
Microwave-Assisted Asinger Synthesis of Thiazolines †


 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
3. Experiment
3.1. Synthesis of Thiazolines under Conventional Conditions—General Procedure
3.2. Synthesis of Thiazolines under Microwave Irradiation Conditions—General Procedure
3.3. 2-(4-Chlorophenyl)-4-methyl-2,5-dihydrothiazole 1
3.4. 2-(2-Chlorophenyl)-4-methyl-2,5-dihydrothiazole 2
3.5. 4-Methyl-2-phenyl-2,5-dihydrothiazole 3
3.6. 4-(4-Methyl-2,5-dihydrothiazol-2-yl)phenol 4
3.7. 4-Methyl-2-(p-tolyl)-2,5-dihydrothiazole 5
3.8. 2-(2,3-Dimethoxyphenyl)-4-methyl-2,5-dihydrothiazole 6
3.9. 2-Methoxy-4-(4-methyl-2,5-dihydrothiazol-2-yl)phenol 7
3.10. 4-Methyl-2-(2-(prop-2-yn-1-yloxy)phenyl)-2,5-dihydrothiazole 8
3.11. 3-Methyl-1-thia-4-azaspiro[4.5]dec-3-ene 9
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaumont, A.C.; Gulea, M.; Levillain, J. Overview of the Chemistry of 2-Thiazolines. Chem. Rev. 2009, 109, 1371–1401. [Google Scholar] [CrossRef] [PubMed]
- Alom, N.E.; Wu, F.; Li, W. One-Pot Strategy for Thiazoline Synthesis from Alkenes and Thioamides. Org. Lett. 2017, 19, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Alsharif, Z.A.; Alam, M. Modular synthesis of thiazoline and thiazole derivatives by using a cascade protocol. RSC Adv. 2017, 7, 32647. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ahmad, A.; Sudhakar, R.; Varshney, H.; Subbarao, N.; Ansari, S.; Rauf, A.; Khan, A.U. Designing, synthesis and antimicrobial action of oxazoline and thiazoline derivatives of fatty acid esters. J. Biomol. Struct. Dyn. 2017, 35, 3412–3431. [Google Scholar] [CrossRef] [PubMed]
- Oniga, O.; Ndongo, J.T.; Moldovan, C.; Tiperciuc, B.; Oniga, S.; Pîrnău, A.; Vlase, L.; Verité, P. Synthesis and antimicrobial activity of some new 2-hydrazone-thiazoline-4-ones. Farmacia 2012, 60, 785–797. [Google Scholar]
- You, S.L.; Razavi, H.; Kelly, J.W. A Biomimetic Synthesis of Thiazolines Using Hexaphenyloxodiphosphonium Trifluoromethanesulfonate. Angew. Chem. Int. Ed. 2003, 42, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.M.F.M.; Sales, E.S.; Livotto, P.R.; Schneider, P.H.; Merlo, A.A. Synthesis of New Family of Thiazoline and Thiazole Esters and Investigation of their Thermal Properties. J. Braz. Chem. Soc. 2014, 25, 1493–1503. [Google Scholar] [CrossRef]
- Diness, F.; Nielsen, D.S.; Fairlie, D.P. Synthesis of the Thiazole−Thiazoline Fragment of Largazole Analogues. J. Org. Chem. 2011, 76, 9845–9851. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Forsyth, C.J. Total synthesis of the marine cyanobacterial cyclodepsipeptide apratoxin A. Proc. Natl. Acad. Sci. USA 2004, 101, 12067–12072. [Google Scholar] [CrossRef]
- Asinger, F. Über die gemeinsame Einwirkung von Schwefel und Ammoniak auf Ketone. Angew. Chem. 1956, 68, 413. [Google Scholar] [CrossRef]
- Asinger, F. Chemiker-Treffen Salzburg. Angew. Chem. 1956, 68, 377. [Google Scholar]
- Asinger, F.; Thiel, M.; Pallas, E. Die gemeinsame einwirkung von schwefel und ammoniak auf diathylketon. Liebigs Ann. Chem. 1957, 602, 37–49. [Google Scholar] [CrossRef]
- Schlüter, T.; Frerichs, N.; Schmidtmann, M.; Martens, J. Consecutive Multicomponent Reactions: Synthesis of 3-Acyl-4-alkynyl-Substituted 1,3-Thiazolidines. Synthesis 2018, 50, 1123–1132. [Google Scholar]
- Rainoldi, G.; Begnini, F.; Silvani, A.; Lesma, G. Efficient Synthesis of Spirooxindole-Fused 3-Thiazoline Derivatives by a One-Pot Asinger-Type Reaction. Synlett 2016, 27, 2831–2835. [Google Scholar]
- Brockmeyer, F.; Schoemaker, R.; Schmidtmann, M.; Martens, J. Multicomponent reaction for the first synthesis of 2,2-dialkyl- and 2-alkyl-2-aralkyl-5,6-diaryl-2H-1,3-thiazines as scaffolds for various 3,4-dihydro-2H-1,3-thiazine derivatives. Org. Biomol. Chem. 2014, 12, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Brockmeyer, F.; van Gerven, D.; Saak, W.; Martens, J. Two Sequential Multicomponent Reactions: Synthesis of Thiazolidin-4-yl-1,3,4-oxadiazoles under Mild Conditions. Synthesis 2014, 46, 1603–1612. [Google Scholar] [CrossRef]
- Zeinab Faghihi, Z.; Oskooie, H.A.; Heravi, M.M.; Tajbakhsh, M.; Shiri, M. A novel analogue of Asinger reaction for the synthesis of thiazinoquinoline derivatives. Monat. Chem. 2017, 148, 315–320. [Google Scholar] [CrossRef]
- Schlemminger, I.; Janknecht, H.H.; Maison, W.; Saak, W.; Martens, J. Synthesis of the First enantiomerically pure 3-thiazolines via Asinger reaction. Tetrahedron. Lett. 2000, 41, 7289–7292. [Google Scholar] [CrossRef]
- Asinger, F.; Offermanns, H. Syntheses with Ketones, Sulfur, and Ammonia or Amines at Room Temperature. Angew. Chem. Int. Ed. 1967, 6, 907–919. [Google Scholar] [CrossRef]
- García-Muñoz, A.; Ortega-Arizmendi, A.I.; García-Carrillo, M.A.; Díaz, E.; Gonzalez-Rivas, N.; Cuevas-Yañez, E. Direct, metal-free synthesis of benzyl alcohols and deuterated benzyl alcohols from p-toluenesulfonylhydrazones using water as solvent. Synthesis 2012, 44, 2237–2242. [Google Scholar]
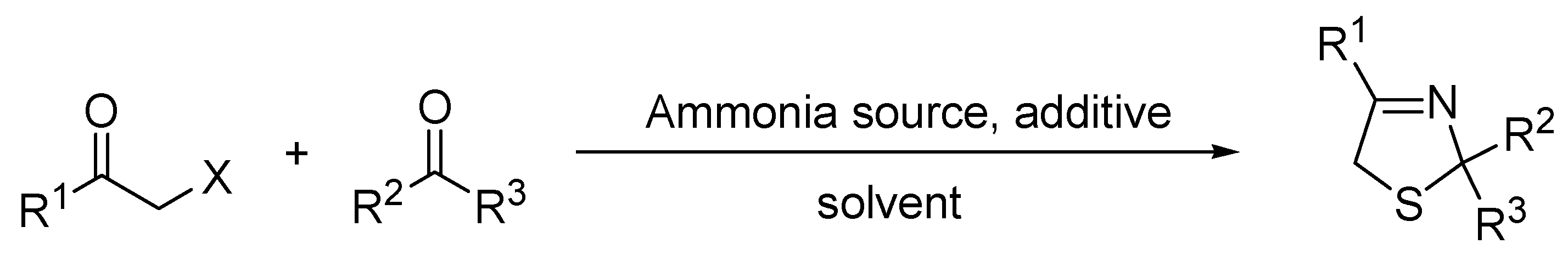
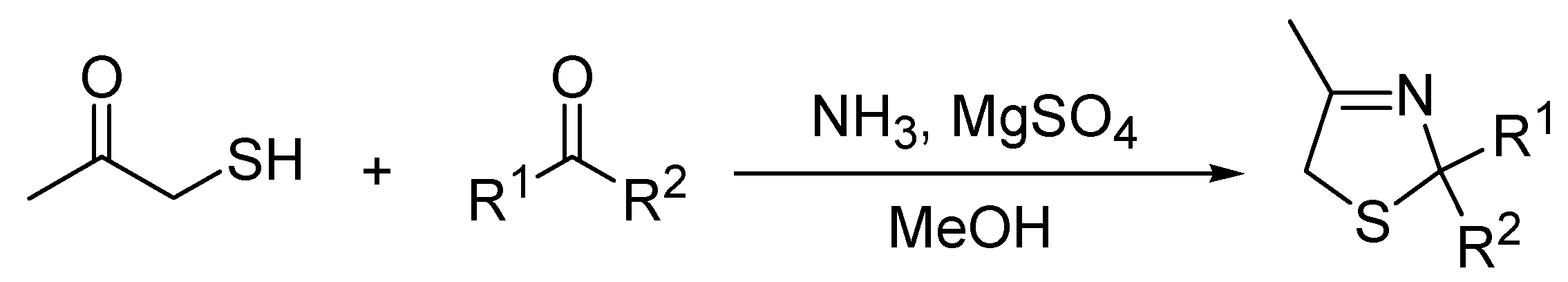

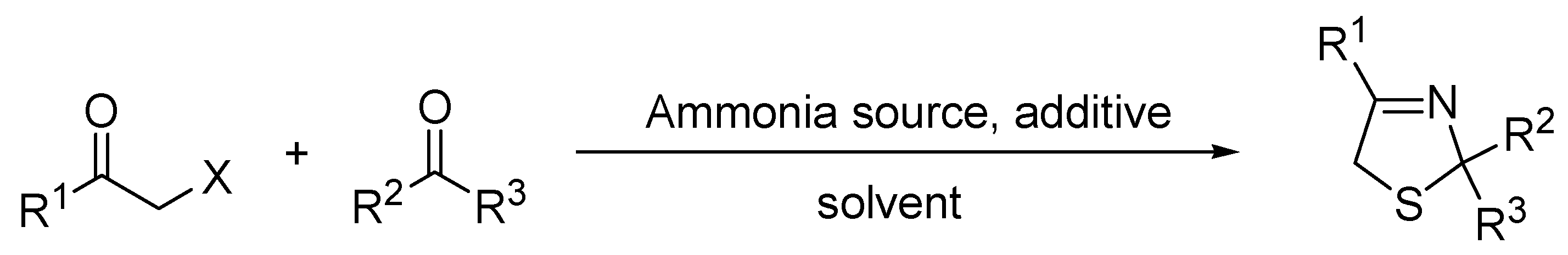{kind=link}
{kind=link}
| Entry | R1 | R2 | R3 | X | Ammonia Source | Solvent | Additive | Reaction Temperature (°C) | Reaction Time (h) | %Yield |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Me | Me | Me | Br | NH4OH/H2O | - | NaSH | R. T. | 24 | <5 |
| 2 | Ph | Me | Me | Br | NH4OH/H2O | - | NaSH | R. T. | 24 | 0 |
| 3 | Me | Ph | Ph | Br | NH4OH/H2O | - | NaSH | R. T. | 24 | 0 |
| 4 | Me | Me | Me | Br | NH4OH/H2O | MeOH | NaSH | R. T. | 24 | 0 |
| 5 | Me | Me | Me | Br | NH4OH/H2O | Acetone | NaSH | R. T. | 24 | 0 |
| 6 | Me | Me | Me | Br | NH4OH/H2O | CH2Cl2 | NaSH | R. T. | 24 | 0 |
| 7 | Me | Me | Me | Br | NH4OH/H2O | THF | NaSH | R. T. | 24 | 0 |
| 8 | Me | Me | Me | Br | NH4OH/H2O | MeOH | NaSH | Reflux | 24 | 0 |
| 9 | Ph | Me | Me | Br | NH4OH/H2O | MeOH | NaSH | Reflux | 24 | 0 |
| 10 | Me | Ph | Ph | Br | NH4OH/H2O | MeOH | NaSH | Reflux | 24 | 0 |
| 11 | Ph | Me | Me | Br | NH4OAc | Acetone | NaSH | R. T. | 48 | 0 |
| 12 | Me | Me | Me | Br | NH4OAc | Acetone | NaSH | R. T. | 48 | 0 |
| 13 | Me | Me | Me | Br | NH4OAc | MeOH | NaSH | R. T. | 48 | 0 |
| 14 | Ph | Me | Me | Br | NH4OAc | MeOH | NaSH | R. T. | 48 | 0 |
| 15 | Ph | Me | Me | Br | NH4OAc | MeOH | NaSH | Reflux | 24 | 0 |
| 16 | Me | Ph | Ph | Br | NH4OAc | AcOH | NaSH | 100 | 24 | 0 |
| 17 | Me | Ph | Ph | Br | NH4OAc | DMF | NaSH | 100 | 24 | 0 |
| 18 | Me | Ph | Ph | Br | NH4OAc | Toluene | NaSH | 100 | 24 | 0 |
| 19 | Me | 4-ClC6H4 | H | Br | NH4OH/H2O | - | NaSH | R. T. | 24 | 8 |
| 20 | Me | 4-ClC6H4 | H | Br | NH4OH/H2O | MeOH | NaSH | R. T. | 24 | 12 |
| 21 | Me | 4-ClC6H4 | H | Br | NH4OH/H2O | MeOH | NaSH | R. T. | 24 | <5 |
| 22 | Me | 4-ClC6H4 | H | Br | NH3/MeOH | MeOH | NaSH | R. T. | 24 | 20 |
| 23 | Me | 4-ClC6H4 | H | Br | NH3/MeOH | MeOH | NaSH, MgSO4 | R. T. | 24 | 35 |
| 24 | Me | 4-ClC6H4 | H | SH | NH3/MeOH | MeOH | MgSO4 | R. T. | 24 | 64 |
| 25 | Me | cyclohexanone | SH | NH3/MeOH | MeOH | MgSO4 | R. T. | 24 | 68 | |
| Compound | R1 | R2 | %Yield at R.T. | %Yield under MW |
|---|---|---|---|---|
| 1 | 4-ClC6H4 | H | 64 | 71 |
| 2 | 2-ClC6H4 | H | 77 | 47 |
| 3 | Ph | H | 33 | 50 |
| 4 | 4-OHC6H4 | H | 55 | 65 |
| 5 | 4-CH3C6H4 | H | 69 | 49 |
| 6 | 2,3-(OCH3)2C6H3 | H | 69 | 75 |
| 7 | (3-OCH3-4-OH)C6H3 | H | 90 | 70 |
| 8 | (2-OCH2C≡CH)C6H4 | H | 88 | 73 |
| 9 | cyclohexanone | 68 | 75 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordillo-Cruz, R.E.; Gonzalez-Reyes, L.; Coporo-Reyes, M.; Zavala-Segovia, N.; Frontana-Uribe, B.A.; García-Eleno, M.A.; Unnamatla, M.V.B.; Cuevas-Yañez, E. Microwave-Assisted Asinger Synthesis of Thiazolines. Chem. Proc. 2021, 3, 27. https://doi.org/10.3390/ecsoc-24-08316
Gordillo-Cruz RE, Gonzalez-Reyes L, Coporo-Reyes M, Zavala-Segovia N, Frontana-Uribe BA, García-Eleno MA, Unnamatla MVB, Cuevas-Yañez E. Microwave-Assisted Asinger Synthesis of Thiazolines. Chemistry Proceedings. 2021; 3(1):27. https://doi.org/10.3390/ecsoc-24-08316
Chicago/Turabian StyleGordillo-Cruz, Raúl Eduardo, Liliana Gonzalez-Reyes, Milton Coporo-Reyes, Nieves Zavala-Segovia, Bernardo A. Frontana-Uribe, Marco A. García-Eleno, M. V. Basavanag Unnamatla, and Erick Cuevas-Yañez. 2021. "Microwave-Assisted Asinger Synthesis of Thiazolines" Chemistry Proceedings 3, no. 1: 27. https://doi.org/10.3390/ecsoc-24-08316
APA StyleGordillo-Cruz, R. E., Gonzalez-Reyes, L., Coporo-Reyes, M., Zavala-Segovia, N., Frontana-Uribe, B. A., García-Eleno, M. A., Unnamatla, M. V. B., & Cuevas-Yañez, E. (2021). Microwave-Assisted Asinger Synthesis of Thiazolines. Chemistry Proceedings, 3(1), 27. https://doi.org/10.3390/ecsoc-24-08316