
A PCR-RFLP Method for Distinguishing Closely Related Common Quail (Coturnix coturnix) and Japanese Quail (Coturnix japonica): Forensics and Conservation Implications

Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Specimen Sampling and DNA Extraction
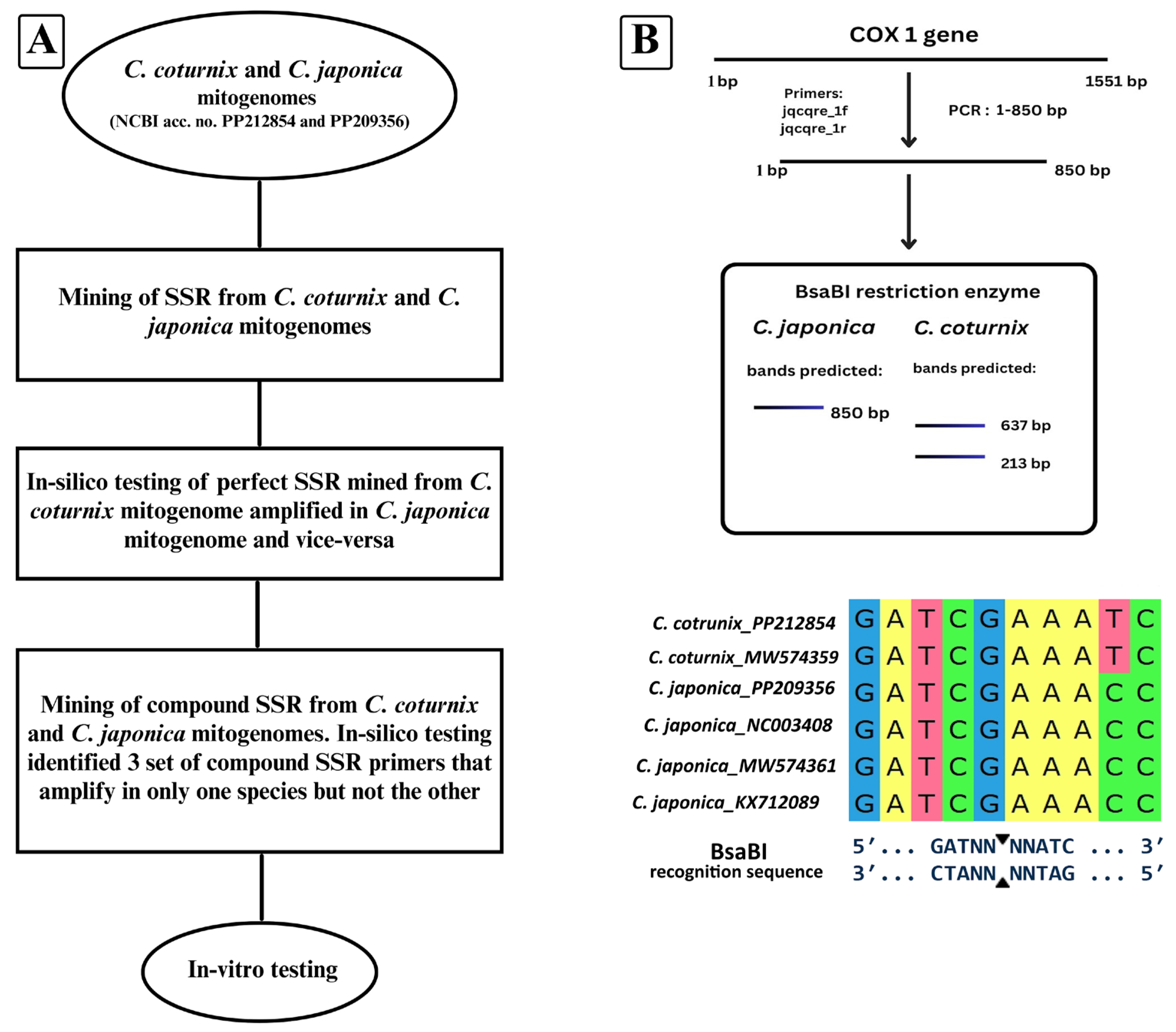
2.2. SSR Mining and In Vitro Analysis
2.3. Development and Testing of PCR-RFLP Assay
3. Results
3.1. Compound SSR Analysis
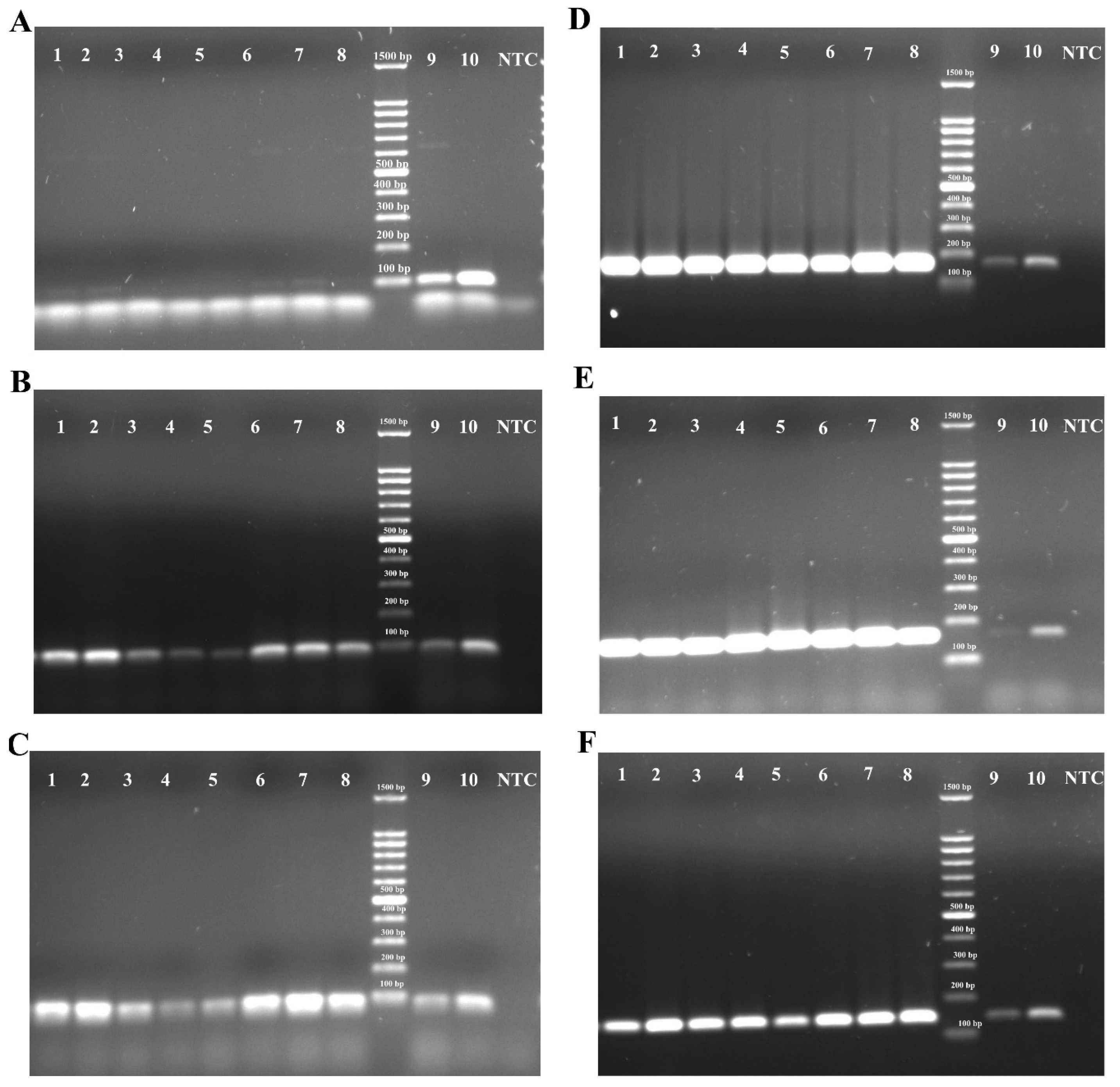
3.2. PCR-RFLP Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnsgard, P.A. The Quails, Partridges, and Francolins of the World; Oxford University Press: Oxford, UK, 1988; ISBN 978-0-19-857193-3. [Google Scholar]
- Minvielle, F. The Future of Japanese Quail for Research and Production. World’s Poult. Sci. J. 2004, 60, 500–507. [Google Scholar] [CrossRef]
- Kawahara-Miki, R.; Sano, S.; Nunome, M.; Shimmura, T.; Kuwayama, T.; Takahashi, S.; Kawashima, T.; Matsuda, Y.; Yoshimura, T.; Kono, T. Next-Generation Sequencing Reveals Genomic Features in the Japanese Quail. Genomics 2013, 101, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Hou, Z.; Fan, G.; Pi, J.; Sun, S.; Chen, J.; Liu, H.; Du, X.; Shen, J.; et al. Population Genomic Data Reveal Genes Related to Important Traits of Quail. GigaScience 2018, 7, giy049. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.M.; Hindle, M.M.; Boitard, S.; Burt, D.W.; Danner, A.F.; Eory, L.; Forrest, H.L.; Gourichon, D.; Gros, J.; Hillier, L.W.; et al. The Quail Genome: Insights into Social Behaviour, Seasonal Biology and Infectious Disease Response. BMC Biol. 2020, 18, 14. [Google Scholar] [CrossRef]
- Dey, P.; Ray, S.D.; Manchi, S.; Pramod, P.; Kochiganti, V.H.S.; Singh, R.P. Whole Genome Sequencing and Microsatellite Motif Discovery of Farmed Japanese Quail (Coturnix japonica): A First Record from India. Proc. Indian Natl. Sci. Acad. 2022, 88, 688–695. [Google Scholar] [CrossRef]
- McGowan, P.J.K.; Kirwan, G.M. Japanese Quail (Coturnix japonica), Version 1.0. Birds of the World 2020. Available online: https://birdsoftheworld.org/bow/species/japqua/1.0/introduction (accessed on 22 May 2025).
- McGowan, P.J.K.; Kirwan, G.M.; de Juana, E.; Boesman, P.F.D. Common Quail (Coturnix coturnix), Version 1.1. Birds of the World 2023. Available online: https://birdsoftheworld.org/bow/species/comqua1/1.1/introduction (accessed on 22 May 2025).
- Stein, R.W.; Brown, J.W.; Mooers, A.Ø. A Molecular Genetic Time Scale Demonstrates Cretaceous Origins and Multiple Diversification Rate Shifts within the Order Galliformes (Aves). Mol. Phylogenet. Evol. 2015, 92, 155–164. [Google Scholar] [CrossRef]
- Wang, N.; Kimball, R.T.; Braun, E.L.; Liang, B.; Zhang, Z. Ancestral Range Reconstruction of Galliformes: The Effects of Topology and Taxon Sampling. J. Biogeogr. 2017, 44, 122–135. [Google Scholar] [CrossRef]
- Dey, P.; Ray, S.D.; Kochiganti, V.H.S.; Pukazhenthi, B.S.; Koepfli, K.-P.; Singh, R.P. Mitogenomic Insights into the Evolution, Divergence Time, and Ancestral Ranges of Coturnix Quails. Genes 2024, 15, 742. [Google Scholar] [CrossRef]
- Ahmed, A. Live Bird Trade in Northern India; TRAFFIC-India: New Delhi, India, 1997. [Google Scholar]
- Wolf, C.; Rentsch, J.; Hübner, P. PCR−RFLP Analysis of Mitochondrial DNA: A Reliable Method for Species Identification. J. Agric. Food Chem. 1999, 47, 1350–1355. [Google Scholar] [CrossRef]
- Rojas, M.; González, I.; Fajardo, V.; Martín, I.; Hernández, P.E.; garcía, T.; Martín, R. Polymerase Chain Reaction-Restriction Fragment Length Polymorphism Authentication of Raw Meats from Game Birds. J. Aoac Int. 2008, 91, 1416–1422. [Google Scholar] [CrossRef]
- Government of India. Prohibition of Farming of Japanese Quails (F. No. 3-3/2011/WL-1). Available online: https://sansad.in/getFile/debatestextmk/15/IX/2012-Final.pdf?source=loksabhadocs (accessed on 22 May 2025).
- Government of India. Removal of Farm Bred Variety of Japanese Quail (F.No.1-15/2013-WL, 2013). Available online: https://forest.kerala.gov.in/kfstorage/2024/11/gazette27032014-22.pdf (accessed on 22 May 2025).
- Boonseub, S.; Tobe, S.S.; Linacre, A.M.T. The Use of Mitochondrial DNA Genes to Identify Closely Related Avian Species. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 275–277. [Google Scholar] [CrossRef]
- Elyasi Gorji, Z.; Izadpanah, M.; Hadi, F.; Zare, M. Mitochondrial Genes as Strong Molecular Markers for Species Identification. Nucleus 2022, 66, 81–93. [Google Scholar] [CrossRef]
- He, H.; Wang, Y.; Qing, Y.; Li, D.; Zhao, X.; Zhu, Q.; Yin, H. Molecular Authentication of Meats from Three Terrestrial Birds Based on Pcr-Rflp Analysis of the Mitochondrial 12S rRNA Gene. Braz. J. Poult. Sci. 2018, 20, 651–656. [Google Scholar] [CrossRef]
- Liu, K.; Wu, M.; Lin, X.; Lonan, P.; Chen, S.; Wu, Y.; Lai, X.; Yu, L.; Zhou, X.; Li, G. Molecular Analysis of Edible Bird’s Nest and Rapid Authentication of Aerodramus fuciphagus from Its Subspecies by PCR-RFLP Based on the Cytb Gene. Anal. Methods 2020, 12, 2710–2717. [Google Scholar] [CrossRef]
- Aranishi, F.; Okimoto, T.; Izumi, S. Identification of Gadoid Species (Pisces, Gadidae) by PCR-RFLP Analysis. J. Appl. Genet. 2005, 46, 69–73. [Google Scholar]
- Canfield, S.; Bowen, B. A Rapid PCR-RFLP Method for Species Identification of the Eastern Pacific Horn Sharks (Genus Heterodontus). Conserv. Genet. Resour. 2021, 13, 79–84. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple Sequences with Complex Evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-Wide Genetic Marker Discovery and Genotyping Using next-Generation Sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Castoe, T.A.; Poole, A.W.; de Koning, A.P.J.; Jones, K.L.; Tomback, D.F.; Oyler-McCance, S.J.; Fike, J.A.; Lance, S.L.; Streicher, J.W.; Smith, E.N.; et al. Rapid Microsatellite Identification from Illumina Paired-End Genomic Sequencing in Two Birds and a Snake. PLoS ONE 2012, 7, e30953. [Google Scholar] [CrossRef]
- Mondal, T.; Dey, P.; Kumari, D.; Ray, S.D.; Quadros, G.; Sastry Kochiganti, V.H.; Singh, R.P. Genome Survey Sequencing and Mining of Genome-Wide Microsatellite Markers in Yellow-Billed Babbler (Turdoides affinis). Heliyon 2023, 9, e12735. [Google Scholar] [CrossRef]
- Grimmett, R.; Inskipp, C.; Inskipp, T. Birds of the Indian Subcontinent: India, Pakistan, Sri Lanka, Nepal, Bhutan, Bangladesh and the Maldives; Bloomsbury Publishing: New Delhi, India, 2016; ISBN 978-1-4081-6265-1. [Google Scholar]
- Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor: New York, NY, USA, 1989; Volume 3, ISBN 978-0-87969-577-4. [Google Scholar]
- Du, L.; Zhang, C.; Liu, Q.; Zhang, X.; Yue, B. Krait: An Ultrafast Tool for Genome-Wide Survey of Microsatellites and Primer Design. Bioinformatics 2018, 34, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Lee, D.; Schulman, A.H. FastPCR Software for PCR, in Silico PCR, and Oligonucleotide Assembly and Analysis. Methods Mol. Biol. 2014, 1116, 271–302. [Google Scholar] [CrossRef] [PubMed]
- Rajendrakumar, P.; Biswal, A.K.; Balachandran, S.M.; Srinivasarao, K.; Sundaram, R.M. Simple Sequence Repeats in Organellar Genomes of Rice: Frequency and Distribution in Genic and Intergenic Regions. Bioinformatics 2007, 23, 1–4. [Google Scholar] [CrossRef]
- Nagpure, N.S.; Rashid, I.; Pathak, A.K.; Singh, M.; Singh, S.P.; Sarkar, U.K. In Silico Analysis of SSRs in Mitochondrial Genomes of Fishes. 2015. Available online: https://pubmed.ncbi.nlm.nih.gov/24660911/ (accessed on 22 May 2025).
- Kuntal, H.; Sharma, V. In Silico Analysis of SSRs in Mitochondrial Genomes of Plants. OMICS 2011, 15, 783–789. [Google Scholar] [CrossRef]
- Filiz, E. SSRs Mining of Brassica Species in Mitochondrial Genomes: Bioinformatic Approaches. Hortic. Environ. Biotechnol. 2013, 54, 548–553. [Google Scholar] [CrossRef]
- Kumar, S.; Kumari, S.; Shanker, A. In Silico Mining of Simple Sequence Repeats in Mitochondrial Genomes of Genus Orthotrichum. J. Sci. Res. 2020, 64, 179–182. [Google Scholar] [CrossRef]
- Rudnick, J.A.; Katzner, T.E.; Bragin, E.A.; DeWOODY, J.A. Species Identification of Birds through Genetic Analysis of Naturally Shed Feathers. Mol. Ecol. Notes 2007, 7, 757–762. [Google Scholar] [CrossRef]
- Zehner, R.; Zimmermann, S.; Mebs, D. RFLP and Sequence Analysis of the Cytochrome b Gene of Selected Animals and Man: Methodology and Forensic Application. Int. J. Leg. Med. 1998, 111, 323–327. [Google Scholar] [CrossRef]
- Derégnaucourt, S.; Guyomarc’h, J.-C.; Spanò, S. Behavioural Evidence of Hybridization (Japanese × European) in Domestic Quail Released as Game Birds. Appl. Anim. Behav. Sci. 2005, 94, 303–318. [Google Scholar] [CrossRef]
- Puigcerver, M.; Vinyoles, D.; Rodríguez-Teijeiro, J.D. Does Restocking with Japanese Quail or Hybrids Affect Native Populations of Common Quail Coturnix Coturnix? Biol. Conserv. 2007, 136, 628–635. [Google Scholar] [CrossRef]
- Smith, S.; Fusani, L.; Boglarka, B.; Sanchez-Donoso, I.; Marasco, V. Lack of Introgression of Japanese Quail in a Captive Population of Common Quail. Eur. J. Wildl. Res. 2018, 64. [Google Scholar] [CrossRef]
- Sanchez-Donoso, I.; Vilà, C.; Puigcerver, M.; Butkauskas, D.; de la Calle, J.R.C.; Morales-Rodríguez, P.A.; Rodríguez-Teijeiro, J.D. Are Farm-Reared Quails for Game Restocking Really Common Quails (Coturnix coturnix)?: A Genetic Approach. PLoS ONE 2012, 7, e39031. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Donoso, I.; Ravagni, S.; Rodríguez-Teijeiro, J.D.; Christmas, M.J.; Huang, Y.; Maldonado-Linares, A.; Puigcerver, M.; Jiménez-Blasco, I.; Andrade, P.; Gonçalves, D.; et al. Massive Genome Inversion Drives Coexistence of Divergent Morphs in Common Quails. Curr. Biol. 2022, 32, 462–469.e6. [Google Scholar] [CrossRef]
- Barilani, M.; Deregnaucourt, S.; Gallego, S.; Galli, L.; Mucci, N.; Piombo, R.; Puigcerver, M.; Rimondi, S.; Rodríguez-Teijeiro, J.D.; Spanò, S.; et al. Detecting Hybridization in Wild (Coturnix c. Coturnix) and Domesticated (Coturnix c. Japonica) Quail Populations. Biol. Conserv. 2005, 126, 445–455. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Direction | Primer Sequence (5′-3′) | Position on Respective Mitogenomes |
|---|---|---|---|
| (Start–End) | |||
| CQ1 | Forward | TCACAGCCCTCCTACTCTCC | 5562–5582 |
| Reverse | TGCTAGTAGGGTGAGGAGGG | ||
| CQ2 | Forward | GGCTTAGCTGGTATGCCCC | 7898–7915 |
| Reverse | TGAGATTAAGGAGCCGATTGAGG | ||
| CQ3 | Forward | CCTCCTTACCCCCATCATCC | 13,065–13,087 |
| Reverse | AGGCGGTTTTAACGGTTTTGG | ||
| JQ1 | Forward | TGCTTGCCGGACATATTTTTACC | 911–946 |
| Reverse | GGTTTGAGGGTATTTGTGCGG | ||
| JQ2 | Forward | CAGCACTTCCTAGGCCTAGC | 7899–7916 |
| Reverse | ACTTTACGTTTTGCTGAGAAGGC | ||
| JQ3 | Forward | TCCTCCTTACCCCCATTATCC | 13,067–13,089 |
| Reverse | GGTAATGATGCTGTCTGTGCC | ||
| jqcqre_1 | Forward | CAACCGATGACTATTTTCAACTAACCA | 6583/6584–7412/7413 |
| Reverse | TCCTACTGTGAATATATGGTGGGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, P.; Sastry, K.V.H.; Singh, R.P. A PCR-RFLP Method for Distinguishing Closely Related Common Quail (Coturnix coturnix) and Japanese Quail (Coturnix japonica): Forensics and Conservation Implications. Birds 2025, 6, 28. https://doi.org/10.3390/birds6020028
Dey P, Sastry KVH, Singh RP. A PCR-RFLP Method for Distinguishing Closely Related Common Quail (Coturnix coturnix) and Japanese Quail (Coturnix japonica): Forensics and Conservation Implications. Birds. 2025; 6(2):28. https://doi.org/10.3390/birds6020028
Chicago/Turabian StyleDey, Prateek, Kochiganti Venkata Hanumat Sastry, and Ram Pratap Singh. 2025. "A PCR-RFLP Method for Distinguishing Closely Related Common Quail (Coturnix coturnix) and Japanese Quail (Coturnix japonica): Forensics and Conservation Implications" Birds 6, no. 2: 28. https://doi.org/10.3390/birds6020028
APA StyleDey, P., Sastry, K. V. H., & Singh, R. P. (2025). A PCR-RFLP Method for Distinguishing Closely Related Common Quail (Coturnix coturnix) and Japanese Quail (Coturnix japonica): Forensics and Conservation Implications. Birds, 6(2), 28. https://doi.org/10.3390/birds6020028