


Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Organic Solvents


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals Used and Preparation of Solutions
2.2. Steady-State Absorption and Fluorescence
2.3. Time-Correlated Single Photon Counting
2.4. Femtosecond and Nanosecond Transient Absorption Spectroscopy
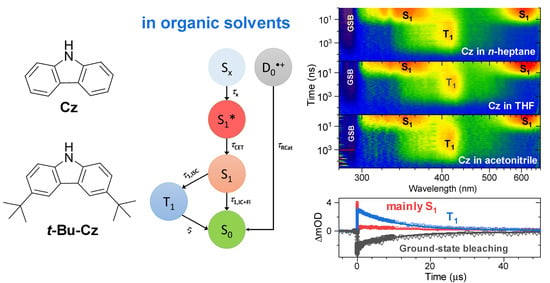
3. Results
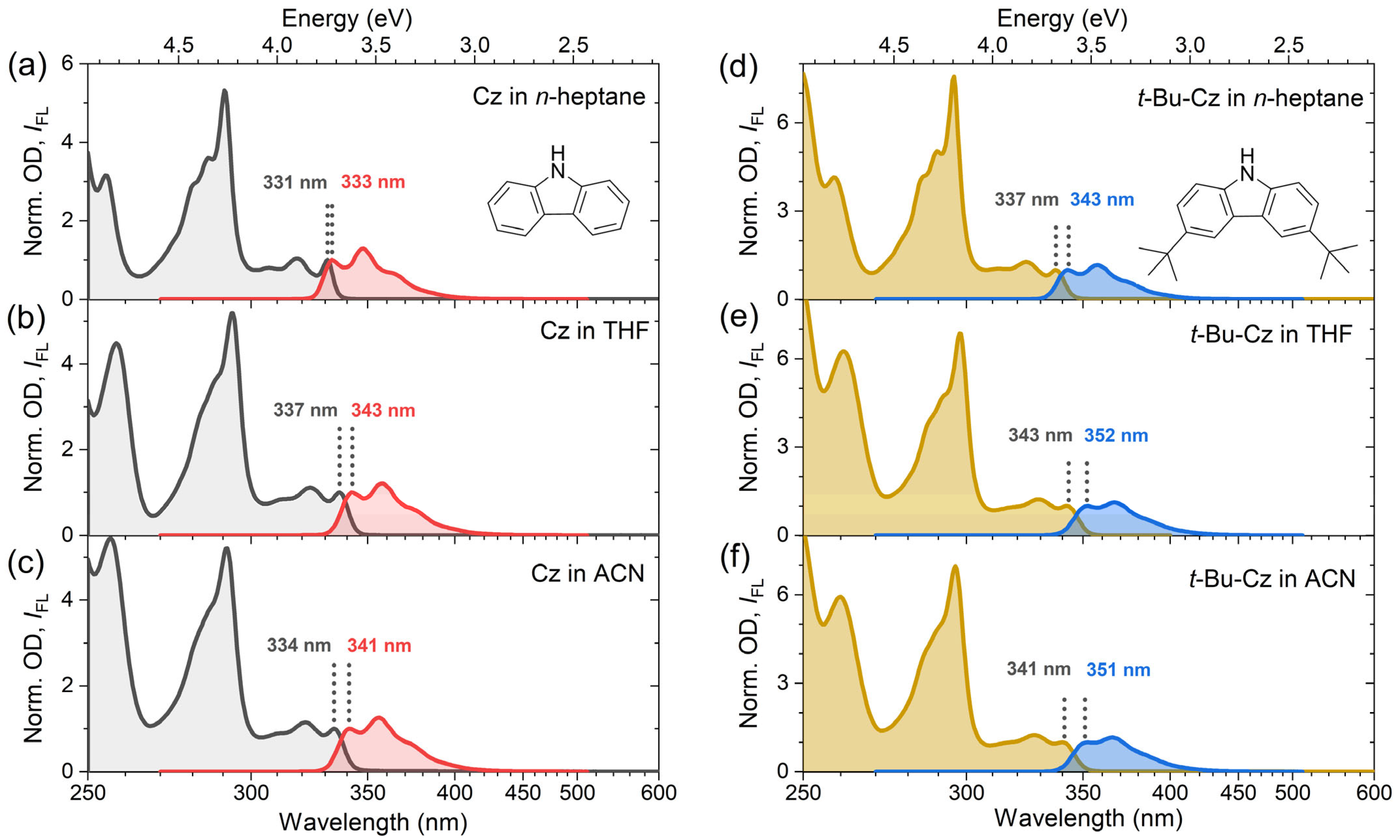
3.1. Steady-State Absorption and Emission of Carbazole and 3,6-Di-Tert-Butyl-Carbazole
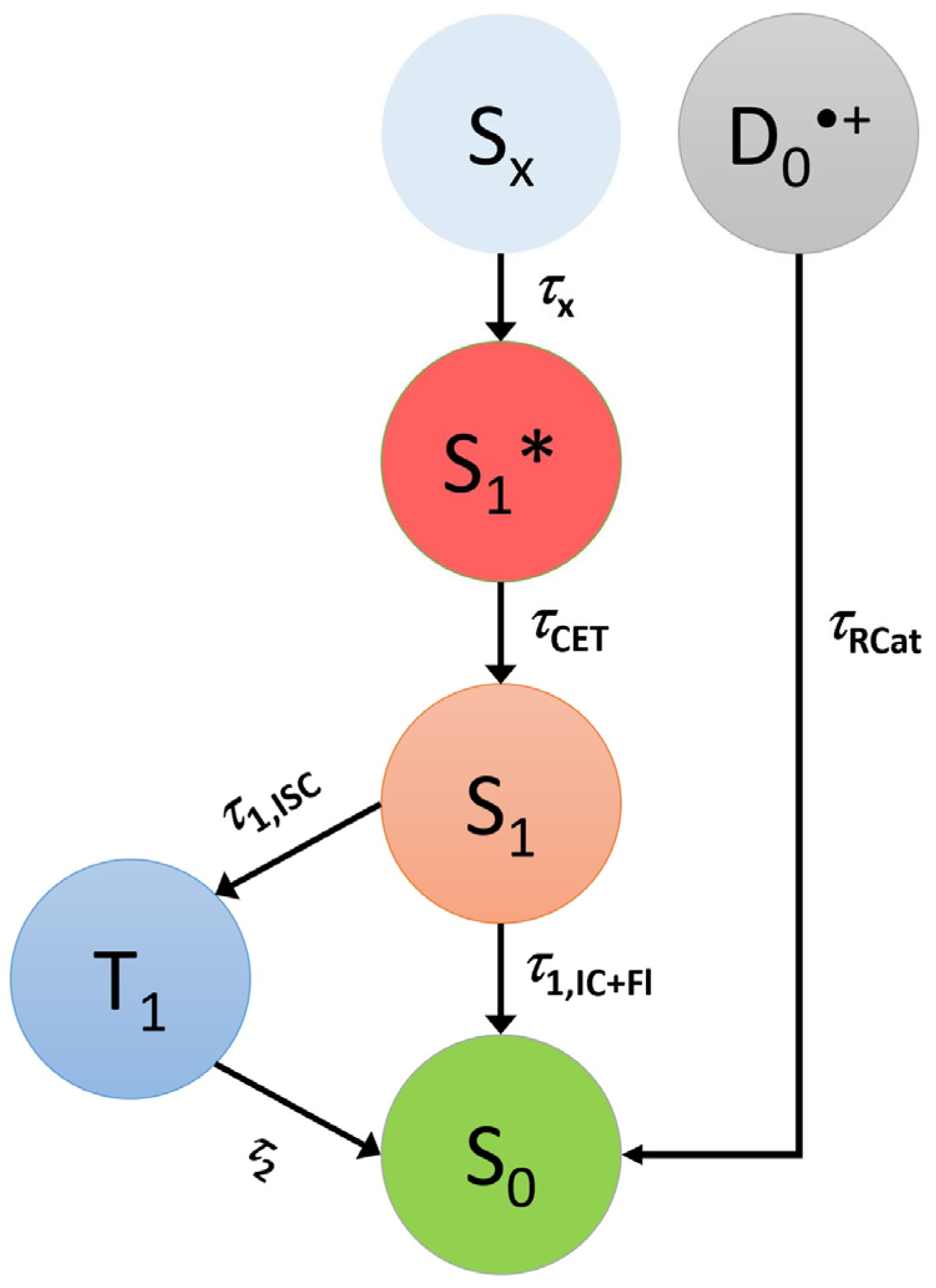
3.2. Transient Absorption Studies and TCSPC Experiments of Carbazole in Organic Solvents
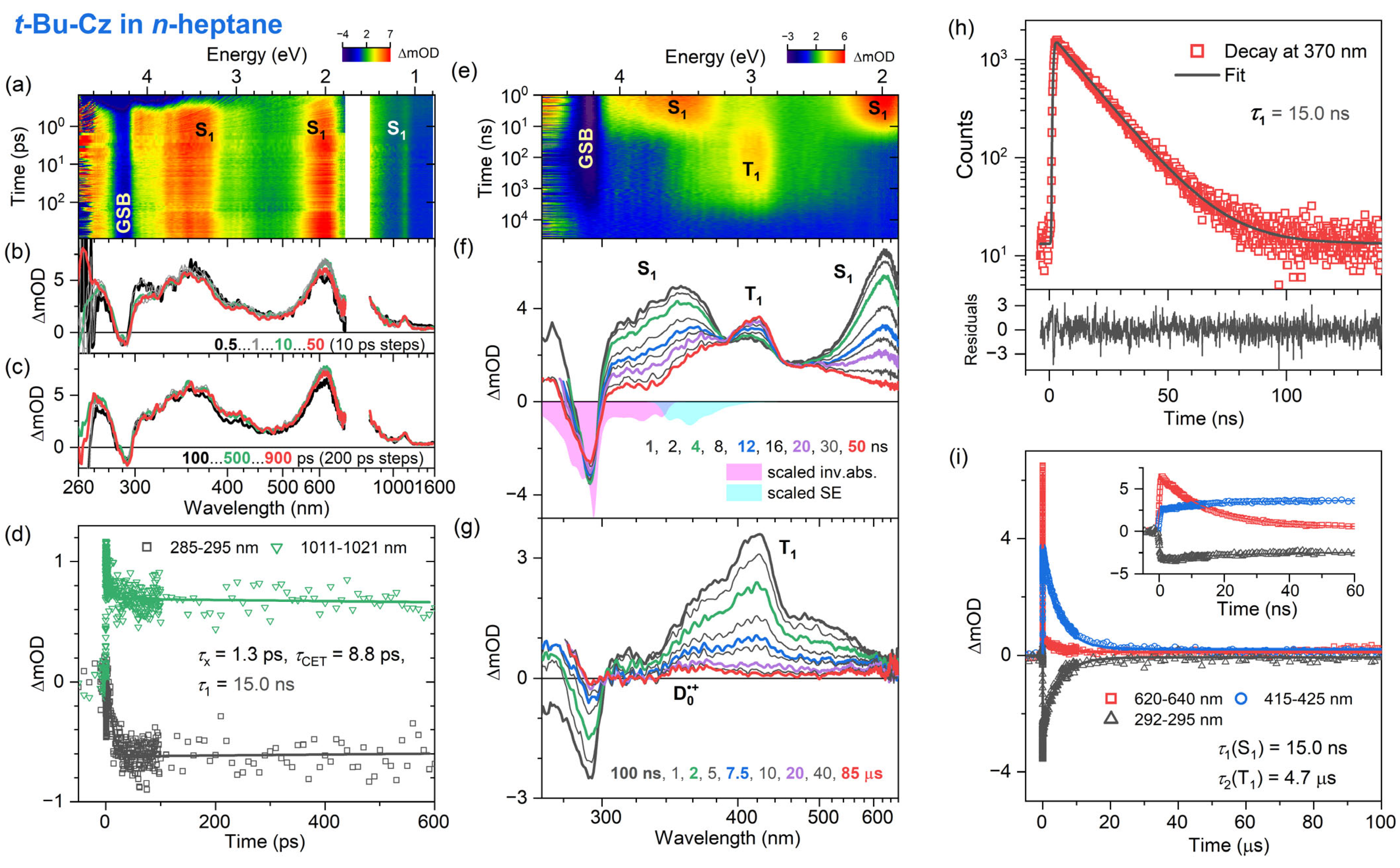
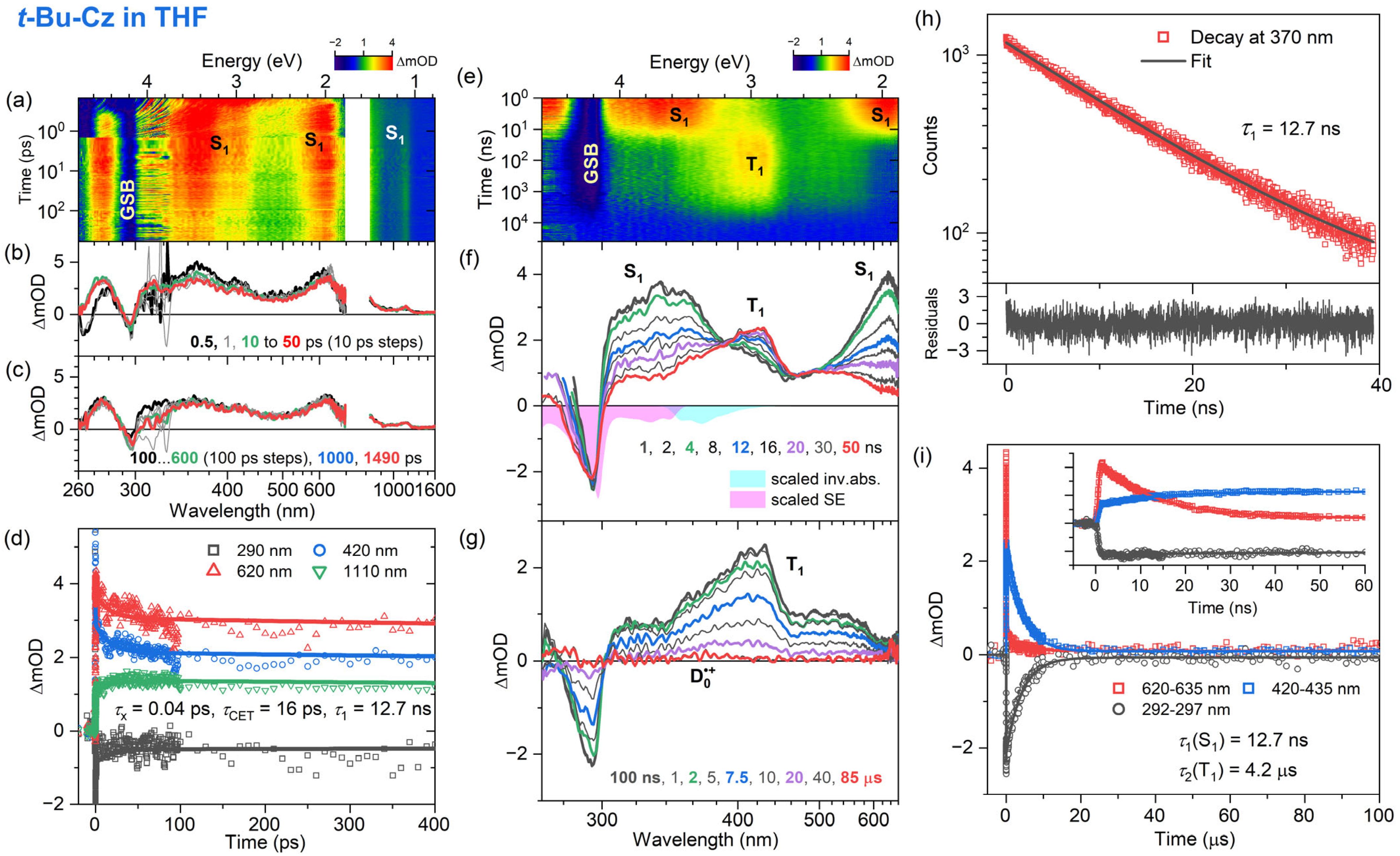
3.3. Transient Absorption and TCSPC Experiments for 3,6-Di-Tert-Butyl Carbazole in Organic Solvents
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blouin, N.; Leclerc, M. Poly(2,7-carbazole)s: Structure-Property Relationships. Acc. Chem. Res. 2008, 41, 1110–1119. [Google Scholar] [CrossRef]
- Beaupré, S.; Leclerc, M. PCDTBT: En route for low cost plastic solar cells. J. Mater. Chem. A 2013, 1, 11097–11105. [Google Scholar] [CrossRef]
- Shizu, K.; Lee, J.; Tanaka, H.; Nomura, H.; Yasuda, T.; Kaji, H.; Adachi, C. Highly efficient electroluminescence from purely organic donor–acceptor systems. Pure Appl. Chem. 2015, 87, 627–638. [Google Scholar] [CrossRef]
- Dumur, F. Carbazole-based polymers as hosts for solution-processed organic light-emitting diodes: Simplicity, efficacy. Org. Electron. 2016, 25, 345–361. [Google Scholar] [CrossRef]
- Ledwon, P. Recent advances of donor-acceptor type carbazole-based molecules for light emitting applications. Org. Electron. 2019, 75, 105422. [Google Scholar] [CrossRef]
- Wex, B.; Kaafarani, B.R. Perspective on carbazole-based organic compounds as emitters and hosts in TADF applications. J. Mater. Chem. C 2017, 5, 8622–8653. [Google Scholar] [CrossRef]
- Kondo, Y.; Yoshiura, K.; Kitera, S.; Nishi, H.; Oda, S.; Gotoh, H.; Sasada, Y.; Yanai, M.; Hatakeyama, T. Narrowband deep-blue organic light-emitting diode featuring an organoboron-based emitter. Nat. Photonics 2019, 13, 678–682. [Google Scholar] [CrossRef]
- Ahn, D.H.; Kim, S.W.; Lee, H.; Ko, I.J.; Karthik, D.; Lee, J.Y.; Kwon, J.H. Highly efficient blue thermally activated delayed fluorescence emitters based on symmetrical and rigid oxygen-bridged boron acceptors. Nat. Photonics 2019, 13, 540–546. [Google Scholar] [CrossRef]
- Hwang, J.; Koh, C.W.; Ha, J.M.; Woo, H.Y.; Park, S.; Cho, M.J.; Choi, D.H. Aryl-Annulated [3,2-a] Carbazole-Based Deep-Blue Soluble Emitters for High-Efficiency Solution-Processed Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes with CIEy < 0.1. ACS Appl. Mater. Interfaces 2021, 13, 61454–61462. [Google Scholar] [PubMed]
- Lee, H.; Braveenth, R.; Muruganantham, S.; Jeon, C.Y.; Lee, H.S.; Kwon, J.H. Efficient pure blue hyperfluorescence devices utilizing quadrupolar donor-acceptor-donor type of thermally activated delayed fluorescence sensitizers. Nat. Commun. 2023, 14, 419. [Google Scholar] [CrossRef]
- Hofkens, J.; Cotlet, M.; Vosch, T.; Tinnefeld, P.; Weston, K.D.; Ego, C.; Grimsdale, A.; Müllen, K.; Beljonne, D.; Brédas, J.L.; et al. Revealing competitive Förster-type resonance energy-transfer pathways in single bichromophoric molecules. Proc. Natl. Acad. Sci. USA 2003, 100, 13146–13151. [Google Scholar] [CrossRef] [PubMed]
- Ruseckas, A.; Ribierre, J.C.; Shaw, P.E.; Staton, S.V.; Burn, P.L.; Samuel, I.D.W. Singlet energy transfer and singlet-singlet annihilation in light-emitting blends of organic semiconductors. Appl. Phys. Lett. 2009, 95, 183305. [Google Scholar] [CrossRef]
- Hedley, G.J.; Schröder, T.; Steiner, F.; Eder, T.; Hofmann, F.J.; Bange, S.; Laux, D.; Höger, S.; Tinnefeld, P.; Lupton, J.M.; et al. Picosecond time-resolved photon antibunching measures nanoscale exciton motion and the true number of chromophores. Nat. Commun. 2021, 12, 1327. [Google Scholar] [CrossRef] [PubMed]
- Haase, N.; Danos, A.; Pflumm, C.; Stachelek, P.; Brütting, W.; Monkman, A.P. Are the rates of dexter transfer in TADF hyperfluorescence systems optically accessible? Mater. Horiz. 2021, 8, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, M.; Lenzer, T.; Oum, K. Understanding Excited-State Relaxation in 1,3-Bis(N-carbazolyl)benzene, a Host Material for Organic Light-Emitting Diodes. J. Phys. Chem. C 2023, 127, 4582–4593. [Google Scholar] [CrossRef]
- Walba, H.; Branch, G.E.K. The Absorption Spectra of Some N-Substituted p-Aminotriphenylmethyl Ions. J. Am. Chem. Soc. 1951, 73, 3341–3348. [Google Scholar] [CrossRef]
- Adams, J.E.; Mantulin, W.W.; Huber, J.R. Effect of Molecular Geometry on Spin-Orbit Coupling of Aromatic Amines in Solution. Diphenylamine, Iminobibenzyl, Acridan, and Carbazole. J. Am. Chem. Soc. 1973, 95, 5477–5481. [Google Scholar] [CrossRef]
- Johnson, G.E. A Spectroscopic Study of Carbazole by Photoselection. J. Phys. Chem. 1974, 78, 1512–1521. [Google Scholar] [CrossRef]
- Auty, A.R.; Jones, A.C.; Phillips, D. Spectroscopy and decay dynamics of jet-cooled carbazole and N-ethylcarbazole and their homocyclic analogues. Chem. Phys. 1976, 103, 163–182. [Google Scholar] [CrossRef]
- Yu, H.; Zain, S.M.; Eigenbrot, I.V.; Phillips, D. Investigation of carbazole derivatives and their van der Waals complexes in the jet by laser-induced fluorescence spectroscopy. J. Photochem. Photobiol. A 1994, 80, 7–16. [Google Scholar] [CrossRef]
- Yi, J.T.; Alvarez-Valtierra, L.; Pratt, D.W. Rotationally resolved S1 ← S0 electronic spectra of fluorene, carbazole, and dibenzofuran: Evidence for Herzberg-Teller coupling with the S2 state. J. Chem. Phys. 2006, 124, 244302. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.R.; Kasha, M. Triplet-Triplet Absorption Studies on Aromatic and Heterocyclic Molecules at 77 °K. J. Chem. Phys. 1967, 47, 3319–3327. [Google Scholar] [CrossRef]
- Fratev, F.; Hermann, H.; Olbrich, G.; Polansky, O.E. Höhere Triplettanregungszustände von Fluoren, Carbazol und Benzologen: CNDO-CI-Berechnungen und Triplett-Triplett-Absorptionsmessungen. Z. Naturforsch. A Phys. Phys. Chem. Kosmophys. 1976, 31, 84–86. [Google Scholar] [CrossRef]
- Johnson, G.E. Intramolecular excimer formation in carbazole double molecules. J. Chem. Phys. 1974, 61, 3002–3008. [Google Scholar] [CrossRef]
- Masuhara, H.; Tohgo, Y.; Mataga, N. Fluorescence quenching processes of carbazole-amine systems as revealed by laser photolysis method. Chem. Lett. 1975, 4, 59–62. [Google Scholar] [CrossRef]
- Martin, M.M.; Ware, W.R. Fluorescence Quenching of Carbazole by Pyridine and Substituted Pyridines. Radiationless Processes in the Carbazole-Amine Hydrogen Bonded Complex. J. Phys. Chem. 1978, 82, 2770–2776. [Google Scholar] [CrossRef]
- Garner, A.; Wilkinson, F. Quenching of triplet states by molecular oxygen and the role of charge-transfer interactions. Chem. Phys. Lett. 1977, 45, 432–435. [Google Scholar] [CrossRef]
- Bigelow, R.W.; Ceasar, G.P. Hydrogen Bonding and N-Alkylation Effects on the Electronic Structure of Carbazole. J. Phys. Chem. 1979, 83, 1790–1795. [Google Scholar] [CrossRef]
- Johnson, G.E. Fluorescence Quenching of Carbazoles. J. Phys. Chem. 1980, 84, 2940–2946. [Google Scholar] [CrossRef]
- Martin, M.M.; Bréhéret, E. Hydrogen Bonding Interaction Effect on Carbazole Triplet State Photophysics. J. Phys. Chem. 1982, 86, 107–111. [Google Scholar] [CrossRef]
- Kikuchi, K.; Yamamoto, S.A.; Kokubun, H. Hydrogen atom transfer reaction from excited carbazole to pyridine. J. Photochem. 1984, 24, 271–283. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Erra-Balsells, R. Electronic spectroscopy of carbazole and N- and C-substituted carbazoles in homogeneous media and in solid matrix. J. Lumin. 2001, 93, 51–74. [Google Scholar] [CrossRef]
- Boo, B.H.; Ryu, S.Y.; Kang, H.S.; Koh, S.G.; Park, C.-J. Time-resolved Fluorescence Studies of Carbazole and Poly(N-vinylcarbazole) for Elucidating Intramolecular Excimer Formation. Bull. Korean Chem. Soc. 2010, 57, 406–411. [Google Scholar] [CrossRef]
- Bayda-Smykaj, M.; Burdzinski, G.; Ludwiczak, M.; Hug, G.L.; Marciniak, B. Early Events in the Photoinduced Electron Transfer between Carbazole and Divinylbenzene in a Silylene-Bridged Donor-Acceptor Compound. J. Phys. Chem. C 2020, 124, 19522–19529. [Google Scholar] [CrossRef]
- Thornton, G.L.; Phelps, R.; Orr-Ewing, A.J. Transient absorption spectroscopy of the electron transfer step in the photochemically activated polymerizations of N-ethylcarbazole and 9-phenylcarbazole. Phys. Chem. Chem. Phys. 2021, 23, 18378–18392. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, Z.; Liu, Y.; Guan, T.; Zhang, Q.; Qin, C.; Jiang, K.; Liu, Y. Transient Absorption Spectroscopy of a Carbazole-Based Room-Temperature Phosphorescent Molecule: Real-Time Monitoring of Singlet-Triplet Transitions. J. Phys. Chem. Lett. 2022, 13, 9381–9389. [Google Scholar] [CrossRef] [PubMed]
- Hiyoshi, R.; Hiura, H.; Sakamoto, Y.; Mizuno, M.; Sakai, M.; Takahashi, H. Time-resolved absorption and time-resolved Raman spectroscopies of the photochemistry of carbazole and N-ethylcarbazole. J. Mol. Struct. 2003, 661–662, 481–489. [Google Scholar] [CrossRef]
- Notsuka, N.; Nakanotani, H.; Noda, H.; Goushi, K.; Adachi, C. Observation of Nonradiative Deactivation Behavior from Singlet and Triplet States of Thermally Activated Delayed Fluorescence Emitters in Solution. J. Phys. Chem. Lett. 2020, 11, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kang, H.; Jeong, J.-E.; Park, S.H.; Koh, C.W.; Kim, C.W.; Woo, H.Y.; Cho, M.J.; Park, S.; Choi, D.H. Ultra-Deep-Blue Aggregation-Induced Delayed Fluorescence Emitters: Achieving Nearly 16% EQE in Solution-Processed Nondoped and Doped OLEDs with CIEy < 0.1. Adv. Funct. Mater. 2021, 31, 2102588. [Google Scholar]
- Morgenroth, M.; Scholz, M.; Guy, L.; Oum, K.; Lenzer, T. Spatiotemporal Mapping of Efficient Chiral Induction by Helicene-Type Additives in Copolymer Thin Films. Angew. Chem. Int. Ed. 2022, 61, e202203075. [Google Scholar] [CrossRef]
- Oum, K.; Lenzer, T.; Scholz, M.; Jung, D.Y.; Sul, O.; Cho, B.J.; Lange, J.; Müller, A. Observation of Ultrafast Carrier Dynamics and Phonon Relaxation of Graphene from the Deep-Ultraviolet to the Visible Region. J. Phys. Chem. C 2014, 118, 6454–6461. [Google Scholar] [CrossRef]
- Flender, O.; Scholz, M.; Klein, J.R.; Oum, K.; Lenzer, T. Excited-state relaxation of the solar cell dye D49 in organic solvents and on mesoporous Al2O3 and TiO2 thin films. Phys. Chem. Chem. Phys. 2016, 18, 26010–26019. [Google Scholar] [CrossRef]
- Dobryakov, A.L.; Kovalenko, S.A.; Weigel, A.; Pérez Lustres, J.L.; Lange, J.; Müller, A.; Ernsting, N.P. Femtosecond pump/supercontinuum-probe spectroscopy: Optimized setup and signal analysis for single-shot spectral referencing. Rev. Sci. Instrum. 2010, 81, 113106. [Google Scholar] [CrossRef] [PubMed]
- Merker, A.; Scholz, M.; Morgenroth, M.; Lenzer, T.; Oum, K. Photoinduced Dynamics of (CH3NH3)4Cu2Br6 Thin Films Indicating Efficient Triplet Photoluminescence. J. Phys. Chem. Lett. 2021, 12, 2736–2741. [Google Scholar] [CrossRef] [PubMed]
- Ljubić, I.; Sabljić, A. CASSCF/CASPT2 and TD-DFT Study of Valence and Rydberg Electronic Transitions in Fluorene, Carbazole, Dibenzofuran, and Dibenzothiophene. J. Phys. Chem. A 2011, 115, 4840–4850. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.R. Classification of Spectra of Cata-Condensed Hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Lohse, P.W.; Bürsing, R.; Lenzer, T.; Oum, K. Exploring 12′-Apo-β-carotenoic-12′-acid as an Ultrafast Polarity Probe for Ionic Liquids. J. Phys. Chem. B 2008, 112, 3048–3057. [Google Scholar] [CrossRef][Green Version]
- Lide, D.R. (Ed.) Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Schwarzer, D.; Troe, J.; Votsmeier, M.; Zerezke, M. Collisional deactivation of vibrationally highly excited azulene in compressed liquids and supercritical fluids. J. Chem. Phys. 1996, 105, 3121–3131. [Google Scholar] [CrossRef]
- Schwarzer, D.; Hanisch, C.; Kutne, P.; Troe, J. Vibrational Energy Transfer in Highly Excited Bridged Azulene-Aryl Compounds: Direct Observation of Energy Flow through Aliphatic Chains and into the Solvent. J. Phys. Chem. A 2002, 106, 8019–8028. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Schanz, R.; Hennig, H.; Ernsting, N.P. Cooling dynamics of an optically excited molecular probe in solution from femtosecond broadband absorption spectroscopy. J. Chem. Phys. 2001, 115, 3256–3273. [Google Scholar] [CrossRef]
- Andersson, P.O.; Gillbro, T. Photophysics and dynamics of the lowest excited singlet state in long substituted polyenes with implications to the very long-chain limit. J. Chem. Phys. 1995, 103, 2509–2519. [Google Scholar] [CrossRef]
- Lenzer, T.; Ehlers, F.; Scholz, M.; Oswald, R.; Oum, K. Assignment of carotene S* state features to the vibrationally hot ground electronic state. Phys. Chem. Chem. Phys. 2010, 12, 8832–8839. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Bréhéret, E.; Tfibel, F.; Lacourbas, B. Two-Photon Stepwise Dissociation of Carbazole in Solution. J. Phys. Chem. 1980, 84, 70–72. [Google Scholar] [CrossRef]
- Shida, T.; Nosaka, Y.; Kato, T. Electronic Absorption Spectra of Some Cation Radicals as Compared with Ultraviolet Photoelectron Spectra. J. Phys. Chem. 1978, 82, 695–698. [Google Scholar] [CrossRef]
- Haink, H.J.; Adams, J.E.; Huber, J.R. The Electronic Structure of Aromatic Amines: Photoelectron Spectroscopy of Diphenylamine, Iminobibenzyl, Acridan and Carbazole. Ber. Bunsenges. Phys. Chem. 1974, 78, 436–440. [Google Scholar] [CrossRef]
- Liptay, W. Electrochromism and Solvatochromism. Angew. Chem. Int. Ed. 1967, 8, 177–188. [Google Scholar] [CrossRef]
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1757. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.J.; Crespo-Otero, R. Excited state mechanisms in crystalline carbazole: The role of aggregation and isomeric defects. J. Mater. Chem. C 2021, 9, 11882–11892. [Google Scholar] [CrossRef]
- Chen, C.; Chong, K.C.; Pan, Y.; Qi, G.; Xu, S.; Liu, B. Revisiting Carbazole: Origin, Impurity, and Properties. ACS Mater. Lett. 2021, 3, 1081–1087. [Google Scholar] [CrossRef]
- Hosokai, T.; Matsuzaki, H.; Nakanotani, H.; Tokumaru, K.; Tsutsui, T.; Furube, A.; Nasu, K.; Nomura, H.; Yahiro, M.; Adachi, C. Evidence and mechanism of efficient thermally activated delayed fluorescence promoted by delocalized excited states. Sci. Adv. 2017, 3, e1603282. [Google Scholar] [CrossRef]
- Godumala, M.; Choi, S.; Cho, M.J.; Choi, D.H. Recent breakthroughs in thermally activated delayed fluorescence organic light emitting diodes containing non-doped emitting layers. J. Mater. Chem. C 2019, 7, 2172–2198. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Solvent | ∆f 1 | λabs0–0 | λfl0–0 | ∆λStokes0–0 | Stokes0–0 |
|---|---|---|---|---|---|---|
| (nm) | (nm) | (nm) | (cm−1) | |||
| Cz | n-Heptane | ≈0 | 331 | 333 | 2 | 193 |
| THF | 0.44 | 337 | 343 | 6 | 525 | |
| Acetonitrile | 0.71 | 334 | 341 | 7 | 614 | |
| t-Bu-Cz | n-Heptane | ≈0 | 337 | 343 | 6 | 519 |
| THF | 0.44 | 343 | 352 | 9 | 745 | |
| Acetonitrile | 0.71 | 341 | 351 | 10 | 835 |
| Cz | fs-TA | TCSPC | ns-TA | |||
|---|---|---|---|---|---|---|
| τx | τCET | τ1 (S1) | τ2 (T1) | Φ (T1) | τRCat | |
| n-Heptane | 0.7 ps | 19 ps | 14.7 ns | 5.9 μs | 55% | >50 μs |
| THF | 0.2 ps | 19 ps | 13.6 ns | 3.4 μs | 51% | >50 μs |
| Acetonitrile | 0.2 ps | 21 ps | 14.3 ns | 10.3 μs | 56% | >50 μs |
| t-Bu-Cz | fs-TA | TCSPC | ns-TA | |||
|---|---|---|---|---|---|---|
| τx | τCET | τ1 (S1) | τ2 (T1) | Φ (T1) | τRCat | |
| n-Heptane | 1.3 ps | 8.9 ps | 15.0 ns | 4.7 μs | 53% | >50 μs |
| THF | 0.04 ps | 16 ps | 12.7 ns | 4.2 μs | 36% | >50 μs |
| Acetonitrile | 0.3 ps | 16 ps | 14.1 ns | 7.5 μs | 54% | >50 μs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knötig, K.M.; Gust, D.; Lenzer, T.; Oum, K. Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Organic Solvents. Photochem 2024, 4, 163-178. https://doi.org/10.3390/photochem4020010
Knötig KM, Gust D, Lenzer T, Oum K. Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Organic Solvents. Photochem. 2024; 4(2):163-178. https://doi.org/10.3390/photochem4020010
Chicago/Turabian StyleKnötig, Konstantin Moritz, Domenic Gust, Thomas Lenzer, and Kawon Oum. 2024. "Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Organic Solvents" Photochem 4, no. 2: 163-178. https://doi.org/10.3390/photochem4020010
APA StyleKnötig, K. M., Gust, D., Lenzer, T., & Oum, K. (2024). Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Organic Solvents. Photochem, 4(2), 163-178. https://doi.org/10.3390/photochem4020010