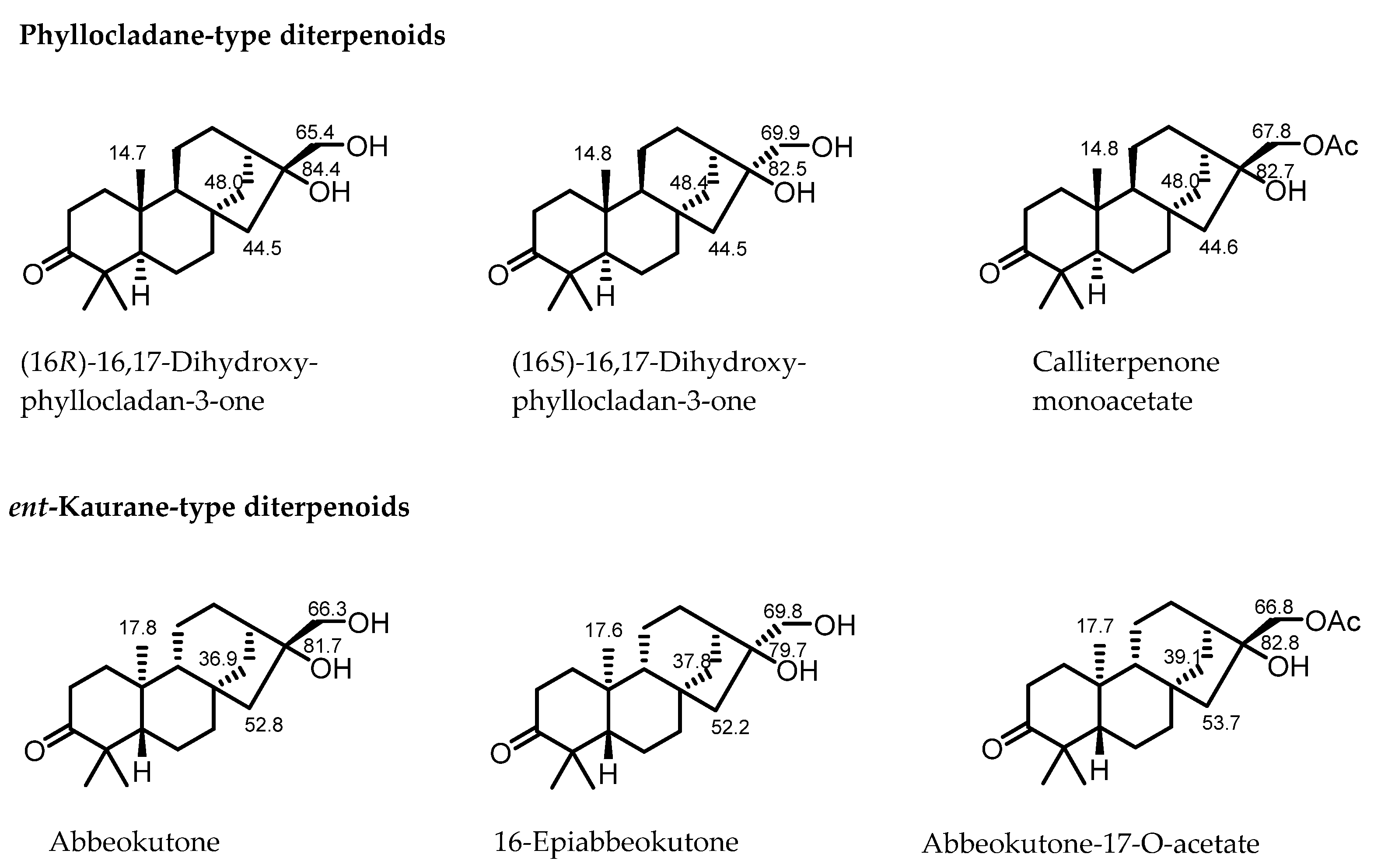
2.1. Spectroscopic Differentiation Between Phyllocladane- and Ent-Kaurane-Type Diterpenoids
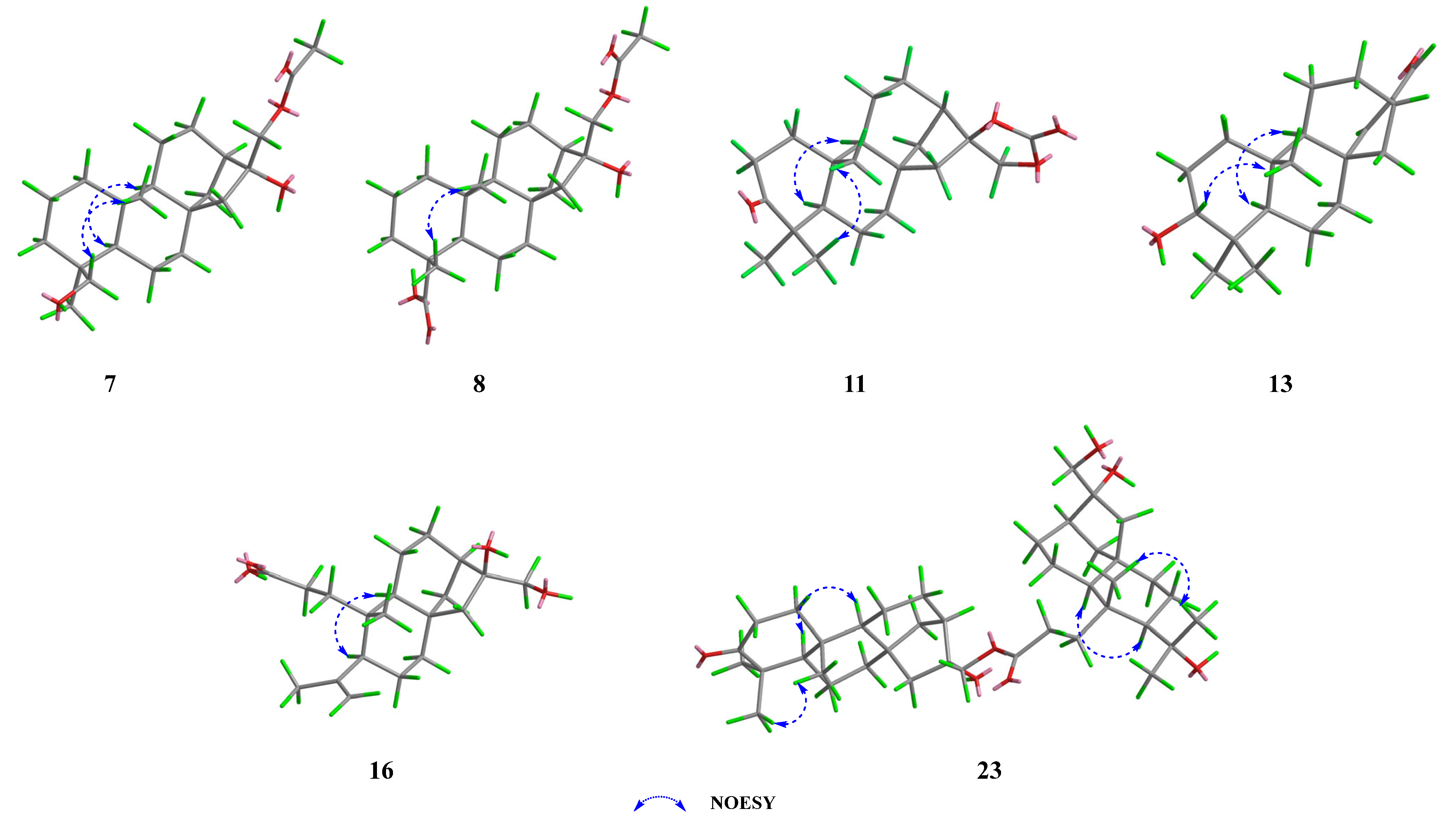
2.2. Structural Elucidation of New Compounds 7–24
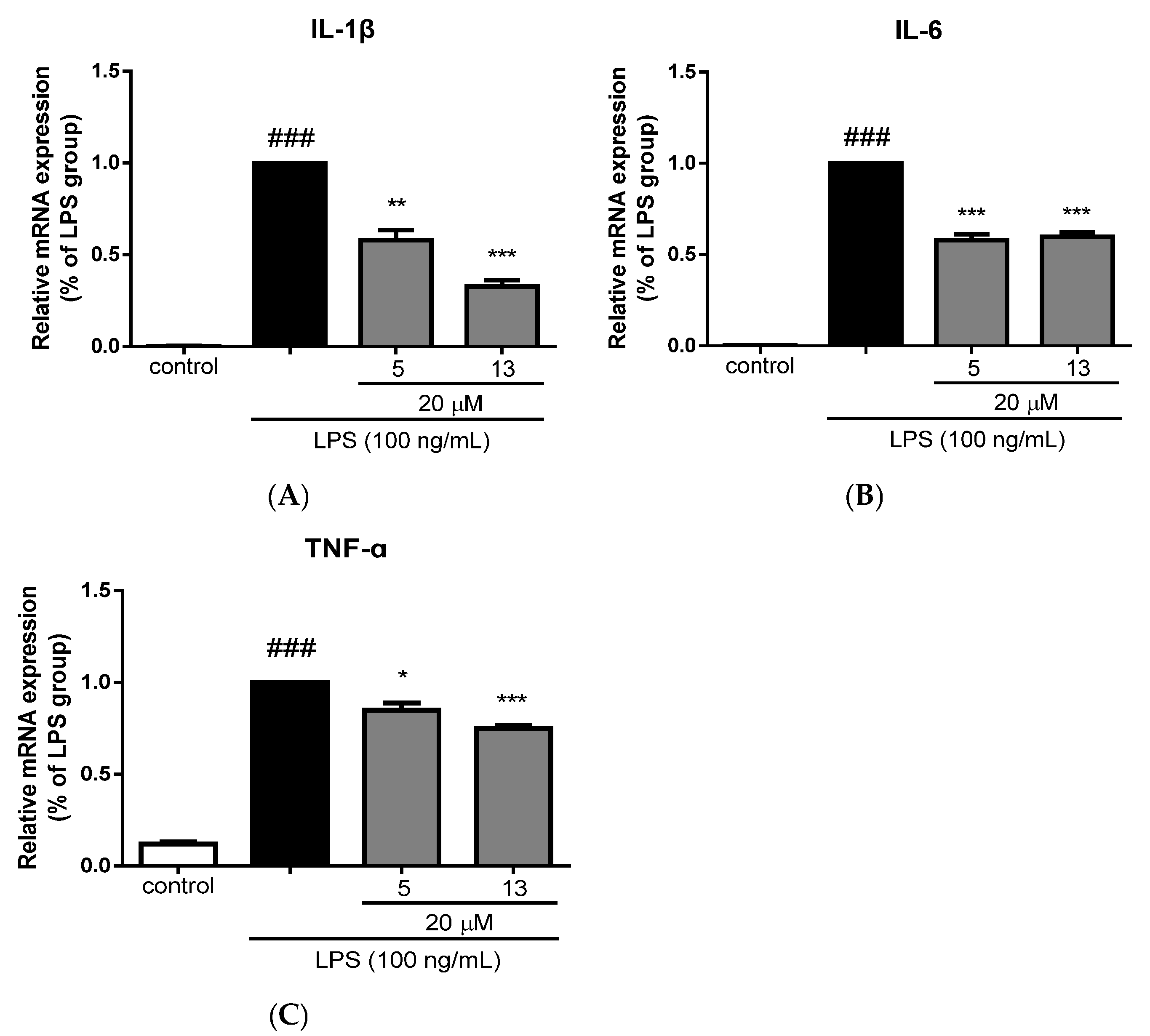
2.3. Anti-neuroinflammatory Activity Evaluation
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.3.1. Calligirlin A (7)
4.3.2. Calligirlin B (8)
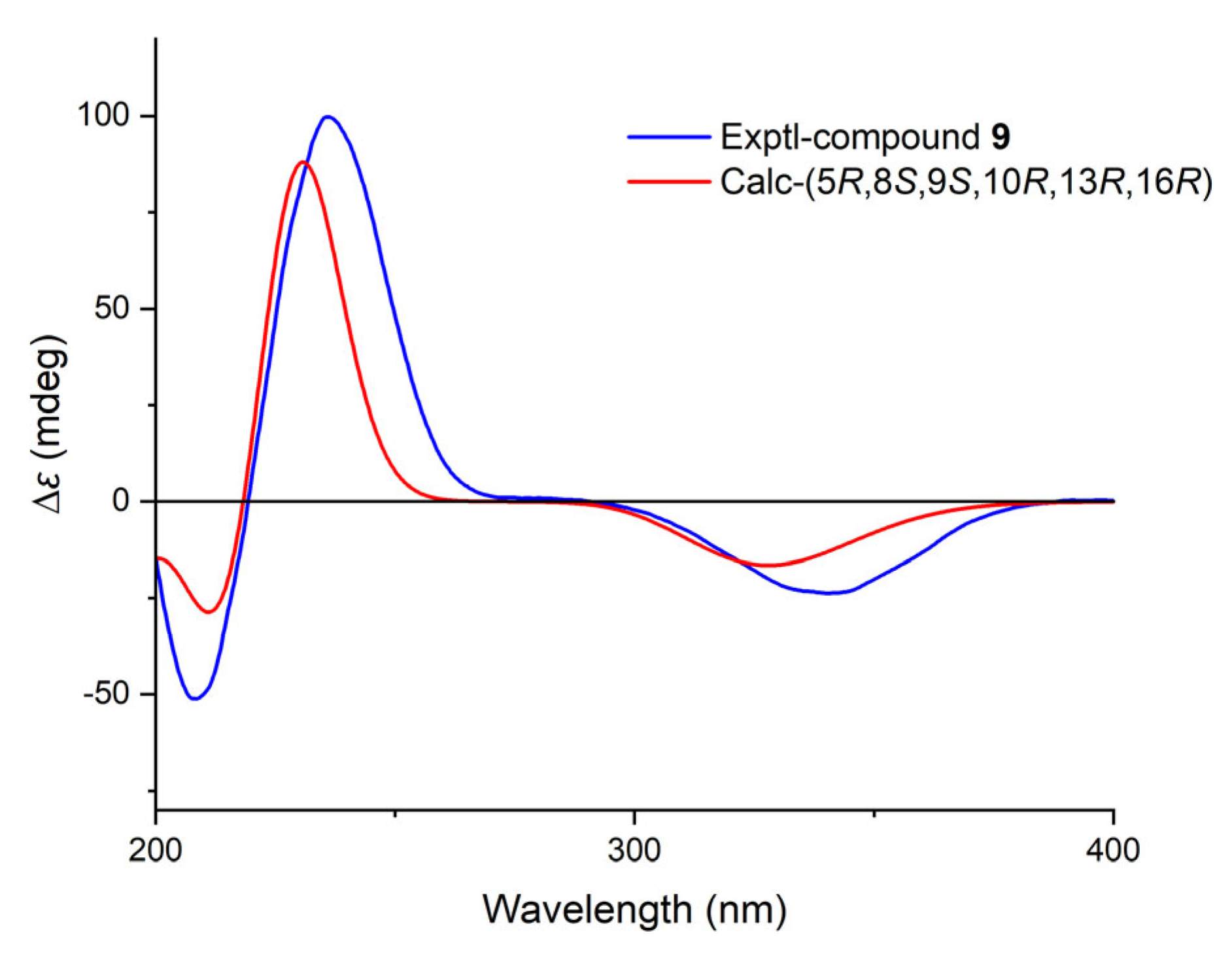
4.3.3. Calligirlin C (9)
4.3.4. Calligirlin D (10)
4.3.5. Calligirlin E (11)
4.3.6. Calligirlin F (12)
4.3.7. Calligirlin G (13)
4.3.8. Calligirlin H (14)
4.3.9. Calligirlin I (15)
4.3.10. Calligirlin J (16)
4.3.11. Calligirlin K (17)
4.3.12. Calligirlin L (18)
4.3.13. Calligirlin M (19)
4.3.14. Calligirlin N (20)
4.3.15. Calligirlin O (21)
4.3.16. Calligirlin P (22)
4.3.17. Calligirlin Q (23)
4.3.18. Calligirlin R (24)
4.4. Computational Section
4.5. X-Ray Crystallographic Analyses
4.5.1. Crystal Data for Compound 1
4.5.2. Crystal Data for Compound 2
4.5.3. Crystal Data for Compound 5
4.5.4. Crystal Data for Compound 7
4.5.5. Crystal Data for Compound 10
4.5.6. Crystal Data for Compound 11
4.5.7. Crystal Data for Compound 12
4.5.8. Crystal Data for Compound 13
4.5.9. Crystal Data for Compound 15
4.6. Pharmacological Activity Assessment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Li, Q.; Shang, K.; Wang, J.; Xu, C.S.; Cai, Z.E.; Yuan, P.; Wang, C.G.; Gu, M.M.; Zhang, Y.; Liao, Z.X. Identification, structural revision and biological evaluation of the phyllocladane-type diterpenoids from Callicarpa longifolia var. floccosa. Tetrahedron 2024, 167, 134304. [Google Scholar] [CrossRef]
- Toyomasu, T.; Niida, R.; Kenmoku, H.; Kanno, Y.; Miura, S.; Nakano, C.; Shiono, Y.; Mitsuhashi, W.; Toshima, H.; Oikawa, H.; et al. Identification of diterpene biosynthetic gene clusters and functional analysis of labdane-related diterpene cyclases in Phomopsis amygdali. Biosci. Biotechnol. Biochem. 2008, 72, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.R.; Shurpali, K.; Gawde, R.L.; Sarkar, D.; Puranik, V.G.; Joshi, S.P. Phyllocladane diterpenes from Anisomeles heyneana. J. Asian Nat. Prod. Res. 2012, 14, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Chang, S.T.; Chang, S.C.; Chang, H.T. Isolation of antibacterial diterpenoids from Cryptomeria japonica bark. Nat. Prod. Res. 2008, 22, 1085–1093. [Google Scholar] [CrossRef]
- Goel, M.K.; Kukreja, A.K.; Singh, A.K.; Khanuja, S.P.S. In vitro plant growth promoting activity of phyllocladane diterpenoids isolated from Callicarpa macrophylla Vahl. in shoot cultures of Rauwolfia serpentina. Nat. Prod. Commun. 2007, 2, 799–802. [Google Scholar] [CrossRef]
- Manríquez-Torres, J.J.; Hernández-Lepe, M.A.; Chávez-Méndez, J.R.; González-Reyes, S.; Serafín-Higuera, I.R.; Rodríguez-Uribe, G.; Torres-Valencia, J.M. Isolation and cytotoxic activity of phyllocladanes from the roots of Acacia schaffneri (Leguminosae). Molecules 2020, 25, 3944. [Google Scholar] [CrossRef]
- Sun, H.D. Diterpenoid Chemistry; Chemical Industry Press: Beijing, China, 2012; ISBN 9787122121394. [Google Scholar]
- Wu, Z.Y.; Raven, P.H.; Hong, D.Y. Flora of China; Science Press (Beijing) & Missouri Botanical Garden Press: Beijing, China, 2011. [Google Scholar]
- Dong, L.; Zhang, L.; Zhang, X.; Liu, M.; Wang, J.; Wang, Y. Two new 3,4-seco-labdane diterpenoids from Callicarpa nudiflora and their inhibitory activities against nitric oxide production. Phytochem. Lett. 2014, 10, 127–131. [Google Scholar] [CrossRef]
- Sun, X.; Liu, F.; Yang, X.; Wang, J.; Dong, B.; Xie, C.; Jin, D.Q.; Zhang, J.; Lee, D.; Ohizumi, Y.; et al. Seco-labdane diterpenoids from the leaves of Callicarpa nudiflora showing nitric oxide inhibitory activity. Phytochemistry 2018, 149, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Z.; Zhu, C.C.; Zhao, Z.X.; Li, X.H.; Xiong, T.Q.; Xia, Y.Y.; Ning, Y. Two new abietane diterpenoids from the caulis and leaves of Callicarpa kochiana. Fitoterapia 2012, 83, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, Y.; Wang, M.; Ren, Q.; Li, S.; Wang, H.; Sun, X.; Jin, D.Q.; Sun, H.; Ohizumi, Y.; et al. Bioactive diterpenoids from the leaves of Callicarpa macrophylla. J. Nat. Prod. 2015, 78, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, S.; Sun, X.; Ma, J.; Liu, F.; Tong, L.; Lee, D.; Ohizumi, Y.; Tuerhong, M.; Guo, Y. Diterpenoids from Callicarpa kwangtungensis and their NO inhibitory effects. Fitoterapia 2016, 113, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, J.; Wang, Q.; Shang, K.; Pu, D.B.; Zhang, R.H.; Li, X.L.; Dai, X.C.; Zhang, X.J.; Xiao, W.L. Clerodane diterpenoids with potential anti-inflammatory activity from the leaves and twigs of Callicarpa cathayana. Chin. J. Nat. Med. 2019, 17, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tang, C.P.; Ke, C.Q.; Shu, R.G.; Ye, Y. 3,4-seco-isopimarane and 3,4-seco-pimarane diterpenoids from Callicarpa nudiflora. Chin. J. Nat. Med. 2021, 19, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G. Study of chemical constituents of two medical plants of Callicarpa Genus. Ph.D Thesis, Guangzhou University of Chinese Medicine, Guangzhou, China, 2019; pp. 72–77. [Google Scholar]
- Gui, L.; Ralph, M.; Peter, R. Chemical transformations of phyllocladane (=13β-Kaurane) diterpenoids. Helv. Chim. Acta 2003, 86, 420–438. [Google Scholar] [CrossRef]
- Singh, A.K.; Agrawal, P.K. 16α, l7-isopropylidenoI-3-oxo-phyllocladane, a diterpenoid from Callicarpa macrophylla. Phytochemistry 1994, 37, 587–588. [Google Scholar] [CrossRef]
- Shao, Y.; Hu, L.H.; Sim, L.Y.; Goh, S.H. Lignanoids and diterpenoids from Callicarpa furfuraceae. Helv. Chim. Acta 2006, 86, 64–71. [Google Scholar] [CrossRef]
- Bohlmannc, F.; Zderob, C.; Turner, B.L. Guaianolides and heliangolides from Hymenopappus newberryi. Phytochemistry 1984, 23, 1055–1058. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Organomet. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, T.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Goto, H.; Obata, S.; Kakayama, N.; Ohta, K. CONFLEX 8, CONFLEX Corporation: Tokyo, Japan, 2017.
- Pescitelli, T.G.; Bruhn, T. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C-14 | C-15 | C-20 | C-17 | |
|---|---|---|---|---|
| Phyllocladane-type | >40 ppm | <50 ppm | About 14–15 ppm | About 65–68 ppm, rel-16R; >69 ppm, rel-16S |
| ent-Kaurane-type | <40 ppm | >50 ppm | About 17 ppm | About 65–68 ppm, rel-16R; >69 ppm, rel-16S |
| No. | 7 a | 8 b | 9 a | 10 a |
|---|---|---|---|---|
| 1 | 0.81 (dd, 4.0, 13.1) 1.60 c | 0.86 (m) 1.69 c | 6.99 (d, 10.2) | 1.37 (m) 1.90 (m) |
| 2 | 1.38 (m, 2H) | 1.40 (m) 1.82 (m) | 5.83 (d, 10.2) | 2.37 (m) 2.49 (m) |
| 3 | 0.92 (m) 1.78 (m) | 1.01 (td, 13.5, 4.2) 2.15 (d, 13.5) | ||
| 5 | 1.10 (m) | 1.08 (m) | 1.62 (m) | 1.35 (m) |
| 6 | 1.63 c (2H) | 1.65 (m) 1.82 (m) | 1.40 (m) 1.58 (m) | 1.37 (m) 1.44 c |
| 7 | 1.49 (m) 1.67 (dt, 13.2, 3.2) | 1.46 (m) 1.69 c | 1.59 (m) 1.74 c | 1.51 (m, 2H) |
| 9 | 0.98 (m) | 1.08 (m) | 1.36 c | 1.09 (m) |
| 11 | 1.20 (m) 1.56 c | 1.19 (m) 1.57 (m) | 1.36 c 1.78 (m) | 1.51 c |
| 12 | 1.43 (m) 1.59 c | 1.44 (m) 1.59 (m) | 1.51 (m) 1.74 c | 1.44 c 1.55 (m) |
| 13 | 1.90 (q, 3.9) | 1.92 (m) | 1.98 (q, 3,9) | 1.94 (m) |
| 14 | 1.07 (m) 2.09 (m) | 1.07 (m) 2.09 (m) | 1.16 (m) 2.14 (ddd, 11.5, 4.8, 2.5) | 0.99 (m) 1.44 c |
| 15 | 1.23 (m) 2.07 (m) | 1.29 (d, 14.6) 2.05 (m) | 1.30 (d, 14.7) 2.05 (dd, 14.7, 2.0) | 0.71 (dd, 13.5, 5.3) 2.20 (m) |
| 16 | 1.94 (m) | |||
| 17 | 4.16 (d, 11.3) 4.24 (d, 11.3) | 4.16 (d, 11.3) 4.23 (d, 11.3) | 3.65 (d, 10.9) 3.79 (d, 10.9) | 3.39 (m, 2H) |
| 18 | 3.41 (d, 10.9) 3.74 (d, 10.9) | 1.23 (s, 3H) | 1.13 (s, 3H) | 1.06 (s, 3H) |
| 19 | 0.96 (s, 3H) | 1.08 (s, 3H) | 1.01 (s, 3H) | |
| 20 | 0.85 (s, 3H) | 0.80 (s, 3H) | 1.09 (s, 3H) | 1.01 (s, 3H) |
| OAc | 2.10 (s, 3H) | 2.10 (s, 3H) |
| No. | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|
| 1 | 39.5 | 39.8 | 158.2 | 38.6 | 38.3 | 38.4 | 36.9 | 38.1 |
| 2 | 18.1 | 19.0 | 125.8 | 34.2 | 34.0 | 34.2 | 27.4 | 34.0 |
| 3 | 35.6 | 38.0 | 205.3 | 218.2 | 217.2 | 218.1 | 79.0 | 216.5 |
| 4 | 38.6 | 44.0 | 44.7 | 47.5 | 47.5 | 47.6 | 39.0 | 47.7 |
| 5 | 57.0 | 57.1 | 53.5 | 55.6 | 55.2 | 55.4 | 55.1 | 55.6 |
| 6 | 20.5 | 22.0 | 20.7 | 21.5 | 20.3 | 21.4 | 19.8 | 19.7 |
| 7 | 41.8 | 41.5 | 40.9 | 40.6 | 39.8 | 40.5 | 36.1 | 37.9 |
| 8 | 43.9 | 43.7 | 44.2 | 44.4 | 44.3 | 43.6 | 49.7 | 36.4 |
| 9 | 56.9 | 56.2 | 50.8 | 56.0 | 55.2 | 56.7 | 53.9 | 50.5 |
| 10 | 37.8 | 38.4 | 39.9 | 37.3 | 37.1 | 37.3 | 37.3 | 37.3 |
| 11 | 19.5 | 19.6 | 19.7 | 20.0 | 21.3 | 19.7 | 19.0 | 25.2 |
| 12 | 26.9 | 26.9 | 26.7 | 32.6 | 26.5 | 28.8 | 24.7 | 216.5 |
| 13 | 44.7 | 44.6 | 44.1 | 46.2 | 43.8 | 47.9 | 36.4 | 50.3 |
| 14 | 48.4 | 48.1 | 48.1 | 47.8 | 48.4 | 51.0 | 54.0 | 55.9 |
| 15 | 44.8 | 44.6 | 45.2 | 36.7 | 47.0 | 33.4 | 156.9 | 37.8 |
| 16 | 82.8 | 82.8 | 84.5 | 36.9 | 93.7 | 38.5 | 147.6 | 31.3 |
| 17 | 68.0 | 67.9 | 65.6 | 67.6 | 70.7 | 179.3 | 190.0 | 22.9 |
| 18 | 65.8 | 29.2 | 28.4 | 26.9 | 26.9 | 26.9 | 28.4 | 26.4 |
| 19 | 27.3 | 181.9 | 21.9 | 21.6 | 21.7 | 21.7 | 15.8 | 21.7 |
| 20 | 15.5 | 13.5 | 18.2 | 15.2 | 15.0 | 15.0 | 15.6 | 13.7 |
| OAc | 171.4, C | 171.5, C | ||||||
| 21.1, CH3 | 21.1, CH3 | |||||||
| C=O | 154.7, C |
| No. | 11 a | 12 b | 13 b | 14 b |
|---|---|---|---|---|
| 1 | 1.42 (m), 1.87 (m) | 1.89 (m) 2.58 c | 1.02 (m) 1.61 c | 1.36 c 1.81 (m) |
| 2 | 2.40 (ddd, 15.9, 7.1, 4.1) 2.51 (m) | 2.37 (ddd, 15.8,7.0, 4.0) 2.53 c | 1.51 (m) 1.62 c | 2.36 (m) 2.57 (ddd, 16.1, 12.2, 6.9) |
| 3 | 3.21 (dd, 11.7, 4.1) | |||
| 5 | 1.39 (m) | 1.38 c | 0.88 (dd, 11.6, 2.2) | 1.36 c |
| 6 | 0.96 (m) 1.67 (m) | 1.48 c(2H) | 1.47 c, 1.64 (m) | 1.51 (m, 2H) |
| 7 | 1.59 (m) 1.72 (m) | 1.50 c 1.63 (m) | 1.42 (dd, 13.2, 4.2) 1.77 (dt, 13.4, 2.9) | 1.25 c 1.60 (m) |
| 9 | 1.19 (m) | 1.14 (m) | 1.18 c | 1.25 c |
| 11 | 1.36 (m) 1.56 (m) | 1.49 c(2H) | 1.56 c 1.15 c | 1.64 (m) 1.84 c |
| 12 | 1.56 (m) 1.77 (m) | 1.47 c 1.82 (m) | 1.46 c 1.58 c | |
| 13 | 2.23 (m) | 3.08 (dt, 12.1, 6.1) | 2.88 (p, 2.8) | 2.16 (m) |
| 14 | 1.29 (dd, 11.7, 2.4) 1.98 (ddd, 11.7, 4.9, 2.4) | 1.26 (dd, 11.1, 2.0) 1.60 (m) | 1.27 (d, 10.3) 1.85 (ddd, 10.3, 5.4, 2.3) | 1.84 c 1.90 (m) |
| 15 | 1.90 (m) 2.28 (dd, 15.2, 2.4) | 1.38 c (2H) | 6.85 (s) | 0.69 (dd, 14.0, 6.6) 2.36 (m) |
| 16 | 2.47 (m) | 2.00 (m) | ||
| 17 | 4.35 (d, 8.8) 4.52 (d, 8.8) | 9.73 (s) | 0.90 (d, 7.0) | |
| 18 | 1.09 (s, 3H) | 1.08 (s, 3H) | 1.00 (s, 3H) | 1.08 (s, 3H) |
| 19 | 1.03 (s, 3H) | 1.04 (s, 3H) | 0.81 (s, 3H) | 1.05 (s, 3H) |
| 20 | 0.97 (s, 3H) | 1.08 (s, 3H) | 0.75 (s, 3H) | 1.14 (s, 3H) |
| No. | 15 a | 16 a | 17 b | 18 c |
|---|---|---|---|---|
| 1 | 1.64 d (2H) | 1.53 (m, 2H) | 1.56 (m, 2H) | 1.52 d |
| 2 | 2.15 (m, 2H) | 2.19 (m) 2.33 (m) | 2.22 (m) 2.37 (m) | 2.18 (m) 2.32 (m) |
| 4 | 1.91 d | |||
| 5 | 1.10 (m) | 2.06 (m) | 1.97 (m) | 1.98 (dd, 12.3, 2.8) |
| 6 | 1.28 d 1.41 (m) | 1.39 (m) 1.70 (m) | 1.39 (m) 1.62 (m) | 1.38 (m) 1.60 (m) |
| 7 | 1.44 d 1.64 d | 1.55 (m, 2H) | 1.50 (m) 1.59 (m) | 1.52 d 1.58 (m) |
| 9 | 1.28 d | 1.31 (m) | 1.23 (m) | 1.20 (m) |
| 11 | 1.35 (m) 1.54 (m) | 1.37 (m) 1.50 (m) | 1.29 (m) 1.48 d | 1.27 (m) 1.49 d |
| 12 | 1.47 d 1.73 (m) | 1.47 (m) 1.72 (m) | 1.48 d | 1.44 (m) 1.68 (m) |
| 13 | 1.89 (m) | 1.91 (m) | 1.92 (m) | 1.92 (m) |
| 14 | 1.10 d 2.10 (ddd, 11.1, 4.7, 2.4) | 1.11 (d, 11.1) 2.10 (m) | 1.10 (d, 11.1) 2.12 (m) | 1.10 (dd, 11.5, 1.9), 2.06 d |
| 15 | 1.20 (d, 14.5) 2.00 (d, 14.5) | 1.22 (d, 14.5) 2.05 (m) | 1.31 (m) 2.07 (d, 15.7) | 1.25 d 2.06 d |
| 17 | 3.57 (d, 11.3) 3.68 (d, 11.3) | 3.58 (d, 11.2) 3.70 (d, 11.2) | 4.18 (d, 11.2) 4.23 (d, 11.2) | 3.62 (d, 11.0) 3.77 (d, 11.0) |
| 18 | 0.93 (d, 6.9, 3H) | 4.67 (s) 4.88 (s) | 4.64 (s) 4.87 (s) | 4.63 (s) 4.87 (s) |
| 19 | 0.81 (d, 6.9, 3H) | 1.75 (s, 3H) | 1.73 (s, 3H) | 1.72 (s, 3H) |
| 20 | 0.87 (s, 3H) | 0.90 (s, 3H) | 0.87 (s, 3H) | 0.86 (s, 3H) |
| OAc | 2.10 (s, 3H) | |||
| C2H5 | 4.10 (q, 7.1, 2H) | |||
| 1.24 (t, 7.1, 3H) |
| No. | 15 a | 16 a | 17 b | 18 b | 19 b | 20 b | 21 b | 22 c |
|---|---|---|---|---|---|---|---|---|
| 1 | 33.5 | 34.3 | 32.8 | 33.0 | 31.9 | 32.1 | 33.5 | 33.4 |
| 2 | 29.3 | 29.3 | 28.0 | 28.6 | 28.0 | 28.5 | 29.2 | 29.2 |
| 3 | 178.6 | 178.2 | 178.3 | 174.3 | 178.8 | 174.5 | 175.4 | 175.3 |
| 4 | 26.7 | 148.8 | 147.2 | 147.3 | 25.2 | 25.2 | 75.9 | 75.8 |
| 5 | 49.3 | 51.9 | 50.8 | 50.7 | 48.1 | 47.6 | 51.8 | 51.8 |
| 6 | 20.9 | 27.5 | 26.2 | 26.2 | 19.9 | 19.9 | 24.6 | 24.6 |
| 7 | 41.7 | 41.3 | 39.9 | 40.1 | 40.4 | 40.6 | 40.8 | 40.6 |
| 8 | 44.8 | 44.6 | 43.7 | 43.6 | 43.9 | 43.8 | 43.9 | 44.0 |
| 9 | 48.9 | 48.7, | 47.6 | 47.5 | 47.6 | 47.6 | 48.4 | 48.4 |
| 10 | 41.1 | 40.6 | 39.6 | 39.7 | 40.4 | 40.3 | 41.8 | 41.8 |
| 11 | 21.0 | 20.9 | 19.8 | 19.9 | 19.9 | 19.9 | 19.2 | 19.1 |
| 12 | 27.9 | 27.8 | 26.9 | 26.9 | 27.0 | 27.0 | 27.1 | 27.1 |
| 13 | 44.9 | 44.8 | 44.6 | 44.0 | 44.6 | 44.0 | 43.9 | 44.5 |
| 14 | 49.5 | 49.2 | 48.2 | 48.4 | 48.3 | 48.5 | 48.2 | 48.1 |
| 15 | 45.2 | 45.1 | 44.5 | 44.7 | 44.6 | 44.7 | 44.5 | 44.5 |
| 16 | 85.6 | 85.6 | 82.7 | 84.5, | 82.6 | 84.5 | 84.4 | 82.6 |
| 17 | 66.2 | 66.2 | 67.9 | 65.8 | 67.9 | 65.8 | 65.8 | 67.9 |
| 18 | 25.2 | 114.2 | 114.0 | 113.9 | 25.0 | 25.0 | 34.2 | 34.2 |
| 19 | 19.5 | 19.8 | 23.8 | 23.9 | 19.1 | 19.1 | 27.8 | 27.2 |
| 20 | 19.3 | 19.4 | 18.8 | 18.9 | 18.7 | 18.8 | 19.4 | 19.3 |
| OAc | 171.4, C | 171.4, C | 171.4, C | |||||
| 21.1, CH3 | 21.1, CH3 | 21.1, CH3 | ||||||
| C2H5 | 60.5, CH2 | 60.5, CH2 | 60.6, CH2 | 60.9, CH2 | ||||
| 14.4, CH3 | 14.4, CH3 | 14.4, CH3 | 14.4, CH3 |
| No. | 19 a | 20 a | 21 a | 22 b |
|---|---|---|---|---|
| 1 | 1.66 (m, 2H) | 1.66 (m, 2H) | 1.64 (m) 2.34 (ddd, 14.9, 10.7, 4.8) | 1.61 c 2.33 (ddd, 15.2, 10.7, 4.8) |
| 2 | 2.21 (m, 2H) | 2.18 (m, 2H) | 2.19 (ddd, 15.0, 10.7, 5.8) 2.45 (ddd, 15.3, 10.6, 4.7) | 2.18 (ddd, 15.0, 10.7, 5.8) 2.45 (ddd, 15.3, 10.7, 4.8) |
| 4 | 1.86 (p, 6.8) | 1.88 (m) | ||
| 5 | 1.02 (m) | 1.23 c | 1.36 c | 1.35 c |
| 6 | 1.20 c 1.42 (m) | 1.23 c | 1.36 c 1.44 c | 1.35 c 1.44 (m) |
| 7 | 1.42 (m) 1.63 (m) | 1.42 c 1.63 (m) | 1.44 c 1.59 (m) | 1.41 (m) 1.60 c |
| 9 | 1.20 c | 1.23 c | 1.23 (m) | 1.22 c |
| 11 | 1.25 c 1.51 (m) | 1.42 c 1.54 (m) | 1.22 c 1.56 (m) | 1.22 c 1.55 c |
| 12 | 1.45 (m) 1.59 (m) | 1.48 (m) 1.70 (m) | 1.67 (m, 2H) | 1.62 c (2H) |
| 13 | 1.92 (m) | 1.93 (m) | 1.91 (q, 3.9) | 1.91 (q, 3.8) |
| 14 | 1.08 (d, 14.5) 2.09 (m) | 1.10 (d, 11.2) 2.08 (m) | 1.07 (m) 2.05 (m) | 1.06 (m) 2.08 c |
| 15 | 1.28 (d, 11.2) 2.02 (m) | 1.26 (m) 2.03 (m) | 1.22 c 2.00 (m) | 1.26 c 2.02 (m) |
| 17 | 4.16 (d, 11.3) 4.23 (d, 11.3) | 3.64 (m) 3.78 (m) | 3.62 (d, 10.9) 3.76 (d, 10.9) | 4.16 (d, 11.3) 4.22 (d, 11.3) |
| 18 | 0.90 (d, 6.8, 3H) | 0.92 (d, 6.8, 3H) | 1.28 (s, 3H) | 1.27 (s, 3H) |
| 19 | 0.78 (d, 6.8, 3H) | 0.80 (d, 6.8, 3H) | 1.22 (s, 3H) | 1.22 (s, 3H) |
| 20 | 0.84 (s, 3H) | 0.86 (s, 3H) | 1.04 (s, 3H) | 1.03 (s, 3H) |
| OAc | 2.11 (s, 3H) | 2.10 (s, 3H) | ||
| C2H5 | 4.14 (q, 7.1, 2H) | 4.11 (q, 7.1, 2H) | 4.11 (q, 7.1, 2H) | |
| 1.28 (t, 7.1, 3H) | 1.25 (t, 7.1, 3H) | 1.25 (t, 7.1, 3H) |
| No. | 23 | 24 | ||
|---|---|---|---|---|
| δC a | δH mult (J in Hz) b | δCc | δH mult (J in Hz) d | |
| 1 | 38.3 | 1.37 * 1.85 (m) | 39.2 | 1.45 * 1.92 * |
| 2 | 34.1 | 2.36 * 2.50 * | 34.9 | 2.40 (m) 2.55 (m) |
| 3 | 217.9 | 220.4 | ||
| 4 | 47.5 | 48.5 | ||
| 5 | 55.4 | 1.36 * | 56.4 | 1.47 * |
| 6 | 21.4 | 1.35 * 1.49 (m) | 22.4 | 1.43 * 1.54 * |
| 7 | 40.6 | 1.54 (m) 1.70 (m) | 41.7 | 1.71 * |
| 8 | 43.7 | 44.9 | ||
| 9 | 55.9 | 1.13 (dd, 12.3, 4.4) | 57.1 | 1.23 (dd, 12.2, 4.7) |
| 10 | 37.4 | 38.3 | ||
| 11 | 19.7 | 1.28 * 1.55 * | 20.8 | 1.41 * 1.61 * |
| 12 | 26.9 | 1.45 * 1.61 * | 27.8 | 1.51 * 1.68 * |
| 13 | 44.6 | 1.94 (m) | 44.7 | 1.90 (m) |
| 14 | 48.0 | 1.09 (m) 2.12 (m) | 49.1 | 1.13 (d, 11.1) 2.18 (m) |
| 15 | 44.5 | 1.28 * 2.05 * | 45.8 | 1.33 * 2.15 (m) |
| 16 | 82.8 | 83.9 | ||
| 17 | 67.6 | 4.19 (s, 2H) | 69.0 | 4.02 (d, 11.3) 4.37 (d, 11.3) |
| 18 | 26.9 | 1.06 (s, 3H) | 27.3 | 1.07 (s, 3H) |
| 19 | 21.7 | 1.01 (s, 3H) | 22.1 | 1.04 (s, 3H) |
| 20 | 14.8 | 0.97 (s, 3H) | 15.3 | 1.02 (s, 3H) |
| 1’ | 33.8 | 1.66 * 2.36 * | 43.0 | 2.51 (m) 2.44 (m) |
| 2’ | 29.2 | 2.20 * 2.51 * | 175.8 | |
| 3’ | 175.5 | 180.7 | ||
| 4’ | 76.1 | miss | ||
| 5’ | 51.9 | 1.38 * | 49.0 | 2.70 (m) |
| 6’ | 24.6 | 1.45 * 1.35 * | 24.8 | 1.44 * 1.56 * |
| 7’ | 40.7 | 1.45 * 1.58 * | 41.8 | 1.58 * |
| 8’ | 43.8 | 44.9 | ||
| 9’ | 48.5 | 1.19 * | 50.4 | 2.00 * |
| 10’ | 41.9 | 43.3 | ||
| 11’ | 19.1 | 1.23 (m) 1.55 * | 20.9 | 1.34 * 1.64 * |
| 12’ | 27.0 | 1.45 * 1.69 * | 21.9 | 1.43 * 1.75 * |
| 13’ | 43.8 | 1.91 (m) | 44.8 | 1.19 (d, 14.5) 1.99 * |
| 14’ | 48.2 | 1.04 * 2.05 (m) | 49.2 | 1.06 (d, 11.7) 2.07 (m) |
| 15’ | 44.4 | 1.20 * 2.00 (m) | 45.4 | 1.97 * |
| 16’ | 84.4 | 85.4 | ||
| 17’ | 65.7 | 3.61 (d, 10.9) 3.75 (d, 10.9) | 66.2 | 3.57 (d, 11.3) 3.69 (d, 11.3) |
| 18’ | 34.4 | 1.27 (s, 3H) | 26.1 | 1.37 (s, 3H) |
| 19’ | 27.6 | 1.21 (s, 3H) | 25.7 | 1.27 (s, 3H) |
| 20’ | 19.4 | 1.03 (s, 3H) | 19.4 | 1.03 (s, 3H) |
| No. | NO Production ± SEM (% of LPS Group) | |
|---|---|---|
| 10 μM | 20 μM | |
| 5 | 95.70 ± 2.77 | 74.52 ± 4.25 ** |
| 10 | 96.95 ± 3.06 | 79.30 ± 0.74 *** |
| 13 | 89.71 ± 0.89 | 73.11 ± 6.20 ** |
| 18 | 93.87 ± 2.71 | 79.84 ± 4.71 ** |
| 19 | 98.12 ± 1.15 | 88.82 ± 4.18 * |
| 20 | 95.15 ± 2.71 | 80.13 ± 3.51 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, X.; Gong, Q.; Xu, Y.; Mu, J.; Tang, C.; Hu, B.; Ke, C.; Yao, S.; Zhang, H.; Ye, Y. Undescribed Phyllocladane-Type Diterpenoids from Callicarpa giraldii Hesse ex Rehd. and Their Anti-Neuroinflammatory Activity. Molecules 2025, 30, 1553. https://doi.org/10.3390/molecules30071553
Liang X, Gong Q, Xu Y, Mu J, Tang C, Hu B, Ke C, Yao S, Zhang H, Ye Y. Undescribed Phyllocladane-Type Diterpenoids from Callicarpa giraldii Hesse ex Rehd. and Their Anti-Neuroinflammatory Activity. Molecules. 2025; 30(7):1553. https://doi.org/10.3390/molecules30071553
Chicago/Turabian StyleLiang, Xu, Qi Gong, Yuting Xu, Jiaxing Mu, Chunping Tang, Bintao Hu, Changqiang Ke, Sheng Yao, Haiyan Zhang, and Yang Ye. 2025. "Undescribed Phyllocladane-Type Diterpenoids from Callicarpa giraldii Hesse ex Rehd. and Their Anti-Neuroinflammatory Activity" Molecules 30, no. 7: 1553. https://doi.org/10.3390/molecules30071553
APA StyleLiang, X., Gong, Q., Xu, Y., Mu, J., Tang, C., Hu, B., Ke, C., Yao, S., Zhang, H., & Ye, Y. (2025). Undescribed Phyllocladane-Type Diterpenoids from Callicarpa giraldii Hesse ex Rehd. and Their Anti-Neuroinflammatory Activity. Molecules, 30(7), 1553. https://doi.org/10.3390/molecules30071553