Abstract
The diagnosis and treatment of lymphoid neoplasms have undergone a continuously progressive positive change in the last three decades, with accelerated progress in the previous decade due to the advent of genomics in cancer diagnosis. Significantly, there has been an increasing emphasis on integrating molecular genetics with clinical, morphologic, immunophenotypic, and cytogenetic evaluation for diagnosis. As we think of moving forward with further advances in the genomics era, it is first helpful to understand our current state of knowledge and how we achieved it in the challenging and complex field of lymphoid neoplasms, which comprise very heterogeneous neoplastic diseases in children and adults, including clinically acute lymphoblastic leukemias (ALLs) arising from precursor lymphoid cells and clinically indolent and aggressive lymphomas arising from mature lymphoid cells. This work aims to provide an overview of the historical evolution and the current state of knowledge to anyone interested in the field of lymphoid neoplasms, including students, physicians, and researchers. Therefore, I discuss this complex topic in three review manuscripts, designated Parts 1–3. In Part 1, I explain the basis of the diagnostic classification of lymphoid neoplasms and its evolution up to the current fifth edition of the World Health Organization classification of hematolymphoid neoplasms, and the crucial importance of diagnostic tumor classifications in achieving and advancing patient care and precision medicine. In the second and third manuscripts, I discuss current diagnostic considerations for B-ALL and T-ALL (Part 2) and common indolent and aggressive mature leukemias/lymphomas (Part 3), including significant updates in the WHO 2022 classification, newly described entities, and concepts, including genetic predisposition to ALLs and lymphomas, and throughout emphasizing the essential integration of molecular genetics with clinical, morphologic (pathologic), immunophenotypic, and cytogenetic evaluation, as is required for precise diagnosis of the type of lymphoma/leukemia in any patient.
1. Introduction
Lymphomas are neoplasms arising from mature B, T, or Natural Killer (NK) lymphocytes. About 90% of lymphomas are of B-cell origin, and 10% are T- and NK-cell neoplasms, as described in Part 1 of this work [1,2]. Lymphomas can affect any site of the body, including lymphoid organs and locations without normal lymphoid tissues, such as the brain. Our collective knowledge of lymphomas has evolved immensely in the last three decades, especially in the previous decade, as with other types of cancer, due to the application of genomics.
This paper represents Part 3 of this work. Part 1 provides a historical overview of lymphoma classifications and the principles of the diagnostic classification of lymphoid neoplasms and includes sections on B- and T-cell development in the bone marrow and the thymus, respectively, germinal center components and the origin of mature B-cell neoplasms, clonality analysis in lymphoid neoplasms, and the crucial role of the diagnostic World Health Organization (WHO) classification in achieving and advancing precision medicine [2]. Following Part 1 as an introduction to both acute lymphoblastic leukemia (ALL) and lymphomas, Part 2 discussed B- and T-ALL as we understand them today in 2023 [3]. Part 3 is focused on the mature B-, T-, and NK-cell lymphomas/leukemias, including in the pediatric and adult age groups. Here, I describe updates in the WHO classification, including a comparison with the International Consensus Classification (ICC) and the evolution of the classification of high-grade B-cell lymphomas, and discuss current epidemiologic, diagnostic, and molecular pathology considerations for common indolent and aggressive mature lymphomas/leukemias in the pediatric and adult age groups, a few rare lymphomas, newly described lymphoma entities, and new concepts, including lymphomas of immune-privileged sites, nodal T follicular helper (TFH) cell lymphomas, Epstein–Barr virus (EBV)+ nodal T- and NK-cell lymphoma, and germline genetic predisposition to lymphoid neoplasms, with emphasis throughout on the integration of molecular genetics with clinical, morphologic, immunophenotypic, and cytogenetic evaluation, as is currently required for precise diagnosis of the specific type of lymphoma in any patient.
2. An Overview of Incidence, Mortality, and Survival Data for the Types of Lymphoid Neoplasms
The International Agency for Research on Cancer provides the Global Cancer Observatory (GLOBOCAN) estimates of cancer incidence and mortality in various cancer types. These estimates are based on population-based national and subnational cancer registries in different countries worldwide. In these global reports, data for lymphoid neoplasms are separately provided for Hodgkin lymphoma (HL), non-Hodgkin lymphoma (NHL), multiple myeloma, and leukemias, as shown in Table 1 [4]. The leukemias include acute and chronic lymphoid and myeloid leukemias. NHL comprises the eleventh most common cancer and the eleventh most common cause of cancer deaths worldwide.

Table 1.
Global incidence and mortality of hematolymphoid neoplasms according to the Global Cancer Observatory estimates in 2020 [4].
In 2020, women in Northern America and Australia/New Zealand had the highest worldwide age-standardized rate (ASR) of 10.0 per 100,000 individuals for NHL. At the same time, men in Australia/New Zealand and Northern America had the highest and second-highest worldwide ASR of 15.3 and 14.2 per 100,000, respectively, for NHL [4].
Table 2 shows the incidence of various types of lymphomas in the USA [5,6,7]. The incidence of the types of NHL varies according to ethnic origin, with the highest rates of NHL, chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), and follicular lymphoma (FL) in non-Hispanic White males [7,8,9]. In contrast, Hispanic males have the highest rates of diffuse large B-cell lymphoma (DLBCL) [7] and B-lymphoblastic lymphoma/leukemia [9]. And T-cell lymphomas are twice as common in non-Hispanic Blacks as in Whites [9].
Table 2.
The incidence rates of various types of lymphomas in the USA based on Morton et al. 2006 [5], Blum et al. 2018 [6], and SEER data [7].
Table 3 shows the relative five-year survival of patients in different age groups with various subtypes of lymphoma, including all races and both sexes, based on the United States Surveillance, Epidemiology, and End Results (SEER) database [6,7,10].
Table 3.
Five-year relative survival of various types of lymphomas in all races, both sexes, based on 2004–2010 SEER data [10] and Blum et al. 2018 [6] and compared with the current 2013–2019 SEER data [7] in the USA.
NHL represents the eighth most common cancer in the USA, with an estimated 80,550 new cases and 20,180 new deaths in 2023. The median age at diagnosis is 68 years. The stage-based incidence of NHL at new diagnosis is as follows: 22% in Stage I, 15% in Stage II, 18% in Stage III, 35% in Stage IV, and 9% with unknown stage. Based on the stage at NHL diagnosis, the 5-year relative survival rates are as follows: 86.2% if Stage 1, 78.9% if Stage II, 73.3% if Stage III, 64.2% if Stage IV, and 70.7% if unknown stage. Based on 2016–2020 data, the death rates for HL, NHL, CLL/SLL, DLBCL, and FL in the USA were 0.3, 5.1, 0.8, 1.7, and 0.4 per 100,000 individuals per year, respectively [7].
3. Mature B-Cell Neoplasms
As shown in Part 1 in Table 3, the mature B-cell neoplasms recognized by the initial World Health Organization (WHO) classification in 2001 increased with each update in 2008 and 2017 [2]. Table 4 compares the neoplasms arising from mature B-cells recognized in the fifth edition of the WHO classification, hereafter referred to as WHO-HAEM5, and the International Consensus Classification (ICC), the creation of which was described in Part 1 [2]. The WHO-HAEM5 classification of lymphoid neoplasms includes a new group, “tumor-like lesions with B-cell predominance,” and therefore, the entire category of mature B-cell neoplasms and tumor-like entities is now named “B-cell lymphoid proliferations and lymphoma,” as depicted in Table 4 [11,12]. B-lymphoid proliferations and lymphomas also include “Precursor B-cell neoplasms” in WHO-HAEM5, but these are not included in Table 4 since they are discussed in Part 2 of this three-part work [3].
Table 4.
Neoplasms arising from mature B-cells in the fifth edition WHO 2022 and International Consensus Classifications [11,12,13].
Lymphomas arising from mature B-cells are heterogeneous in clinical behavior and include lymphomas referred to as indolent, aggressive, or highly aggressive. They may be discovered in a routine examination or present as lymphadenopathy or tissue mass. Small B-cell lymphomas, except mantle cell lymphoma, are often regarded as indolent lymphomas and include chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), follicular lymphomas, marginal zone lymphomas, and lymphoplasmacytic lymphoma. Indolent lymphomas may transform into aggressive lymphoma, a category now explicitly included in the WHO classification.
3.1. Lymphomas in the Pediatric and Adult Age Groups
Lymphomas may affect children, adolescents, young adults (AYA), and older adults.
Hodgkin lymphomas have a prominent bimodal age peak. Non-Hodgkin lymphomas have a low incidence in children. In contrast, the incidence increases progressively with age from the AYA group to adults older than 75 years, as shown in Table 5 [14]. Non-Hodgkin lymphomas are about 2 to 3 times more common in boys than in girls, more common in White children than Black children, and are uncommon before the age of five years [15]. Lymphomas are the third most common type of cancer in children aged 0–14 years, following leukemias and brain cancer as the two most common cancer types in that age group. However, in adolescents aged 15–19 years, lymphomas are the second most common, after brain and other central nervous system tumors as the most common and before leukemias as the third most common type of cancer [16].
Table 5.
Incidence of Hodgkin and non-Hodgkin lymphomas distributed by patient age in the USA [14].
In contrast with adults, high-grade lymphomas may occur more often in children in the setting of inherited genetic cancer predisposition syndromes (reviewed in [17]). The most common types of lymphomas in children are, in contrast with adults, high-grade mature B-cell lymphomas (Burkitt lymphoma is most common, followed by diffuse large B-cell lymphoma [DLBCL]), T lymphoblastic lymphoma, and anaplastic large-cell lymphoma (ALCL) as the most common mature T-cell lymphoma; the lower-risk lymphomas, such as follicular lymphoma and marginal zone lymphoma occur with increasing age in children [18,19]. High-grade B-cell lymphomas in children are often curable, treatment responses in children are usually better than those in adults for the same type of non-Hodgkin lymphoma, and recurrences are uncommon in children; if they occur, a cure is infrequent [18].
Brentuximab vedotin, an anti-CD30 antibody-drug conjugate directed against the malignant cells in classic Hodgkin lymphoma (CHL), showed a lower risk of disease progression, death, or noncomplete response [20] and five-year progression-free survival of 82%. These results were achieved by substituting bleomycin with brentuximab vedotin in combination with doxorubicin, vinblastine, and dacarbazine chemotherapy, with febrile neutropenia and peripheral neuropathy as adverse effects of therapy [21]. In children with high-risk CHL, brentuximab vedotin has similarly shown superior efficacy, a 59% lower risk of an event or death, and no increase in toxic effects at three years [22]. Challenges in pediatric non-Hodgkin lymphomas include defining the value of prognostic factors, such as early response in the radiologic and measurable residual disease evaluation, applying new technologies to improve risk stratification, and developing innovative therapies in the first-line setting and relapse [18].
3.2. Bruton’s Tyrosine Kinase Inhibitors as an Example of Precision Medicine
Bruton’s tyrosine kinase (BTK), discovered due to its impaired function in inherited X-linked agammaglobulinemia in 1993, is a member of a family of cytoplasmic tyrosine kinases termed TEC kinases, which include TEC, ITK, RLK, BMX, and BTK [23]. BTK is expressed in B lymphocytes, myeloid cells, mast cells, and platelets and is downregulated in T-cells, which express other TEC kinases, ITK, RLK, and TEC [23,24]. The BTK gene encodes for the BTK protein, which is located downstream of the B-cell receptor and CD19 pathways in B-cells. The BTK protein is critical for B-cell signaling and proliferation in normal, neoplastic, and autoreactive B-cells. BTK signaling is also implicated in autoimmune diseases [25], which are well-known to be risk factors for developing several types of lymphomas (reviewed in [26,27]). BTK is also a downstream target in non-hematopoietic cells (reviewed in [28]). Still, it is currently most well-known for its role in B-cell signaling due to the development of BTK inhibitors, which have transformed the treatment landscape of several types of mature B-cell neoplasms in the last decade.
Ibrutinib, a first-generation covalent BTK inhibitor, covalently binds to the C481 residue in BTK’s kinase domain to irreversibly inhibit BTK. Ibrutinib is currently approved by the United States Food and Drug Administration (FDA) for the treatment of patients with CLL/SLL, mantle cell lymphoma (second-line therapy), marginal zone lymphoma (second-line therapy), and Waldenstrom’s macroglobulinemia (WM) and chronic graft versus host disease (second-line therapy) [29]. Acalabrutinib and zanubrutinib are examples of second-generation covalent BTK inhibitors, and both are approved for the treatment of CLL/SLL and mantle cell lymphoma (second-line therapy) [30,31,32]. Zanubrutinib is also approved to treat patients with marginal zone lymphoma (second-line therapy) and WM, similar to ibrutinib [31]. In January 2023, the non-covalent BTK inhibitor, pirtobrutinib, was FDA-approved to treat relapsed or refractory mantle cell lymphoma after ≥2 lines of therapy that include a previous BTK inhibitor [33], while it continues to be in clinical trials for patients with CLL/SLL.
The BTK inhibitors briefly described above represent one example of novel therapies in mature B-cell neoplasms, particularly CLL [34,35,36,37]. Current treatment paradigms based on our vastly increased understanding of the molecular mechanisms underlying tumors in general and specifically hematolymphoid neoplasms in the last decade mandate a precise diagnosis of lymphomas, including the subtype, which is where it becomes critically important to have a common worldwide language for tumor classification and for it to be easily understood to be applied reproducibly by pathologists and clinicians everywhere. Therefore, this section will briefly overview the most commonly encountered types of lymphoma/leukemia and discuss significant classification updates that have emerged from advances in our understanding of these lymphomas.
3.3. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
CLL is the most common chronic leukemia in Western countries, showing a male predominance with a median age of 72 years at diagnosis [36]. For likely genetic but yet unknown reasons, CLL is 5–10 times less common in Asians than in individuals of European descent, as reviewed in [38]. According to the United States SEER data, the age-adjusted incidence of CLL in 2020 per 100,000 individuals was 2.8 in females and 5.3 in males, with a decreasing trend reported from 2014 to 2019 [14]. In 2023, CLL is expected to comprise almost one-third (31.8%) of all new leukemia cases (n = 59,610), with an expected 18,740 new CLL cases in 12,130 males and 6610 females, with a male-to-female ratio of 1.8:1. The estimated number of deaths due to CLL in 2023 is 4490, comprising less than 20% of all deaths (n = 23,710) due to leukemias [39]. The disease is currently incurable and has a highly variable clinical course, ranging from an indolent disease that does not require treatment for many years to an active, progressive disease [36].
Most CLL patients have a preceding asymptomatic monoclonal B-cell lymphocytosis (MBL) phase [40]. MBL has been observed in up to 4% of healthy individuals in the general population who harbor increased numbers of monoclonal B-cells with an immunophenotype characteristic of CLL, even with absolute lymphocyte counts less than 5 × 109/L [41,42]. As shown in Table 4, in WHO-HAEM5, both MBL and CLL/SLL are included in the family “pre-neoplastic and neoplastic small lymphocytic proliferations.” Three subtypes of MBL are now recognized in WHO-HAEM5: (A) low count MBL or clonal B-cell expansion, with a clonal CLL/SLL-phenotype B-cell count below 0.5 × 109/L and with no other features diagnostic of a B-lymphoproliferative disorder; (B) CLL/SLL-type MBL, with a monoclonal CLL/SLL-phenotype B-cell count ≥0.5 × 109/L, total B-cell count <5 × 109/L, and no other features diagnostic of CLL/SLL; and (C) non-CLL/SLL-type MBL, with any monoclonal non-CLL/SLL phenotype B-cell expansion and no symptoms or features diagnostic of another mature B-cell neoplasm; the majority of these cases have features consistent with a marginal zone origin [11]. The thresholds in MBL types A and B are arbitrary but based on population studies compared with hospital hematology cases with prior or current lymphocytosis for MBL type A [11,43]. The count for type B is based on the substantially lower likelihood (hazard ratio = 0.32, p = 0.04) of MBL type B individuals requiring treatment compared with low count, Rai stage 0 CLL patients with B-cell counts between 5.0 and 10.0 × 109/L (n = 94), and patients with Rai stage 0 CLL with an absolute lymphocyte count greater than 10.0 × 109/L (n = 219; p = 0.0003) [11,44].
Some CLL patients have a family history of lymphoproliferative disorders, and susceptibility loci have been identified by genome-wide association studies [45,46]. An inherited polygenic risk is also present in individuals of European ancestry for developing MBL [47]. However, the causes of familial CLL are not yet understood. Rare germline ATM variants have been reported in CLL, associated with somatic ATM mutations; whether they have a role in familial CLL is not yet known [48,49].
Biologically, the cell of origin in CLL is being investigated. There is evidence that aberrant hematopoietic stem cells may lead to stem cell skewing towards a B-cell lineage and, eventually, the emergence of clonal B-cell populations in CLL. Experimentally in mice, stem cells from CLL patients developed into oligoclonal B-cells similar to MBL (Kikushige et al. 2011) [50]. The mutational status of the IGH variable region gene (IGHV) impacts prognosis in CLL, with IGHV unmutated CLL having a worse prognosis than IGHV-mutated CLL (CLL-International Prognostic Index [CLL-IPI] 2016) [51]. Gene expression profiling studies have shown that IGHV-unmutated CLL is derived from unmutated mature CD5+ B-cells and IGHV-mutated CLL is derived from CD5+, CD27+ post-germinal center B-cells [52]. Continuous antigen stimulation via the B-cell receptor and the cellular microenvironment appears to lead to MBL; with continuing antigenic stimulus and proliferation, additional genetic alterations are acquired, leading to the development of CLL, as reviewed in [53,54].
The immunoglobulin repertoire in CLL is biased and characterized by the presence of subsets of patients with closely homologous or stereotyped, complementarity-determining region 3 (CDR3) sequences [55,56]. The CDR3 sequence in each lymphocyte represents a unique “clonotype” for that lymphoid cell, and this sequence is formed with the rearrangement of the antigen receptor genes, as described in Part 1 [2]. The term clonotype contrasts with the term clone, which indicates identical cells usually arising from a single cell. The presence of identical or stereotyped B-cell receptor immunoglobulins in unrelated and geographically distant CLL patients is evidence of the significant role of antigen selection in the biology of CLL, likely following binding to self- or non-self-antigens. Interestingly, identical or stereotyped patient subsets show similar clinical features and are designated as numbered subsets. Subsets 1, 2, and 8 are very aggressive, each comprising less than 3% of all CLL with time-to-first treatment earlier than two years; subset 8 has the highest risk of Richter’s transformation [56]. Since the CDR3 sequence needs to be determined, this analysis requires next-generation sequencing (NGS).
Significantly, skewing of the B-cell receptor IGH gene repertoire to a dominant clonotype was detected by NGS up to 16 years before diagnosis in patients with CLL transformed to aggressive lymphoma, indicating that even high-risk CLL can have a prolonged indolent preclinical stage [57]. This finding suggests that individuals at risk of developing a lymphoid neoplasm, such as immune deficiencies and immune dysregulation, could be screened by NGS to detect early lymphoma.
Further, a consistent IGHV mutation status and cytogenetic aberrations were present among multiple members of three Dutch families, indicating shared genetic features in familial CLL [58]. Therefore, in addition to intense interest in studying the “immunome,” which represents the complete set of unique clonotypes in the genome, there is now a growing demand for NGS technology analysis of the mutational status of B-cell receptor IGH variable region genes in CLL, since NGS can provide much more immunogenetics information for the clinical management of patients with pre-neoplastic states and overt CLL disease [59,60].
The somatic mutational landscape of CLL shows alterations in several cellular pathways, including NOTCH1, B-cell receptor, and the nuclear factor kappa B (NFκB) signaling, DNA damage/cell cycle, RNA/ribosome processing, and mitogen-activated protein (MAP) kinase pathways (without the BRAF p.V600E mutation, which is present in other lymphoid neoplasms, including hairy cell leukemia) [61,62,63,64]. A recent whole genome sequencing analysis of 485 CLL patients enrolled in clinical trials as part of the United Kingdom’s 100,000 Genomes Project showed novel findings, including putative drivers in non-coding regions within regulatory elements for potentially druggable target genes (NOTCH1, DTX1, NFKBIZ, NTRK2, and BACH2) and chromosomal translocations with breakpoints in WDHD1 and CTNND2::ARHGAP18 [65]. The study identified several associations, including:
- (1)
- SETD2/del3p21.31, del9p21.3, and gains of chr17q21.31 are associated with relapsed/refractory disease and TP53 disruption.
- (2)
- MED12 and DDX3X mutations are associated with unmutated IGHV CLL.
- (3)
- B-cell receptor immunoglobulin subset 2, which represents about 3% of all CLL and is known to be associated with a poor prognosis, is linked to the putative driver, FAM50A.
- (4)
- IGHV3-21 gene rearrangement is enriched for FAM50A, ATM/del11q22, SF3B1 mutations, and chr21q21.3-q22.3 gains.
Further, shorter telomeres were associated with p53 pathway alterations, relapsed/refractory disease, and worse progression-free survival. By integrating 186 distinct recurrent genomic alterations in their cohort, the authors defined five genomic subgroups associated with response to therapy. While the results require validation, the study highlighted the potential of whole genome sequencing to inform risk stratification in CLL [65].
3.3.1. CLL Diagnosis and Prognosis
CLL/SLL is a neoplasm composed of predominantly small mature B lymphocytes with few admixed medium-sized or larger cells (prolymphocytes); the neoplastic cells typically express surface CD5 and CD23 with dim surface expression of light chain-restricted immunoglobulin. CLL/SLL is considered one disease entity, with CLL and SLL diagnosed based on the level of involvement of peripheral blood and tissues. Before, and according to the WHO 2001 classification, CLL was defined by peripheral blood absolute lymphocyte count >5 × 109/L. By WHO 2008 criteria, CLL diagnosis required a monoclonal B-cell count of >5 × 109/L, in the absence of disease-related symptoms or cytopenias, with the clonal B-cells having characteristic morphologic and immunophenotypic features of CLL, and those criteria remain unchanged; the increased B-cell count should be sustained for at least three months [66].
SLL is diagnosed when the B-cell count is <5 × 109/L, but there is lymphadenopathy, splenomegaly, or other extramedullary involvement due to a neoplastic B-cell infiltrate similar to the neoplastic cells in CLL. The histopathologic features of CLL/SLL in tissues show a neoplastic infiltrate comprised predominantly of small mature B-cells admixed with fewer medium-sized or larger cells. The larger cells, termed “paraimmunoblasts” in tissues, are characteristically identified as clusters within “proliferation centers,” also called “pseudofollicles,” in lymph nodes and tissue specimens involved by CLL/SLL and help to diagnose CLL/SLL.
By immunophenotyping, the neoplastic cells in CLL/SLL typically express CD19, dim CD20, CD5, variable CD11c, CD23, CD43, CD45, CD200, and dim surface immunoglobulin, and the neoplastic cells are negative for CD10, CD79b, FMC7, CD25, and CD103. In a significant harmonization effort, the required diagnostic markers by flow cytometry included CD19, CD5, CD20, CD23, kappa, and lambda light chain immunoglobulin [67].
Progression in CLL/SLL may be suggested by the presence of ≥15% prolymphocytes in peripheral blood (prolymphocytoid progression), and care must be taken to exclude mantle cell lymphoma in these cases. Large, confluent proliferation centers in tissue sections or bone marrow core biopsies also indicate a more aggressive disease. An expanded proliferation center broader than one 20× microscopic field or high proliferation indices, defined as >2.4 mitoses per proliferation center or >40% Ki67-positive cells per proliferation center, predict a poor outcome [68]. Of note, B-cell prolymphocytic leukemia is now eliminated from the WHO 2022 classification since it is considered a progression of CLL [11].
CLL/SLL is clinically and genetically heterogeneous. In 2000, Döhner et al. found the chromosomal abnormalities, deletion of 13q14 as a sole abnormality, normal karyotype, trisomy 12, deletion of 11q22 (ATM), and deletion of 17p13 (TP53) to have a prognosis ranging from the best, for 13q as a sole abnormality, to the worst for 17p deletion [69]. Since then, interphase FISH for these abnormalities has been routinely performed at diagnosis. Chromosomal banding analysis using novel mitogens for culture and FISH are complementary methods. A complex karyotype in CLL classically refers to ≥3 clonal abnormalities. However, the highest risk is present with ≥5 abnormalities. The chromosomal microarray may also detect these abnormalities [70,71]. In addition, the presence of unmutated variable regions of the IGH gene, with unmutated defined as ≥98% identical with the germline sequence [72,73], TP53 mutations and a complex karyotype or genomic complexity have an adverse prognosis and affect treatment decisions [34,35,36,37]. Mutations in NOTCH1 (10–15%), ATM (10–15%), SF3B1 (10%), TP53 (5–10%), and BIRC3 (5%) are most frequent at diagnosis, and these mutations have clinical correlations (reviewed in [12,34,54]).
TP53 deletions occur with chromosomal 17p deletion, and TP53 mutations may occur in the absence of cytogenetic abnormalities so that FISH alone will not detect all clinically relevant TP53 alterations [66,74]. In addition, TP53 mutations may occur with disease evolution. Therefore, a comprehensive analysis of TP53 genetic alterations [75] is required at the time of diagnosis and disease progression. In contrast, the mutational status of the IGH variable region gene does not change during the disease course, and this analysis only needs to be performed once for each patient; analysis for the #2 subset configuration is essential for predictive purposes. In addition, testing to demonstrate a complex karyotype and analysis for BTK, PLCG2, and BCL2 mutations, which can arise during therapy, is desirable by WHO-HAEM5 [12]. In addition to the #2 subset analysis, the European Research Initiative on CLL (ERIC) recommends analysis for the #8 subset since it is associated with the highest risk for Richter’s transformation [76].
In cases of Richter’s transformation, it is essential to establish whether the transformed lymphoma is clonally related or unrelated to the neoplastic clone in the earlier CLL phase since, if unrelated, the prognosis is better, and the management is different than for the clonally related transformation, which has a dismal prognosis [77].
3.3.2. Other Relevant Diagnostic Aspects in B-Cell Neoplasms in Relation to CLL in WHO-HAEM5
CLL/SLL represents one of the “small B-cell neoplasms”, and in that respect, this section briefly describes a few relevant diagnostic considerations and WHO-HAEM5 updates.
WHO-HAEM5 Eliminated B-Prolymphocytic Leukemia
The elimination of B-cell prolymphocytic leukemia (B-PLL) by WHO-HAEM5 raised questions, which were answered by the editors of the WHO classification [78], briefly described here. Mantle cell lymphoma with nucleolated cells resembling prolymphocytes was already excluded from B-prolymphocytic leukemia by the revised fourth edition WHO classification. The two categories of CD5+ B-cell lymphoid proliferations with >15% prolymphocytes previously included (1) atypical CLL (CLL/PLL) with ≤55% prolymphocytes and (2) B-PLL with >55% prolymphocytes. The 55% cutoff distinguishing atypical PLL and B-PLL was arbitrary, and the >55% prolymphocytes category was also heterogeneous in morphology, immunophenotype, and genetics, with the genetics showing a strong resemblance to poor-risk CLL without transformation to DLBCL-type Richter’s transformation [78,79]. These findings supported WHO-HAEM5 classifying such cases as prolymphocytoid transformation of CLL [78]. As shown in Table 4, the ICC continues to recognize B-PLL as a distinct entity to be diagnosed only after rigorously excluding other lymphoid neoplasms, particularly transformation of CLL, mantle cell lymphoma, and marginal zone lymphoma [13].
Splenic B-Cell Lymphomas/Leukemias in WHO-HAEM5
Primary splenic lymphomas, defined as lymphomas confined to the spleen and splenic hilar lymph nodes, are very rare. Most splenic lymphomas/leukemias represent secondary involvement of the spleen by a generalized primary disease, including large B-cell lymphoma (most common) and small B-cell neoplasms, including CLL/SLL, mantle cell lymphoma, follicular lymphoma, splenic marginal zone lymphoma (SMZL), lymphoplasmacytic lymphoma, hairy cell leukemia [HCL], T-cell neoplasms, and CHL [80]. These primary lymphoma/leukemia diseases causing spleen involvement and splenomegaly are well-characterized, including recent characterization of the genomics of SMZL [81], the differential diagnosis of which from other small B-cell neoplasms can be difficult to establish.
The essential WHO-HAEM5 diagnostic criteria for SMZL [12] are:
- (1)
- Small B-cell lymphoma involving bone marrow, peripheral blood, or both, composed of small lymphoid cells with villous processes;
- (2)
- Neoplastic cells express pan-B-cell markers, IgM, and IgD and are negative for BCL6, annexin A1, CD103, cyclin D1, SOX11, and LEF1;
- (3)
- Other splenic and nodal B-cell lymphomas should be excluded; and
- (4)
- Clinical or imaging studies should show splenomegaly.
The desirable WHO-HAEM5 criteria are negativity for CD5 and CD10 in the neoplastic cells [12].
HCL is readily diagnosed by the characteristic peripheral blood findings, i.e., monocytopenia and small to medium-sized mature lymphoid cells with round, oval, or kidney-shaped nuclei, indistinct or absent nucleoli, and variably abundant cytoplasm with wispy projections, bone marrow aspirate, and trephine biopsy morphologic findings in conjunction with the clinical and immunophenotypic features, including bright expression of surface immunoglobulin, CD11c, CD20, CD22, CD25, and CD103 on the neoplastic B-cells. Further, the BRAF p.V600E mutation as the genetic cause of HCL was established in 2011 [82], providing another manner to confirm the diagnosis, distinguish HCL from other HCL-like neoplastic diseases, and allow targeted therapeutic choices with BRAF inhibitors for HCL patients [83]. In contrast with the B-cell lymphomas involving the white pulp mentioned above [80], HCL, HCL variant, and splenic diffuse red pulp small B-cell lymphoma involve the red pulp predominantly.
The new WHO-HAEM5 group, “splenic B-cell lymphoma with prominent nucleoli,” is meant to include CD5-negative splenic lymphomas that could not be grouped with other more common diseases [78]. This new group is not meant to be a definite entity. It has been introduced as a placeholder until additional evidence allows the precise classification of the entities (some cases of splenic marginal zone lymphoma, CD5-negative B-PLL cases, and HCL variant) currently included in this group [11,12,78].
3.4. Follicular Lymphoma
Follicular lymphoma comprises the second most common form of lymphoma in the Western parts of the world, after DLBCL. In the USA, the incidence of FL is 3.4/100,000, less than that of CLL/SLL (5.1/100,000) and DLBCL (6.9/100,000), and greater than that of mantle cell lymphoma (0.8/100,000) and marginal zone lymphoma (1.8/100,000) [9]. The disease is currently incurable. Most FLs are clinically indolent, with a long natural history for most patients. However, about 20% of patients with FL progress early in their disease course within 24 months of starting therapy, and these patients have poor outcomes [84]. In addition, 3% of FL patients transform into aggressive lymphoma yearly, with a continuing risk for 15 years [85].
Biologically, the origin of the malignant transformation of FL is being investigated. Many healthy individuals carry a t(14;18) chromosomal translocation in their circulating B-cells in the peripheral blood; this chromosomal translocation is present in about 85% of all FL cases but is clearly insufficient to cause neoplastic transformation. FL presumably arises from a mature B-cell that has previously acquired the t(14;18) translocation and undergone additional oncogenic changes in the germinal center that lead to malignant transformation [86]. Histologically, FL comprises neoplastic centrocytes and centroblasts, with the lymphoma showing at least a partially follicular pattern. Immunophenotypically, the neoplastic cells express B lineage markers, CD19, CD20, PAX5, CD22, and CD79a, and they express BCL2 protein, in contrast with reactive B-cells that are negative for BCL2 by immunohistochemistry. The neoplastic cells in FL also express germinal center-associated markers CD10, BCL6, GCET1, HGAL (GCET2), LMO2, AID, MEF2B, and stathmin, with variable sensitivity and specificity of each of the markers. In rare cases negative for CD10 and BCL6, the additional germinal center-associated markers help support the diagnosis of FL [12].
NGS studies have identified the most common mutations in FL to be epigenetic in chromatin-modifying genes. These include mutations in genes involved with posttranslational histone modification, such as the histone H3 lysine 4 (H3K4) methyltransferases KMT2D and KTM2C, the histone acetyltransferases CREBBP and EP300, and the histone H3 lysine 27 (H3K27) methyltransferase EZH2 (enhancer of zester homolog 2). Other mutations that occur in lower frequency include the Switch/sucrose nonfermentable (SWI/SNF) complex components and genes in the HIST1H1/2 linker histone family [87]. Interestingly, FL neoplastic cells appear to acquire these mutations in the same cell to remain in a germinal center differentiation state [87,88]. An inhibitor of EZH2, tazemetostat, is FDA-approved for treating relapsed FL after two or more lines of therapy in EZH2-mutated FL and independent of EZH2 mutation status if there is no other available therapeutic option [89].
3.4.1. Grading in Follicular Lymphoma
In 1994, the REAL classification introduced grading of FL into grades 1, 2, 3A, and 3B [90] based on criteria proposed in 1983 for counting centroblasts in ten high-power microscopic fields [91]. The WHO 2001 classification divided grade 3 FL into grades 3A (centrocytes present) and 3B (centrocytes absent) [92]. The REAL classification was based on the Kiel classification, compared in [93] (Table 3A, pp. 325–326). Still, the introduction of FL grading by the REAL and WHO classifications was in contrast with the Kiel classification due to the following reasons cited by the Kiel group: “unclear clinical relevance and lack of reproducibility of stratification based on the numbers of blasts in the neoplastic follicles, including about 50% reproducibility among different experienced hematopathologists for this grading system” [94] (p. 12). As stated in the cited reference, the Kiel group believed that “in our opinion, it is of major importance to distinguish between diffusely distributed blasts and solid sheets. The latter (corresponding to grade 3b) indicates a transformation into a diffuse large B-cell lymphoma, which should be noted in the report” [94] (p. 12). In WHO 2008, FL grades 1 and 2 were merged as low grade (0–15 centroblasts per high-power microscopic field), and grade 3A needed to be distinguished from grade 3B. This grading system continued in the revised fourth edition in 2017 but is no longer mandatory and considered optional in WHO-HAEM5 for similar and continued reasons cited earlier, including lack of reproducibility due to technical reasons, inter-observer variability in counting centroblasts, and differences in sizes of high-power microscopic fields [11,95,96]. Challenges in FL grading were noted to be present in the SWOG study that showed a 10-year overall survival of 79% for FL grades 1/2 compared to 68% for FL grade 3A (p = 0.0990) [97]. In contrast with WHO-HAEM5, the ICC has continued to recommend grading to study further in the context of current clinical therapies [13].
In WHO-HAEM5, FL grades 1, 2, and 3A are considered one disease representing classical FL (cFL), which is separated from grade 3B, termed follicular large-cell lymphoma [11]. The ICC recognizes the similarities between grades 1, 2, and 3A and suggests the use of ancillary tests to aid in distinguishing grade 3A (BCL2-rearranged and CD10-positive neoplastic cells) from grade 3B [13]. Interestingly, an analysis of United States SEER data for FL grades 1, 2, and 3, with grades 3A and 3B lumped together as grade 3 in SEER data, correlated with clinical outcomes showed that, despite all difficulties in grading, grading does impact clinical outcome in FL [98]. More reproducible methods for grading FL, such as digital pathology and artificial intelligence, applied to clinical outcomes with current treatments and prognostic factors defined only in the last decade, may provide a more precise understanding of the significance and clinical relevance of grading in FL.
3.4.2. Other Diagnostic Considerations in Follicular Lymphoma
Since FL grade 3B often coexists with DLBCL, if diagnosing a follicular large-cell lymphoma or FL grade 3B, FISH testing for BCL2 and MYC rearrangement is recommended to rule out a large B-cell lymphoma with dual BCL2 and MYC gene rearrangements [12]. In a young patient, pediatric or young adult, a diagnosis of FL grade 3B should prompt interferon regulatory factor 4, IRF4, gene rearrangement studies to distinguish from large B-cell lymphoma with an IRF4 rearrangement (LBCL-IRF4), which has a very different (favorable) prognosis [12]. LBCL-IRF4 was a provisional entity in 2017 and was upgraded to a definite entity by WHO-HAEM5 and ICC. LBCL-IRF4 may also occur in adults. This differential diagnosis should also be considered with strong immunohistochemical positivity for IRF4/multiple myeloma 1 (MUM1) protein in the neoplastic cells [12].
Patients with LBCL-IRF4 typically present with localized involvement of the head and neck region, most commonly tonsils and cervical lymph node enlargement. The essential diagnostic criteria by WHO-HAEM5 for LBCL-IRF4 are:
- (1)
- Intermediate or large-cell morphology;
- (2)
- Follicular, diffuse, or follicular and diffuse growth pattern;
- (3)
- Mature B-cell phenotype with co-expression of B-cell lymphoma 6 (BCL6) and MUM1 proteins, and
- (4)
- IRF4 translocation.
In the proper clinical setting, in combination with a typical immunophenotype, the diagnosis is allowed without an IRF4 rearrangement as “not molecularly confirmed.” The desirable WHO-HAEM5 criteria include evidence of the IG::IRF4 translocation and absence of BCL2 and MYC gene rearrangement [12].
Pediatric-type follicular lymphoma (PTFL) is also typically seen in childhood and young adults but may also occur in adults, usually aged <40 years, most often 2–25 years [12], but cases up to the age of 60 years have been reported [99]. This entity was introduced in the WHO 2017 classification [100]. PTFL has an excellent prognosis with usually conservative management. The disease is often localized to the head and neck region. Still, unlike LBCL-IRF4, the neoplastic cells in PTFL show a follicular pattern, which may be serpiginous, often with a rim of reactive lymphoid tissue at the periphery, and importantly, a diffuse component must be absent. The neoplastic cells are large and blastoid with a high proliferative fraction, but the large cells may be mistaken for large cells of FL, grade 3. The neoplastic cells express B-cell antigens. Although BCL2 protein is not usually expressed, it can be expressed by immunohistochemistry in the absence of a BCL2 gene rearrangement. In addition to the above features, the diagnosis requires evidence of B-cell monoclonality by immunophenotyping or genetics, absence of gene rearrangements in BCL2, BCL6, and MYC, and absence of strong, uniform MUM1 protein expression and absence of IRF4 rearrangement. Markedly expansile follicles and mutations in MAP2K1 and TNFRSF14 are desirable criteria for diagnosis [12]. In contrast with classic FL, the PTFL genome is characterized by low complexity, and the chromatin-modifying mutations typically seen in classic FL, mentioned above, are rare in PTFL [99,101]. Recently, pediatric nodal marginal zone lymphoma was shown to be similar to PTFL in its clinical and genomic profile [102]; watchful waiting after complete resection is the therapy of choice for both [103].
The WHO-HAEM5 has introduced “FL with uncommon features” as a new subgroup of FL, including two FL types: one with blastoid or large centrocyte variant and the other with a predominantly diffuse pattern. The latter type in WHO-HAEM5 overlaps with the FL subtype, BCL2-rearrangement-negative, CD23+ follicle center lymphoma, newly introduced by the ICC as a provisional entity [12,13,104]. Conversely, the ICC has introduced testicular FL as a distinct type of FL [13], while WHO-HAEM5 includes it among FL. Testicular FL occurs in the testes in young males and, in six cases, was limited to the testes, completely resected by unilateral orchiectomy, and there were no relapses with a long-term follow-up; histology showed the presence of diffuse and follicular areas [105], although the clinical features were reminiscent of a PTFL.
3.5. In Situ Mantle Cell Neoplasia in Relation to Overt Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is a mature B-cell lymphoma that is often aggressive, representing the classic form involving lymph nodes. However, the overt neoplasm includes an indolent, primarily non-nodal, or leukemic form.
In situ mantle cell neoplasia (or in situ mantle cell neoplasm) (ISMCN) is an earlier form of the disease that may precede, coexist with, or occur after the development of an overt MCL (reviewed in [106]). The relationships between in situ mantle cell neoplasia and the two overt forms of MCL are depicted in Figure 1 (figure previously published in open access publication [106]). The genetic hallmark of MCL is the t(11;14)(q13;q32) translocation, which leads to CCND1 juxtaposed with the IGH locus, causing overexpression of cyclin D1, a cell cycle regulator. Rare cyclin D1-negative MCL cases may harbor CCND2 or CCND3 gene rearrangements.
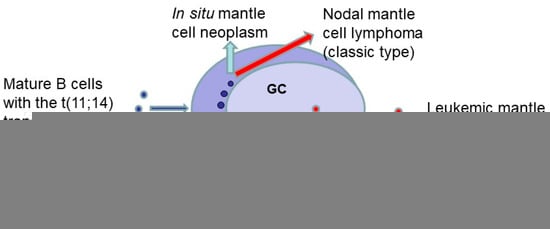
Figure 1.
Schematic showing the two main routes for the development of overt mantle cell lymphoma (MCL) from an in situ mantle cell neoplasm. Mature B-cells harboring the t(11;14) translocation (depicted by small blue round shapes) colonize in the inner part of the non-expanded mantle zone (MZ) in secondary lymphoid follicles to form an in situ mantle cell neoplasm. The classic nodal form of MCL usually arises from mantle zone B-cells that have not traversed the germinal center (GC). Rarely, in situ, neoplastic mantle cells may be present in the follicle center (depicted by small red round shapes in the GC). The non-nodal leukemic form of MCL may arise from in situ mantle cell neoplastic B-cells that have traversed the germinal center (figure modified from a previous publication [106]).
The diagnosis of in situ mantle cell neoplasia requires showing the presence of cyclin D1-positive B-cells (CD20+) in non-expanded or slightly expanded mantle zones of secondary lymphoid follicles in their natural position, as depicted in Figure 1. The diagnosis is more often made in patients with newly diagnosed overt MCL in whom a retrospective examination of a prior reactive-appearing lymph node or lymphoid tissue biopsy shows IS-MCN. In 672 cases examined in four retrospective studies of reactive or resected lymph nodes due to cancer [106], ISMCN was not detected or was very rare, identified in 0.58% (2/341) cases in one study [106,107]. Minimal infiltration by cyclin D1 positive-neoplastic mantle cells was identified at extranodal sites in previous lymphoid tissues predating an overt MCL diagnosis in 16.2% (6/37) cases of overt MCL [108].
Nevertheless, the actual incidence of in situ mantle cell neoplasia, especially how often it occurs before the development of an overt MCL, is currently unknown since its diagnosis requires a biopsy for histopathologic examination, and a biopsy is only performed with an enlarged lymph node, and in situ mantle cell neoplasia could be present in non-enlarged lymph nodes before the development of overt MCL.
The clinicopathologic features of 31 reported cases of histologically identifiable IS-MCN were reviewed at the individual patient level, including the ISMCN background and the outcomes reported in 16 publications [106,107,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123]. The findings relevant to the background and outcomes of ISMCN are summarized in Table 6, indicating that a high percentage, at least 77% (24/31), of ISMCN patients were associated with overt lymphoma (concurrent or subsequent after 1–20 years), treated for lymphoma, or died [106]. The remaining seven patients included four with <1 year or no follow-up, while 9.6% (3/31) patients did not develop overt lymphoma at 3, 5, and 16 years of follow-up [106]. ISMCN was associated with Castleman disease, hyaline vascular type, and multicentric HHV8-positive in 3/31 cases [106,109,122,123].
Table 6.
The outcomes of 31 reported cases of in situ mantle cell neoplasia from 17 publications (references given in the table), adapted from reference [106].
Of note, by WHO-HAEM5, the diagnosis of ISMCN warrants rigorous clinical staging and careful follow-up to exclude overt MCL [12].
The t(11:14) translocation characteristic of overt MCL was also present in the examined cases of ISMCN. However, this translocation may also be present in other lymphoid neoplasms and has also been identified in healthy individuals at a lower incidence than the t(14;18) translocation in healthy individuals, as previously reviewed [106,124,125,126].
Overt MCL was first described in 1982 as a follicular variant of intermediate lymphocytic lymphoma (ILL) [127]. ILL represented lymphomas with features intermediate between the poorly differentiated and well-differentiated types of lymphomas in the Rappaport classification, described in Part 1 [2] and [93]). Lymphomas with a “mantle zone” histologic pattern were observed while studying the clinicopathologic features of patients with ILL, wherein wide mantles of slightly atypical cells were noted around normal germinal centers, also with focal, diffuse areas, by morphologic examination; importantly, the clinical outcomes showed that these were aggressive lymphomas [128]. The BCL1 chromosomal rearrangement was described in 1990 [129,130]. In 1992, American and European lymphoma pathologists described MCL [131], which was subsequently included in the REAL and WHO classifications in 1994 and 2001, respectively.
The essential WHO-HAEM5 criteria for the diagnosis of cyclin D1-positive MCL require (1) lymphoma cells of B-lineage (CD20+ and usually CD5+), (2) the morphology of the classic variant (monomorphic and centrocyte-like) or less often variant morphology, and (3) cyclin D1-positivity, detection of CCND1 rearrangement, or both; SOX11 expression positivity is desirable. For cyclin D1-negative MCL, the essential WHO-HAEM5 criteria require (1) B-lineage lymphoma cells and morphology similar to cyclin D1-positive MCL, (2) an immunophenotype consistent with MCL, including SOX11 expression, and (3) absence of cyclin D1 expression and CCND1 rearrangement; CCND2 rearrangement is desirable [12].
The essential WHO-HAEM5 diagnostic criteria for the leukemic non-nodal MCL require (1) a typical clinical presentation (lymphocytosis, mostly asymptomatic with absent or insignificant nodal involvement), (2) usually monomorphic small to medium-sized cells of B-lineage (CD20+), and (3) cyclin D1 positivity, detection of CCND1 rearrangement, or both. SOX11 is commonly negative, and this evaluation is desirable [12].
The blastoid and pleomorphic variants, illustrated in reference [132], are usually associated with higher mitotic and proliferative indices [12]. Evaluating Ki67 and TP53 expression by immunohistochemistry and TP53 mutational status may help define higher-risk MCL patients [12,132]. Ki67 > 30% is the currently accepted cutoff for patients, with Ki67 > 30% associated with worse outcomes; high TP53 expression is considered to be 50% TP53-positive lymphoma cells and is associated with poor overall survival (median two years) [12].
Genetically, overt MCL involves dysregulated cell cycle and DNA damage response pathways. In a systematic meta-analysis of 32 studies, including the nodal and the leukemic forms of MCL, published from 2006 to 2019, excluding review articles, Hill et al. detailed the findings for gene mutations in overt MCL by analyzing 2127 MCL patients and 2173 samples included in the analyzed studies [106,133]. As per this meta-analysis, in MCL tumor or bone marrow samples at diagnosis or baseline, the most frequent genetic abnormalities occurred in the ATM (43.5%), TP53 (26.8%), CDKN2A (23.9%), CCND1 (20.2%), NSD2 (15.0%), KMT2A (8.9%), S1PR1 (8.6%), and CARD11 (8.5%) genes. Cytogenetic methods detected aberrations in IGH (38.4%) and MYC (20.8%) in the same tumor specimens [106,133]. However, the steps to the development of an overt MCL from an ISMCN are not yet understood [106]. The reader is referred to a comprehensive review from 2022 of the molecular pathogenesis and other current clinical aspects in MCL [134].
MCL has been reported to occur in families with other members having lymphoproliferative disorders, including CLL. Still, the prevalence of familial MCL is currently unknown, and the germline causes of familial cases have only rarely been described [106]. Of note, familial lymphoproliferative disorders that included CLL before MCL was described might have included instances of MCL. Specifically, the analysis for familial risk among 153,115 Swedish patients with hematologic malignancies diagnosed between 1958 and 2015 included 18,521 CLL patients, including 8,043 (43.4%; 8043/18,521) CLL patients diagnosed during the same period when MCL was not yet recognized [135], as previously reviewed [106].
Two Spanish patients were described from a Spanish study of 85 MCL patients, with a third MCL patient, with all three having family members with another lymphoid neoplasm [136]. One patient in that report was initially diagnosed with CLL and subsequently diagnosed with MCL after re-reviewing the previous pathology. In one family, typical MCL was diagnosed at age 77 in one male patient, and his daughter developed a blastoid MCL at a younger (43 years) age, consistent with anticipation in familial MCL [136]. Germline mutations have been described in rare cases, including a 15 bp deletion in CHEK2 in an overt MCL in one of seven examined patients with lymphoma [137], a heterozygous abnormality of ATM in one of four MCL patients [138], and germline mutations in ATM and CHEK2 in seven and two MCL cases, respectively [139]; however, a personal history of cancer other than MCL or a family history of cancer was unavailable for these cases [137,138,139]. In one Chinese family, Wang et al. determined the germline basis of familial MCL in the proband with overt MCL to be maternally inherited Lynch syndrome with co-segregation of an MLH1 variant and a history of DLBCL in the father and follicular lymphoma in one sibling. The index patient with MCL also developed a colonic adenocarcinoma due to mismatch DNA repair defects [140]. Germline mutations in MCL need to be studied, as well as early and, when possible, preclinical stages of MCL by methods other than tissue biopsies.
3.6. Lymphoplasmacytic Lymphoma
Lymphoplasmacytic lymphoma (LPL) is an uncommon mature B-cell neoplasm composed of small lymphocytes, lymphoplasmacytoid lymphocytes, and plasma cells involving the bone marrow primarily and, less frequently, the spleen and lymph nodes, and rarely other organs, such as the CNS. Morphologically, the neoplastic infiltrate in LPL shows admixed mast cells, and intranuclear cytoplasmic immunoglobulin inclusions termed Dutcher bodies may be identified, which help to confirm the diagnosis. Waldenström macroglobulinemia is a clinicopathologic diagnosis requiring the presence of LPL infiltrating the bone marrow associated with monoclonal IgM protein [141].
In contrast, IgM monoclonal gammopathy of unknown significance (MGUS) requires the presence of a monoclonal IgM protein and absence of bone marrow involvement on histologic examination by clinical criteria [141]; WHO-HAEM5 requires less than 10% bone marrow infiltration by clonal lymphoplasmacytic cells for a diagnosis of IgM MGUS, the presence of a serum monoclonal IgM protein of <3g/dl, and no evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder [12]. IgM MGUS can progress to WM or other B-cell lymphoproliferative disorders, with an estimated probability of progression of 1.5 to 3.0% per year [142]. It is not possible to distinguish between IgM MGUS and WM based on the IgM level since this level is usually not greater than three g/dL in MGUS and is less than three g/dL in most patients with WM [141].
The MYD88 p.L265P mutation is present in >90% of patients with WM. MYD88 is an adaptor protein recruited to the activated Toll-like and interleukin-1 receptor (IL-1R) complex as a homodimer, which then complexes with IL-1R-associated kinase (IRAK) 4 to activate IRAK1 and tumor necrosis factor receptor-associated factor 6, leading to NFκB activation via IκBα phosphorylation [142,143]. The MYD88 p.L265P is a gain-of-function mutation that gives the mutated cells a survival advantage [141]. The MYD88 p.L265P mutation activates NFκB signaling, as described above, and triggers BTK signaling through the SRC family member HCK, and both HCK and BTK are targeted by ibrutinib [144,145]. In addition, in 30–40% of patients with WM, CXCR4 gene mutations are present similar to those seen in the Warts, Hypogammaglobulinaemia, Infections, and Myelokathexis (WHIM) syndrome [146]. CXCR4 mutations are associated with higher levels of bone marrow involvement and serum IgM protein, symptomatic hyperviscosity in patients with CXCR4 nonsense mutations, and resistance to ibrutinib [147,148].
The clinical task force recommendations for the diagnosis and initial evaluation of patients with WM include molecular testing of the bone marrow aspirate for MYD88 mutations. However, routine testing for CXCR4 mutations was not mandated [149]. Rare non-p.L265P MYD88 mutations have been reported by Sanger sequencing. MYD88 wildtype WM patients are rare, and an alternative clinicopathologic diagnosis is common in patients suspected to have WM with wildtype MYD88. The rare WM patients with MYD88 wildtype have a high incidence of associated DLBCL and significantly shorter survival than MYD88-mutated WM [150]. The MYD88 wildtype WM patients harbor NFκB activating mutations downstream of BTK and IRAK, overlapping with somatic mutations found in DLBCL [151]. Interestingly, integrating epigenomic transcriptomic and genomic profiling analyses in WM has revealed the neoplastic cells in WM to have either a memory B-cell or a plasma cell type of differentiation, which have correlations with the MYD88 and CXCR4 mutational status, as illustrated in the cited publication [152].
The essential diagnostic criteria for LPL by WHO-HAEM5 are >10% involvement of the bone marrow cellularity by the neoplastic infiltrate comprised of small lymphocytes, plasmacytoid cells, and plasma cells (similar to WHO 2017), with the following immunophenotype of LPL cells: IgM+, CD19+, CD20+, CD22+, CD25+, CD10−, CD23−, CD103−, CD138+/− [12]. The desirable criteria are the detection of MYD88 p.L265P and CXCR4 somatic mutations and monoclonal IgM by serum electrophoresis and immunofixation [12]. Slightly different from WHO-HAEM5, the ICC criteria also allow a diagnosis of WM with less than 10% bone marrow infiltration by a clonal lymphoplasmacytic infiltrate [13].
Of note, MYD88 mutations are readily detectable by allele-specific PCR, and this method was found to be superior to the NGS method for detecting these mutations. In a study of 391 patients with MYD88 p.L265P identified by allele-specific PCR in CD19-selected bone marrow, allele-specific PCR identified 96% of the patients with MYD88 L265P mutations using unselected bone marrow. However, NGS identified only 66% of those patients [153]. The MYD88 mutation can also be detected in clinical samples other than bone marrow in WM patients, including peripheral blood, cerebrospinal fluid, and pleural effusions. Cell-free DNA analysis has also detected MYD88 p.L265P and CXCR5 p.S338X mutations in WM patients [154,155].
In rare cases, LPL may be associated with a non-IGM protein (IgG or IgA) or no monoclonal protein. The non-IgM LPL patients have a higher frequency of lymph node, splenic, and extranodal involvement and may also harbor MYD88 p.L265P and CXCR4 mutations [156].
Further, almost 20% of WM patients are known to have a familial predisposition, which affects treatment outcomes [157]. The median bone marrow involvement at diagnosis was higher (50%) in patients with familial WM who had a first-line relative with WM or plasma cell myeloma compared to non-familial WM or WM patients with a B-cell malignancy in first-line relatives [158]. Variants in the LAPTM5 and HCLS1 genes have been identified in familial WM [159].
3.7. Immunophenotypic Features Characteristic for Specific Mature B-Cell Lymphomas/Leukemias Composed of Small to Medium-Sized Neoplastic Cells
The diagnosis of lymphoma/leukemia is most often made by histologic examination of a tissue biopsy or by morphologic evaluation and flow cytometric immunophenotyping (FCI) of peripheral blood or bone marrow involved in leukemia. The histologic features must be evaluated before obtaining immunohistochemical (IHC) stains, which must always be selected in a panel based on the differential diagnosis from the cytomorphologic, histologic, and clinical features. Table 7 shows the characteristic immunophenotypic features of the neoplastic cells in the small B-cell neoplasms discussed in the previous sections. This table includes surface antigens evaluated by FCI or IHC stains. Of note, there is variability in the panels used for diagnosis, and the antigens shown in the table may not all be necessary for diagnosis in any individual case.
Table 7.
Immunophenotypic features of the neoplastic cells in mature B-cell leukemias/lymphomas composed of small to medium-sized cells.
It is essential to remember that the expression of any single antigen by FCI or immunohistochemistry is not diagnostic of any specific type of lymphoid neoplasm. Besides morphologic features, BCL2 protein expression by immunohistochemistry helps distinguish follicular lymphoma from reactive follicular hyperplasia. This distinction requires a CD3 stain to evaluate the numbers and distribution of CD3+ T-cells, which are positive for BCL2, while reactive B-cells are negative for BCL2. The BCL2 stain is also required to confirm the presence of suspected in situ follicular neoplasia in lymphoid tissue sections. An in situ follicular neoplasm is diagnosed when intensely-stained BCL2-positive B-cells are seen in the germinal centers in otherwise reactive nodal or extranodal lymphoid tissues. In addition, CD43 positivity alone must never be used as the criterion to diagnose a lymphoma since reactive cells can also be positive for CD43. The interpretation of a panel of antigens examined by FCI or immunohistochemistry must be made in the context of clinical, cytomorphologic, and histopathologic features, which can be supplemented by molecular genetic techniques as needed for diagnosis.
3.8. Aggressive Mature B-Cell Lymphomas
Large B-cell lymphomas comprise a very heterogeneous group of lymphomas in clinical and molecular genetic features; they are defined by the size of the neoplastic (lymphoma) cells being large. As shown in Table 4, WHO-HAEM5 recognizes 17 specific types of large B-cell lymphomas in addition to DLBCL, not otherwise specified (NOS), which is diagnosed when a DLBCL cannot be classified into any of the specific types. DLBCL NOS is the most common among all large B-cell lymphomas [12] and lymphomas in the USA [1,9]; the specific types are less common.
Most large B-cell lymphomas are aggressive lymphomas that arise de novo or as lymphomas transformed from indolent lymphomas; the latter are newly classified as a separate group in WHO-HAEM5. The transformed lymphomas most often consist of large-cell lymphomas arising from CLL/SLL, FL, or marginal zone lymphoma. However, they can also have blastoid or intermediate-sized morphology or may represent an entirely different lymphoma, such as classic Hodgkin lymphoma. A DLBCL can also occur as an unrelated aggressive lymphoma in a patient with a previous indolent lymphoma. Therefore, it is essential to establish the clonal nature of the transformed lymphoma as similar (or not similar) to the previous indolent lymphoma since clonally related transformed lymphomas usually have a worse prognosis, and clonal relatedness versus unrelatedness lead to different management.
Conversely, aggressive lymphomas may be composed of large-sized cells, such as in DLBCL, medium-sized cells in Burkitt lymphoma, or small to medium-sized neoplastic cells in mantle cell lymphoma, as discussed earlier. This section will discuss Burkitt lymphoma first since it is the prototypic aggressive lymphoma, followed by other types of aggressive lymphomas.
3.8.1. Burkitt Lymphoma
Burkitt lymphoma (BL) is a highly aggressive, rapidly growing lymphoma with a short doubling time, characterized as endemic in Africa, sporadic, and immunodeficiency-associated forms [163]. BL involves extranodal sites, including the abdomen, particularly the ileocecal region, the maxillary and mandibular regions in the endemic form, the orbit, Waldeyer’s ring, thyroid, breasts, ovaries, and testes. A leukemic presentation may occur in immunocompromised patients [12,164]. The three forms of BL have similar micro RNA expression profiles, confirming that all three forms represent the same disease that is different from DLBCL [165]. BL is the most common pediatric malignancy in sub-Saharan Africa and also affects adolescents and adults associated with human immunodeficiency virus infection [166]. Endemic BL occurs in children at peak ages between 5 and 9 years in regions with endemic Plasmodium falciparum malaria, where Epstein–Barr virus (EBV) is associated with BL in >90% of cases. In contrast, in other geographic regions, EBV is associated with only 30% of sporadic BL [167]. In germinal center B-cells, immunoglobulin gene somatic mutation and class switch recombination is initiated by the activation-induced cytidine deaminase (AID) enzyme [168]. Chronic malarial and EBV infection causes B-cell hyperplasia with aberrantly increased activity of AID, leading to widespread genomic instability in the germinal center B-cells formed during the chronic infection [169], which increases the possibility of a chromosomal translocation involving MYC and an immunoglobulin gene, the genetic hallmark of BL. Although BL has a very aggressive natural history of disease, appropriate treatment specific to BL leads to a cure in most patients. Therefore, prompt diagnosis and distinction from other mature aggressive B-cell lymphomas, including DLBCL, is essential.
The historical background for BL, including the discovery of the diagnostic genetic abnormalities in BL, was described in Part 1 [2]. In 80% of BL cases, MYC is juxtaposed to the IGH gene in the t(8;14)(q24;q32) translocation or, less commonly, to IGL or IGK in the t(8;22)(q24;q11) and t(2;8)(p12;q24) translocations, resulting in constitutive MYC expression. However, the site of chromosomal breakpoints and structural alterations differ between endemic and sporadic BL [170]; see references in [12]. In 2006, gene expression profiling accurately distinguished BL from DLBCL and identified cases of BL not identifiable by routine genetic techniques, termed molecularly-defined BL (mBL) [171,172].
In 2012, three studies described the mutational landscape of BL [173,174,175]. BL harbors mutations in the transcription factor 3 (TCF3) or inhibitor of DNA binding 3 (ID3), a negative regulator of TCF3, to cause tonic B-cell receptor signaling that activates the phosphatidylinositol 3-kinase (PI3K) pathway in the lymphoma cells, seen in 70% of sporadic BL cases. Alternatively, oncogenic cyclin D3 (CCND3) mutations that drive cell cycle progression by producing highly stable cyclin D3 isoforms are present in 38% of sporadic BL cases [173]. ID3, TCF3, or both mutations were also present in HIV-associated BL (67% cases), and endemic BL (40% cases), and these mutations were rare in other lymphoid cancers [173].
WGS, whole exome, and transcriptome sequencing of four pediatric sporadic prototypical BLs with IG::MYC translocation and corresponding germline samples identified seven recurrently somatically mutated genes. These genes included TP53, MYC, and SMARCA4, previously known to be mutated in BL, FBXO11, and DDX3X12, also involved in other B-cell lymphomas, RHOA, and ID3. ID3 mutations were also present in 68% (36/53) of other mBL cases. Of note, ID3 mutations were strongly enriched with lower patient age, negative BCL2, positive BCL6, and high Ki-67 index by immunohistochemistry, absence of t(14;18) and BCL6 breaks, low genetic complexity, and low IGHV mutation, and showed a significantly favorable prognosis [174].
In the third study, exome sequencing of 51 primary BL tumors, 14 with paired normal tissue, and 8 BL cell lines showed recurrent mutations in 70 genes, most frequently MYC (40%) and ID3 (34%); other frequently mutated genes included ARID1A, SMARCA4, TP53, PIK3R1, and NOTCH1 [175]. In 2015, RNA sequencing of 20 endemic BL samples showed lower frequencies of mutations in MYC, ID3, TCF3, DDX3X, CCND3, and TP53, as compared to their reported recurrence in sporadic BL, and higher mutational frequencies in ARID1A, RHOA, and CCNF [176].
Further, several studies showed differences between EBV+ and EBV-negative BL [176,177,178,179]. In 2005, endemic and HIV-related BLs showed higher IGHV mutation rates than sporadic BL and also showed signs of antigen selection, which were absent in sporadic BL; these differences were even more evident between EBV+ and EBV-negative BL when compared regardless of geographic location or HIV status [177]. The presence of EBV was almost mutually exclusive with mutations in TCF/ID3, considered driver mutations for sporadic BL [176]. In 2019, WGS and transcriptomic analysis supported the link between EBV and aberrant AID activity since EBV+ BL showed a striking genome-wide increase in aberrant somatic hypermutation and had significantly fewer driver mutations, especially among genes with roles in apoptosis. In addition, IGHV4-34, known to produce autoreactive antibodies, and IGKV3-20, were overrepresented in the lymphoma cells [178].
Moreover, in a 2022 study, mutations in ID3, TCF3, and CCND3 and the expression of sex-determining region Y-box transcription factor 11 (SOX11) were significantly decreased with patient age at diagnosis. In contrast, EBV was more frequently detected in adult patients. Further, irrespective of age, EBV-positive sporadic BL showed significantly less frequent mutations in ID3/TCF3/CCND3 but more often mutations of G protein subunit alpha 13 (GNA13) and forkhead box O1 (FOXO1) compared to EBV-negative BL [179].
The collective evidence described above indicates that EBV status provides a better separation of the differences in biologic heterogeneity, irrespective of geographical location and patient age (pediatric versus adult). Therefore, WHO-HAEM5 recommends this distinction in the diagnosis [11,12], which is easy to perform since the characterization of EBV positivity or negativity in lymphomas is readily obtained in most clinical laboratories. A 2023 study showed shared features between pediatric and adult BL and confirmed the vital role of EBV status in distinguishing the molecular pathogenesis of BL [180].
Diagnostically, BL arises from proliferating centroblasts in the dark zone of the germinal center and is characterized by the presence of MYC translocations with the IGH, IGK, or IGL gene; see review [181]. Histologically, BL is composed of a diffuse monomorphous infiltrate of medium-sized, not large, lymphoid cells with round nuclei, finely clumped and dispersed nuclear chromatin, multiple peripherally located nucleoli, and a moderate quantity of deeply basophilic, often vacuolated cytoplasm. Mitoses are frequent. The neoplastic cells are densely packed with a “squared-off” appearance, and numerous tingible body macrophages engulfing fragments of apoptotic neoplastic cells cause a “starry-sky” appearance. The neoplastic cells express CD19, CD20, CD79a, CD22, and PAX5 (pan B-cell antigens), CD10 (strong expression), and BCL6, CD38, HGAL, and MEF2B (other germinal center-associated antigens); GCET is variably expressed, and the neoplastic cells are consistently negative for LMO2. Cyclin D1 must be absent for differential diagnosis with blastoid mantle cell lymphoma. The proliferative rate (Ki67) is >95%, and BCL2 protein expression is typically absent. The cytogenetics in BL typically shows a simple karyotype with the primary abnormality causing the MYC translocation. Notably, the cytogenetics findings are not complex at initial diagnosis; if multiple cytogenetics abnormalities are found, the diagnosis should be questioned, and additional tests are needed. Nevertheless, routine cytogenetics and molecular genetics analysis may not detect molecular abnormalities in all cases of BL.
The WHO-HAEM5 essential diagnostic criteria for BL require the morphologic features with CD20+ CD10+ neoplastic cells, absent or rarely weakly expressed BCL2, Ki67 index >95%, usually strong MYC expression in >80% of cells, demonstration of MYC breakage or IG::MYC translocation, or both MYC expression and translocation [12]. The WHO-HAEM5 desirable criteria include a starry-sky pattern, cohesive growth pattern, BCL6 positivity, TdT negativity, CD38 positivity, and the exclusion of BCL2 and BCL6 rearrangements, which are mainly required to be excluded in adult BL [12].
For resource-poor settings such as in sub-Saharan Africa, where BL is frequent, and FISH is not readily available, a diagnostic algorithm has been proposed that requires characteristic morphologic and immunophenotypic features, CD20+, CD10+, BCL2 negative or weak, CD38+ (clone SPC32), CD44 negative (clone DF1485), MYC ≥ 80% (using the Y69 antibody), and Ki67 > 95% [12,182]. If the characteristic morphologic and immunophenotypic features are absent, the differential diagnosis with other aggressive non-Burkitt (large-cell) mature B-cell lymphomas requires further workup. Distinguishing B-ALL, in which the neoplastic cells may be small to medium-sized and, regardless of size, have finely dispersed nuclear chromatin with indistinct nucleoli, from the differential diagnosis of BL requires integration of all clinical, pathologic, and genetic features [183].
3.8.2. How the Classification of Aggressive Non-Burkitt (or large) B-Cell Lymphomas Has Evolved
As shown in Table 3 in Part 1 [2], the aggressive B-cell lymphomas in WHO 2001 included BL, DLBCL, four specific types of large B-cell lymphomas (mediastinal thymic large B-cell lymphoma, intravascular large B-cell lymphoma, primary effusion lymphoma, and lymphomatoid granulomatosis), and mantle cell lymphoma. All these except BL and mantle cell lymphoma are large-cell lymphomas. At that time, all large-cell lymphomas except the specific types were lumped into one diagnosis of DLBCL. Figure 2 depicts the evolution of the classification of large B-cell lymphomas in relation to BL and CHL. With advances in our understanding of lymphomas, the classification updates in 2008 and 2017 separated additional specific types of large-cell lymphomas. The cases that could not be classified as any specific type were termed DLBCL, not otherwise specified (NOS). In addition, patients who did not meet the BL or DLBCL NOS criteria were now grouped into a heterogeneous group termed unclassifiable with features intermediate between BLBCL and BL, as depicted in Figure 2.
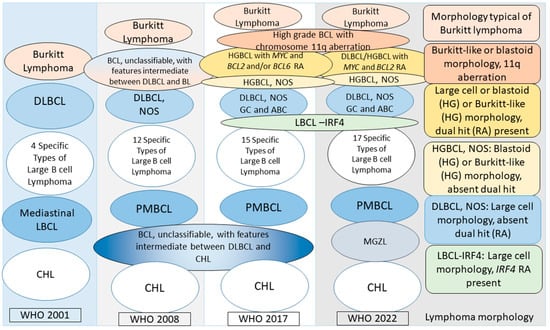
Figure 2.
Evolution of the current classification of large B-cell lymphomas in relation to Burkitt lymphoma and classical Hodgkin lymphoma. From left to right, the ovals in the first four columns represent lymphoma entities in the WHO 2001, 2008, 2017, and WHO 2022 classifications. The specific types of large B-cell lymphomas exclude DLBCL and are shown in Table 3 in Part 1 [2]. WHO 2001 recognized four specific types of large B-cell lymphomas; WHO 2008 recognized eight additional specific types: T-cell/histiocyte-rich large B-cell lymphoma, primary DLBCL of the central nervous system, primary cutaneous DLBCL, leg type, EBV+ DLBCL of the elderly (provisional), DLBCL associated with chronic inflammation, ALK+ large B-cell lymphoma, plasmablastic lymphoma, large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease, and two unclassifiable types: B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL, and B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL. Further, DLBCL was now termed DLBCL, NOS. WHO 2017 mandated specifying DLBCL, NOS, as a germinal center (GC) B-cell subtype or activated B-cell (ABC) subtype. The following additional specific subtypes were introduced in 2017: fibrin-associated DLBCL, HHV8+ DLBCL, NOS (provisional), HHV8+ germinotrophic lymphoproliferative disorder, and Burkitt-like lymphoma with chromosome 11q aberration (provisional). Further, the WHO 2008 B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL, was changed to high-grade B-cell lymphoma (HGBCL), with two subtypes: HGBCL with MYC and BCL2 and/or BCL6 rearrangements, and HGBCL, NOS. WHO 2022 recognized large/HGBCL with MYC and BCL2 rearrangements and retained HGBCL, NOS. The ICC recognizes HGBCL with MYC and BCL2 rearrangements and also continues recognizing HGBCL with MYC and BCL6 rearrangements as a provisional entity (not shown in the figure). The previous B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL, is recognized by WHO 2022 and the ICC as mediastinal grey zone lymphoma. The fifth column (far right) highlights the morphology of the lymphoma cells in the six depicted types of lymphomas in WHO 2022 and the ICC. Note that LBCL-IRF4 is not an aggressive lymphoma but has large-cell morphology and can show a diffuse growth pattern. WHO, World Health Organization; DLBCL, diffuse large B-cell lymphoma; NOS, not otherwise specified; BL, Burkitt lymphoma; HGBCL, high-grade B-cell lymphoma, RA, rearrangement; GC, germinal center subtype; ABC, activated B-cell subtype; LBCL, large B-cell lymphoma; LBCL-IRF4, large B-cell lymphoma with IRF4 rearrangement; PMBCL, primary mediastinal large B-cell lymphoma; BCL, B-cell lymphoma; CHL, classical Hodgkin lymphoma; MGZL, mediastinal grey zone lymphoma; ICC, International Consensus Classification.
Patients with DLBCL are treated with R-CHOP (anti-CD20 (rituximab) combined with chemotherapy (CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisolone)) as the standard-of-care first-line therapy. About 60% of patients are cured; the remainder are either refractory to primary treatment or relapse [184,185]. Identifying upfront at the time of diagnosis the patients who have a high chance of cure and those at high risk for being refractory to treatment or for relapse continues to be a significant research goal, which includes defining different subsets of patients with specific types of diseases so that each could be treated appropriately; this is a major goal of cancer classification and, as applied to large-cell lymphomas, is depicted in Figure 2.
3.8.3. DLBCL/High-Grade B-Cell Lymphoma with MYC and BCl2 Rearrangements
The revised fourth edition WHO 2017 classification created the high-grade B-cell lymphoma group with MYC and BCL2 and/or BCL6 rearrangement. This group included WHO 2008 subsets of cases from the unclassifiable group (with features intermediate between DLBCL and BL) and DLBCL NOS. Therefore, diagnosing these cases required FISH on virtually all cases of the two WHO 2008 groups, including DLBCL NOS. These lymphomas were termed double-hit or triple-hit lymphomas. In 2022, both WHO-HAEM5 and ICC recognized DLBCL/high-grade B-cell lymphoma with MYC and BCl2 rearrangements as a distinct type due to its homogeneous genetic features. The previously included subtype with rearranged MYC and BCL6 was considered heterogeneous with available evidence and, therefore, deleted in WHO-HAEM5 while the ICC continued to recognize it as a provisional entity.
Enormous progress has been made in understanding the molecular pathogenesis of DLBCL in the last 25 years. In 2000, DLBCL was classified into the germinal center and activated B-cell subtypes and an unclassified type by gene expression profiling, with the germinal center subtype DLBCL patients showing a better overall survival than the activated B-cell subtype [186]. Subsequently, the Hans algorithm using CD10, BCL6, and MUM1 immunohistochemical stains on formalin-fixed, paraffin-embedded tissues [187] has been most commonly used as a surrogate for defining a germinal center or activated B-cell DLBCL. This cell-of-origin classification for distinguishing the two subtypes of DLBCL was included in the revised fourth edition WHO update [100,188]. In the meanwhile, molecular genetic studies showed alterations in genes involved with chromatin modification in follicular lymphoma and germinal center subtype of DLBCL [189,190,191,192,193,194,195,196], as reviewed in [197,198], and chronic B-cell receptor signaling with NFκB pathway activation in the activated B-cell subtype of DLBCL [193,194,199,200,201].
In the last five years, three groups from the National Cancer Institute (NCI) [202,203] and Dana Farber Cancer Center (DFCC) in the USA [204] and the UK Hematologic Malignancy Research Network (HMRN) [205] independently characterized 5–7 subgroups in DLBCL with distinct genetic profiles showing characteristic co-occurring mutations in specific genes. Notably, there was significant overlap with similar profiles of mutations in each genetic group among all three studies [202,203,204,205], and the mutational profile in each genetic group in DLBCL resembled that of other (specific) mature B-cell lymphomas [206].
Despite the significant advances in molecular genetics briefly summarized above in DLBCL, there is current evidence for one DLBCL genetic subgroup within the NCI EZB group (or cluster 3 in DFCC and BCL2 group in HMRN) harboring dual MYC and BCL2 gene rearrangements. This group of lymphomas is included in WHO-HAEM5, with or without the presence of BCL6 translocations and with or without TdT expression [12]. The dual MYC- and BCL2-rearranged cases showed mutations characteristic of follicular lymphoma (BCL2, KMT2D, CREBBP, EZH2, and TNFRSF14), and some patients had a history or concurrent diagnosis of follicular lymphoma [207]. Interestingly, 65% of BCL2-rearranged cases showed the characteristic genetic mutations of follicular lymphoma in the absence of a history or concurrent diagnosis of follicular lymphoma, suggesting transformation from an occult follicular lymphoma [207]. These lymphomas usually comprise the germinal center subtype by the cell-or-origin classification of DLBCL [208]. The presence of IG::MYC translocations, TdT expression, and MYC hotspot mutation may indicate a higher risk [12,207,209].
Intriguingly, many, but not all, of these MYC and BCL2 dual-hit DLBCL cases show a molecularly-defined high-grade signature harbored in about 9% of DLBCL patients with a poor prognosis, as identified by Sha et al. [210]. Of note, the dual-hit cases without the molecularly-defined high-grade signature did not show outcomes worse than the other germinal center B-cell-like DLBCL cases [207,210]. Conversely, only 48.6% and 36.1% of the molecularly-defined high-grade group were MYC-rearranged and double-hit (MYC and BCL2 rearranged), respectively, indicating that identifying this molecularly-defined high-grade signature doubled the size of the poor prognosis cases [210]. Interestingly, at the same time, Ennishi et al. reported similar findings with a gene expression signature for double-hit lymphomas predicting a poor clinical outcome, and only half of those double-hit-signature-positive cases harbored the actual MYC and BCL2 gene rearrangements [211].
Of note, these findings in the above two studies [210,211] are reminiscent of the diagnostic situation with Ph-like B-ALL, described in Part 2 [3], and similarly suggest that attempts should be made to identify these dual MYC- and BCL2-rearrangement-like cases with a molecularly-defined high-risk signature and a poor outcome.
The essential diagnostic criteria in WHO-HAEM5 are morphology and phenotype consistent with an aggressive B-cell lymphoma and evidence of concurrent MYC and BCL2 rearrangements, with or without BCL6 rearrangement. The presence of a germinal center phenotype and determining the TdT protein expression status and the MYC fusion partner are desirable in WHO-HAEM5 [12].
3.8.4. High-Grade B-Cell Lymphoma with Chromosome 11q Aberration
High-grade B-cell lymphoma with chromosome 11q aberration (HGBCL-11q) in WHO-HAEM5 was introduced provisionally in WHO 2017 as Burkitt-like B-cell lymphoma with chromosome 11q aberration. These are very rare lymphomas originally described by studying aggressive lymphomas with morphologic, immunophenotypic, and gene expression profiles of BL but without the hallmark MYC translocation of BL [212]. The defining genetic feature of this lymphoma is a combined pattern of proximal chromosomal gains and telomeric loss at 11q23. The telomeric loss shows a copy-number-neutral loss of heterozygosity. This telomeric loss is considered to be characteristic of this type of lymphoma since a proximal gain of 11q23 can be seen in other high-grade lymphomas, including BL and high-grade B-cell lymphoma with MYC translocation. Large-cell lymphomas may also show this cytogenetic aberration, and it is not clear yet whether these cases are the same biologically as the WHO-HAEM5-described entity [12]. Despite the similarities with BL, HGBCL-11q is molecularly distinct. It does not harbor genetic alterations frequently present in BL (genes in the ID3-TCF3 axis or the SWI/SNF complex) or in the germinal-center-derived B-cell lymphomas, e.g., KMT2D or CREBBP [213]; the latter mutations were noted in another study in cases with the 11q aberration and large-cell morphology [214].
The disease occurs in childhood and young adults with a median age at diagnosis of 14 or 15.5 years (overall range, 4–52 years) in two series of cases and shows a male predominance with a male-to-female ratio of 2.75:1 [213,214]. The disease may be localized and show nodal involvement of the head and neck region, but it may also be extranodal and present in the abdomen [214]. A characteristic finding on morphologic evaluation is the presence of coarse 5–9 apoptotic fragments phagocytosed inside the “starry-sky” macrophages in contrast with 1–2 apoptotic bodies in the macrophages in BL [215], which can help confirm the diagnosis [216]. Flow cytometry can show the lymphoma cells to express CD16 and CD56 or CD8 expression and the absence of CD38 (high) expression, which are highly characteristic of HGBL-11q [12,217].
The essential diagnostic criteria by WHO-HAEM5 are lymphoma with an intermediate/blastoid or Burkitt-like morphology, typical immunophenotype (B-cell markers+, CD10+, BCL6+, BCL2-negative), chromosome 11q-gain/loss, telomeric loss or telomeric loss of heterozygosity (LOH) pattern, and exclusion of an MYC translocation. Expression of CD56 in the absence of high expression of CD38 by flow cytometry is a desirable criterion for diagnosing HGBCL-11q [12].
3.8.5. Primary Large B-Cell Lymphoma of Immune-Privileged Sites
Primary large B-cell lymphoma of immune-privileged sites is included as a new group by WHO-HAEM5. These lymphomas include primary large B-cell lymphomas that arise in the central nervous system (CNS), vitreoretina, or the testes in immunocompetent individuals, excluding lymphoma arising in the dura or the choroid and those that spread to these immune-privileged sites from other body sites [11,12].
Primary large B-cell lymphoma of the CNS (PCNS-LBCL) presents as a mass, solitary or multiple, in the brain parenchyma. If this lymphoma occurs in an immunocompromised individual, which is more frequent than in immunocompetent individuals, it would be classified as a lymphoma arising in immune deficiency/dysregulation, which is included in another new group in WHO-HAEM5, as shown in Table 4.
In immunodeficient individuals, PCNS-LBCL occurs at a younger age, in the 30s, predominantly in males, as multiple masses, and it is frequently associated with viral infections (EBV and HIV); this lymphoma is an acquired immune deficiency syndrome (AIDS)-defining illness. In contrast, PCNS-LBCL in immunocompetent individuals presents as a solitary mass in the brain parenchyma in older individuals aged >60 years, with an equal incidence in males and females [218]. Among immunocompetent individuals, primary CNS lymphoma rates increased significantly in men (1.7%/year) and women (1.6%/year) aged >65 years from 1992 to 2011 in the USA but remained stable in younger individuals [219].
The principal differential diagnosis for PCNS-LBCL is secondary CNS involvement from another body site [220]. However, in addition to PCNS-LBCL in the immune-privileged sites group, several other primary lymphomas may rarely arise in the CNS. These include highly aggressive lymphomas, such as primary CNS BL and primary CNS T-cell lymphoma, and lymphomas with clinically indolent behavior, such as primary CNS parenchymal small B-cell lymphoma and primary dural lymphoma [221]. Of note, intravascular large B-cell lymphoma (IVBCL), a highly aggressive lymphoma that frequently affects the CNS, does not present with a mass in the CNS in contrast with PCNS-LBCL and other primary CNS lymphomas; IVBCL is briefly described separately.
The neoplastic cells in PCNS-LBCL, primary vitreoretinal (PVR-LBCL), and primary testicular large B-cell lymphoma are similar to a late germinal center mature B-cell phenotype with ongoing somatic hypermutations [12,222]. PCNS-LBCL and PVR-LBCL preferentially show IGHV4-34 gene rearrangement, which is common in autoimmune diseases [223,224,225]. Genetically, these lymphomas show recurrent MYD88 and CD79B hotspot mutations among other mutations in the Toll-like receptor and NFκB pathways and inactivated CDKN2A and belong to the MCD, C5, and MYD88 genetic groups of DLBCL described in Section 3.8.3 [202,203,204,205].
Nonetheless, recent evidence indicates that ~65% of PCNS-LBCL represent the MCD subtype, and ~35% of PCNS-LBCL may represent one of the other non-MCD groups [226]. The MCD subtype included recurrent mutations in MYD88 and CD79B, among other mutations affecting the JAK/STAT, Toll-like receptor, and NFκB pathways [226]. The non-MCD types included the EZB group, or germinal-center-like DLBCL, with deletions in CREBB, a hallmark of follicular lymphoma [195], which would suggest that PCNS-LBCL might represent an occult follicular lymphoma first presenting as a CNS lymphoma [226].
Interestingly, kataegis, a phenomenon associated with a promiscuous non-IGH activity of the enzyme activation-induced cytidine deaminase (AID) in B-cells [227], was observed in PCNS-LBCL with numerous mutations in IGH, IGK, IGL, and non-IG genes. While most mutations overlapped between PCNS-DLBCL and the activated B-cell types of DLBCL, mutations in BTG2, GRHPR, OSBPL10, and ZNF860 were significantly more frequent in PCNS-DLBCL [226]. In contrast with PCNS-LBCL in the WHO-HAEM5 immune-privileged sites group, which are EBV-negative as mentioned above, EBV+ PCNS-LBCLs show a different mutational profile and do not harbor MYD88 and CD79B mutations [227].
The diagnosis of PCNS-LBCL requires a tissue biopsy, which should ideally be performed before giving steroids, if clinically feasible since steroids cause lysis of neoplastic cells and increased numbers of macrophages and may lead to a false-negative biopsy [12,218]. Histologically, the neoplastic cells are medium- to large-sized CD20+ B-cells that infiltrate diffusely with characteristic perivascular infiltration and splitting of vessel walls, which is most evident at the tumor periphery. Necrosis and high mitotic activity are present. The lymphoma cells are intermingled with reactive bystander cells consisting of small mature T and B lymphocytes, macrophages, activated microglial cells, and reactive astrocytes [228]. In addition to mature B-cell antigens, CD20, CD22, CD79a, CD19, and PAX5, the lymphoma cells express IRF4/MUM1, BCL2, BCL6, and IGM, have a high (>80–90%) Ki67 proliferation index, and are negative for CD38 and CD138 [12]. CD10 is not usually expressed, and its expression should suggest the possibility of a secondary CNS lymphoma. EBV is also negative in PCNS-LBCL in immunocompetent individuals; its expression should lead to suspecting an immune deficiency/dysregulation state [12,229].
Intraocular lymphomas include lymphomas arising in the vitreoretina (PVR-LBCL) or the uvea, with the latter including choroidal, iridal, or ciliary body lymphoma [230]. The PVR-LBCLs are protected by a blood–brain barrier [230]; in contrast, choroidal lymphomas do not have that protection [231]. Therefore, PVR-LBCLs alone are classified as lymphomas of immune-privileged sites [12]. PVR-LBCL may occur concurrently with, before, or after a PCNS-LBCL. The diagnosis may masquerade as uveitis and is often delayed for this reason. Close collaboration between the ophthalmologist and a pathologist with experience in ocular cytology is necessary for evaluating these samples; refer to excellent reviews [230,231].
PVR-LBCL must be distinguished from secondary involvement by systemic lymphoma and a PCNS-LBCL. The lymphoma cells in PVR-LBCL involve and are contained within the vitreous, retina, and subretinal space anterior to the Bruch membrane [230,231]. In contrast with PVR-LBCL, which are challenging to diagnose, choroidal lymphomas, which occur in a non-immune-privileged site, are low-grade lymphomas typically diagnosed by a fine needle aspiration biopsy, and patients respond to low-dose radiotherapy [231].
MYD88 and CD79B mutations are recurrent in all three lymphomas in immune-privileged sites mentioned above. Detecting these mutations may allow a precise diagnosis in cases where clonality may not be ascertained or in samples with low cellularity [12]. Especially in PVR-LBCL samples with low cellularity wherein the MYD88 p.L265P mutation occurs in 60–80% of cases and can be readily identified by a single gene molecular assay, this assay can be very helpful in establishing a diagnosis [231]. The WHO-HAEM5 essential diagnostic criteria require a large B-cell lymphoma primarily confined to the CNS, vitreoretina, or testes at presentation, exclusion of secondary involvement by other entities of large B-cell lymphoma, and exclusion of immune deficiency/dysregulation-related settings. The desirable WHO-HAEM5 findings include a post-germinal center B-cell phenotype (MUM1+, BCL6+, CD10-negative), absence of EBV (in >97% of cases), and showing a clonal B-cell population or an MYD88 or CD79B hotspot, or both mutations if histology is not definitive, such as in corticosteroid-treated PCNS-LBCL or PVR-LBCL [12].
3.8.6. Intravascular Large B-Cell Lymphoma
Intravascular large B-cell lymphoma (IVBCL) is a rare, highly aggressive B-cell lymphoma that has often been diagnosed at autopsy due to an unsuspected diagnosis during life. There are no classification changes, but the entity is briefly described to continue spreading awareness to enable diagnosis during life and prompt treatment. The disease often presents with neurologic symptoms or a skin rash [232,233]. However, in most instances, the neurologic symptoms resemble a stroke, and there is no mass in the CNS, which is usually expected in lymphoma involving the CNS. Notably, in five Japanese patients, a CNS mass due to lymphoma was present with features typical of IVBCL [234]. IVBCL may precede or follow other lymphomas, including small lymphocytic lymphoma, follicular lymphoma, gastric MALT lymphoma, DLBCL NOS, and lymphoplasmacytic lymphoma [235]. Of note, in the Japanese IVBCL patients with a CNS mass, if the IVBCL had not been diagnosed, the CNS lymphoma would have been interpreted as a PCNS-LBCL [234], indicating that the disease biology and inter-relationships between these types of lymphomas remain to be understood.
IVBCL appears to have a higher incidence, accounting for 1% of all B-cell lymphomas, in Japan than in Western countries, but this could be due to a greater awareness of IVBCL in Japan [232,236]. Fever, general malaise, neurological symptoms, and dyspnea were reported most frequently in Japanese patients, while skin eruptions were observed in only 6% of patients [236]. Lymphadenopathy is also usually not observed in IVBCL, although newer radiologic methods have shown the presence of lymphadenopathy in some patients.
IVBCL comprised 88% (n = 651) of all intravascular lymphoma (IVL) (n = 740) patients reported over 50 years in 431 publications between 1959 and 2011; the remaining patients included 6% (n = 45) with T-cell intravascular lymphoma and 2% (n = 12) with NK-cell lymphoma. Post-mortem diagnosis was most frequent (60%, n = 250) with CNS involvement by IVL, followed by skin (8%, n = 21), bone marrow and spleen (11%, n = 28), and lung (7%, n = 17) involvement by IVL [237].
In a multi-institutional U.S. study of 54 IVBCL patients, the most common presenting symptoms were non-specific, including fever, night sweats, or weight loss, followed by neurologic symptoms (altered mental status, weakness, headache, paresthesia, vision changes, or seizure) in 41% (18/43), skin involvement (rash, cutaneous nodules, and hemangiomas) in 21% (n = 9), respiratory symptoms (shortness of breath, cough, and respiratory failure) in 21% (n = 9) of patients, and rarely, gastrointestinal symptoms (abdominal pain, and ascites) and endocrine abnormalities (panhypopituitarism, and goiter) [238].
The lack of specific symptoms is due to the unique biology of this disease, which is still not well-understood, as recently reviewed [236]. The lymphoma cells are located and grow inside small vessels and capillaries and can be found in virtually any organ in the body. The occlusion of the capillary vessels by the lymphoma causes progressive rapid deterioration and death if the diagnosis is not made.
The only definitive way to diagnose IVBCL is a surgical biopsy of involved tissue, and often, a bone marrow or random skin biopsy will allow a diagnosis. The diagnosis may be based on a small number of cells in single, or very few, tiny foci in the bone marrow, and the lymphoma cells are easier to recognize by the evaluation of morphology in a trephine core biopsy compared with the bone marrow aspirate smears or aspirate clot (unpublished observation). Of note, a bone marrow biopsy provided the diagnosis in 45% of patients in a single institution cohort; at diagnosis, anemia, leucopenia, and thrombocytopenia were present in 91%, 30%, and 45% of those patients, respectively [239]. In the U.S. multi-institutional study, the bone marrow was involved by lymphoma in 60% (n = 26) of patients, but it was the diagnostic biopsy site in only ten (23%) patients [238].
Immunohistochemically, the lymphoma cells show strong positivity for CD20, which is the basis for the treatment with rituximab. The lymphoma cells showed positivity for BCL2, BCL6, MYC, and MUM1 in 100% (19/19), 61.9% (13/21), 71.4% (5/7), and 77% (17/22) of patients, respectively [239]. While BCL6 was rearranged in one patient, BCL2 or MYC was not rearranged in six examined patients [239].
Most significantly, cell-free DNA (cfDNA) studies have shown promising results for the diagnosis and even monitoring of patients with IVBCL [240,241]. Cell-free DNA is present at a higher concentration in patients with IVBCL compared with DLBCL patients and healthy controls. Whole exome sequencing of cfDNA in patients with IVBCL showed similar genetic mutations as in the bone marrow involved by IVBCL [240]. IVBCL showed mutations in MYD88 (57%), CD79B (67%), SETD1B (57%), and HLAB (57%). Rearrangements involving the 3’ untranslated region of programmed cell death ligands 1 and 2 (PDL1/PDL2) were identified in 38% (n = 8) of IVLBCL, suggesting the presence of immune evasion via PD-L1/PD-L2 overexpression in IVBCL, which could be used therapeutically [240].
Of note, the MYD88 p.L265P mutation, found in most cases of Waldenström macroglobulinemia [142,143] and also present in other lymphomas, including those occurring in immune-privileged sites as described in the previous section, was detected by digital droplet PCR assays in 50.0% (5/10) of cfDNA samples and 52.8% (19/36) of tumor-tissue-derived DNA samples from 16/27 (59.3%) patients with IVBCL [241]. Targeted sequencing showed MYD88 p.L265P (44%) and CD79B p.Y196 mutations (26%) in 25 patients with IVBCL [242].
3.9. Immunophenotypic Features Characteristic of Mature B-Cell Lymphomas/Leukemias Composed of Medium-Sized to Large Cells
Table 8 summarizes the immunophenotypic findings by FCI and IHC stains performed in lymphomas described in the previous sections.
Table 8.
Antigen expression by FCI or IHC stains in the neoplastic cells in mature B-cell lymphomas composed of medium-sized to large cells.
The Hans algorithm is used most commonly by immunohistochemistry as a surrogate for gene expression profiling to classify DLBCL as germinal center B-cell (GCB) and activated B-cell (ABC) types [187]. This algorithm uses three IHC stains, CD10, BCL6, and MUM1. Each antigen is considered positive if 30% or more neoplastic cells in DLBCL stain positive. If the lymphoma cells are CD10+, the subtype is GCB. If the neoplastic cells are CD10-negative and BCL6+, then MUM1 expression determines the subtype. If the neoplastic cells are CD10-, BCL6+, and MUM1+, the subtype is ABC. If the neoplastic cells are CD10-, BCL6+, and MUM1-negative, the subtype is GCB [187]. The lymphoma cells in DLBCL may be positive for CD5 and indicate an inferior prognosis to CD5-negative DLBCL [246].
LIM-domain only 2 (LMO2), a cysteine-rich protein containing two zinc-binding LIM domains, is expressed in all tissues except mature T-cells. LMO2 is upregulated in germinal center B-cells. Its expression in DLBCL has been associated with increased genomic instability and double-stranded DNA breaks [247,248]. In cases classified by the Hans algorithm, LMO2 expression has been shown to correlate with the genetic features of DLBCL. In that study, LMO2 was considered positive if more than 30% of lymphoma cells showed nuclear positivity; MYC was considered positive if more than 40% of lymphoma cells showed nuclear positivity [243]. The GCB cases of DLBCL with negative or low (<30%) expression of LMO2 were significantly enriched in the subtype with dual MYC and BCL2 gene rearrangements [243]. High (>30%) LMO2 expression was associated with isolated BCL2 translocations; it was suggested that LMO2 expression might be downregulated or lost after MYC rearrangements in aggressive cases of DLBCL.
3.10. Primary Mediastinal Lymphomas
Lymphomas comprise 50–60% of all mediastinal malignancies in children and adults, including ~65% of non-Hodgkin lymphomas. The majority of mediastinal involvement by non-Hodgkin lymphoma is due to secondary involvement of the mediastinum from systemic disease. Primary mediastinal non-Hodgkin lymphomas comprise only about 5% of all mediastinal lymphomas. They are defined as the involvement of mediastinal lymph nodes, thymus, and/or extranodal mediastinal organs (heart, lung, pleura, and pericardium) by lymphoma without evidence of systemic disease at presentation [249]. T lymphoblastic lymphoma, primary mediastinal (thymic) large B-cell lymphoma (PMBCL), and primary mediastinal “nonthymic” DLBCL represent the most common types of non-Hodgkin lymphomas [249]. Knowledge of the anatomic site of the tumor mass and the biopsy or fluid sample examined is essential for precise diagnostic classification of lymphomas arising in the mediastinum. Therefore, Table 9, modified from the cited reference [249], shows the types of primary mediastinal lymphomas based on their anatomical location in the mediastinum.
Table 9.
Primary mediastinal lymphomas classified according to the primary anatomical site presentation (modified from [249]).
Mediastinal Grey Zone Lymphoma: In the Middle of the Spectrum between Classic Hodgkin Lymphoma and Primary Mediastinal Large B-CELL lymphoma
The term “grey zone lymphoma” was first introduced in 1998 when three cases of mediastinal lymphomas with features of CHL and DLBCL were discussed at a workshop conducted by 12 expert hematopathologists for borderline cases between CHL and non-Hodgkin lymphomas [250]. In 2005, mediastinal grey zone lymphomas (MGZLs) were described as the “missing link” between CHL and primary mediastinal large B-cell lymphoma (PMBCL) [251] and included in the WHO 2008 and 2017 classifications as a provisional entity. Both WHO-HAEM5 and ICC confirmed MGZL as a definite entity replacing the previous provisional entity “B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classic Hodgkin lymphoma.” MGZL represents the mediastinal lymphoma with features intermediate between PMBCL and CHL nodular sclerosis. MGZL is considered to be a part of the spectrum of the mediastinal lymphomas, PMBCL, and CHL, particularly the nodular sclerosis subtype, and shows clinical, pathologic, immunophenotypic, and genetic overlap with nodular sclerosis CHL and PMBCL.
PMBCL, nodular sclerosis CHL in its first age peak, and MGZL occur in young individuals aged <40 years. In Western countries, females predominate in PMBCL and CHL but not in MGZL, which is more frequent in males [251,252]. The treatment approaches differ between nodular sclerosis CHL and MGZL, so the diagnostic distinction is necessary [253]. The differential diagnosis is critically based on pathologic features, especially the integration of morphologic and immunophenotypic features of the neoplastic cells in these three mediastinal lymphomas, all occurring as a mediastinal mass. Therefore, the diagnostic criteria for each entity are briefly described in this section.
The diagnostic criteria for CHL have remained the same since nodular lymphocyte predominant Hodgkin lymphoma was separated from CHL in the WHO 2001 classification. CHL is characterized by the presence of a few neoplastic Hodgkin or Reed–Sternberg (HRS) cells scattered in a background of non-neoplastic polymorphous inflammatory cells required for the neoplasm to grow. The nodular sclerosis subtype is characterized by nodules of the cellular infiltrate described above, typically with the “lacunar” variants of Reed–Sternberg cells and surrounded by fibroblast-poor collagen bands. Foci of necrosis may be present. The HRS cells have a “crippled” B-cell phenotype and express weak PAX5, with its stain intensity less than normal B-cells, and CD30, with strong membrane and Golgi region positivity, and the HRS cells usually express CD15 at least focally. The HRS cells are negative for CD45, negative or variably positive for CD20, negative for CD79a, OCT2, and BOB1, and consistently express MUM1. The HRS cells are most often negative for T-cell markers but may show aberrant positivity in rare cases. EBV-infected HRS cells express EBV latent membrane protein 1 (LMP1) and EBNA1 without EBNA2, characteristic of type II EBV latency; EBV positivity is seen in ~40% of cases [12].
PMBCL was first described in 1980 in 17 patients with a mediastinal mass, comprising 9% of 184 DLBCL patients. The PMBCL patients showed rapid onset symptoms due to mass effect and pathologically diffusely infiltrating lymphoma [254]. PMBCL was included in the REAL classification in 1994 as a specific subtype of DLBCL and is now a well-characterized entity [255,256]. The presumed cell of origin is the thymic B-cell, first described in the human thymus in 1987 [257] and subsequently described by immunohistochemistry as B-cells with dendritic processes (asteroid B-cells) [258]. The gene expression profiles [259,260] and the genetic abnormalities in PMBCL are similar to those in CHL [259,260,261,262,263]. PMBCL may occur before, concurrently with, or after the development of nodular sclerosis CHL [251] and can occur in children [264]. There is a female predominance in Western countries, but male predominance (3:1 male-to-female ratio) has also been noted [265,266].
Histologically, in contrast with CHL, PMBCL is characterized by diffusely infiltrating large neoplastic mature B-cells, often separated into clusters by fine fibrosis or sclerosis, often termed “compartmentalizing” fibrosis. The medium- to large-sized lymphoma cells have clear to pale cytoplasm, express CD45, and show strong and uniform expression of B-cell antigens and transcription factors, including CD20, CD79a, PAX5, OCT2, and BOB1. This expression by immunohistochemistry in PMBCL contrasts with the HRS cells, which have downregulated the B-cell program, which, if expressed, show non-uniform and variable intensity and staining of the neoplastic cells in HRS cells. The neoplastic cells in PMBCL frequently express CD30 (weak and heterogeneous positivity) and MUM1 and variably express BCL6 and BCL2. Positivity for CD15 and EBV may be seen in rare cases [12]. The lymphoma cells in PMBCL are positive for CD23, MAL (myelin and lymphocyte protein), CD200, PD-L1, and PD-L2, which, in addition to CD30, can help distinguish from DLBCL NOS [12].
In low-resource settings, careful attention to the staining pattern with only three stains, CD20, CD3, and CD30, in conjunction with morphologic evaluation, has been reported to help distinguish most cases of CHL and PMBCL [265]. However, a minimum of six stains, CD45, CD20, PAX5, CD3, CD30, and CD15, are often helpful to diagnose CHL.
The essential diagnostic criteria for PMBCL by WHO-HAEM5 require a large B-cell lymphoma in the anterior mediastinum and mature B-cell phenotype, accompanied by at least partial expression of CD23 and CD30 in the neoplastic cells. The desirable criteria are distinctive stromal sclerosis, expression of at least one of the markers, MAL, CD200, PD-L1, and PD-L2, and copy number gain or rearrangement of the CD274/PDCD1LG2 locus, rearrangement involving CIITA (C2TA), or both genetic abnormalities, which are recurrently found in PMBCL [12].
Extra-mediastinal DLBCLs without mediastinal involvement have been shown to harbor gene expression profiles similar to those of PMBCL [267]. In addition, copy number gains at the 9p24.1 chromosome locus were shown in 10% of DLBCL cases, including amplification of 9p24.1 with high levels of PD-L1, PD-L2, and JAK2 expression in seven cases without a mediastinal mass, similar to 9p24.1 amplification that may occur in up to 75% of PMBCL [268]. These extra-mediastinal cases are classified as DLBCL NOS by WHO-HAEM5 [12]. PD-L1 inhibitors are emerging as new treatment approaches in relapsed/refractory PMBCL [269]. PMBCL can be familial and has been reported in one family with three siblings having PMBCL and one cousin having extranodal DLBCL [270].
Notably, the diagnosis of MZBL is recommended to be made only after excluding all other possible neoplasms, especially nodular sclerosis CHL. Still, an accurate diagnosis of MZBL is challenging. An expert review of 68 cases diagnosed as MGZL at 15 North American academic centers showed only 26 (38%) confirmed as MGZL. Of note, 64% of the reclassified cases were nodular sclerosis CHL (n = 27, including 10 cases of grade 2 nodular sclerosis), showing that the distinction between CHL and MGZL can be very challenging, and the reproducibility of MGZL diagnosis is low even among expert hematopathologists. The other reclassified cases included lymphocyte predominant Hodgkin lymphoma (n = 4), DLBCL (n = 4), EBV+ DLBCL (n = 3), PMBCL (n = 2), lymphocyte rich CHL (n = 1), and B-cell lymphoma, NOS (n = 1) [271]. The diagnosis of MGZL is restricted to mediastinal cases in WHO-HAEM5 and ICC [12,13], in contrast with WHO 2017, wherein extra-mediastinal cases were also included. Most extra-mediastinal DLBCL cases with features of MZBL are now recommended to be classified as DLBCL NOS [12,13].
The diagnosis of MGZL is based on a mismatch between the histologic features and the immunophenotypic profile of the neoplastic cells [249,250,251,252,253,272,273]. MGZL shows either of the following combinations of histology and immunophenotype: (1) histologic features of nodular sclerosis CHL, but the immunophenotype is that of PMBCL, termed CHL-like MGZL; or (2) the neoplasm shows histologic features of PMBCL, but the immunophenotypic profile resembles that of CHL, termed PMBCL-like MGZL. An intermediate form with features between the above two described combinations, CHL-like and PMBCL-like, has also been described.
In the largest study thus far of 139 MGZL, 17% of patients without mediastinal involvement at diagnosis were older than those with mediastinal tumors, with a median age of 56 vs. 39 years [272]. The 139 gray zone lymphoma cases included 62% (86/139) classified as CHL-like and 38% (53/139) as PMBCL-like. Of note, this study described four groups, zero to three, in the spectrum of gray zone lymphomas and reported transitional areas within the lymphoma biopsies [272]. In a 2020 study from the NCI, the proportions of these three combinations, CHL-like, PMBCL-like, and intermediate, were significantly different, i.e., 25% (5/20) CHL-like, 25% (5/20) PMBCL-like, 5% (1/20) composite CHL and PMBCL, and 45% (9/20) intermediate [273], further indicating the possible challenges in identifying this very rare lymphoma. The only prospective study thus far has shown dose-adjusted etoposide, doxorubicin, and cyclophosphamide with vincristine, prednisone, and rituximab (DA-EPOCH-R) alone to be effective in MGZL. Still, MGZL has an inferior outcome compared to PMBCL [274].
The ICC requires a high density of tumor cells by morphologic evaluation and at least two immunophenotypically strongly positive B-cell markers to diagnose CHL-like MZBL [13]. Notably, CD20 positivity, as the only aberrant marker in nodular sclerosis CHL, was one of the reasons for the misdiagnosis of CHL as MGZL [271]. The essential diagnostic criteria for CHL-like MGZL in WHO-HAEM5 require the following:
- (1)
- A confluent growth of pleomorphic cells within a variably abundant microenvironment and dense fibrotic stroma;
- (2)
- Uniform strong expression of CD20 and uniform strong PAX5 and uniform and strong expression of at least one additional B-cell marker, CD19, CD79a, BOB1, and OCT2;
- (3)
- CD30 positive expression, with varying intensity.
The essential diagnostic criteria for PMBL-like MGZL require (1) monomorphic sheets of medium to large neoplastic cells within a variably dense fibrotic stroma and (2) CD30 strong and uniform positive expression and partial or complete loss of B-cell markers or strong CD15 expression [12]. The desirable WHO-HAEM5 criteria are (1) as the complex histological features are often not reliably identifiable in core needle biopsies, a larger biopsy is required for the diagnosis, and (2) the absence of EBV [12]. Composite or synchronous and sequential or metachronous lymphomas comprised of CHL and PMBCL or DLBCL should not be classified as MGZL [12,251,275]. Molecular genetic and cytogenetic findings in MGZL are not specific to MGZL and confirm the shared biology with CHL and PMBCL [276].
Gene expression profiling of MGZL also showed similarities with CHL and PMBCL, with the tumor microenvironment showing increased macrophages in MGZL compared to CHL and PMBCL [277]. Whole exome sequencing analysis with subsequent targeted sequencing of MGZL cases with a mediastinal mass showed a mutational profile similar to that of CHL and PMBCL, with the most frequent alterations in SOCS1 (45%), B2M (45%), TNFAIP3 (35%), GNA13 (35%), LRRN3 (32%), and NFKBIA (29%) [278]. In contrast, TP53, BCL2, and CREBBP mutations were found in grey zone lymphomas without a mediastinal mass [278]. Further, gene expression profiling and mutational analysis of EBV+ DLBCL with grey zone lymphoma-like morphology identified this EBV+ DLBCL entity to be distinct from MGZL, indicating that these cases should be classified as EBV+ DLBCL [13,277,278].
3.11. Immunohistochemical Features of Hodgkin Lymphomas, Peripheral T-Cell Lymphomas, and Other Lymphomas That May Be in the Differential Diagnosis
Immunohistochemistry is required to diagnose classic Hodgkin lymphoma, peripheral T-cell lymphomas, and other lymphomas in the differential diagnosis. Table 10 shows the results of a panel of antigens detected by immunohistochemistry on the neoplastic cells in these lymphomas. In CHL, the neoplastic cells are the Reed–Sternberg (RS) cells and variants. In nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), the neoplastic cells are lymphohistiocytic (L&H) cells or lymphocyte predominant (LP) cells, also called “popcorn cells.” In interpreting the IHC stains, it is essential to correlate the results with the clinical history and the histologic features in hematoxylin and eosin (H&E)-stained tissue sections.
Table 10.
Immunohistochemical features of Hodgkin lymphomas, peripheral T-cell lymphomas, and other lymphomas in the differential diagnosis.
When interpreting stains, the intensity and the pattern of staining on the neoplastic cells must be noted in reaching a definitive diagnosis. For example, in CHL, CD20, if positive, variably stains the scattered RS cells in a heterogeneous manner; this staining is variable in the number of CD20+ RS cells and in the intensity of staining, and this variability in CD20 staining is characteristic of CHL. The nodular pattern in the lymphocyte-rich type of CHL can resemble NLPHL on histologic sections; immunohistochemistry is very helpful to diagnose this type of CHL, in conjunction with morphologic features [280].
4. Mature T-Cell and NK-Cell Neoplasms
Mature T- and Natural Killer (NK)-cell neoplasms are derived from mature, post-thymic T-cells and NK-cells and comprise a diverse group of rare neoplasms with a wide range of clinical behavior, including highly aggressive lymphomas. The specific types of these neoplasms in WHO 2001, 2008, and 2017 classifications were shown in Table 4 in Part 1 [2]. Mature T-cell and NK-cell neoplasms classified by WHO 2008 comprised 5–9% of all mature non-Hodgkin lymphomas, including ~4% peripheral T-cell lymphomas and ~1% mycosis fungoides [9]. Table 11 shows the new WHO-HAEM5 category named T-cell and NK-cell lymphoid proliferations and lymphomas, which includes benign tumor-like lesions with T-cell predominance, precursor T-cell neoplasms (not shown in this table), and the mature T-cell and NK-cell neoplasms, compared with mature T-cell and NK-cell neoplasms in the ICC [11,12,13]. The mature T- and NK-cell neoplasms are classified based on various variables, including the primary site of presentation in the peripheral blood and bone marrow (leukemic), skin (cutaneous), gastrointestinal tract (intestinal), and the liver and spleen (hepatosplenic)—all extranodal sites, as shown in Table 11. Of note, there is a marked geographic variation in these neoplasms, and the occurrence and frequency of specific neoplasms are dependent on the geographic regions [281].
Table 11.
T- and NK-cell neoplasms in the fifth edition WHO 2022 and ICC [11,12,13].
4.1. Name Changes in WHO-HAEM5 and ICC Compared with the WHO 2017 Classification
Table 12 shows the name changes in WHO-HAEM5 and the ICC compared with the WHO 2017 classification. Mature T-cell lymphomas involving lymph nodes are clinically aggressive neoplasms and included angioimmunoblastic T-cell lymphoma (AITL) and anaplastic large-cell lymphoma (ALCL) as specific entities in WHO 2001, with peripheral T-cell lymphoma (PTCL), unspecified or not otherwise specified (PTCL NOS), as the remaining heterogeneous group of nodal mature T-cell lymphomas. In a large international study, PTCL NOS and AITL were the two most frequent mature T-cell neoplasms [281]. This section will briefly describe nodal T-follicular helper cell lymphomas, ALCL, EBV+ nodal T- and NK-cell lymphoma, and PTCL NOS, which remains a diagnosis of exclusion. ALCL consists of systemic (ALK+ and ALK-negative), breast-implant-associated, and cutaneous ALCL; cutaneous ALCL is not described as it is best described with cutaneous lymphoproliferative disorders.
Table 12.
Name changes in WHO-HAEM5 and ICC compared with the revised fourth edition for mature T-cell and NK-cell lymphomas and Hodgkin lymphomas.
4.2. Nodal T-Follicular Helper (TFH) Cell Lymphoma
Nodal follicular T-cell lymphoma is the new WHO-HAEM5 name unifying three entities of nodal T-cell lymphoma with the phenotype and gene expression signatures of T-follicular helper (TFH) cells [11,12,282]. AITL was included as a specific entity in the REAL classification in 1994 and comprised the most frequent (18.5%) specific type in a cohort of 1153 T- and NK-cell neoplasms [281]. The other two entities in this group, follicular type and not otherwise specified, were included provisionally in WHO 2017 after a subset of PTCL NOS cases showed TFH-associated markers similar to AITL [283,284]. While the features of AITL are well-defined, including variable occurrence in different geographic locations [285,286], the other two entities have not yet been studied as much as AITL, and there are overlapping morphologic, clinical, and genetic features between these lymphomas [287,288].
Notably, there are treatment implications for the nodal T-follicular helper cell lymphomas group. These lymphomas frequently harbor mutations in ten-eleven translocation-2 (TET2), DNA methyl transferase-3A (DNMT3A), and isocitrate dehydrogenase-2 (IDH2), which affect DNA methylation and silence tumor suppressor genes; therefore, effective treatment options include epigenetic modifying and hypomethylating agents [289,290,291].
Nodal follicular T-cell lymphoma, angioimmunoblastic type, abbreviated AITL for simplicity, is diagnosed by integrating clinical, laboratory, characteristic histopathologic, and immunophenotypic findings. Histologically, three patterns have been described with partial or complete obliteration of nodal architecture, expanded CD21+ (or CD23+) follicular dendritic cell (FDC) meshworks, prominent high endothelial venules (HEVs), and small to medium-sized TFH marker-positive lymphoma cells with clear cytoplasm admixed with polymorphous inflammatory cells; a tumor-cell rich pattern may lack HEVs [12,292,293]. The lymphoma cells express PD1 (CD279), ICOS, BCL6, CXCL13, and CD10 at varying levels of sensitivity and sensitivity. PD1 and ICOS are more sensitive than CD10 and CXCL13, while CD10 and CXCL13 are more specific than PD1 and ICOS. Strong PD1 staining is more specific [12]. AITL frequently harbors mutations in Ras Homolog Family Member A, RHOA (p.G17V), TET2, IDH2 (p.R172), DNMT3A, VAV1, and PLCG1 [12,288,294,295,296]. Mutations in RHOA and IDH2 appear to be specific to the TFH lymphomas and are absent in PTCL NOS [288,296]. RHOA p.G17V mutation analysis may be valuable in the early detection of AITL and PTCL with TFH features [297]. IDH2 p.R172-mutated AITL shows specific features, including medium- to large-sized neoplastic cells with clear cytoplasm and a TFH phenotype with CD10 and CXCL3 immunohistochemical positivity [298,299].
The essential WHO-HAEM5 diagnostic criteria for the diagnosis of typical patterns 2, 3, and tumor-rich AITL require the following:
- (1)
- Nodal disease,
- (2)
- CD4+, occasionally CD4-negative, CD8-negative atypical lymphoid cells,
- (3)
- Extrafollicular FDC expansion, and
- (4)
- HEV hyperplasia, which is mild in tumor-cell-rich cases.
The desirable criteria are expression of >2 TFH markers, including strong PD1, clonal T-cell receptor (TCR) gene rearrangement, mutation involving RHOA p.G17V (NP_001655.1) or IDH2 p.R172, or both clonally rearranged TCR and the mutations, and EBV+ B-cells [12].
The essential WHO-HAEM5 diagnostic criteria to diagnose partial nodal involvement (patterns 1–2) require (1) nodal disease, (2) perifollicular CD4+, occasionally CD4-negative, and CD8-negative atypical T-cells that express >2 TFH markers, including strong PD1, and (3) clonal TCR gene rearrangement, mutation involving RHOA p.G17V or IDH2 p.R172, or both clonally rearranged TCR and the mutations. If the diagnostic criteria are not fulfilled due to insufficient sampling, re-biopsy is recommended [12].
Nodal TFH cell lymphoma, follicular-type, may represent a morphologic pattern of the broader group of TFH lymphomas instead of a distinct entity [12,288]. The essential WHO-HAEM5 diagnostic criteria require a follicular growth pattern (follicular lymphoma-like or progressive transformation of germinal center (PTGC)-like), no (absent) extrafollicular FDC expansion, and CD4+, occasionally CD4-negative, and CD8-negative atypical T-cells that express >2 TFH markers, including strong PD1. The desirable WHO-HAEM5 criteria are lack of a polymorphous infiltrate, HEV hyperplasia, and clonal TCR gene rearrangement [12].
Nodal TFH lymphoma, NOS, shows overlap with the tumor-rich pattern of AITL but lacks a prominent polymorphous inflammatory background, HEV hyperplasia, and extrafollicular FDC expansion. The less TFH-specific markers, PD1 and ICOS, are more often positive in these cases. The essential WHO-HAEM5 diagnostic criteria for nodal TFH lymphoma NOS require (1) nodal disease with effaced architecture/T-zone pattern by a morphologically atypical, immunophenotypically aberrant atypical T-cell infiltrate that is CD4+ and CD8-negative and expresses at least two TFH markers, including strong PD1, or both morphologically atypical and immunophenotypically aberrant T-cell infiltrate; and (2) lack of extrafollicular FDC hyperplasia, perifollicular distribution of neoplastic T-cells, and follicular growth pattern. The desirable criteria are clonal TCR gene rearrangement, mutation involving RHOA p.G17V, or both [12].
The T-follicular helper (TFH) phenotype was defined by WHO 2017 by the expression of at least two or preferably three TFH markers. The ICC recommends a five-marker immunohistochemistry panel to identify the TFH phenotype since establishing a TFH phenotype is critical to diagnose the follicular and NOS types of TFH lymphomas [13]. An immunohistochemistry panel of five TFH markers, CD10, BCL6, PD1, CXCL13, and ICOS, reclassified 41% of PTCL NOS cases to PTCL with TFH phenotype [300].
4.3. Anaplastic Large-Cell Lymphoma
The monoclonal antibody, Ki-1, was found to be specific for HRS cells in all examined cases of Hodgkin lymphoma in 1982 [301]. Subsequently, ALCL was first described in 1985 after applying Ki-1 (CD30) to a series of non-neoplastic and lymphoma tissues. Ki-1 was strongly expressed in 45 cases of diffuse large-cell lymphoma previously diagnosed as malignant histiocytosis or carcinoma due to marked pleomorphism in tumor cells; 35 of those cases expressed T-cell antigens, including 9 with co-expressed B-cell antigens, 7 expressing only B-cell-related antigens, and three lacking T- and B-cell markers [301]. Ki-1 was also expressed in variable numbers of cells in all cases of lymphomatoid papulosis and angioimmunoblastic lymphadenopathy, later termed AILT, and in 28% of cases of PTCL. The neoplastic cells in these lymphoid neoplasms resembled HRS cells in CHL and consistently expressed lymphoid markers in the absence of markers of other lineages [302]. The nature of the neoplastic cells as lymphoid was further confirmed by clonal rearrangements of antigen receptor genes [302,303,304]. These lymphomas were then termed Ki-1-positive ALCL.
In the late 1980s, the t(2;5)(p23;q35) translocation was described in a subset of ALCLs. Subsequently, in 1994, the nucleolar phosphoprotein NPM gene located at 5q35 was shown to fuse with the previously unidentified gene at 2p23 that encodes for the receptor tyrosine kinase anaplastic lymphoma kinase (ALK) [305]. The resulting chimeric NPM::ALK gene, formed in the t(2;5) translocation due to the ALK gene now under the control of the NPM promoter, is transcribed to produce an 80-kd chimeric protein causing constitutive activation of ALK [305], first detected by the ALK monoclonal antibody in 1997 [306]. Variant ALK translocations were recognized in ~15% of ALCL with only cytoplasmic ALK staining, occasionally with granular positivity, compared to nuclear and cytoplasmic staining in NPM::ALK translocations [305,306,307,308,309].
ALCL was introduced in the REAL classification as anaplastic large-cell lymphoma, CD30+, T-, and null-cell types. The WHO 2008 classification recognized ALK+ ALCL as a distinct entity and provisionally included ALK-negative ALCL, which was subsequently included as a separate entity by WHO 2017. In 2022, DUSP22-rearranged ALCL was recognized as a specific genetic type of ALK-negative ALCL by WHO-HAEM5 and ICC.
The chimeric ALK protein is present only in neoplastic cells. Of note, the presence of the chimeric ALK protein does not indicate that the neoplastic cell is lymphoid since ALK gene abnormalities also occur in many other non-lymphoid neoplasms. WHO-HAEM5 defines ALK-positive ALCL as a CD30+ mature T-cell lymphoma with aberrant expression of the ALK protein secondary to ALK gene rearrangements. The essential WHO-HAEM5 diagnostic criteria require ALK expression in lymphoma cells and a strong uniform expression of CD30 in lymphoma cells [12]. A characteristic sinusoidal pattern of nodal involvement by lymphoma cells is often present, as illustrated in reference [279].
The essential WHO-HAEM5 diagnostic criteria require the following:
- (1)
- Complete or partial infiltration of lymph node or extranodal tissue by large pleomorphic cells with lobated nuclei, distinct nucleoli, including “hallmark cells,”
- (2)
- Uniform strong expression of CD30,
- (3)
- Absence of ALK protein expression or ALK rearrangement, and
- (4)
- Negativity for EBV. The desirable criteria include expression of T-cell markers and cytotoxic markers, albeit with frequent losses, and clonal rearrangement of the TCR gene [12].
ALK-negative ALCL has a worse prognosis than ALK+ ALCL when treated by conventional chemotherapy, but it is better than that of PTCL NOS [310]. In 2014, ALK-negative ALCL was shown to be a genetically heterogeneous disease with varying outcomes after standard therapy. In that study, DUSP22-rearranged and TP53-rearranged types of ALK-negative ALCL comprised 30% and 8% of all ALK-negative ALCL, respectively [311]. Of note, the 5-year overall survival rates in that study were 90% for DUSP22-rearranged ALCL, 85% for ALK+ ALCL, 17% for TP63-rearranged ALCL, and 42% for ALCL cases lacking all three genetic markers (p < 0.0001) [311]. Although all ALCL share similar histologic growth patterns, DUSP22-rearranged ALCL is more likely to show doughnut-shaped cells, less likely to show pleomorphic cells, and very likely to show a sheet-like growth pattern [312].
Significantly, the genome of ALK+ ALCL is less complex than that of ALK-negative ALCL, which indicates the strong potential of the ALK chimeric fusion to cause lymphoma [279,313]. Indeed, in January 2021, the ALK inhibitor, crizotinib, was FDA-approved for pediatric patients aged ≥1 year and young adults with relapsed or refractory systemic ALK+ ALCL, which could potentially improve clinical outcomes [314].
Notably, gene expression profiling showed a similar signature of 14 genes expressed by ALK+ ALCL and ALK- ALCL, indicating a shared ALCL profile that also distinguished ALK-negative ALCL from PTCL NOS [315]. Subsequently, gene expression profiling was able to separate ALK+ ALCL from ALK-negative ALCL [316,317]. Interestingly, ALK+ ALCL were enriched for the expression of hypoxia-inducible factor 1 alpha (HIF1α) target genes, IL10-induced genes, and H-ras/K-ras-induced genes compared with ALK-negative ALCL [317]. ALK-negative ALCL expressed TNSFR8 (CD30), BATF3, and TMOD1 [316,317], and compared to PTCL NOS, ALK-negative ALCL were enriched for the expression of MYC, IRF4, and proliferation and mTOR pathway genes [317]. Clinically, the anti-CD30 antibody-drug conjugate, brentuximab vedotin, is effective as a single agent in treating relapsed or refractory systemic ALCL [318] and is being studied to treat patients with PTCL NOS [319].
Breast-implant-associated ALCL is a mature CD30+ T-cell lymphoma that arises in relation to a breast implant and is usually confined by a fibrous capsule [12]. It usually presents as an effusion-associated fibrous capsule surrounding the implant and, less frequently, as a mass [320]. Treatment with capsulectomy and implant removal results in remission in most patients with a fibrous capsule-limited disease. In contrast, the disease may be highly aggressive and even fatal in patients presenting with a mass [320]. Tumor spread beyond the capsule is associated with a higher risk of lymph node involvement, which is associated with decreased overall survival [321]. The essential WHO-HAEM5 diagnostic criteria include (1) the presence of breast implant, (2) CD30+ lymphoma cells with anaplastic features, and (3) proven T-cell lineage supported by expression of one or more T-lineage markers, clonal TCR gene rearrangement, or both. The desirable WHO-HAEM5 criterion is the identification of lymphoma cells on the luminal side of the capsule in correctly oriented sections [12]. Clonal TCR rearrangement is present in >80% of cases, and breast-implant-associated ALCL is consistently negative for gene rearrangements involving ALK (2p23), DUSP22 (6p25.3), and TP63 (3q28), all of which can be detected by FISH [12]. The differential diagnosis of breast-implant-associated ALCL includes systemic lymphomas involving the breast, with DLBCL and extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue being the most common [322]. Rare cases of EBV+ fibrin-associated large B-cell lymphomas described around breast implants must also be excluded [12,323,324].
4.4. EBV+ Nodal T- and NK-Cell Lymphoma
EBV+ nodal T- and NK-cell lymphoma was included as a variant of PTCL in WHO 2017 and is now included as a definite entity in WHO-HAEM5 and provisionally in ICC [11,12,13]. It is a rare lymphoma of EBV+ cytotoxic T- or NK-cells that occurs mainly in East Asia and typically presents with lymphadenopathy at an advanced stage in adults. Bone marrow and the liver may be involved. The lymphoma is composed of EBV+ monomorphous large centroblastoid cells with a cytotoxic phenotype, with T-cell more common than NK-cell lineage. This lymphoma is a systemic disease with an aggressive clinical course and a CD8+, CD56-negative immunophenotype distinct from extranodal NK/T-cell lymphoma and PTCL NOS [325]. In contrast with extranodal NK/T-cell lymphoma, there is a lack of nasopharyngeal involvement, lack of prominent angiocentric growth and necrosis, and less frequent extranodal involvement. The outcome is worse than NK/T-cell lymphoma and PTCL, with a median overall survival of 2.8 to 8 months compared with 26–76 months for extranodal NK-/T-cell lymphoma and 16–20 months for PTCL NOS [12,325].
The essential WHO-HAEM5 criteria require (1) a cytotoxic T-cell or NK-cell lymphoma, (2) EBV-encoded RNA (EBER) present in the majority of neoplastic cells, (3) tumor primarily localized within lymph nodes but may involve a limited number of extranodal sites; no nasal involvement, and (4) exclusion of immune-deficiency-associated T- and NK-cell lymphoproliferative diseases, extranodal NK/T-cell lymphoma, systemic EBV+ lymphoproliferative diseases of childhood, and aggressive NK-cell leukemia with progression or secondary involvement of lymph nodes [12].
4.5. Peripheral T-Cell Lymphoma, Not Otherwise Specified (PTCL NOS)
PTCL NOS is a mature T-cell lymphoma that cannot be assigned to any specific mature T-cell lymphoma entity. Therefore, it is a diagnosis of exclusion and shows heterogeneity in pathologic and genomic features. Clinically, it affects primarily adults and has an aggressive clinical course [281].
Two molecular groups were identified by gene expression profiling in 2014 before the non-AITL nodal TFH lymphomas were included in the WHO 2017 classification. These two groups were characterized by high expression of either GATA3 (33%; 40/121) or TBX21 (49%; 59/121); the remaining 22 PTCL NOS cases were unclassified [317]. GATA3 and TBX21 are master transcriptional regulators of T-helper 2 and T-helper 1/cytotoxic T-cell differentiation. The GATA3 subgroup showed marginal enrichment for mTOR- and MYC-related gene signatures, also seen in ALK-negative ALCL in this same study [317], and significant enrichment of phosphatidylinositol 3 kinase (PI3K)-induced gene signatures [317]. The TBX21 subgroup showed significant enrichment of IFN α/β/γ-regulated gene signatures, a CD8+ T-cell profile, and NFκB pathway signatures. In the TBX21 subgroup, a subset with cytotoxic profiles showed a poor prognosis [317]. The GATA3 subgroup was significantly associated with poor overall survival. In addition, high expression of cytotoxic gene signature within the TBX21 group showed poor clinical outcomes [317].
The study in 2019 [326] also showed the two molecular subgroups, with the GATA3 subgroup characterized by greater genomic complexity and frequent loss or mutation of tumor suppressor genes targeting the CDKN2A/B-TP53 axis and PTEN-PI3K pathways. As would be expected, loss of CDKN2A correlated with poor prognosis, which was seen in PTCL NOS and the GATA3 subgroup [326]. The GATA3 subgroup showed co-occurring gains or amplifications of STAT3 and MYC. Both subgroups showed copy number aberrations affecting metabolic processes regulating RNA/protein degradation and T-cell receptor signaling [326].
TP53 mutations co-occurred with the loss of CDNK2A and were present in the DNA binding and tetramerization domains, including the hotspot TP53 p.R175H. The presence of TP53 mutations and copy number loss leading to aberrant TP53 signaling was significantly associated with the GATA3 subgroup of PTCL [326]. Both subgroups harbored mutations in other DNA repair or TP53 signaling genes, ATM and TRRAP [326]. As mentioned earlier, PTCL NOS are usually negative for RHOA p.G17V and IDH2 p.R172 mutations, which are often present in nodal TFH cell lymphomas. An immunohistochemical algorithm using antibodies against the transcriptional factors, GATA3, TBX21, and their target proteins, CCR4 and CXCR3, respectively, has been shown to correlate with the two molecular subgroups [12,327].
The histologic and cytologic features of PTCL NOS are highly variable. There is partial or complete effacement of the nodal architecture by a paracortical or diffuse infiltrate of medium- to large-sized lymphoma cells, often with an inflammatory background. HRS-like cells may be present. The inflammatory background is much more frequent in the TBX21 subtype than in the GATA3 subtype [12,327]. The lymphoma cells express pan T-cell antigens and are more often CD4+ than CD8+ and frequently show decreased expression of CD5 and CD7. The expression of a single TFH marker is acceptable for the diagnosis of PTCL NOS. However, cases with diffuse positivity for two or more TFH markers should be diagnosed as nodal TFH cell lymphoma. Scattered Epstein–Barr virus-encoded RNA (EBER)-positive B-cells may be present. CD30 is expressed in 25% of cases, but the positivity is non-uniform and variable, not strong like in ALCL. CD15 may be positive in ~5% of cases and may be co-expressed with CD30 [12]. The differential diagnosis of PTCL NOS may include non-neoplastic conditions such as Kikuchi’s lymphadenitis and other lymphomas, including CHL, T-cell rich large B-cell lymphoma, and nodal marginal zone lymphoma. Careful integration of the clinical, histopathologic, and immunophenotypic features usually resolves the diagnosis of lymphoma versus non-neoplastic states. In difficult cases or specific diagnostic situations, the molecular genetic analysis would be helpful for precise diagnosis.
The essential WHO-HAEM5 criteria for a diagnosis of PTCL NOS require the following:
- (1)
- The presence of an abnormal T-cell infiltrate, which is morphologically or immunophenotypically aberrant, monoclonal by ancillary studies, or both;
- (2)
- The tumor cells are negative or express only one TFH marker (to differentiate from nodal TFH cell lymphomas), and the neoplasm only shows EBER positivity in scattered B-cells (to differentiate from EBV+ nodal T- and NK-cell lymphoma);
- (3)
- The exclusion of other nodal or extranodal mature T- and NK-cell lymphomas, i.e., ALK+ ALCL, ALK-negative ALCL, adult T-cell leukemia/lymphoma, extranodal NK/T-cell lymphoma.
The desirable WHO-HAEM5 criteria are the presence of clonal TCR gene rearrangements and establishing the biological designation of PTCL-TBX21 and PTCL-GATA3 [12].
5. Genetic Predisposition and Constitutional Inherited Syndromes
Genetic predisposition to lymphoid neoplasms, including ALL and lymphomas, occurs in several constitutional inherited cancer predisposition syndromes and as non-syndromic germline predisposition. These constitutional syndromes include Li–Fraumeni syndrome, constitutional mismatch repair deficiency syndrome, Bloom syndrome, Werner syndrome, ataxia telangiectasia syndrome, Nijmegen breakage syndrome, the RASopathies (including juvenile myelomonocytic leukemia), and Down syndrome (trisomy 21). The interested reader is referred to a recent review, including these inherited syndromes in the context of hematologic and lymphoid neoplasm; the brief content in this section (#5) is adapted from that review [17].
An exome sequencing study of 91 patients with lymphoma in the USA showed germline variants in 8% (7/91), including in DLBCL (n = 2), follicular lymphoma (n = 2), Hodgkin lymphoma (n = 1), cutaneous T-cell lymphoma (n = 1), and cutaneous T-cell lymphoma and cutaneous B-cell lymphoma (n = 1) [328]. Of note, more than one-third of somatically mutated genes in DLBCL have been shown to harbor germline mutations in DLBCL [329]. These authors reported 49 genes with germline variants in patients with DLBCL, 35 of which were involved in DNA repair [329]. Germline mutations were also reported in 12 genes related to immunodeficiency, including LIG4 (DNA ligase IV), NHEJ1 (XLF), DCLRE1C (Artemis), NBN (Nibrin), WAS, PIK3CD, PIK3R1, SH2D1A (SAP), IFNGR1, STAT3, perforin-encoding PRF1, and FAS [329].
A significant risk association of non-Hodgkin lymphomas was reported with single nucleotide polymorphisms at the ATM locus [330]. In 2022, in a Japanese study of 2066 lymphoma patients and 38,153 cancer-free controls, samples from 1982 lymphoma patients and 37,592 controls were further analyzed for 27 cancer-predisposing genes. Pathogenic variants were identified in ATM, BRCA1, BRCA2, and TP53 in 1.6% of lymphoma patients, and these variants were associated with a higher risk of MCL [331]. For a systematic review of the genetic predisposition to developing lymphomas in primary immunodeficiencies and immune dysregulation disorders, refer to cited reference [332].
6. Conclusions
Accurate diagnosis and precise classification critically impact the treatment of patients with lymphoid neoplasms. This goal has always been achieved by the careful, collaborative work of countless pathologists, clinicians, geneticists, and researchers, all of whom have entirely changed the landscape of lymphoid neoplasms in the last thirty years. Morphologic evaluation has been crucial in identifying new and specific lymphoma entities. In the last decade, advances in genomics and technology applied to lymphoblastic leukemias and lymphomas have transformed our understanding of the underlying biology of these neoplasms in children and adults, which is now reflected in the current diagnostic classifications. The fifth edition WHO classification of hematolymphoid neoplasms, which follows the work of countless individuals for all of the previous classifications briefly described in this review, is a truly remarkable effort by numerous individuals worldwide and will most likely serve as a treasure trove of information for any practicing pathologist serving patients with hematolymphoid neoplasms globally.
Precise diagnostic classification established by including molecular genetic tests in the diagnostic workup is now critical for ALL. The role of morphologic evaluation in ALL is mainly limited to visually identifying leukemic cells or blasts. In contrast, morphologic evaluation of a tissue biopsy in conjunction with immunophenotypic and molecular genetic analysis currently remains the cornerstone of lymphoma diagnosis. Several aspects deserving further research are discussed in Part 1 [2] and the sections devoted to the specific types of lymphoid neoplasms in this review. With a continued and increasingly more robust understanding of lymphoid neoplasms, liquid biopsies, which can identify molecular abnormalities that might be impossible to detect in a focal tissue biopsy of a heterogeneous neoplasm, are expected to have more significant clinical utility in patients with lymphoid neoplasms. Germline mutations need to be studied further in lymphoid neoplasms. The rapid shift to precisely dissect the biology of lymphoid neoplasms needs to be translated to clinical patient care in the community, resource-poor regions and institutions in developed countries, and low- and middle-income countries for better patient outcomes worldwide.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| ABC | activated B-cell |
| AESOP | adenopathy and extensive skin patch overlying a plasmacytoma |
| AID | activation-induced cytidine deaminase |
| AIDS | acquired immune deficiency syndrome |
| AITL | angioimmunoblastic T-cell lymphoma |
| ALCL | anaplastic large-cell lymphoma |
| ALK | anaplastic lymphoma kinase |
| ALL | acute lymphoblastic leukemia |
| ASR | age-standardized rate |
| AYA | adolescents and young adults |
| BL | Burkitt lymphoma |
| B-PLL | B-cell prolymphocytic leukemia |
| BTK | Bruton’s tyrosine kinase |
| CDR3 | complementarity-determining region 3 |
| CHL | classic Hodgkin lymphoma |
| CHOP | cyclophosphamide, doxorubicin, vincristine, and prednisolone |
| CLL | chronic lymphoid leukemia |
| CNS | central nervous system |
| DLBCL | diffuse large B cell lymphoma |
| DLBCL NOS | diffuse large B-cell lymphoma, not otherwise specified |
| EBV | Epstein–Barr virus |
| FCI | flow cytometric immunophenotyping |
| FDA | Food and Drug Administration |
| FDC | Follicular dendritic cells |
| FISH | Fluorescence in situ hybridization |
| FL | follicular lymphoma |
| FOXO1 | forkhead box O1 |
| GC | germinal center |
| GCB | germinal center B-cell |
| GI | gastrointestinal |
| GLOBOCAN | Global Cancer Observatory |
| GNA13 | G protein subunit alpha 13 |
| H&E | Hematoxylin and eosin |
| HCL | hairy cell leukemia |
| HEVs | high endothelial venules |
| HGBCL | high grade B-cell lymphoma |
| HGBCL-11q | high-grade B-cell lymphoma with chromosome 11q aberration |
| HL | Hodgkin lymphoma |
| HMRN | Hematologic Malignancy Research Network |
| HRS | Hodgkin or Reed–Sternberg |
| ICC | International Consensus Classification |
| IDD | immune deficiency or dysregulation |
| IHC | immunohistochemical |
| ILL | intermediate lymphocytic lymphoma |
| IRAK | IL-1R-associated kinase |
| ISMCN | in situ mantle cell neoplasia (or in situ mantle cell neoplasm) |
| IVBCL | intravascular large B-cell lymphoma |
| L&H | lymphohistiocytic |
| LBCL-IRF4 | large B-cell lymphoma with an IRF4 rearrangement |
| LMO2 | LIM-domain only 2 |
| LMP1 | latent membrane protein 1 |
| LOH | loss of heterozygosity |
| LPD | lymphoproliferative disorder |
| LPL | lymphoplasmacytic lymphoma |
| MALT | mucosa-associated lymphoid tissue |
| MBL | monoclonal B-cell lymphocytosis |
| MCL | mantle cell lymphoma |
| MGUS | monoclonal gammopathy of undetermined significance |
| MGZL | mediastinal gray (or grey) zone lymphoma |
| MZ | mantle zone |
| MZL | marginal zone lymphoma |
| NGS | next-generation sequencing |
| NFκB | nuclear factor kappa B |
| NHL | non-Hodgkin lymphoma |
| NK | Natural Killer |
| NLPHL | nodular lymphocyte predominant Hodgkin lymphoma |
| NOS | not otherwise specified |
| PCNS-LBCL | Primary large B-cell lymphoma of the CNS |
| PMBCL | primary mediastinal large B-cell lymphoma |
| POEMS | polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes |
| PTCL | peripheral T-cell lymphoma |
| PTCL NOS | peripheral T-cell lymphoma, not otherwise specified |
| PTFL | pediatric-type follicular lymphoma |
| PTLD | post-transplant lymphoproliferative disorder |
| PVR-LBCL | primary vitreoretinal large B-cell lymphoma |
| REAL | Revised European American Lymphoma Classification |
| SEER | Surveillance, Epidemiology, and End Results |
| SLL | small lymphocytic lymphoma |
| SMZL | splenic marginal zone lymphoma |
| SOX11 | sex-determining region Y-box transcription factor 11 |
| TEMPI | telangiectasias, erythrocytosis with elevated erythropoietin, monoclonal gammopathy, perinephric fluid collections, intrapulmonary shunting |
| TFH | T-follicular helper |
| WHO | World Health Organization |
| WHO-HAEM5 | Fifth edition of WHO classification for hematolymphoid tumors |
| WM | Waldenström macroglobulinemia |
References
- The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood 1997, 89, 3909–3918. [Google Scholar] [CrossRef]
- Kansal, R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 1: Lymphoid Neoplasms. Lymphatics 2023, 1, 55–76. [Google Scholar] [CrossRef]
- Kansal, R. Diagnosis and Molecular Pathology of Lymphoblastic Leukemias and Lymphomas in the Era of Genomics and Precision Medicine: Historical Evolution and Current Concepts—Part 2: B-/T-Cell Acute Lymphoblastic Leukemias. Lymphatics 2023, 1, 118–154. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [CrossRef]
- Blum, K.A.; Keller, F.G.; Castellino, S.; Phan, A.; Flowers, C.R. Incidence and outcomes of lymphoid malignancies in adolescent and young adult patients in the United States. Br. J. Haematol. 2018, 183, 385–399. [Google Scholar] [CrossRef]
- The United States National Cancer Institute Surveillance, Epidemiology, and End Results Program Database. Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/ (accessed on 25 June 2023).
- Al-Hamadani, M.; Habermann, T.M.; Cerhan, J.R.; Macon, W.R.; Maurer, M.J.; Go, R.S. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: A longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am. J. Hematol. 2015, 90, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef]
- The United States National Cancer Institute Surveillance, Epidemiology, and End Results Program Database. Browse the SEER Cancer Statistics Review (CSR) 1975–2011. Table 19.29. All Lymphoid Neoplasms with Detailed Non-Hodgkin Lymphoma Subtypes. Available online: https://seer.cancer.gov/archive/csr/1975_2011/browse_csr.php?sectionSEL=19&pageSEL=sect_19_table.29.html (accessed on 23 June 2023).
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Hematolymphoid Tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2022. (WHO Classification of Tumours Series, 5th ed.; vol. 11). Available online: https://https://tumourclassification.iarc.who.int/home (accessed on 16 May 2023).
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- SEER*Explorer: An Interactive Website for SEER Cancer Statistics [Internet]. Surveillance Research Program, National Cancer Institute; 19 April 2023. Data Source(s): SEER Incidence Data, November 2022 Submission (1975–2020), SEER 22 Registries. Available online: https://seer.cancer.gov/statistics-network/explorer/ (accessed on 14 May 2023).
- American Cancer Society. About Non-Hodgkin Lymphoma in Children. Available online: https://www.cancer.org/cancer/types/childhood-non-hodgkin-lymphoma/about.html (accessed on 14 May 2023).
- National Cancer Institute. Cancer in Children and Adolescents. Available online: https://www.cancer.gov/types/childhood-cancers/child-adolescent-cancers-fact-sheet#r2 (accessed on 14 May 2023).
- Kansal, R. Germline predisposition in hematologic malignancies. In Comprehensive Hematology and Stem Cell Research; Rezaei, N., Ed.; Elsevier: Amsterdam, The Netherlands, 2023; submitted. [Google Scholar]
- Minard-Colin, V.; Brugières, L.; Reiter, A.; Cairo, M.S.; Gross, T.G.; Woessmann, W.; Burkhardt, B.; Sandlund, J.T.; Williams, D.; Pillon, M.; et al. Non-Hodgkin Lymphoma in Children and Adolescents: Progress Through Effective Collaboration, Current Knowledge, and Challenges Ahead. J. Clin. Oncol. 2015, 33, 2963–2974. [Google Scholar] [CrossRef] [PubMed]
- Sandlund, J.T.; Martin, M.G. Non-Hodgkin lymphoma across the pediatric and adolescent and young adult age spectrum. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.J.; Długosz-Danecka, M.; Connors, J.M.; Alekseev, S.; Illés, Á.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; et al. Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. Lancet Haematol. 2021, 8, e410–e421. [Google Scholar] [CrossRef]
- Castellino, S.M.; Pei, Q.; Parsons, S.K.; Hodgson, D.; McCarten, K.; Horton, T.; Cho, S.; Wu, Y.; Punnett, A.; Dave, H.; et al. Brentuximab Vedotin with Chemotherapy in Pediatric High-Risk Hodgkin’s Lymphoma. N. Engl. J. Med. 2022, 387, 1649–1660. [Google Scholar] [CrossRef]
- Conley, M.E.; Dobbs, A.K.; Farmer, D.M.; Kilic, S.; Paris, K.; Grigoriadou, S.; Coustan-Smith, E.; Howard, V.; Campana, D. Primary B cell immunodeficiencies: Comparisons and contrasts. Annu. Rev. Immunol. 2009, 27, 199–227. [Google Scholar] [CrossRef]
- Smith, C.I.; Baskin, B.; Humire-Greiff, P.; Zhou, J.N.; Olsson, P.G.; Maniar, H.S.; Kjellén, P.; Lambris, J.D.; Christensson, B.; Hammarström, L.; et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J. Immunol. 1994, 152, 557–565. [Google Scholar] [CrossRef]
- Neys, S.F.H.; Rip, J.; Hendriks, R.W.; Corneth, O.B.J. Bruton’s Tyrosine Kinase Inhibition as an Emerging Therapy in Systemic Autoimmune Disease. Drugs 2021, 81, 1605–1626. [Google Scholar] [CrossRef]
- Baecklund, E.; Smedby, K.E.; Sutton, L.A.; Askling, J.; Rosenquist, R. Lymphoma development in patients with autoimmune and inflammatory disorders--what are the driving forces? Semin. Cancer Biol. 2014, 24, 61–70. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Shabani, M.; Tabary, M.; Rayzan, E.; Rezaei, N. Lymphoma in the setting of autoimmune diseases: A review of association and mechanisms. Crit. Rev. Oncol. Hematol. 2020, 150, 102945. [Google Scholar] [CrossRef]
- Grassilli, E.; Cerrito, M.G.; Lavitrano, M. BTK, the new kid on the (oncology) block? Front. Oncol. 2022, 12, 944538. [Google Scholar] [CrossRef] [PubMed]
- Imbruvica (Ibrutinib) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/205552s030,210563s006lbl.pdf (accessed on 25 March 2023).
- Calquence (Acalabrutinib) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/210259s009lbl.pdf (accessed on 25 March 2023).
- National Cancer Institute. Zanubrutinib’s Approval Improves Targeted Treatment for CLL. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2023/fda-zanubrutinib-cll-sll (accessed on 25 March 2023).
- Brukinsa (Zanubrutinib) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213217s005lbl.pdf (accessed on 25 March 2023).
- Food and Drug Administration. Drug Approvals and Databases. 27 January 2023. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pirtobrutinib-relapsed-or-refractory-mantle-cell-lymphoma (accessed on 25 March 2023).
- Rai, K.R.; Jain, P. Chronic lymphocytic leukemia (CLL)-Then and now. Am. J. Hematol. 2016, 91, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Schiattone, L.; Ghia, P.; Scarfò, L. The evolving treatment landscape of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2019, 31, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Treatment of Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2020, 383, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Albiol, N.; Arguello-Tomas, M.; Moreno, C. The road to chemotherapy-free treatment in chronic lymphocytic leukaemia. Curr. Opin. Oncol. 2021, 33, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.M.; Li, J.Y.; Gale, R.P.; Huang, X.J. The mystery of chronic lymphocytic leukemia (CLL): Why is it absent in Asians and what does this tell us about etiology, pathogenesis and biology? Blood Rev. 2015, 29, 205–213. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Landgren, O.; Albitar, M.; Ma, W.; Abbasi, F.; Hayes, R.B.; Ghia, P.; Marti, G.E.; Caporaso, N.E. B-cell clones as early markers for chronic lymphocytic leukemia. N. Engl. J. Med. 2009, 360, 659–667. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Green, M.J.; Kuzmicki, A.; Kennedy, B.; Fenton, J.A.; Evans, P.A.; O’Connor, S.J.; Richards, S.J.; Morgan, G.J.; Jack, A.S.; et al. Monoclonal B lymphocytes with the characteristics of “indolent” chronic lymphocytic leukemia are present in 3.5% of adults with normal blood counts. Blood 2002, 100, 635–639. [Google Scholar] [CrossRef]
- Marti, G.E.; Rawstron, A.C.; Ghia, P.; Hillmen, P.; Houlston, R.S.; Kay, N.; Schleinitz, T.A.; Caporaso, N.; International Familial CLL Consortium. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br. J. Haematol. 2005, 130, 325–332. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Shanafelt, T.; Lanasa, M.C.; Landgren, O.; Hanson, C.; Orfao, A.; Hillmen, P.; Ghia, P. Different biology and clinical outcome according to the absolute numbers of clonal B-cells in monoclonal B-cell lymphocytosis (MBL). Cytom. B Clin. Cytom. 2010, 78 (Suppl. S1), S19–S23. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Kay, N.E.; Rabe, K.G.; Call, T.G.; Zent, C.S.; Maddocks, K.; Jenkins, G.; Jelinek, D.F.; Morice, W.G.; Boysen, J.; et al. Brief report: Natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J. Clin. Oncol. 2009, 27, 3959–3963. [Google Scholar] [CrossRef]
- Goldin, L.R.; Pfeiffer, R.M.; Li, X.; Hemminki, K. Familial risk of lymphoproliferative tumors in families of patients with chronic lymphocytic leukemia: Results from the Swedish Family-Cancer Database. Blood 2004, 104, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Slager, S.L.; Caporaso, N.E.; de Sanjose, S.; Goldin, L.R. Genetic susceptibility to chronic lymphocytic leukemia. Semin. Hematol. 2013, 50, 296–302. [Google Scholar] [CrossRef]
- Kleinstern, G.; Weinberg, J.B.; Parikh, S.A.; Braggio, E.; Achenbach, S.J.; Robinson, D.P.; Norman, A.D.; Rabe, K.G.; Boddicker, N.J.; Vachon, C.M.; et al. Polygenic risk score and risk of monoclonal B-cell lymphocytosis in caucasians and risk of chronic lymphocytic leukemia (CLL) in African Americans. Leukemia 2022, 36, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Tiao, G.; Improgo, M.R.; Kasar, S.; Poh, W.; Kamburov, A.; Landau, D.A.; Tausch, E.; Taylor-Weiner, A.; Cibulskis, C.; Bahl, S.; et al. Rare germline variants in ATM are associated with chronic lymphocytic leukemia. Leukemia 2017, 31, 2244–2247. [Google Scholar] [CrossRef]
- Lampson, B.L.; Gupta, A.; Tyekucheva, S.; Mashima, K.; Petráčková, A.; Wang, Z.; Wojciechowska, N.; Shaughnessy, C.J.; Baker, P.O.; Fernandes, S.M.; et al. Rare Germline ATM Variants Influence the Development of Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2023, 41, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [PubMed]
- International CLL-IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): A meta-analysis of individual patient data. Lancet Oncol. 2016, 17, 779–790. [Google Scholar] [CrossRef]
- Seifert, M.; Sellmann, L.; Bloehdorn, J.; Wein, F.; Stilgenbauer, S.; Dürig, J.; Küppers, R. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J. Exp. Med. 2012, 209, 2183–2198. [Google Scholar] [CrossRef]
- Sutton, L.A.; Rosenquist, R. Deciphering the molecular landscape in chronic lymphocytic leukemia: Time frame of disease evolution. Haematologica 2015, 100, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Belessi, C.; Moreno, C.; Boudjograh, M.; Guida, G.; Smilevska, T.; Belhoul, L.; Stella, S.; Stavroyianni, N.; Crespo, M.; et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood 2007, 109, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, K.; Agathangelidis, A.; Rosenquist, R.; Ghia, P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia 2017, 31, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, P.M.; Hosnijeh, F.S.; Späth, F.; Hengeveld, P.J.; Agathangelidis, A.; Saleh, M.; Casabonne, D.; Benavente, Y.; Jerkeman, M.; Agudo, A.; et al. High-risk subtypes of chronic lymphocytic leukemia are detectable as early as 16 years prior to diagnosis. Blood 2022, 139, 1557–1563. [Google Scholar] [CrossRef]
- Kolijn, P.M.; Muggen, A.F.; Ljungström, V.; Agathangelidis, A.; Wolvers-Tettero, I.L.M.; Beverloo, H.B.; Pál, K.; Hengeveld, P.J.; Darzentas, N.; Hendriks, R.W.; et al. Consistent B Cell Receptor Immunoglobulin Features Between Siblings in Familial Chronic Lymphocytic Leukemia. Front. Oncol. 2021, 11, 740083. [Google Scholar] [CrossRef]
- Davi, F.; Langerak, A.W.; de Septenville, A.L.; Kolijn, P.M.; Hengeveld, P.J.; Chatzidimitriou, A.; Bonfiglio, S.; Sutton, L.A.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin gene analysis in chronic lymphocytic leukemia in the era of next generation sequencing. Leukemia 2020, 34, 2545–2551. [Google Scholar] [CrossRef]
- Hengeveld, P.J.; Levin, M.D.; Kolijn, P.M.; Langerak, A.W. Reading the B-cell receptor immunome in chronic lymphocytic leukemia: Revelations and applications. Exp. Hematol. 2021, 93, 14–24. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Robbe, P.; Ridout, K.E.; Vavoulis, D.V.; Dréau, H.; Kinnersley, B.; Denny, N.; Chubb, D.; Appleby, N.; Cutts, A.; Cornish, A.J.; et al. Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features. Nat. Genet. 2022, 54, 1675–1689. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Kreuzer, K.A.; Soosapilla, A.; Spacek, M.; Stehlikova, O.; Gambell, P.; McIver-Brown, N.; Villamor, N.; Psarra, K.; Arroz, M.; et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom. B Clin. Cytom. 2018, 94, 121–128. [Google Scholar] [CrossRef]
- Giné, E.; Martinez, A.; Villamor, N.; López-Guillermo, A.; Camos, M.; Martinez, D.; Esteve, J.; Calvo, X.; Muntañola, A.; Abrisqueta, P.; et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica 2010, 95, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef]
- Baliakas, P.; Espinet, B.; Mellink, C.; Jarosova, M.; Athanasiadou, A.; Ghia, P.; Kater, A.P.; Oscier, D.; Haferlach, C.; Stamatopoulos, K. Cytogenetics in Chronic Lymphocytic Leukemia: ERIC Perspectives and Recommendations. Hemasphere 2022, 6, e707. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jäger, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. TP53 aberrations in chronic lymphocytic leukemia: An overview of the clinical implications of improved diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Tausch, E.; Rossi, D.; Sutton, L.A.; Soussi, T.; Zenz, T.; Kater, A.P.; Niemann, C.U.; Gonzalez, D.; Davi, F.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia 2018, 32, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Agathangelidis, A.; Chatzidimitriou, A.; Chatzikonstantinou, T.; Tresoldi, C.; Davis, Z.; Giudicelli, V.; Kossida, S.; Belessi, C.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: The 2022 update of the recommendations by ERIC, the European Research Initiative on CLL. Leukemia 2022, 36, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Favini, C.; Talotta, D.; Almasri, M.; Andorno, A.; Rasi, S.; Adhinaveni, R.; Kogila, S.; Awikeh, B.; Schipani, M.; Boggione, P.; et al. Clonally unrelated Richter syndrome are truly de novo diffuse large B-cell lymphomas with a mutational profile reminiscent of clonally related Richter syndrome. Br. J. Haematol. 2022, 198, 1016–1022. [Google Scholar] [CrossRef]
- Siebert, R.; Schuh, A.; Ott, G.; Cree, I.A.; Du, M.Q.; Ferry, J.; Hochhaus, A.; Naresh, K.N.; Solary, E.; Khoury, J.D. Response to the Comments from the Groupe Francophone de Cytogénétique Hématologique (GFCH) on the 5th edition of the World Health Organization classification of haematolymphoid tumors. Leukemia 2023, 37, 1170–1172. [Google Scholar] [CrossRef]
- Chapiro, E.; Pramil, E.; Diop, M.; Roos-Weil, D.; Dillard, C.; Gabillaud, C.; Maloum, K.; Settegrana, C.; Baseggio, L.; Lesesve, J.F.; et al. Genetic characterization of B-cell prolymphocytic leukemia: A prognostic model involving MYC and TP53. Blood 2019, 134, 1821–1831. [Google Scholar] [CrossRef]
- Kansal, R.; Ross, C.W.; Singleton, T.P.; Finn, W.G.; Schnitzer, B. Histopathologic features of splenic small B-cell lymphomas. A study of 42 cases with a definitive diagnosis by the World Health Organization classification. Am. J. Clin. Pathol. 2003, 120, 335–347. [Google Scholar] [CrossRef]
- Bonfiglio, F.; Bruscaggin, A.; Guidetti, F.; di Bergamo, L.T.; Faderl, M.; Spina, V.; Condoluci, A.; Bonomini, L.; Forestieri, G.; Koch, R.; et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood 2022, 139, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef]
- Tiacci, E.; Pettirossi, V.; Schiavoni, G.; Falini, B. Genomics of Hairy Cell Leukemia. J. Clin. Oncol. 2017, 35, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Dixon, J.G.; Le-Rademacher, J.; Hoster, E.; Hochster, H.S.; Hiddemann, W.; Marcus, R.; Kimby, E.; Herold, M.; Sebban, C.; et al. Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 2022, 139, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Al-Tourah, A.J.; Gill, K.K.; Chhanabhai, M.; Hoskins, P.J.; Klasa, R.J.; Savage, K.J.; Sehn, L.H.; Shenkier, T.N.; Gascoyne, R.D.; Connors, J.M. Population-based analysis of incidence and outcome of transformed non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008, 26, 5165–5169. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Primers 2019, 5, 83. [Google Scholar] [CrossRef]
- Green, M.R. Chromatin modifying gene mutations in follicular lymphoma. Blood 2018, 131, 595–604. [Google Scholar] [CrossRef]
- Küppers, R.; Stevenson, F.K. Critical influences on the pathogenesis of follicular lymphoma. Blood 2018, 131, 2297–2306. [Google Scholar] [CrossRef]
- von Keudell, G.; Salles, G. The role of tazemetostat in relapsed/refractory follicular lymphoma. Ther. Adv. Hematol. 2021, 12, 20406207211015882. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef]
- Mann, R.B.; Berard, C.W. Criteria for the cytologic subclassification of follicular lymphomas: A proposed alternative method. Hematol. Oncol. 1983, 1, 187–192. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. (Eds.) World Health Organization Classification of Haematopoietic Tumours. In Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2001. [Google Scholar]
- Kansal, R. The World Health Organization (WHO) Classification of Tumors with Emphasis on the Classification of Hematolymphoid Neoplasms. In Precision Medicine: Where Are We and Where Are We Going? Nova Science Publishers, Inc.: New York, NY, USA, 2023; pp. 315–416. [Google Scholar]
- Feller, A.C.; Diebold, J. History of lymphoma classification. In Histopathology of Nodal and Extranodal Non-Hodgkin’s Lymphomas Based on the WHO Classification, 3rd ed.; Completely Revised and Updated Edition; Keller, A.C., Diebold, J., Paulli, M., Le Tourneau, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 1–13. [Google Scholar]
- Metter, G.E.; Nathwani, B.N.; Burke, J.S.; Winberg, C.D.; Mann, R.B.; Barcos, M.; Kjeldsberg, C.R.; Whitcomb, C.C.; Dixon, D.O.; Miller, T.P.; et al. Morphological subclassification of follicular lymphoma: Variability of diagnoses among hematopathologists, a collaborative study between the Repository Center and Pathology Panel for Lymphoma Clinical Studies. J. Clin. Oncol. 1985, 3, 25–38. [Google Scholar] [CrossRef]
- Cree, I.A.; Tan, P.H.; Travis, W.D.; Wesseling, P.; Yagi, Y.; White, V.A.; Lokuhetty, D.; Scolyer, R.A. Counting mitoses: SI(ze) matters! Mod. Pathol. 2021, 34, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Rimsza, L.M.; Li, H.; Braziel, R.M.; Spier, C.M.; Persky, D.O.; Dunlap, J.; LeBlanc, M.; Bartlett, N.; Leonard, J.P.; Smith, S.M.; et al. Impact of histological grading on survival in the SWOG S0016 follicular lymphoma cohort. Haematologica 2018, 103, e151–e153. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Gooley, T.; Loeb, K.; Soma, L.; Smith, S.D.; Gopal, A.; Naresh, K.N. The impact of histological grade on outcomes in follicular lymphoma: An analysis of patients in the SEER database in the context of evolving disease classification and treatment. Br. J. Haematol. 2022, 199, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Louissaint, A., Jr.; Schafernak, K.T.; Geyer, J.T.; Kovach, A.E.; Ghandi, M.; Gratzinger, D.; Roth, C.G.; Paxton, C.N.; Kim, S.; Namgyal, C.; et al. Pediatric-type nodal follicular lymphoma: A biologically distinct lymphoma with frequent MAPK pathway mutations. Blood 2016, 128, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Bosman, F.T., Lakhani, S.R., Jaffe, E.S., Ohgaki, H., Eds.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Schmidt, J.; Gong, S.; Marafioti, T.; Mankel, B.; Gonzalez-Farre, B.; Balagué, O.; Mozos, A.; Cabeçadas, J.; van der Walt, J.; Hoehn, D.; et al. Genome-wide analysis of pediatric-type follicular lymphoma reveals low genetic complexity and recurrent alterations of TNFRSF14 gene. Blood 2016, 128, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Salmeron-Villalobos, J.; Egan, C.; Borgmann, V.; Müller, I.; Gonzalez-Farre, B.; Ramis-Zaldivar, J.E.; Nann, D.; Balagué, O.; López-Guerra, M.; Colomer, D.; et al. A unifying hypothesis for PNMZL and PTFL: Morphological variants with a common molecular profile. Blood Adv. 2022, 6, 4661–4674. [Google Scholar] [CrossRef]
- Attarbaschi, A.; Abla, O.; Arias Padilla, L.; Beishuizen, A.; Burke, G.A.A.; Brugières, L.; Bruneau, J.; Burkhardt, B.; d’Amore, E.S.G.; Klapper, W.; et al. Rare non-Hodgkin lymphoma of childhood and adolescence: A consensus diagnostic and therapeutic approach to pediatric-type follicular lymphoma, marginal zone lymphoma, and nonanaplastic peripheral T-cell lymphoma. Pediatr. Blood Cancer 2020, 67, e28416. [Google Scholar] [CrossRef]
- Nann, D.; Ramis-Zaldivar, J.E.; Müller, I.; Gonzalez-Farre, B.; Schmidt, J.; Egan, C.; Salmeron-Villalobos, J.; Clot, G.; Mattern, S.; Otto, F.; et al. Follicular lymphoma t(14;18)-negative is genetically a heterogeneous disease. Blood Adv. 2020, 4, 5652–5665. [Google Scholar] [CrossRef]
- Lones, M.A.; Raphael, M.; McCarthy, K.; Wotherspoon, A.; Terrier-Lacombe, M.J.; Ramsay, A.D.; Maclennan, K.; Cairo, M.S.; Gerrard, M.; Michon, J.; et al. Primary follicular lymphoma of the testis in children and adolescents. J. Pediatr. Hematol. Oncol. 2012, 34, 68–71. [Google Scholar] [CrossRef]
- Kansal, R. In Situ Mantle Cell Neoplasia. Compendium of Cancer Genome Aberrations (CCGA), Cancer Genomics Consortium (CGC). Available online: https://www.ccga.io/index.php/In_Situ_Mantle_Cell_Neoplasia (accessed on 26 July 2022).
- Bermudez, G.; González de Villambrosía, S.; Martínez-López, A.; Batlle, A.; Revert-Arce, J.B.; Cereceda Company, L.; Ortega Bezanilla, C.; Piris, M.A.; Montes-Moreno, S. Incidental and Isolated Follicular Lymphoma In Situ and Mantle Cell Lymphoma In Situ Lack Clinical Significance. Am. J. Surg. Pathol. 2016, 40, 943–949. [Google Scholar] [CrossRef]
- Adam, P.; Schiefer, A.I.; Prill, S.; Henopp, T.; Quintanilla-Martínez, L.; Bösmüller, H.C.; Chott, A.; Fend, F. Incidence of preclinical manifestations of mantle cell lymphoma and mantle cell lymphoma in situ in reactive lymphoid tissues. Mod. Pathol. 2012, 25, 1629–1636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carvajal-Cuenca, A.; Sua, L.F.; Silva, N.M.; Pittaluga, S.; Royo, C.; Song, J.Y.; Sargent, R.L.; Espinet, B.; Climent, F.; Jacobs, S.A.; et al. In situ mantle cell lymphoma: Clinical implications of an incidental finding with indolent clinical behavior. Haematologica 2012, 97, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Healey, B.M.; Pinkus, G.S.; Kuo, F.C.; Dal Cin, P.; Kutok, J.L. Mantle cell lymphoma arising within primary nodal marginal zone lymphoma: A unique presentation of two uncommon B-cell lymphoproliferative disorders. Cancer Genet. Cytogenet. 2006, 171, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Aqel, N.; Barker, F.; Patel, K.; Naresh, K.N. In-situ mantle cell lymphoma--a report of two cases. Histopathology 2008, 52, 256–260. [Google Scholar] [CrossRef]
- Roullet, M.R.; Martinez, D.; Ma, L.; Fowler, M.H.; McPhail, E.D.; Judkins, A.; Arber, D.A.; Bagg, A. Coexisting follicular and mantle cell lymphoma with each having an in situ component: A novel, curious, and complex consultation case of coincidental, composite, colonizing lymphoma. Am. J. Clin. Pathol. 2010, 133, 584–591. [Google Scholar] [CrossRef]
- Demurtas, A.; Aliberti, S.; Bonello, L.; Di Celle, P.F.; Cavaliere, C.; Barreca, A.; Novero, D.; Stacchini, A. Usefulness of multiparametric flow cytometry in detecting composite lymphoma: Study of 17 cases in a 12-year period. Am. J. Clin. Pathol. 2011, 135, 541–555. [Google Scholar] [CrossRef]
- Papathomas, T.G.; Venizelos, I.; Dunphy, C.H.; Said, J.W.; Wang, M.L.; Campo, E.; Swerdlow, S.H.; Chan, J.C.; Bueso-Ramos, C.E.; Weisenburger, D.D.; et al. Mantle cell lymphoma as a component of composite lymphoma: Clinicopathologic parameters and biologic implications. Hum. Pathol. 2012, 43, 467–480. [Google Scholar] [CrossRef]
- Subtil, A.; Xu, Z. Follicular lymphoma with composite in situ mantle cell neoplasia. Blood 2019, 133, 2460. [Google Scholar] [CrossRef]
- Bassarova, A.; Tierens, A.; Lauritzsen, G.F.; Fosså, A.; Delabie, J. Mantle cell lymphoma with partial involvement of the mantle zone: An early infiltration pattern of mantle cell lymphoma? Virchows Arch. 2008, 453, 407–411. [Google Scholar] [CrossRef]
- Neto, A.G.; Oroszi, G.; Protiva, P.; Rose, M.; Shafi, N.; Torres, R. Colonic in situ mantle cell lymphoma. Ann. Diagn. Pathol. 2012, 16, 508–514. [Google Scholar] [CrossRef]
- Teixeira Mendes, L.S.; Wotherspoon, A. The relationship between overt and in-situ lymphoma: A retrospective study of follicular and mantle cell lymphoma cases. Histopathology 2016, 68, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Edlefsen, K.L.; Greisman, H.A.; Yi, H.S.; Mantei, K.M.; Fromm, J.R. Early lymph node involvement by mantle cell lymphoma limited to the germinal center: Report of a case with a novel “follicular in situ” growth pattern. Am. J. Clin. Pathol. 2011, 136, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Espinet, B.; Solé, F.; Pedro, C.; Garcia, M.; Bellosillo, B.; Salido, M.; Florensa, L.; Camacho, F.I.; Baró, T.; Lloreta, J.; et al. Clonal proliferation of cyclin D1-positive mantle lymphocytes in an asymptomatic patient: An early-stage event in the development or an indolent form of a mantle cell lymphoma? Hum. Pathol. 2005, 36, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Koletsa, T.; Markou, K.; Ouzounidou, S.; Tsiompanou, F.; Karkavelas, G.; Kostopoulos, I. In situ mantle cell lymphoma in the nasopharynx. Head Neck 2013, 35, E333–E337. [Google Scholar] [CrossRef]
- Dobrea, C.; Mihai, M.; Dănăilă, E.; Găman, A.; Coriu, D.; Ursuleac, I. “In situ” mantle cell lymphoma associated with hyaline-vascular Castleman disease. Rom. J. Morphol. Embryol. 2011, 52 (Suppl. S3), 1147–1151. [Google Scholar]
- Zanelli, M.; Stingeni, L.; Zizzo, M.; Martino, G.; Sanguedolce, F.; Marra, A.; Crescenzi, B.; Pileri, S.A.; Ascani, S. HHV8-Positive Castleman Disease and In Situ Mantle Cell Neoplasia within Dermatopathic Lymphadenitis, in Longstanding Psoriasis. Diagnostics 2021, 11, 1150. [Google Scholar] [CrossRef]
- Hirt, C.; Schüler, F.; Dölken, L.; Schmidt, C.A.; Dölken, G. Low prevalence of circulating t(11;14)(q13;q32)-positive cells in the peripheral blood of healthy individuals as detected by real-time quantitative PCR. Blood 2004, 104, 904–905. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.; Raghavan, S.C. Prevalence and analysis of t(14;18) and t(11;14) chromosomal translocations in healthy Indian population. Ann. Hematol. 2010, 89, 35–43. [Google Scholar] [CrossRef]
- Lecluse, Y.; Lebailly, P.; Roulland, S.; Gac, A.C.; Nadel, B.; Gauduchon, P. t(11;14)-positive clones can persist over a long period of time in the peripheral blood of healthy individuals. Leukemia 2009, 23, 1190–1193. [Google Scholar] [CrossRef]
- Weisenburger, D.D.; Kim, H.; Rappaport, H. Mantle-zone lymphoma: A follicular variant of intermediate lymphocytic lymphoma. Cancer 1982, 49, 1429–1438. [Google Scholar] [CrossRef]
- Weisenburger, D.D.; Nathwani, B.N.; Diamond, L.W.; Winberg, C.D.; Rappaport, H. Malignant lymphoma, intermediate lymphocytic type: A clinicopathologic study of 42 cases. Cancer 1981, 48, 1415–1425. [Google Scholar] [CrossRef]
- Williams, M.E.; Westermann, C.D.; Swerdlow, S.H. Genotypic characterization of centrocytic lymphoma: Frequent rearrangement of the chromosome 11 bcl-1 locus. Blood 1990, 76, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, L.J.; Van Krieken, J.H.; Jaffe, E.S.; Raffeld, M. Association of bcl-1 rearrangements with lymphocytic lymphoma of intermediate differentiation. Blood 1990, 76, 2086–2090. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.M.; Chan, J.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Gatter, K.; Grogan, T.M.; Harris, N.L.; Isaacson, P.G.; Jaffe, E.S.; et al. Mantle cell lymphoma. A proposal for unification of morphologic, immunologic, and molecular data. Am. J. Surg. Pathol. 1992, 16, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O.; Longo, D.L. Mantle-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 2495–2506. [Google Scholar] [CrossRef]
- Hill, H.A.; Qi, X.; Jain, P.; Nomie, K.; Wang, Y.; Zhou, S.; Wang, M.L. Genetic mutations and features of mantle cell lymphoma: A systematic review and meta-analysis. Blood Adv. 2020, 4, 2927–2938. [Google Scholar] [CrossRef]
- Jain, P.; Wang, M.L. Mantle cell lymphoma in 2022-A comprehensive update on molecular pathogenesis, risk stratification, clinical approach, and current and novel treatments. Am. J. Hematol. 2022, 97, 638–656. [Google Scholar] [CrossRef]
- Sud, A.; Chattopadhyay, S.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Houlston, R.S.; Hemminki, K. Analysis of 153 115 patients with hematological malignancies refines the spectrum of familial risk. Blood 2019, 134, 960–969. [Google Scholar] [CrossRef]
- Tort, F.; Camacho, E.; Bosch, F.; Harris, N.L.; Montserrat, E.; Campo, E. Familial lymphoid neoplasms in patients with mantle cell lymphoma. Haematologica 2004, 89, 314–319. [Google Scholar]
- Hangaishi, A.; Ogawa, S.; Qiao, Y.; Wang, L.; Hosoya, N.; Yuji, K.; Imai, Y.; Takeuchi, K.; Miyawaki, S.; Hirai, H. Mutations of Chk2 in primary hematopoietic neoplasms. Blood 2002, 99, 3075–3077. [Google Scholar] [CrossRef]
- Camacho, E.; Hernández, L.; Hernández, S.; Tort, F.; Bellosillo, B.; Beà, S.; Bosch, F.; Montserrat, E.; Cardesa, A.; Fernández, P.L.; et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood 2002, 99, 238–244. [Google Scholar] [CrossRef]
- Nadeu, F.; Martin-Garcia, D.; Clot, G.; Díaz-Navarro, A.; Duran-Ferrer, M.; Navarro, A.; Vilarrasa-Blasi, R.; Kulis, M.; Royo, R.; Gutiérrez-Abril, J.; et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood 2020, 136, 1419–1432. [Google Scholar] [CrossRef]
- Wang, X.; Song, Y.; Chen, W.; Ding, N.; Liu, W.; Xie, Y.; Wang, Y.; Zhu, J.; Zeng, C. Germline variants of DNA repair genes in early onset mantle cell lymphoma. Oncogene 2021, 40, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L.; Morra, E.; Pangalis, G.A.; San Miguel, J.F.; Branagan, A.R.; et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef]
- Treon, S.P.; Hunter, Z.R. A new era for Waldenstrom macroglobulinemia: MYD88 L265P. Blood 2013, 121, 4434–4436. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, Y.; Liu, X.; Xu, L.; Cao, Y.; Manning, R.J.; Patterson, C.J.; Buhrlage, S.J.; Gray, N.; Tai, Y.T.; et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Buhrlage, S.J.; Tan, L.; Liu, X.; Chen, J.; Xu, L.; Tsakmaklis, N.; Chen, J.G.; Patterson, C.J.; Brown, J.R.; et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood 2016, 127, 3237–3252. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Manning, R.J.; Tripsas, C.; Patterson, C.J.; Sheehy, P.; et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 2014, 123, 1637–1646. [Google Scholar] [CrossRef]
- Treon, S.P.; Cao, Y.; Xu, L.; Yang, G.; Liu, X.; Hunter, Z.R. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014, 123, 2791–2796. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Garcia-Sanz, R.; Hatjiharissi, E.; Kyle, R.A.; Leleu, X.; McMaster, M.; Merlini, G.; Minnema, M.C.; Morra, E.; Owen, R.G.; et al. Recommendations for the diagnosis and initial evaluation of patients with Waldenström Macroglobulinaemia: A Task Force from the 8th International Workshop on Waldenström Macroglobulinaemia. Br. J. Haematol. 2016, 175, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Gustine, J.; Xu, L.; Manning, R.J.; Tsakmaklis, N.; Demos, M.; Meid, K.; Guerrera, M.L.; Munshi, M.; Chan, G.; et al. MYD88 wild-type Waldenstrom Macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br. J. Haematol. 2018, 180, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Jimenez, C.; Chan, G.G.; Chen, J.; Liu, X.; Munshi, M.; et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018, 2, 2937–2946. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Treon, S.P. Epigenomics in Waldenström macroglobulinemia. Blood 2020, 136, 527–529. [Google Scholar] [CrossRef]
- Kofides, A.; Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Munshi, M.; Liu, X.; Guerrera, M.L.; Leventoff, C.R.; White, T.P.; et al. Diagnostic Next-generation Sequencing Frequently Fails to Detect MYD88L265P in Waldenström Macroglobulinemia. Hemasphere 2021, 5, e624. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Jia, M.N.; Cai, H.; Qiu, Y.; Zhou, D.B.; Li, J.; Cao, X.X. Detection of the MYD88L265P and CXCR4S338X mutations by cell-free DNA in Waldenström macroglobulinemia. Ann. Hematol. 2020, 99, 1763–1769. [Google Scholar] [CrossRef]
- Demos, M.G.; Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Kofides, A.; Munshi, M.; Liu, X.; Guerrera, M.L.; Leventoff, C.R.; White, T.P.; et al. Cell-free DNA analysis for detection of MYD88L265P and CXCR4S338X mutations in Waldenström macroglobulinemia. Am. J. Hematol. 2021, 96, E250–E253. [Google Scholar] [CrossRef]
- Varettoni, M.; Boveri, E.; Zibellini, S.; Tedeschi, A.; Candido, C.; Ferretti, V.V.; Rizzo, E.; Doni, E.; Merli, M.; Farina, L.; et al. Clinical and molecular characteristics of lymphoplasmacytic lymphoma not associated with an IgM monoclonal protein: A multicentric study of the Rete Ematologica Lombarda (REL) network. Am. J. Hematol. 2019, 94, 1193–1199. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.; Hanzis, C.; Ioakimidis, L.; Patterson, C.J.; Manning, R.J.; Sheehy, P.; Turnbull, B.; Hunter, Z.R. Familial disease predisposition impacts treatment outcome in patients with Waldenström macroglobulinemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 433–437. [Google Scholar] [CrossRef]
- Treon, S.P.; Hunter, Z.R.; Aggarwal, A.; Ewen, E.P.; Masota, S.; Lee, C.; Santos, D.D.; Hatjiharissi, E.; Xu, L.; Leleu, X.; et al. Characterization of familial Waldenstrom’s macroglobulinemia. Ann. Oncol. 2006, 17, 488–494. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Shi, J.; Chiarini, M.; Perilla-Glen, A.; Manier, S.; Glavey, S.; Aljawai, Y.; Mishima, Y.; Kawano, Y.; et al. Exome sequencing reveals recurrent germ line variants in patients with familial Waldenström macroglobulinemia. Blood 2016, 127, 2598–2606. [Google Scholar] [CrossRef]
- Abboudi, Z.; Patel, K.; Naresh, K.N. Cyclin D1 expression in typical chronic lymphocytic leukaemia. Eur. J. Haematol. 2009, 83, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Miquel, R.; Krenacs, L.; Sorbara, L.; Raffeld, M.; Jaffe, E.S. Primary nodal marginal zone lymphomas of splenic and MALT type. Am. J. Surg. Pathol. 1999, 23, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Weiss, L.M.; Chang, K.L.; Arber, D.A. Frequency of CD43 expression in non-Hodgkin lymphoma. A survey of 742 cases and further characterization of rare CD43+ follicular lymphomas. Am. J. Clin. Pathol. 1999, 111, 488–494. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Magrath, I. Epidemiology: Clues to the pathogenesis of Burkitt lymphoma. Br. J. Haematol. 2012, 156, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Liu, H.; Miranda, R.N.; Golardi, N.; Billah, S.; Medeiros, L.J.; Jaso, J.M. Burkitt lymphoma presenting as a mass in the thyroid gland: A clinicopathologic study of 7 cases and review of the literature. Hum. Pathol. 2016, 56, 101–108. [Google Scholar] [CrossRef]
- Lenze, D.; Leoncini, L.; Hummel, M.; Volinia, S.; Liu, C.G.; Amato, T.; De Falco, G.; Githanga, J.; Horn, H.; Nyagol, J.; et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia 2011, 25, 1869–1876. [Google Scholar] [CrossRef]
- Gopal, S.; Gross, T.G. How I treat Burkitt lymphoma in children, adolescents, and young adults in sub-Saharan Africa. Blood 2018, 132, 254–263. [Google Scholar] [CrossRef]
- Moormann, A.M.; Bailey, J.A. Malaria—How this parasitic infection aids and abets EBV-associated Burkitt lymphomagenesis. Curr. Opin. Virol. 2016, 20, 78–84. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef]
- Pelicci, P.G.; Knowles, D.M., 2nd; Magrath, I.; Dalla-Favera, R. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 2984–2988. [Google Scholar] [CrossRef]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Fu, K.; Wright, G.W.; Lam, L.T.; Kluin, P.; Boerma, E.J.; Greiner, T.C.; Weisenburger, D.D.; Rosenwald, A.; Ott, G.; et al. Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med. 2006, 354, 2431–2442. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct Viral and Mutational Spectrum of Endemic Burkitt Lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed]
- Bellan, C.; Lazzi, S.; Hummel, M.; Palummo, N.; de Santi, M.; Amato, T.; Nyagol, J.; Sabattini, E.; Lazure, T.; Pileri, S.A.; et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood 2005, 106, 1031–1036. [Google Scholar] [CrossRef]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Richter, J.; John, K.; Staiger, A.M.; Rosenwald, A.; Kurz, K.; Michgehl, U.; Ott, G.; Franzenburg, S.; Kohler, C.; Finger, J.; et al. Epstein-Barr virus status of sporadic Burkitt lymphoma is associated with patient age and mutational features. Br. J. Haematol. 2022, 196, 681–689. [Google Scholar] [CrossRef]
- Thomas, N.; Dreval, K.; Gerhard, D.S.; Hilton, L.K.; Abramson, J.S.; Ambinder, R.F.; Barta, S.; Bartlett, N.L.; Bethony, J.; Bhatia, K.; et al. Genetic subgroups inform on pathobiology in adult and pediatric Burkitt lymphoma. Blood 2023, 141, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Naresh, K.N.; Lazzi, S.; Santi, R.; Granai, M.; Onyango, N.; Leoncini, L. A refined approach to the diagnosis of Burkitt lymphoma in a resource-poor setting. Histopathology 2022, 80, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Deeb, G.; Barcos, M.; Wetzler, M.; Brecher, M.L.; Block, A.W.; Stewart, C.C. Precursor B lymphoblastic leukemia with surface light chain immunoglobulin restriction: A report of 15 patients. Am. J. Clin. Pathol. 2004, 121, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W. Relapsed/refractory diffuse large B-cell lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 498–505. [Google Scholar] [CrossRef]
- Davies, A. Tailoring front-line therapy in diffuse large B-cell lymphoma: Who should we treat differently? Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 284–294. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef]
- Jiang, Y.; Dominguez, P.M.; Melnick, A.M. The many layers of epigenetic dysfunction in B-cell lymphomas. Curr. Opin. Hematol. 2016, 23, 377–384. [Google Scholar] [CrossRef]
- Lunning, M.A.; Green, M.R. Mutation of chromatin modifiers; An emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J. 2015, 5, e361. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Young, R.M.; Staudt, L.M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013, 12, 229–243. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Morin, R.D.; Arthur, S.E.; Hodson, D.J. Molecular profiling in diffuse large B-cell lymphoma: Why so many types of subtypes? Br. J. Haematol. 2022, 196, 814–829. [Google Scholar] [CrossRef]
- Cucco, F.; Barrans, S.; Sha, C.; Clipson, A.; Crouch, S.; Dobson, R.; Chen, Z.; Thompson, J.S.; Care, M.A.; Cummin, T.; et al. Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia 2020, 34, 1329–1341. [Google Scholar] [CrossRef]
- Scott, D.W.; King, R.L.; Staiger, A.M.; Ben-Neriah, S.; Jiang, A.; Horn, H.; Mottok, A.; Farinha, P.; Slack, G.W.; Ennishi, D.; et al. High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 2018, 131, 2060–2064. [Google Scholar] [CrossRef]
- Rosenwald, A.; Bens, S.; Advani, R.; Barrans, S.; Copie-Bergman, C.; Elsensohn, M.H.; Natkunam, Y.; Calaminici, M.; Sander, B.; Baia, M.; et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 2019, 37, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Sha, C.; Barrans, S.; Cucco, F.; Bentley, M.A.; Care, M.A.; Cummin, T.; Kennedy, H.; Thompson, J.S.; Uddin, R.; Worrillow, L.; et al. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J. Clin. Oncol. 2019, 37, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Ennishi, D.; Jiang, A.; Boyle, M.; Collinge, B.; Grande, B.M.; Ben-Neriah, S.; Rushton, C.; Tang, J.; Thomas, N.; Slack, G.W.; et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 190–201. [Google Scholar] [CrossRef]
- Salaverria, I.; Martin-Guerrero, I.; Wagener, R.; Kreuz, M.; Kohler, C.W.; Richter, J.; Pienkowska-Grela, B.; Adam, P.; Burkhardt, B.; Claviez, A.; et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 2014, 123, 1187–1198. [Google Scholar] [CrossRef]
- Wagener, R.; Seufert, J.; Raimondi, F.; Bens, S.; Kleinheinz, K.; Nagel, I.; Altmüller, J.; Thiele, H.; Hübschmann, D.; Kohler, C.W.; et al. The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood 2019, 133, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Farre, B.; Ramis-Zaldivar, J.E.; Salmeron-Villalobos, J.; Balagué, O.; Celis, V.; Verdu-Amoros, J.; Nadeu, F.; Sábado, C.; Ferrández, A.; Garrido, M.; et al. Burkitt-like lymphoma with 11q aberration: A germinal center-derived lymphoma genetically unrelated to Burkitt lymphoma. Haematologica 2019, 104, 1822–1829. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Kalmbach, S.; Wagener, R.; Staiger, A.M.; Hüttl, K.; Mottok, A.; Bens, S.; Traverse-Glehen, A.; Fontaine, J.; Siebert, R.; et al. A Diagnostic Approach to the Identification of Burkitt-like Lymphoma With 11q Aberration in Aggressive B-Cell Lymphomas. Am. J. Surg. Pathol. 2021, 45, 356–364. [Google Scholar] [CrossRef]
- Yu, Y.T.; Takeuchi, K.; Baba, S.; Chang, K.C. Morphologically Suspected Burkitt-like Lymphoma With 11q Aberrations Confirmed by Fluorescence In Situ Hybridization. Am. J. Surg. Pathol. 2022, 46, 576–577. [Google Scholar] [CrossRef]
- Rymkiewicz, G.; Grygalewicz, B.; Chechlinska, M.; Blachnio, K.; Bystydzienski, Z.; Romejko-Jarosinska, J.; Woroniecka, R.; Zajdel, M.; Domanska-Czyz, K.; Martin-Garcia, D.; et al. A comprehensive flow-cytometry-based immunophenotypic characterization of Burkitt-like lymphoma with 11q aberration. Mod. Pathol. 2018, 31, 732–743. [Google Scholar] [CrossRef]
- Arnold, L.M.; D’Agostino, E.; Thomas, A.A.; DeWitt, J.C. Tumors of the Central Nervous System. In Precision Medicine: Where Are We and Where Are We Going? Kansal, R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2023. [Google Scholar]
- Shiels, M.S.; Pfeiffer, R.M.; Besson, C.; Clarke, C.A.; Morton, L.M.; Nogueira, L.; Pawlish, K.; Yanik, E.L.; Suneja, G.; Engels, E.A. Trends in primary central nervous system lymphoma incidence and survival in the U.S. Br. J. Haematol. 2016, 174, 417–424. [Google Scholar] [CrossRef]
- Ferreri, A.J.M. Secondary CNS lymphoma: The poisoned needle in the haystack. Ann. Oncol. 2017, 28, 2335–2337. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.; Kim, S.J.; Kim, K.; Lee, J.I.; Lim, D.H.; Lee, D.J.; Baek, K.K.; Lee, H.Y.; Han, B.; Uhm, J.E.; et al. Primary CNS lymphoma other than DLBCL: A descriptive analysis of clinical features and treatment outcomes. Ann. Hematol. 2011, 90, 1391–1398. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Brunn, A.; Bentink, S.; Basso, K.; Lim, W.K.; Klapper, W.; Schaller, C.; Reifenberger, G.; Rubenstein, J.; Wiestler, O.D.; et al. Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia 2008, 22, 400–405. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Purschke, F.; Küppers, R.; Deckert, M. Immunoglobulin repertoire of primary lymphomas of the central nervous system. J. Neuropathol. Exp. Neurol. 2014, 73, 1116–1125. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Küppers, R.; Schlüter, D.; Spieker, T.; Van Roost, D.; Schaller, C.; Reifenberger, G.; Wiestler, O.D.; Deckert-Schlüter, M. Primary central nervous system lymphomas are derived from germinal-center B cells and show a preferential usage of the V4-34 gene segment. Am. J. Pathol. 1999, 155, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Belhouachi, N.; Xochelli, A.; Boudjoghra, M.; Lesty, C.; Cassoux, N.; Fardeau, C.; Tran, T.H.C.; Choquet, S.; Sarker, B.; Houillier, C.; et al. Primary vitreoretinal lymphomas display a remarkably restricted immunoglobulin gene repertoire. Blood Adv. 2020, 4, 1357–1366. [Google Scholar] [CrossRef]
- Radke, J.; Ishaque, N.; Koll, R.; Gu, Z.; Schumann, E.; Sieverling, L.; Uhrig, S.; Hübschmann, D.; Toprak, U.H.; López, C.; et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat. Commun. 2022, 13, 2558. [Google Scholar] [CrossRef]
- Casellas, R.; Basu, U.; Yewdell, W.T.; Chaudhuri, J.; Robbiani, D.F.; Di Noia, J.M. Mutations, kataegis and translocations in B cells: Understanding AID promiscuous activity. Nat. Rev. Immunol. 2016, 16, 164–176. [Google Scholar] [CrossRef]
- Gandhi, M.K.; Hoang, T.; Law, S.C.; Brosda, S.; O’Rourke, K.; Tobin, J.W.D.; Vari, F.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 2021, 137, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Deckert, M.; Brunn, A.; Montesinos-Rongen, M.; Terreni, M.R.; Ponzoni, M. Primary lymphoma of the central nervous system—A diagnostic challenge. Hematol. Oncol. 2014, 32, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.E.; Damato, B. Understanding intraocular lymphomas. Clin. Exp. Ophthalmol. 2008, 36, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Pulido, J.S.; Johnston, P.B.; Nowakowski, G.S.; Castellino, A.; Raja, H. The diagnosis and treatment of primary vitreoretinal lymphoma: A review. Int. J. Retin. Vitr. 2018, 4, 18. [Google Scholar] [CrossRef]
- Zuckerman, D.; Seliem, R.; Hochberg, E. Intravascular lymphoma: The oncologist’s “great imitator”. Oncologist 2006, 11, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Fonkem, E.; Dayawansa, S.; Stroberg, E.; Lok, E.; Bricker, P.C.; Kirmani, B.; Wong, E.T.; Huang, J.H. Neurological presentations of intravascular lymphoma (IVL): Meta-analysis of 654 patients. BMC Neurol. 2016, 16, 9. [Google Scholar] [CrossRef]
- Imai, H.; Kajimoto, K.; Taniwaki, M.; Miura, I.; Hatta, Y.; Hashizume, Y.; Watanabe, M.; Shiraishi, T.; Nakamura, S. Intravascular large B-cell lymphoma presenting with mass lesions in the central nervous system: A report of five cases. Pathol. Int. 2004, 54, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, M.; Campo, E.; Nakamura, S. Intravascular large B-cell lymphoma: A chameleon with multiple faces and many masks. Blood 2018, 132, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Kiyoi, H. Current progress and future perspectives of research on intravascular large B-cell lymphoma. Cancer Sci. 2021, 112, 3953–3961. [Google Scholar] [CrossRef]
- Fonkem, E.; Lok, E.; Robison, D.; Gautam, S.; Wong, E.T. The natural history of intravascular lymphomatosis. Cancer Med. 2014, 3, 1010–1024. [Google Scholar] [CrossRef]
- Geer, M.; Roberts, E.; Shango, M.; Till, B.G.; Smith, S.D.; Abbas, H.; Hill, B.T.; Kaplan, J.; Barr, P.M.; Caimi, P.; et al. Multicentre retrospective study of intravascular large B-cell lymphoma treated at academic institutions within the United States. Br. J. Haematol. 2019, 186, 255–262. [Google Scholar] [CrossRef]
- Seegobin, K.; Li, Z.; Alhaj Moustafa, M.; Majeed, U.; Wang, J.; Jiang, L.; Kuhlman, J.; Menke, D.; Li, K.; Kharfan-Dabaja, M.A.; et al. Clinical characteristics, prognostic indicators, and survival outcomes in intravascular lymphoma: Mayo Clinic experience (2003–2018). Am. J. Hematol. 2022, 97, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Yoshida, K.; Suzuki, Y.; Iriyama, C.; Inoue, Y.; Sanada, M.; Kataoka, K.; Yuge, M.; Takagi, Y.; Kusumoto, S.; et al. Frequent genetic alterations in immune checkpoint-related genes in intravascular large B-cell lymphoma. Blood 2021, 137, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Suehara, Y.; Sakata-Yanagimoto, M.; Hattori, K.; Nanmoku, T.; Itoh, T.; Kaji, D.; Yamamoto, G.; Abe, Y.; Narita, K.; Takeuchi, M.; et al. Liquid biopsy for the identification of intravascular large B-cell lymphoma. Haematologica 2018, 103, e241–e244. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.M.R.; Jansen, P.M.; Willemze, R.; Vermeer, M.H.; Cleton-Jansen, A.M.; Somers, S.F.; Veelken, H.; van Eijk, R.; Kraan, W.; Kersten, M.J.; et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood 2018, 131, 2086–2089. [Google Scholar] [CrossRef]
- Chapman, J.; Verdun, R.E.; Lossos, I.S. Low LIM-domain only 2 (LMO2) expression in aggressive B cell lymphoma correlates with MYC and MYC/BCL2 rearrangements, especially in germinal center cell-type tumors. Leuk. Lymphoma 2021, 62, 2547–2550. [Google Scholar] [CrossRef]
- Salaverria, I.; Philipp, C.; Oschlies, I.; Kohler, C.W.; Kreuz, M.; Szczepanowski, M.; Burkhardt, B.; Trautmann, H.; Gesk, S.; Andrusiewicz, M.; et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 2011, 118, 139–147. [Google Scholar] [CrossRef]
- Chen, L.; Al-Kzayer, L.F.; Liu, T.; Kobayashi, N.; Nakazawa, Y.; Koike, K. IFR4/MUM1-positive lymphoma in Waldeyer ring with co-expression of CD5 and CD10. Pediatr. Blood Cancer 2017, 64, 311–314. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Seto, M.; Okamoto, M.; Ichinohasama, R.; Nakamura, N.; Yoshino, T.; Suzumiya, J.; Murase, T.; Miura, I.; Akasaka, T.; et al. De novo CD5+ diffuse large B-cell lymphoma: A clinicopathologic study of 109 patients. Blood 2002, 99, 815–821. [Google Scholar] [CrossRef]
- Parvin, S.; Ramirez-Labrada, A.; Aumann, S.; Lu, X.; Weich, N.; Santiago, G.; Cortizas, E.M.; Sharabi, E.; Zhang, Y.; Sanchez-Garcia, I.; et al. LMO2 Confers Synthetic Lethality to PARP Inhibition in DLBCL. Cancer Cell 2019, 36, 237–249.e6. [Google Scholar] [CrossRef]
- Natkunam, Y.; Zhao, S.; Mason, D.Y.; Chen, J.; Taidi, B.; Jones, M.; Hammer, A.S.; Hamilton Dutoit, S.; Lossos, I.S.; Levy, R. The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 2007, 109, 1636–1642. [Google Scholar] [CrossRef]
- Piña-Oviedo, S.; Moran, C.A. Primary Mediastinal Nodal and Extranodal Non-Hodgkin Lymphomas: Current Concepts, Historical Evolution, and Useful Diagnostic Approach: Part 1. Adv. Anat. Pathol. 2019, 26, 346–370. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, T.; Jaffe, E.S.; Delsol, G.; deWolf-Peeters, C.; Gascoyne, R.D.; Georgii, A.; Harris, N.L.; Kadin, M.E.; MacLennan, K.A.; Poppema, S.; et al. Workshop report on Hodgkin’s disease and related diseases (‘grey zone’ lymphoma). Ann. Oncol. 1998, 9 (Suppl. S5), S31–S38. [Google Scholar] [CrossRef] [PubMed]
- Traverse-Glehen, A.; Pittaluga, S.; Gaulard, P.; Sorbara, L.; Alonso, M.A.; Raffeld, M.; Jaffe, E.S. Mediastinal gray zone lymphoma: The missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am. J. Surg. Pathol. 2005, 29, 1411–1421. [Google Scholar] [CrossRef]
- García, J.F.; Mollejo, M.; Fraga, M.; Forteza, J.; Muniesa, J.A.; Pérez-Guillermo, M.; Pérez-Seoane, C.; Rivera, T.; Ortega, P.; Piris, M.A. Large B-cell lymphoma with Hodgkin’s features. Histopathology 2005, 47, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Dunleavy, K.; Eberle, F.C.; Pittaluga, S.; Wilson, W.H.; Jaffe, E.S. Primary mediastinal large B-cell lymphoma, classic Hodgkin lymphoma presenting in the mediastinum, and mediastinal gray zone lymphoma: What is the oncologist to do? Curr. Hematol. Malig. Rep. 2011, 6, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.K.; Levine, A.; Taylor, C.R.; Boswell, W.; Rossman, S.; Feinstein, D.I.; Lukes, R.J. Primary mediastinal lymphoma in adults. Am. J. Med. 1980, 68, 509–514. [Google Scholar] [CrossRef]
- Cazals-Hatem, D.; Lepage, E.; Brice, P.; Ferrant, A.; d’Agay, M.F.; Baumelou, E.; Brière, J.; Blanc, M.; Gaulard, P.; Biron, P.; et al. Primary mediastinal large B-cell lymphoma. A clinicopathologic study of 141 cases compared with 916 nonmediastinal large B-cell lymphomas, a GELA (“Groupe d’Etude des Lymphomes de l’Adulte”) study. Am. J. Surg. Pathol. 1996, 20, 877–888. [Google Scholar] [CrossRef]
- Dunleavy, K. Primary mediastinal B-cell lymphoma: Biology and evolving therapeutic strategies. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 298–303. [Google Scholar] [CrossRef]
- Isaacson, P.G.; Norton, A.J.; Addis, B.J. The human thymus contains a novel population of B lymphocytes. Lancet 1987, 2, 1488–1491. [Google Scholar] [CrossRef]
- Hofmann, W.J.; Momburg, F.; Möller, P. Thymic medullary cells expressing B lymphocyte antigens. Hum. Pathol. 1988, 19, 1280–1287. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Monti, S.; Kutok, J.L.; Cattoretti, G.; Neuberg, D.; De Leval, L.; Kurtin, P.; Dal Cin, P.; Ladd, C.; Feuerhake, F.; et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879. [Google Scholar] [CrossRef]
- Joos, S.; Otaño-Joos, M.I.; Ziegler, S.; Brüderlein, S.; du Manoir, S.; Bentz, M.; Möller, P.; Lichter, P. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 1996, 87, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Küpper, M.; Ohl, S.; von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Möller, P.; Pfreundschuh, M.; Trümper, L.; et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Bentz, M.; Barth, T.F.; Brüderlein, S.; Bock, D.; Schwerer, M.J.; Baudis, M.; Joos, S.; Viardot, A.; Feller, A.C.; Müller-Hermelink, H.K.; et al. Gain of chromosome arm 9p is characteristic of primary mediastinal B-cell lymphoma (MBL): Comprehensive molecular cytogenetic analysis and presentation of a novel MBL cell line. Genes Chromosomes Cancer 2001, 30, 393–401. [Google Scholar] [CrossRef]
- Oschlies, I.; Burkhardt, B.; Salaverria, I.; Rosenwald, A.; d’Amore, E.S.; Szczepanowski, M.; Koch, K.; Hansmann, M.L.; Stein, H.; Möller, P.; et al. Clinical, pathological and genetic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas in children. Haematologica 2011, 96, 262–268. [Google Scholar] [CrossRef]
- Aggarwal, R.; Rao, S.; Dhawan, S.; Bhalla, S.; Kumar, A.; Chopra, P. Primary mediastinal lymphomas, their morphological features and comparative evaluation. Lung India 2017, 34, 19–24. [Google Scholar] [CrossRef]
- al-Sharabati, M.; Chittal, S.; Duga-Neulat, I.; Laurent, G.; Mazerolles, C.; al-Saati, T.; Brousset, P.; Delsol, G. Primary anterior mediastinal B-cell lymphoma. A clinicopathologic and immunohistochemical study of 16 cases. Cancer 1991, 67, 2579–2587. [Google Scholar] [CrossRef]
- Yuan, J.; Wright, G.; Rosenwald, A.; Steidl, C.; Gascoyne, R.D.; Connors, J.M.; Mottok, A.; Weisenburger, D.D.; Greiner, T.C.; Fu, K.; et al. Identification of Primary Mediastinal Large B-cell Lymphoma at Nonmediastinal Sites by Gene Expression Profiling. Am. J. Surg. Pathol. 2015, 39, 1322–1330. [Google Scholar] [CrossRef]
- Wang, Y.; Wenzl, K.; Manske, M.K.; Asmann, Y.W.; Sarangi, V.; Greipp, P.T.; Krull, J.E.; Hartert, K.; He, R.; Feldman, A.L.; et al. Amplification of 9p24.1 in diffuse large B-cell lymphoma identifies a unique subset of cases that resemble primary mediastinal large B-cell lymphoma. Blood Cancer J. 2019, 9, 73. [Google Scholar] [CrossRef]
- Savage, K.J. Primary mediastinal large B-cell lymphoma. Blood 2022, 140, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Saarinen, S.; Kaasinen, E.; Karjalainen-Lindsberg, M.L.; Vesanen, K.; Aavikko, M.; Katainen, R.; Taskinen, M.; Kytölä, S.; Leppä, S.; Hietala, M.; et al. Primary mediastinal large B-cell lymphoma segregating in a family: Exome sequencing identifies MLL as a candidate predisposition gene. Blood 2013, 121, 3428–3430. [Google Scholar] [CrossRef] [PubMed]
- Pilichowska, M.; Pittaluga, S.; Ferry, J.A.; Hemminger, J.; Chang, H.; Kanakry, J.A.; Sehn, L.H.; Feldman, T.; Abramson, J.S.; Kritharis, A.; et al. Clinicopathologic consensus study of gray zone lymphoma with features intermediate between DLBCL and classical HL. Blood Adv. 2017, 1, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Copie-Bergman, C.; Damotte, D.; Ben-Neriah, S.; Burroni, B.; Cornillon, J.; Lemal, R.; Golfier, C.; Fabiani, B.; Chassagne-Clément, C.; et al. Gray-zone Lymphoma Between cHL and Large B-Cell Lymphoma: A Histopathologic Series From the LYSA. Am. J. Surg. Pathol. 2019, 43, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, S.; Nicolae, A.; Wright, G.W.; Melani, C.; Roschewski, M.; Steinberg, S.; Huang, D.; Staudt, L.M.; Jaffe, E.S.; Wilson, W.H. Gene Expression Profiling of Mediastinal Gray Zone Lymphoma and Its Relationship to Primary Mediastinal B-cell Lymphoma and Classical Hodgkin Lymphoma. Blood Cancer Discov. 2020, 1, 155–161. [Google Scholar] [CrossRef]
- Wilson, W.H.; Pittaluga, S.; Nicolae, A.; Camphausen, K.; Shovlin, M.; Steinberg, S.M.; Roschewski, M.; Staudt, L.M.; Jaffe, E.S.; Dunleavy, K. A prospective study of mediastinal gray-zone lymphoma. Blood 2014, 124, 1563–1569. [Google Scholar] [CrossRef]
- Aussedat, G.; Traverse-Glehen, A.; Stamatoullas, A.; Molina, T.; Safar, V.; Laurent, C.; Michot, J.M.; Hirsch, P.; Nicolas-Virelizier, E.; Lamure, S.; et al. Composite and sequential lymphoma between classical Hodgkin lymphoma and primary mediastinal lymphoma/diffuse large B-cell lymphoma, a clinico-pathological series of 25 cases. Br. J. Haematol. 2020, 189, 244–256. [Google Scholar] [CrossRef]
- Eberle, F.C.; Salaverria, I.; Steidl, C.; Summers, T.A., Jr.; Pittaluga, S.; Neriah, S.B.; Rodriguez-Canales, J.; Xi, L.; Ylaya, K.; Liewehr, D.; et al. Gray zone lymphoma: Chromosomal aberrations with immunophenotypic and clinical correlations. Mod. Pathol. 2011, 24, 1586–1597. [Google Scholar] [CrossRef]
- Sarkozy, C.; Chong, L.; Takata, K.; Chavez, E.A.; Miyata-Takata, T.; Duns, G.; Telenius, A.; Boyle, M.; Slack, G.W.; Laurent, C.; et al. Gene expression profiling of gray zone lymphoma. Blood Adv. 2020, 4, 2523–2535. [Google Scholar] [CrossRef]
- Sarkozy, C.; Hung, S.S.; Chavez, E.A.; Duns, G.; Takata, K.; Chong, L.C.; Aoki, T.; Jiang, A.; Miyata-Takata, T.; Telenius, A.; et al. Mutational landscape of gray zone lymphoma. Blood 2021, 137, 1765–1776. [Google Scholar] [CrossRef]
- Kansal, R.; Sait, S.N.; Block, A.W.; Ward, P.M.; Kelly, F.L.; Cheney, R.T.; Czuczman, M.; Brecher, M.L.; Barcos, M. Extra copies of chromosome 2 are a recurring aberration in ALK-negative lymphomas with anaplastic morphology. Mod. Pathol. 2005, 18, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Singleton, T.P.; Ross, C.W.; Finn, W.G.; Padmore, R.F.; Schnitzer, B. Follicular hodgkin lymphoma: A histopathologic study. Am. J. Clin. Pathol. 2002, 117, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Vose, J.; Armitage, J.; Weisenburger, D.; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- de Leval, L.; Rickman, D.S.; Thielen, C.; de Reynies, A.; Huang, Y.L.; Delsol, G.; Lamant, L.; Leroy, K.; Brière, J.; Molina, T.; et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood 2007, 109, 4952–4963. [Google Scholar] [CrossRef]
- Agostinelli, C.; Hartmann, S.; Klapper, W.; Korkolopoulou, P.; Righi, S.; Marafioti, T.; Piccaluga, P.P.; Patsouris, E.; Hansmann, M.L.; Lennert, K.; et al. Peripheral T cell lymphomas with follicular T helper phenotype: A new basket or a distinct entity? Revising Karl Lennert’s personal archive. Histopathology 2011, 59, 679–691. [Google Scholar] [CrossRef]
- Mourad, N.; Mounier, N.; Brière, J.; Raffoux, E.; Delmer, A.; Feller, A.; Meijer, C.J.; Emile, J.F.; Bouabdallah, R.; Bosly, A.; et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood 2008, 111, 4463–4470. [Google Scholar] [CrossRef]
- Chiba, S.; Sakata-Yanagimoto, M. Advances in understanding of angioimmunoblastic T-cell lymphoma. Leukemia 2020, 34, 2592–2606. [Google Scholar] [CrossRef]
- Huang, Y.; Moreau, A.; Dupuis, J.; Streubel, B.; Petit, B.; Le Gouill, S.; Martin-Garcia, N.; Copie-Bergman, C.; Gaillard, F.; Qubaja, M.; et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am. J. Surg. Pathol. 2009, 33, 682–690. [Google Scholar] [CrossRef]
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica 2017, 102, e148–e151. [Google Scholar] [CrossRef]
- Ghione, P.; Faruque, P.; Mehta-Shah, N.; Seshan, V.; Ozkaya, N.; Bhaskar, S.; Yeung, J.; Spinner, M.A.; Lunning, M.; Inghirami, G.; et al. T follicular helper phenotype predicts response to histone deacetylase inhibitors in relapsed/refractory peripheral T-cell lymphoma. Blood Adv. 2020, 4, 4640–4647. [Google Scholar] [CrossRef] [PubMed]
- Falchi, L.; Ma, H.; Klein, S.; Lue, J.K.; Montanari, F.; Marchi, E.; Deng, C.; Kim, H.A.; Rada, A.; Jacob, A.T.; et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: A multicenter phase 2 study. Blood 2021, 137, 2161–2170. [Google Scholar] [CrossRef]
- Krug, A.; Tari, G.; Saidane, A.; Gaulard, P.; Ricci, J.E.; Lemonnier, F.; Verhoeyen, E. Novel T Follicular Helper-like T-Cell Lymphoma Therapies: From Preclinical Evaluation to Clinical Reality. Cancers 2022, 14, 2392. [Google Scholar] [CrossRef]
- Attygalle, A.; Al-Jehani, R.; Diss, T.C.; Munson, P.; Liu, H.; Du, M.Q.; Isaacson, P.G.; Dogan, A. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood 2002, 99, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Attygalle, A.D.; Cabeçadas, J.; Gaulard, P.; Jaffe, E.S.; de Jong, D.; Ko, Y.H.; Said, J.; Klapper, W. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward—Report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology 2014, 64, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef]
- Rodríguez, M.; Alonso-Alonso, R.; Tomás-Roca, L.; Rodríguez-Pinilla, S.M.; Manso-Alonso, R.; Cereceda, L.; Borregón, J.; Villaescusa, T.; Córdoba, R.; Sánchez-Beato, M.; et al. Peripheral T-cell lymphoma: Molecular profiling recognizes subclasses and identifies prognostic markers. Blood Adv. 2021, 5, 5588–5598. [Google Scholar] [CrossRef]
- Dobson, R.; Du, P.Y.; Rásó-Barnett, L.; Yao, W.Q.; Chen, Z.; Casa, C.; Ei-Daly, H.; Farkas, L.; Soilleux, E.; Wright, P.; et al. Early detection of T-cell lymphoma with T follicular helper phenotype by RHOA mutation analysis. Haematologica 2022, 107, 489–499. [Google Scholar] [CrossRef]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef]
- Steinhilber, J.; Mederake, M.; Bonzheim, I.; Serinsöz-Linke, E.; Müller, I.; Fallier-Becker, P.; Lemonnier, F.; Gaulard, P.; Fend, F.; Quintanilla-Martinez, L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2R172 mutations. Mod. Pathol. 2019, 32, 1123–1134. [Google Scholar] [CrossRef]
- Basha, B.M.; Bryant, S.C.; Rech, K.L.; Feldman, A.L.; Vrana, J.A.; Shi, M.; Reed, K.A.; King, R.L. Application of a 5 Marker Panel to the Routine Diagnosis of Peripheral T-Cell Lymphoma With T-Follicular Helper Phenotype. Am. J. Surg. Pathol. 2019, 43, 1282–1290. [Google Scholar] [CrossRef]
- Schwab, U.; Stein, H.; Gerdes, J.; Lemke, H.; Kirchner, H.; Schaadt, M.; Diehl, V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 1982, 299, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, N.T.; Stein, H.; Gatter, K.C.; Wainscoat, J.S.; Crick, J.; Al Saati, T.; Falini, B.; Delsol, G.; Mason, D.Y. Genotypic analysis of large cell lymphomas which express the Ki-1 antigen. Histopathology 1987, 11, 733–740. [Google Scholar] [CrossRef]
- Foss, H.D.; Anagnostopoulos, I.; Araujo, I.; Assaf, C.; Demel, G.; Kummer, J.A.; Hummel, M.; Stein, H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood 1996, 88, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugières, L.; Terrier-Lacombe, M.J.; Haralambieva, E.; Pulford, K.; Pileri, S.; Morris, S.W.; et al. ALK-positive lymphoma: A single disease with a broad spectrum of morphology. Blood 1998, 91, 2076–2084. [Google Scholar] [CrossRef]
- Falini, B.; Bigerna, B.; Fizzotti, M.; Pulford, K.; Pileri, S.A.; Delsol, G.; Carbone, A.; Paulli, M.; Magrini, U.; Menestrina, F.; et al. ALK expression defines a distinct group of T/null lymphomas (“ALK lymphomas”) with a wide morphological spectrum. Am. J. Pathol. 1998, 153, 875–886. [Google Scholar] [CrossRef]
- Falini, B.; Pulford, K.; Pucciarini, A.; Carbone, A.; De Wolf-Peeters, C.; Cordell, J.; Fizzotti, M.; Santucci, A.; Pelicci, P.G.; Pileri, S.; et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999, 94, 3509–3515. [Google Scholar]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef]
- King, R.L.; Dao, L.N.; McPhail, E.D.; Jaffe, E.S.; Said, J.; Swerdlow, S.H.; Sattler, C.A.; Ketterling, R.P.; Sidhu, J.S.; Hsi, E.D.; et al. Morphologic Features of ALK-negative Anaplastic Large Cell Lymphomas with DUSP22 Rearrangements. Am. J. Surg. Pathol. 2016, 40, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Zettl, A.; Rüdiger, T.; Konrad, M.A.; Chott, A.; Simonitsch-Klupp, I.; Sonnen, R.; Müller-Hermelink, H.K.; Ott, G. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am. J. Pathol. 2004, 164, 1837–1848. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Drug Approvals and Databases. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizotinib-children-and-young-adults-relapsed-or-refractory-systemic-anaplastic-large (accessed on 9 April 2023).
- Piva, R.; Agnelli, L.; Pellegrino, E.; Todoerti, K.; Grosso, V.; Tamagno, I.; Fornari, A.; Martinoglio, B.; Medico, E.; Zamò, A.; et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J. Clin. Oncol. 2010, 28, 1583–1590. [Google Scholar] [CrossRef]
- Iqbal, J.; Weisenburger, D.D.; Greiner, T.C.; Vose, J.M.; McKeithan, T.; Kucuk, C.; Geng, H.; Deffenbacher, K.; Smith, L.; Dybkaer, K.; et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010, 115, 1026–1036. [Google Scholar] [CrossRef]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017, 130, 2709–2717. [Google Scholar] [CrossRef]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Trümper, L.; Iyer, S.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann. Oncol. 2022, 33, 288–298. [Google Scholar] [CrossRef]
- Miranda, R.N.; Aladily, T.N.; Prince, H.M.; Kanagal-Shamanna, R.; de Jong, D.; Fayad, L.E.; Amin, M.B.; Haideri, N.; Bhagat, G.; Brooks, G.S.; et al. Breast implant-associated anaplastic large-cell lymphoma: Long-term follow-up of 60 patients. J. Clin. Oncol. 2014, 32, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ferrufino-Schmidt, M.C.; Medeiros, L.J.; Liu, H.; Clemens, M.W.; Hunt, K.K.; Laurent, C.; Lofts, J.; Amin, M.B.; Ming Chai, S.; Morine, A.; et al. Clinicopathologic Features and Prognostic Impact of Lymph Node Involvement in Patients with Breast Implant-associated Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2018, 42, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Medeiros, L.J.; Clemens, M.W.; Ferrufino-Schmidt, M.C.; Pina-Oviedo, S.; Miranda, R.N. Breast implant-associated anaplastic large cell lymphoma: A review. Mod. Pathol. 2019, 32, 166–188. [Google Scholar] [CrossRef]
- Medeiros, L.J.; Marques-Piubelli, M.L.; Sangiorgio, V.F.I.; Ruiz-Cordero, R.; Vega, F.; Feldman, A.L.; Chapman, J.R.; Clemens, M.W.; Hunt, K.K.; Evans, M.G.; et al. Epstein-Barr-virus-positive large B-cell lymphoma associated with breast implants: An analysis of eight patients suggesting a possible pathogenetic relationship. Mod. Pathol. 2021, 34, 2154–2167. [Google Scholar] [CrossRef] [PubMed]
- Mescam, L.; Camus, V.; Schiano, J.M.; Adélaïde, J.; Picquenot, J.M.; Guille, A.; Bannier, M.; Ruminy, P.; Viailly, P.J.; Jardin, F.; et al. EBV+ diffuse large B-cell lymphoma associated with chronic inflammation expands the spectrum of breast implant-related lymphomas. Blood 2020, 135, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Yamashita, D.; Nakamura, S. Nodal EBV+ cytotoxic T-cell lymphoma: A literature review based on the 2017 WHO classification. J. Clin. Exp. Hematop. 2020, 60, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Heavican, T.B.; Bouska, A.; Yu, J.; Lone, W.; Amador, C.; Gong, Q.; Zhang, W.; Li, Y.; Dave, B.J.; Nairismägi, M.L.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef]
- Amador, C.; Greiner, T.C.; Heavican, T.B.; Smith, L.M.; Galvis, K.T.; Lone, W.; Bouska, A.; D’Amore, F.; Pedersen, M.B.; Pileri, S.; et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood 2019, 134, 2159–2170. [Google Scholar] [CrossRef]
- Scott, A.J.; Tokaz, M.C.; Jacobs, M.F.; Chinnaiyan, A.M.; Phillips, T.J.; Wilcox, R.A. Germline variants discovered in lymphoma patients undergoing tumor profiling: A case series. Fam. Cancer 2021, 20, 61–65. [Google Scholar] [CrossRef]
- Leeksma, O.C.; de Miranda, N.F.; Veelken, H. Germline mutations predisposing to diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e532. [Google Scholar] [CrossRef]
- Rendleman, J.; Antipin, Y.; Reva, B.; Adaniel, C.; Przybylo, J.A.; Dutra-Clarke, A.; Hansen, N.; Heguy, A.; Huberman, K.; Borsu, L.; et al. Genetic variation in DNA repair pathways and risk of non-Hodgkin’s lymphoma. PLoS ONE 2014, 9, e101685. [Google Scholar] [CrossRef]
- Usui, Y.; Iwasaki, Y.; Matsuo, K.; Endo, M.; Kamatani, Y.; Hirata, M.; Sugano, K.; Yoshida, T.; Matsuda, K.; Murakami, Y.; et al. Association between germline pathogenic variants in cancer-predisposing genes and lymphoma risk. Cancer Sci. 2022, 113, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Riaz, I.B.; Faridi, W.; Patnaik, M.M.; Abraham, R.S. A Systematic Review on Predisposition to Lymphoid (B and T cell) Neoplasias in Patients with Primary Immunodeficiencies and Immune Dysregulatory Disorders (Inborn Errors of Immunity). Front. Immunol. 2019, 10, 777. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).