
Exploring the Volatility, Phase Transitions, and Solubility Properties of Five Halogenated Benzaldehydes

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
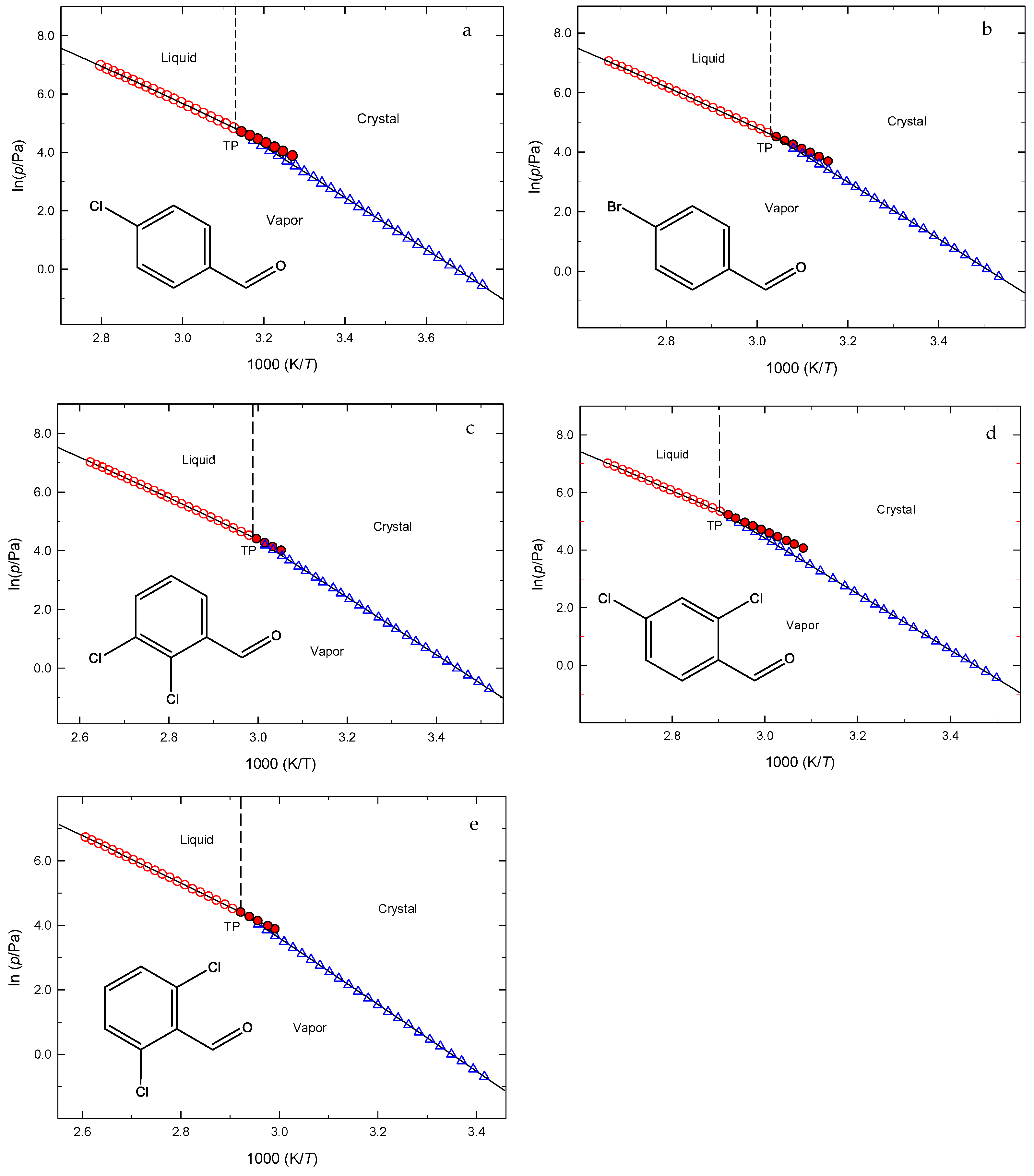
2.1. Volatility and Phase Transitions Study
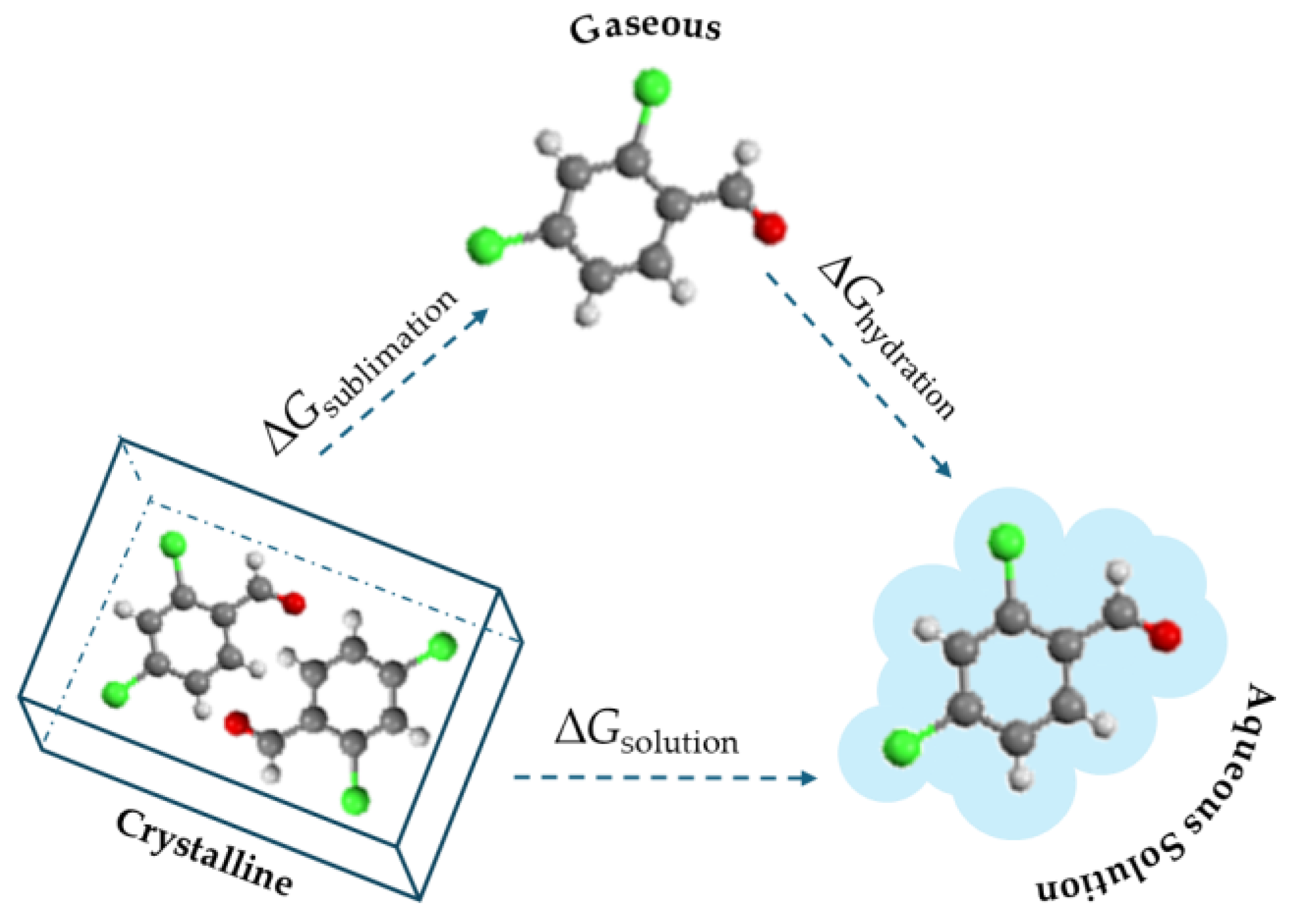
2.2. Solubility Measurements
3. Discussion
3.1. Vapor Pressures Study
3.2. Solubility Properties
3.3. Estimation Methods
4. Materials and Methods
4.1. Compounds and Purity Analysis
4.2. Thermal Analysis
4.2.1. Melting Temperatures and Enthalpies
4.2.2. Crystalline Heat Capacities
4.3. Volatility Study
4.4. Solubility Experiments
5. Conclusions
- Melting Properties: The melting temperatures and molar enthalpies for all five compounds, as well as their crystalline isobaric molar heat capacities, were determined using differential scanning calorimetry.
- Vapor Pressure Analysis: Vapor pressure measurements conducted across a range of temperatures allowed for the determination of molar enthalpies, entropies, and Gibbs energies of sublimation and vaporization. Corresponding phase diagrams, including the (p, T) coordinates for the triple points, were also obtained.
- Volatility Assessment: Sublimation vapor pressure measurements rank the volatilities as: 4-Chlorobenzaldehyde > 4-Bromobenzaldehyde > 2,4-Dichlorobenzaldehyde ~ 2,3-Dichlorobenzaldehyde > 2,6-Dichlorobenzaldehyde. These differences are mainly influenced by molecular weight, intermolecular interactions, and crystal packing. The higher volatility of 4-chlorobenzaldehyde is due to its weaker intermolecular forces and lower molecular weight, while stronger interactions in 2,6-dichlorobenzaldehyde reduce its volatility.
- Aqueous Solubility Measurements: The water solubilities of the five halogenated benzaldehydes follow the order: 4-Chlorobenzaldehyde > 4-Bromobenzaldehyde > 2,6-Dichlorobenzaldehyde > 2,3-Dichlorobenzaldehyde ~ 2,4-Dichlorobenzaldehyde. These differences were influenced by the position and number of halogen substituents, affecting molecular polarity and steric hindrance. The higher solubility of 4-chlorobenzaldehyde was due to chlorine’s smaller size and greater electronegativity, while the lower solubility of dichlorinated isomers resulted from increased steric hindrance and reduced polarity.
- Gibbs Energy and Henry’s Constants: By combining volatility and solubility data, the Gibbs energy of hydration and Henry’s law constant were calculated for each compound. The Henry’s law constant indicated that 2,3- and 2,4-dichlorobenzaldehydes had the highest potential for volatilization from water, while 2,6-dichlorobenzaldehyde had the lowest tendency to escape into the atmosphere
- Formyl Group Contribution: Using an estimation model, the contributions of the formyl group (-CHO) to key physicochemical properties of the substituted benzenes were estimated.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohamed, M.A.F.; Benjamin, I.; Okon, G.A.; Ahmad, I.; Khan, S.A.P.M.; Patel, H.; Agwamba, E.C.; Louis, H. Insights into in-vitro studies and molecular modelling of the antimicrobial efficiency of 4-chlorobenzaldehyde and 4-methoxybenzaldehyde derivatives. J. Biomol. Struct. Dyn. 2023, 42, 6042–6064. [Google Scholar] [CrossRef]
- Bakale, R.P.; Naik, G.N.; Machakanur, S.S.; Mangannavar, C.V.; Muchchandi, I.S.; Gudasi, K.B. Synthesis, characterization, spectroscopic properties and antihypertensive activity of 4-chlorobenzaldehyde hydralazine hydrazone and its metal complexes. J. Iran. Chem. Soc. 2023, 20, 147–154. [Google Scholar] [CrossRef]
- Senthamarai, T.; Chandrashekhar, V.G.; Gawande, M.B.; Kalevaru, N.V.; Zbořil, R.; Kamer, P.C.J.; Jagadeesh, R.V.; Beller, M. Ultra-small cobalt nanoparticles from molecularly-defined Co–salen complexes for catalytic synthesis of amines. Chem. Sci. 2020, 11, 2973–2978. [Google Scholar] [CrossRef]
- Casa, S.; Henary, M. Synthesis and Applications of Selected Fluorine-Containing Fluorophores. Molecules 2021, 26, 1160. [Google Scholar] [CrossRef]
- Elliott, M.; Janes, N.F.; Elliott, R.L. Pesticides. US4668701A, 26 May 1987. [Google Scholar]
- Schacht, E.; Desmarets, G.; Bogaert, Y. Polymeric Pesticides. Makromol. Chem. 1978, 179, 837–840. [Google Scholar] [CrossRef]
- Wang, S.-X.; Tan, Z.-C.; Di, Y.-Y.; Xu, F.; Zhang, H.-T.; Sun, L.-X.; Zhang, T. Heat capacity and thermodynamic properties of 2,4-dichlorobenzaldehyde. J. Chem. Thermodyn. 2004, 36, 393–399. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, X.; Han, Y.; Zang, H.; Zhu, C.; Shin, W. A Kind of Synthesis Technique of 2,3 Dichlorobenzaldehyde. CN107032968A 11 August 2017. [Google Scholar]
- Zhang, H.; Yang, Z.; Zhang, L.; Yue, W.; Zhu, Y.; Zhang, X. Inhibition performance of halogen-substituted benzaldehyde thiosemicarbazones as corrosion inhibitors for mild steel in hydrochloric acid solution. RSC Adv. 2022, 12, 30611–30625. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yan, W.; Chen, K.; Hung, C.T.; Liu, L.-L.; Wu, P.-H.; Huang, S.-J.; Liu, S.-B. Heteropolyacidbased ionic liquids as effective catalysts for the synthesis of benzaldehyde glycol acetal. Appl. Catal. A Gen. 2014, 485, 149–156. [Google Scholar] [CrossRef]
- Ribeiro-Claro, P.J.A.; Amorim da Costa, A.M.; Vueba, M.L.; Pina, M.E.; Amado, A.M. Para-halogenated benzaldehyde molecules included in cyclodextrins: A combined spectroscopic and thermal analysis. J. Raman Spectrosc. 2006, 37, 472–479. [Google Scholar] [CrossRef]
- Bernardes, M.F.F.; Pazin, M.; Pereira, L.C.; Dorta, D.J. Impact of Pesticides on Environmental and Human Health. In Toxicology Studies—Cells, Drugs and Environment; IntechOpen: London, UK, 2015; pp. 195–233. [Google Scholar] [CrossRef]
- Keshavarz, M.H.; Rezaei, M.; Hosseini, S.H. A simple approach for prediction of Henry’s law constant of pesticides, solvents, aromatic hydrocarbons, and persistent pollutants without using complex computer codes and descriptors. Process Saf. Environ. Prot. 2022, 162, 867–877. [Google Scholar] [CrossRef]
- Matos, G.D.R.; Kyu, D.Y.; Loeffler, H.H.; Chodera, J.D.; Shirts, M.R.; Mobley, D.L. Approaches for calculating solvation free energies and enthalpies demonstrated with an update of the freesolv database. J. Chem. Eng. Data 2017, 62, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.J.S.; Almeida, A.R.R.P. A new approach for the estimation of sublimation enthalpies and vapor pressures of crystalline benzene derivatives. Struct. Chem. 2013, 24, 2001–2016. [Google Scholar] [CrossRef]
- Monte, M.J.S.; Almeida, A.R.R.P. Estimations of the thermodynamic properties of halogenated benzenes as they relate to their environment mobility. Chemosphere 2017, 189, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.C.W.; Glew, D.N. Evaluation of thermodynamic functions from equilibrium constants. Trans. Faraday Soc. 1966, 62, 539–547. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Acree, W., Jr.; Chickos, J.S. Phase transition enthalpy measurements of organic and organometallic compounds. sublimation, vaporization and fusion enthalpies from 1880 to 2015. Part 1. C1–C10. J. Phys. Chem. Ref. Data 2016, 45, 033101. [Google Scholar] [CrossRef]
- Johnson, R.D., III. NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 16a; NIST Chemistry Web Book, SRD 69. 2013. Available online: https://cccbdb.nist.gov/vibscalejustx.asp (accessed on 3 February 2025).
- Cervinka, C.; Fulem, M.; Ruzicka, K. Evaluation of uncertainty of ideal-gas entropy and heat capacity calculations by density functional theory (DFT) for molecules containing symmetrical internal rotors. J. Chem. Eng. Data 2013, 58, 1382–1390. [Google Scholar] [CrossRef]
- Furukawa, G.T.; McCoskey, R.E.; King, G.J. Calorimetric properties of benzoic acid from 0° to 410° K. J. Res. Natl. Inst. 1951, 47, 256–261. [Google Scholar]
- Levi, L.; Nicholls, R.V.V. Formation of Styrenes by Pyrolysis of Aromatic Heterocyclic Aldehyde- Aliphatic Acid Anhydride Mixtures on Morden Bentonite. Ind. Eng. Chem. 1958, 50, 1005–1008. [Google Scholar] [CrossRef]
- Acree, W.E. Thermodynamic properties of organic compounds: Enthalpy of fusion and melting point temperature compilation. Thermochim. Acta 1991, 189, 37–56. [Google Scholar] [CrossRef]
- Plato, C.; Glasgow, A.R., Jr. Differential scanning calorimetry as a general method for determining the purity and heat of fusion of high-purity organic chemicals. Application to 95 compounds. Anal. Chem. 1969, 41, 330–336. [Google Scholar] [PubMed]
- van de Lande, L.M.F. The action of sodium methylate on some derivatives of m-dichlorobenzene. Recl. Trav. Chim. Pays-Bas 1932, 51, 98–113. [Google Scholar] [CrossRef]
- Stork, G.; White, W.N. The Stereochemistry of the SN2′ Reaction II. J. Am. Chem. Soc. 1956, 78, 4609–4619. [Google Scholar] [CrossRef]
- Palmer, D.S.; McDonagh, J.L.; Mitchell, J.B.O.; van Mourik, T.; Fedorov, M.V. First-Principles Calculation of the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules. J. Chem. Theory Comput. 2012, 8, 3322–3337. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.; van Mourika, T.; Mitchell, J.B.O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed]
- Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef]
- Modarresi, H.; Modarress, H.; Dearden, J.C. Henry’s law constant of hydrocarbons in airewater system: The cavity ovality effect on the nonelectrostatic contribution term of solvation free energy. SAR QSAR Environ. Res. 2005, 16, 461–482. [Google Scholar] [CrossRef]
- Available online: http://ec.europa.eu/enterprise/sectors/chemicals/reach/index_en.htm (accessed on 11 February 2025).
- Oshin, K.D.; Cavalier, A.R.; Zeller, M. CCDC 1429514: Experimental Crystal Structure Determination. CSD Commun. 2005. CSD Entry: YICFAR01. [Google Scholar] [CrossRef]
- Ndima, L.; Cuthbertson, J.; Hosten, E.C.; Betz, R. Crystal structure of 4-bromobenzaldehyde—Complete redetermination at 200 K, C7H5BrO. Z. Kristallogr. 2021, 236, 143–145. [Google Scholar] [CrossRef]
- Solanko, K.A.; Bond, A.D. Intermolecular interactions and unexpected isostructurality in the crystal structures of the dichlorobenzaldehyde isomers. Acta Cryst. 2011, B67, 437–445. [Google Scholar] [CrossRef]
- 4-Chlorobenzaldehyde. In SAFETY DATA SHEET According to Regulation (EC), Version 6.6; No. 1907/2006. Sigma-Aldrich; Available online: https://www.sigmaaldrich.com/PT/en/sds/aldrich/112216?srsltid=AfmBOopZimZ-OncbjPHzL4EZT68vGtrQiI0IlOwwKcUArktKNFGyGakf (accessed on 5 February 2025).
- Available online: https://www.chemicalbook.com/ChemicalProductProperty_EN_CB2316334.htm (accessed on 5 February 2025).
- Available online: https://www.thegoodscentscompany.com/data/rw1098371.html (accessed on 5 February 2025).
- Available online: https://www.ambeed.com/products/104-88-1.html (accessed on 5 February 2025).
- NETZSCH Analyzing & Testing DSC 204 F1 Phoenix. Calibration Set. Selb, Germany. 2022. Available online: https://analyzing-testing.netzsch.com/en/products/differential-scanning-calorimeter-dsc-differential-thermal-analyzer-dta/dsc-204-f1-phoenix (accessed on 20 December 2024).
- Sabbah, R.; El Watik, L. New reference materials for the calibration (temperature and energy) of differential thermal analysers and scanning calorimeters. J. Therm. Anal. 1992, 38, 855–863. [Google Scholar] [CrossRef]
- Sabbah, R.; Xu-Wu, A.; Chickos, J.S.; Planas Leitão, M.L.; Roux, M.V.; Torres, L.A. Reference materials for calorimetry and differential thermal analysis. Thermochim. Acta 1999, 331, 93–204. [Google Scholar] [CrossRef]
- Della Gatta, G.; Richarson, M.J.; Sarge, S.M.; Stølen, S. Standards, calibration, and guidelines in microcalorimetry part 2. Calibration standards for differential scanning calorimetry. Pure Appl. Chem. 2006, 78, 1455–1476. [Google Scholar] [CrossRef]
- Roux, M.V.; Temprado, M.; Chickos, J.S.; Nagano, Y. Critically evaluated thermochemical properties of polycyclic aromatic hydrocarbons. J. Phys. Chem. Ref. Data 2008, 37, 1855–1996. [Google Scholar] [CrossRef]
- Chang, S.S.; Bestul, A.B. Heat capacity and thermodynamic properties of o-terphenyl crystal, glass, and liquid. J. Chem. Phys. 1972, 56, 503–516. [Google Scholar] [CrossRef]
- ASTM E1269; Standard Test Method for Determining Specific Heat Capacity by Differential Scanning Calorimetry. ASTM International: Conshohocken, PA, USA, 2024.
- DIN 51007; Thermal Analysis (TA)—Differential Thermal Analysis (DTA) and Differential Scanning Calorimetry (DSC)—General Basics. Deutsches Institut für Normung (DIN): Berlin, Germany, 2019.
- ISO 11357; Plastics—Differential scanning calorimetry (DSC)—Part 1: General principles (ISO 11357-1:2016). International Organization for Standardization (ISO): Geneva, Switzerland, 2023.
- Monte, M.J.S.; Santos, L.M.N.B.F.; Fulem, M.; Fonseca, J.M.S.; Sousa, C.A.D. New static apparatus and vapor pressure of reference materials: Naphthalene, benzoic acid, benzophenone and ferrocene. J. Chem. Eng. Data 2006, 51, 757–766. [Google Scholar] [CrossRef]
- Yalkowsky, S.H.; He, Y.; Jain, P. Handbook of Aqueous Solubility Data, 2nd ed.; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: Abingdon, UK, 2010. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| T/K | p/Pa | 100Δp/p b | T/K | p/Pa | 100Δp/p b | T/K | p/Pa | 100Δp/p b |
|---|---|---|---|---|---|---|---|---|
| 4-Chlorobenzaldehyde | ||||||||
| Crystalline phase | ||||||||
| 267.45 | 0.57 | 0.4 | 285.16 | 4.49 | 0.1 | 303.05 | 28.05 | 0.4 |
| 269.33 | 0.72 | 0.5 | 287.22 | 5.65 | 0.7 | 305.04 | 34.26 | 1.4 |
| 271.42 | 0.93 | 0.5 | 289.23 | 6.93 | −0.3 | 306.98 | 40.71 | 0.5 |
| 273.29 | 1.15 | −0.7 | 291.18 | 8.49 | −0.5 | 309.01 | 49.06 | 0.3 |
| 275.39 | 1.48 | −0.3 | 293.07 | 10.45 | 0.7 | 310.96 | 58.06 | −0.6 |
| 277.25 | 1.84 | −0.3 | 295.14 | 12.75 | −0.5 | 312.92 | 69.28 | −0.6 |
| 279.32 | 2.33 | −0.4 | 297.14 | 15.67 | 0.0 | 314.91 | 82.74 | −0.6 |
| 281.20 | 2.90 | 0.2 | 299.12 | 19.07 | 0.0 | |||
| 283.26 | 3.60 | −1.2 | 300.99 | 23.01 | 0.4 | |||
| Liquid phase c | ||||||||
| 305.70 | 48.17 d | 0.3 | 325.71 | 183.0 | −0.4 | 343.52 | 520.0 | 0.3 |
| 307.91 | 56.22 d | −0.1 | 327.66 | 206.8 | −0.2 | 345.50 | 578.0 | 0.2 |
| 309.86 | 65.09 d | 0.8 | 329.70 | 235.5 | 0.4 | 347.47 | 640.9 | 0.0 |
| 311.92 | 75.03 d | 0.7 | 331.69 | 264.4 | 0.1 | 347.50 | 641.0 | −0.1 |
| 313.90 | 86.06 d | 0.8 | 333.66 | 298.2 | 0.5 | 349.45 | 711.8 | 0.1 |
| 315.83 | 96.33 d | −0.9 | 335.64 | 336.0 | 0.9 | 351.43 | 788.7 | 0.0 |
| 317.92 | 110.0 d | −1.5 | 337.61 | 375.5 | 0.7 | 353.30 | 864.6 | −0.4 |
| 319.87 | 125.4 | −1.1 | 339.57 | 415.3 | −0.3 | 355.30 | 958.0 | −0.2 |
| 321.85 | 143.8 | −0.2 | 339.57 | 416.0 | −0.1 | 357.33 | 1058 | −0.4 |
| 323.75 | 162.2 | −0.2 | 341.56 | 468.0 | 0.6 | |||
| 4-Bromobenzaldehyde | ||||||||
| Crystalline phase | ||||||||
| 283.09 | 0.83 | −0.3 | 298.95 | 4.98 | −0.6 | 314.77 | 24.88 | 0.4 |
| 285.23 | 1.07 | −0.4 | 301.06 | 6.27 | 0.0 | 316.79 | 29.92 | −0.4 |
| 287.05 | 1.33 | 0.1 | 302.93 | 7.60 | −0.3 | 318.78 | 36.33 | 0.4 |
| 289.17 | 1.71 | 0.7 | 304.86 | 9.21 | −0.9 | 320.66 | 43.34 | 0.7 |
| 291.17 | 2.13 | −0.1 | 306.87 | 11.43 | 0.2 | 322.62 | 51.66 | 0.4 |
| 292.95 | 2.63 | 1.0 | 308.76 | 13.82 | 0.2 | 324.68 | 61.49 | −0.8 |
| 295.00 | 3.25 | −0.5 | 310.84 | 17.02 | 0.5 | |||
| 297.08 | 4.12 | 0.5 | 312.78 | 20.28 | −0.9 | |||
| Liquid phase c | ||||||||
| 316.76 | 39.76 d | −1.0 | 336.48 | 148.9 | −0.1 | 356.26 | 463.6 | −0.4 |
| 318.77 | 46.26 d | −0.2 | 338.43 | 168.4 | 0.2 | 358.21 | 513.4 | −0.6 |
| 320.73 | 53.22 d | 0.1 | 340.46 | 189.9 | 0.0 | 360.13 | 573.1 | 0.3 |
| 322.70 | 60.73 d | −0.2 | 342.44 | 213.5 | −0.1 | 362.14 | 628.6 | −0.8 |
| 324.68 | 69.79 d | 0.2 | 344.41 | 240.4 | 0.2 | 364.12 | 701.8 | 0.1 |
| 326.64 | 79.36 d | 0.0 | 346.34 | 266.7 | −0.6 | 366.10 | 775.8 | 0.1 |
| 328.63 | 90.64 d | 0.1 | 348.34 | 300.1 | −0.2 | 368.08 | 860.4 | 0.6 |
| 330.60 | 103.9 | 1.0 | 350.32 | 333.2 | −0.9 | 370.05 | 946.6 | 0.5 |
| 332.51 | 117.8 | 1.3 | 352.34 | 375.3 | −0.2 | 371.99 | 1026 | −0.8 |
| 334.45 | 131.9 | 0.4 | 354.27 | 418.5 | 0.1 | 373.98 | 1147 | 0.8 |
| 2,3-Dichlorobenzaldehyde | ||||||||
| Crystalline phase | ||||||||
| 284.21 | 0.49 | −0.7 | 302.18 | 3.78 | −1.2 | 319.59 | 21.97 | −0.5 |
| 286.16 | 0.63 | 0.9 | 303.89 | 4.58 | −0.2 | 321.86 | 27.35 | 0.0 |
| 288.13 | 0.78 | −1.2 | 305.78 | 5.63 | 0.7 | 323.56 | 31.92 | −0.4 |
| 290.09 | 0.99 | −0.3 | 308.08 | 7.12 | 0.4 | 325.70 | 39.35 | 0.9 |
| 292.06 | 1.26 | 1.0 | 309.86 | 8.45 | −0.7 | 327.48 | 45.52 | −0.7 |
| 294.04 | 1.58 | 1.1 | 312.03 | 10.58 | 0.0 | 329.63 | 56.32 | 1.3 |
| 296.22 | 2.00 | 0.2 | 313.92 | 12.78 | 0.1 | 331.62 | 65.87 | −0.6 |
| 298.17 | 2.47 | −0.3 | 315.60 | 15.22 | 1.1 | |||
| 299.97 | 3.02 | 0.2 | 317.96 | 18.67 | −1.3 | |||
| Liquid phase c | ||||||||
| 327.59 | 54.84 d | −1.1 | 347.44 | 189.1 | 0.6 | 367.35 | 569.4 | 0.0 |
| 329.66 | 62.05 d | 0.4 | 349.41 | 214.0 | −0.2 | 369.27 | 630.4 | −0.4 |
| 331.60 | 71.02 d | −0.2 | 351.50 | 240.6 | 0.1 | 371.17 | 695.2 | −0.6 |
| 333.65 | 80.84 d | 0.3 | 353.48 | 267.2 | 0.9 | 373.23 | 767.3 | −0.2 |
| 335.54 | 91.33 | 0.3 | 355.36 | 297.6 | 0.7 | 375.13 | 843.4 | −0.3 |
| 337.55 | 103.4 | 0.6 | 357.43 | 335.9 | 0.0 | 377.17 | 930.5 | −0.2 |
| 339.54 | 117.4 | 0.4 | 359.33 | 371.6 | 0.3 | 379.07 | 1011 | 0.6 |
| 341.49 | 133.6 | −0.4 | 361.39 | 419.6 | −0.8 | 381.03 | 1104 | 1.0 |
| 343.48 | 150.6 | −0.3 | 363.27 | 461.9 | −0.4 | |||
| 345.45 | 169.9 | −0.5 | 365.34 | 516.4 | −0.6 | |||
| 2,4-Dichlorobenzaldehyde | ||||||||
| Crystalline phase | ||||||||
| 285.78 | 0.64 | 0.8 | 305.23 | 5.65 | −0.3 | 327.66 | 50.25 | 0.1 |
| 287.68 | 0.80 | 0.3 | 307.12 | 6.86 | −0.6 | 329.75 | 60.90 | 0.6 |
| 289.73 | 1.02 | 0.4 | 308.88 | 8.29 | 0.2 | 331.73 | 72.78 | 0.9 |
| 291.36 | 1.23 | 0.1 | 310.95 | 10.12 | −0.8 | 333.66 | 85.89 | 0.6 |
| 293.15 | 1.50 | −0.6 | 313.23 | 12.78 | −0.3 | 335.72 | 102.0 | 0.1 |
| 294.83 | 1.81 | −0.9 | 315.25 | 15.58 | −0.4 | 337.64 | 119.1 | −0.7 |
| 296.85 | 2.29 | 0.0 | 317.80 | 20.05 | 0.1 | 339.73 | 141.9 | −0.8 |
| 298.81 | 2.85 | 0.2 | 320.56 | 26.18 | 0.4 | 341.66 | 166.2 | −1.0 |
| 301.19 | 3.67 | −0.4 | 322.93 | 32.84 | 0.9 | |||
| 303.16 | 4.55 | −0.1 | 325.24 | 40.77 | 1.2 | |||
| Liquid phase c | ||||||||
| 324.34 | 57.72 d | −0.2 | 342.29 d | 184.1 | 0.6 | 359.46 | 479.4 | −1.0 |
| 326.40 | 66.54 d | 0.1 | 344.36 | 207.6 | 0.2 | 361.34 | 533.0 | −0.4 |
| 328.22 | 75.25 d | 0.2 | 346.33 | 233.6 | 0.4 | 363.54 | 603.2 | 0.5 |
| 330.22 | 85.67 d | −0.1 | 348.33 | 261.6 | 0.1 | 365.63 | 668.2 | 0.0 |
| 332.21 | 97.72 d | 0.1 | 349.66 | 282.0 | 0.0 | 367.44 | 733.6 | 0.1 |
| 334.17 | 110.6 d | −0.1 | 351.41 | 310.8 | −0.2 | 369.34 | 810.6 | 0.5 |
| 336.16 | 125.6 d | 0.0 | 353.17 | 342.8 | −0.3 | 371.29 | 885.4 | −0.3 |
| 338.15 | 141.4 d | −0.5 | 355.39 | 390.3 | 0.4 | 373.62 | 998.4 | 0.3 |
| 340.44 | 163.3 d | −0.2 | 357.59 | 437.4 | −0.1 | 375.74 | 1100 | −0.2 |
| 2,6-Dichlorobenzaldehyde | ||||||||
| Crystalline phase | ||||||||
| 292.72 | 0.50 | −0.3 | 308.53 | 3.07 | 0.4 | 324.42 | 15.66 | 0.4 |
| 294.67 | 0.63 | −0.6 | 310.48 | 3.74 | −0.8 | 326.34 | 18.87 | 0.5 |
| 296.72 | 0.81 | 0.3 | 312.49 | 4.61 | −1.2 | 328.34 | 22.67 | −0.2 |
| 298.56 | 1.00 | −0.1 | 314.41 | 5.68 | −0.4 | 330.30 | 27.33 | 0.0 |
| 300.60 | 1.28 | 1.1 | 316.45 | 7.02 | −0.2 | 332.29 | 32.89 | 0.0 |
| 302.53 | 1.58 | 0.3 | 318.40 | 8.62 | 0.4 | 334.27 | 39.51 | 0.2 |
| 304.58 | 1.97 | −0.6 | 320.45 | 10.52 | −0.2 | 336.23 | 46.91 | −0.4 |
| 306.53 | 2.49 | 1.3 | 322.40 | 12.77 | −0.2 | 338.20 | 56.28 | 0.2 |
| Liquid phase c | ||||||||
| 334.28 | 48.38 d | 0.6 | 352.10 | 151.3 | 0.5 | 369.87 | 415.4 | 0.9 |
| 335.87 | 53.45 d | −0.2 | 354.09 | 168.0 | −0.9 | 371.82 | 456.8 | 0.1 |
| 338.22 | 62.48 d | −0.3 | 356.04 | 190.2 | 0.0 | 373.82 | 506.8 | 0.0 |
| 340.16 | 71.24 d | 0.0 | 358.03 | 212.4 | −0.5 | 375.77 | 559.6 | −0.1 |
| 342.24 | 81.73 d | 0.3 | 359.98 | 239.9 | 0.4 | 377.79 | 620.3 | −0.1 |
| 344.17 | 91.46 | −0.8 | 361.98 | 266.2 | −0.5 | 379.71 | 682.3 | −0.2 |
| 346.13 | 103.7 | 0.0 | 363.92 | 297.7 | −0.1 | 381.71 | 755.4 | 0.0 |
| 348.09 | 118.4 | 0.2 | 365.92 | 333.0 | 0.1 | 383.62 | 827.1 | −0.2 |
| 350.07 | 133.6 | 0.3 | 367.87 | 372.6 | 0.7 | |||
| ΔT | θ | p (Equation (1)) | (Equation (2)) | σr a | |||
|---|---|---|---|---|---|---|---|
| K | K | kJ·mol−1 | Pa | kJ·mol−1 | J·K−1·mol−1 | J·K−1·mol−1 | |
| 4-Chlorobenzaldehyde | |||||||
| Crystalline phase | |||||||
| 267.45 to 314.91 | 298.15 | 21.47 ± 0.01 | 17.3 | 73.3 ± 0.2 | 173.8 ± 0.7 | 32.1 ± 9.5 b | 0.0062 |
| 291.18 c | 22.68 ± 0.01 | 8.54 | 73.6 ± 0.1 | ||||
| 319.43 d | 17.79 ± 0.02 | 123 | 72.6 ± 0.6 | ||||
| Liquid phase e | |||||||
| 305.70 to 357.33 | 298.15 | 20.34 ± 0.03 | 27.3 | 56.7 ± 0.6 | 122.0 ± 2.0 | 71.2 ± 9.6 b | 0.0053 |
| 331.52 c | 16.39 ± 0.01 | 262 | 54.4 ± 0.1 | ||||
| 319.43 d | 17.79 ± 0.02 | 123 | 55.2 ± 0.2 | ||||
| 4-Bromobenzaldehyde | |||||||
| Crystalline phase | |||||||
| 283.09 to 324.68 | 298.15 | 24.76 ± 0.01 | 4.59 | 79.4 ± 0.2 | 183.3 ± 0.7 | 36.9 ± 13.5 b | 0.0058 |
| 303.88 c | 23.71 ± 0.01 | 8.40 | 79.2 ± 0.2 | ||||
| 330.02 d | 18.98 ± 0.03 | 99.1 | 78.2 ± 0.7 | ||||
| Liquid phase e | |||||||
| 316.76 to 373.98 | 298.15 | 22.94 ± 0.05 | 9.57 | 61.2 ± 0.7 | 128.3 ± 2.4 | 79.6 ± 7.9 b | 0.0057 |
| 345.37 c | 17.16 ± 0.01 | 254 | 57.4 ± 0.1 | ||||
| 330.02 d | 18.98 ± 0.03 | 99.1 | 58.6 ± 0.3 | ||||
| 2,3-Dichlorobenzaldehyde | |||||||
| Crystalline phase | |||||||
| 284.21 to 331.62 | 298.15 | 26.30 ± 0.01 | 2.47 | 81.2 ± 0.3 | 184.1 ± 1.0 | 28.7 ± 14.0 b | 0.0081 |
| 307.92 c | 24.50 ± 0.01 | 6.98 | 81.0 ± 0.2 | ||||
| 334.82 d | 19.60 ± 0.03 | 87.6 | 80.2 ± 0.4 | ||||
| Liquid phase e | |||||||
| 327.59 to 381.03 | 298.15 | 24.18 ± 0.08 | 5.81 | 62.7 ± 1.1 | 129.2 ± 3.7 | 71.8 ± 9.6 b | 0.0056 |
| 354.31 c | 17.29 ± 0.01 | 282 | 58.6 ± 0.1 | ||||
| 334.82 d | 19.60 ± 0.03 | 87.6 | 60.0 ± 0.4 | ||||
| 2,4-Dichlorobenzaldehyde | |||||||
| Crystalline phase | |||||||
| 285.78 to 341.66 | 298.15 | 26.13 ± 0.01 | 2.64 | 81.5 ± 0.2 | 185.7 ± 0.7 | 35.4 ± 7.7 b | 0.0063 |
| 313.72 c | 23.25 ± 0.01 | 13.5 | 81.0 ± 0.1 | ||||
| 343.90 d | 17.75 ± 0.02 | 201 | 80.0 ± 0.5 | ||||
| Liquid phase e | |||||||
| 324.34 to 375.74 | 298.15 | 23.40 ± 0.05 | 7.95 | 61.9 ± 0.7 | 129.1 ± 2.4 | 74.1 ± 6.8 b | 0.0037 |
| 350.04 c | 17.02 ± 0.01 | 289 | 58.0 ± 0.1 | ||||
| 343.90 d | 17.75 ± 0.02 | 201 | 58.5 ± 0.1 | ||||
| 2,6-Dichlorobenzaldehyde | |||||||
| Crystalline phase | |||||||
| 292.72 to 338.20 | 298.15 | 28.66 ± 0.01 | 0.95 | 85.9 ± 0.4 | 192.0 ± 1.3 | 31.7 ± 11.8 b | 0.0058 |
| 315.46 c | 25.35 ± 0.01 | 6.35 | 85.4 ± 0.1 | ||||
| 342.99 d | 20.14 ± 0.02 | 85.7 | 84.5 ± 0.7 | ||||
| Liquid phase e | |||||||
| 334.28 to 383.62 | 298.15 | 25.95 ± 0.10 | 2.84 | 66.2 ± 1.2 | 135.0 ± 4.0 | 78.9 ± 9.6 b | 0.0045 |
| 358.95 c | 18.19 ± 0.01 | 225 | 61.4 ± 0.1 | ||||
| 342.99 d | 20.14 ± 0.02 | 85.7 | 62.7 ± 0.3 | ||||
| 4-Chloro Benzaldehyde | 4-Bromo Benzaldehyde | 2,3-Dichloro Benzaldehyde | 2,4-Dichloro Benzaldehyde | 2,6-Dichloro Benzaldehyde | |
|---|---|---|---|---|---|
| a | 87.963 | 47.890 | 141.52 | 97.926 | 79.188 |
| b | −0.068938 | 0.33078 | −0.32612 | 0.032624 | 0.13591 |
| c | 1.0454 × 10−3 | 1.7586 × 10−4 | 1.3994 × 10−3 | 7.4315 × 10−4 | 6.6381 × 10−4 |
| R2 a | 0.9996 | 0.9996 | 0.9997 | 0.9996 | 0.9995 |
| σ b | 0.20 | 0.21 | 0.17 | 0.22 | 0.24 |
| Temperature range/K | 221.3–292.1 | 221.3–299.6 | 241.4–309.6 | 241.4–314.6 | 241.4–312.1 |
| 4-Chloro Benzaldehyde | 4-Bromo Benzaldehyde | 2,3-Dichloro Benzaldehyde | 2,4-Dichloro Benzaldehyde | 2,6-Dichloro Benzaldehyde |
|---|---|---|---|---|
| a | ||||
| 127.9 ± 3.8 | 129.7 ± 3.9 | 143.9 ± 4.3 | 144.1 ± 4.3 | 144.2 ± 4.3 |
| Experimental (DSC, this work) b | ||||
| 160.3 ± 4.1 | 162.1 ± 2.8 | 168.7 ± 5.1 | 173.7 ± 1.9 | 178.7 ± 1.7 |
| Estimated c | ||||
| 151.5 ± 17.0 | 154.2 ± 17.0 | 170.7 ± 17.0 | 170.7 ± 17.0 | 170.7 ± 17.0 |
| −32.4 ± 5.6 | −32.4 ± 4.8 | −24.8 ± 6.7 | −29.6 ± 4.7 | −34.5 ± 4.6 |
| −23.6 ± 17.4 | −24.5 ± 17.4 | −26.8 ± 17.5 | −26.6 ± 17.5 | −26.5 ± 17.5 |
| Experimental (Equation (1)) d | ||||
| −32.1 ± 9.5 | −36.9 ± 13.5 | −28.7 ± 14.0 | −35.4 ± 7.7 | −31.7 ± 11.8 |
| Ttp | Tmelting a | b | b | Method c/(Ref.) |
|---|---|---|---|---|
| K | K | kJ·mol−1 | J·K−1·mol−1 | |
| 4-Chlorobenzaldehyde | ||||
| 319.85 ± 0.92 | 18.05 ± 0.80 a | 56.4 ± 2.5 | DSC/this work | |
| 319.43 | 17.4 ± 0.62 | VP/this work | ||
| 319.85 | [23] | |||
| 4-Bromobenzaldehyde | ||||
| 330.52 ± 0.93 | 18.79 ± 0.76 a | 56.9 ± 2.3 | DSC/this work | |
| 330.02 | 19.6 ± 0.8 | VP/this work | ||
| 334.2 | 22.6 | [24] | ||
| 329.15 | [25] | |||
| 2,3-Dichlorobenzaldehyde | ||||
| 334.66 ± 0.92 | 20.24 ± 0.79 a | 60.5 ± 2.4 | DSC/this work | |
| 334.82 | 20.2 ± 0.9 | VP/this work | ||
| 2,4-Dichlorobenzaldehyde | ||||
| 344.11 ± 0.92 | 21.95 ± 0.76 a | 63.8 ± 2.2 | DSC/this work | |
| 343.90 | 21.4 ± 0.5 | VP/this work | ||
| 347.2 | 20.47 | [7] | ||
| 345 | [26] | |||
| 2,6-Dichlorobenzaldehyde | ||||
| 343.11 ± 0.93 | 21.50 ± 0.78 a | 62.7 ± 2.3 | DSC/this work | |
| 342.99 | 21.8 ± 0.75 | VP/this work | ||
| 343 | [27] | |||
| Property | 4-Chloro Benzaldehyde | 4-Bromo Benzaldehyde | 2,3-Dichloro Benzaldehyde | 2,4-Dichloro Benzaldehyde | 2,6-Dichloro Benzaldehyde |
|---|---|---|---|---|---|
| S/mol·L−1 | (8.832 ± 0.035) × 10−3 | (3.713 ± 0.045) × 10−3 | (0.8529 ± 0.0035) × 10−3 | (0.8209 ± 0.0070) × 10−3 | (1.128 ± 0.0028) × 10−3 |
| /kJ·mol−1 | −17.7 | −18.8 | −16.7 | −16.5 | −19.8 |
| −2.71 | −2.90 | −2.53 | −2.50 | −3.07 | |
| 1.95 | 1.26 | 2.95 | 3.16 | 0.851 |
| Property (Y) | Contribution of -CHO Group for Property Y | Equation n° |
|---|---|---|
| Tm/K | 73.2 ± 12.5 | (Equation (8)) |
| /kJ·mol−1 | 10.4 ± 0.9 | (Equation (9)) |
| /kJ·mol−1 | 9.9 ± 1.0 | (Equation (10)) |
| log10(pcr/Pa) | −1.82 ± 0.17 | (Equation (11)) |
| log10(pl/Pa) | −1.77 ± 0.21 | (Equation (12)) |
| log10(Scr/mol·m−3 ) | 0.37 ± 0.09 | (Equation (13)) |
| /kJ·mol−1 | −12.2 ± 1.4 | (Equation (14)) |
| −2.15 ± 0.24 | (Equation (15)) |
| Compound | CASNR | Minimum Initial Purity a | Final Mass Fraction Purity | Analysis Method b | % Water Content c |
|---|---|---|---|---|---|
| 4-Chlorobenzaldehyde | 104-88-1 | 0.97 | 0.9978 | GC (FID) | 0.02 ± 0.01 |
| 4-Bromobenzaldehyde | 1122-91-4 | 0.99 | 0.9992 | 0.03 ± 0.01 | |
| 2,3-Dichlorobenzaldehyde | 6334-18-5 | 0.99 | 0.9983 | 0.02 ± 0.01 | |
| 2,4-Dichlorobenzaldehyde | 874-42-0 | 0.99 | 0.9991 | 0.02 ± 0.01 | |
| 2,6-Dichlorobenzaldehyde | 83-38-5 | 0.99 | 0.9990 | 0.04 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, A.R.R.P.; Pinheiro, B.D.A.; León, G.P.; Postolnyi, B.; Araújo, J.P.; Monte, M.J.S. Exploring the Volatility, Phase Transitions, and Solubility Properties of Five Halogenated Benzaldehydes. Molecules 2025, 30, 1551. https://doi.org/10.3390/molecules30071551
Almeida ARRP, Pinheiro BDA, León GP, Postolnyi B, Araújo JP, Monte MJS. Exploring the Volatility, Phase Transitions, and Solubility Properties of Five Halogenated Benzaldehydes. Molecules. 2025; 30(7):1551. https://doi.org/10.3390/molecules30071551
Chicago/Turabian StyleAlmeida, Ana R. R. P., Bruno D. A. Pinheiro, Gastón P. León, Bogdan Postolnyi, João P. Araújo, and Manuel J. S. Monte. 2025. "Exploring the Volatility, Phase Transitions, and Solubility Properties of Five Halogenated Benzaldehydes" Molecules 30, no. 7: 1551. https://doi.org/10.3390/molecules30071551
APA StyleAlmeida, A. R. R. P., Pinheiro, B. D. A., León, G. P., Postolnyi, B., Araújo, J. P., & Monte, M. J. S. (2025). Exploring the Volatility, Phase Transitions, and Solubility Properties of Five Halogenated Benzaldehydes. Molecules, 30(7), 1551. https://doi.org/10.3390/molecules30071551