
Revealing the Ion Chemistry Occurring in High Kinetic Energy-Ion Mobility Spectrometry: A Proof of Principle Study

 , , , and
, , , and
Abstract
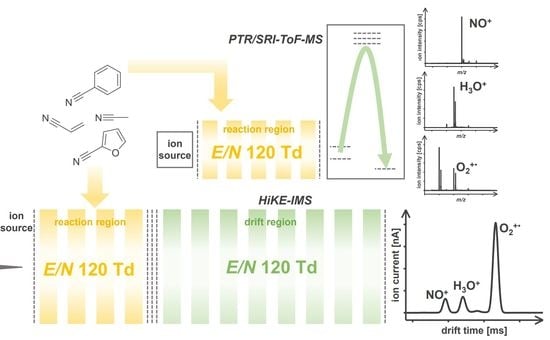
:
1. Introduction
2. Materials and Methods
2.1. HiKE-IMS Dimensions and Operational Details
2.2. PTR/SRI-ToF-MS Operational Details
2.3. Nitrile Compounds
2.4. Gas Standard Production
3. Results and Discussion
3.1. Reagent Ions
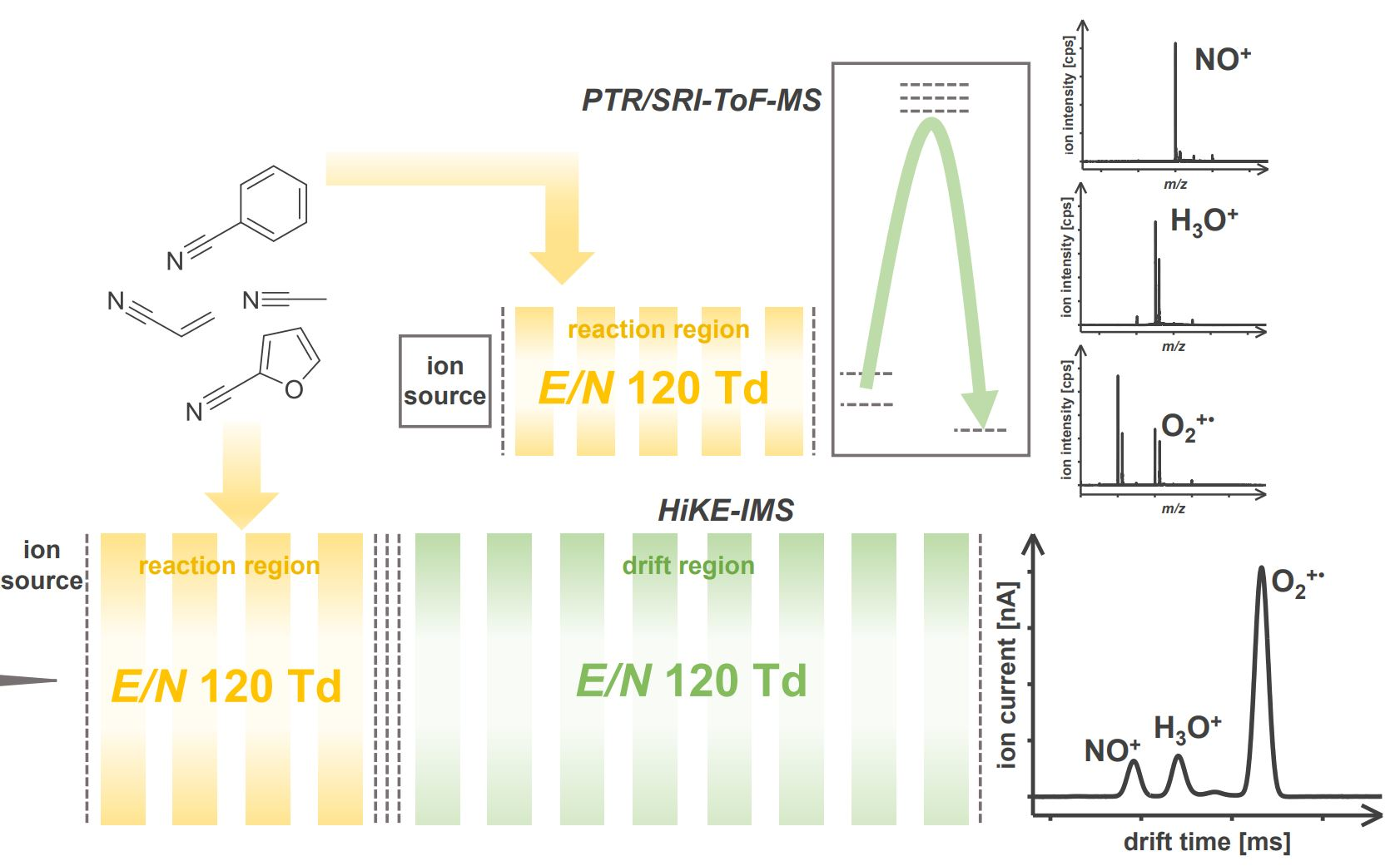
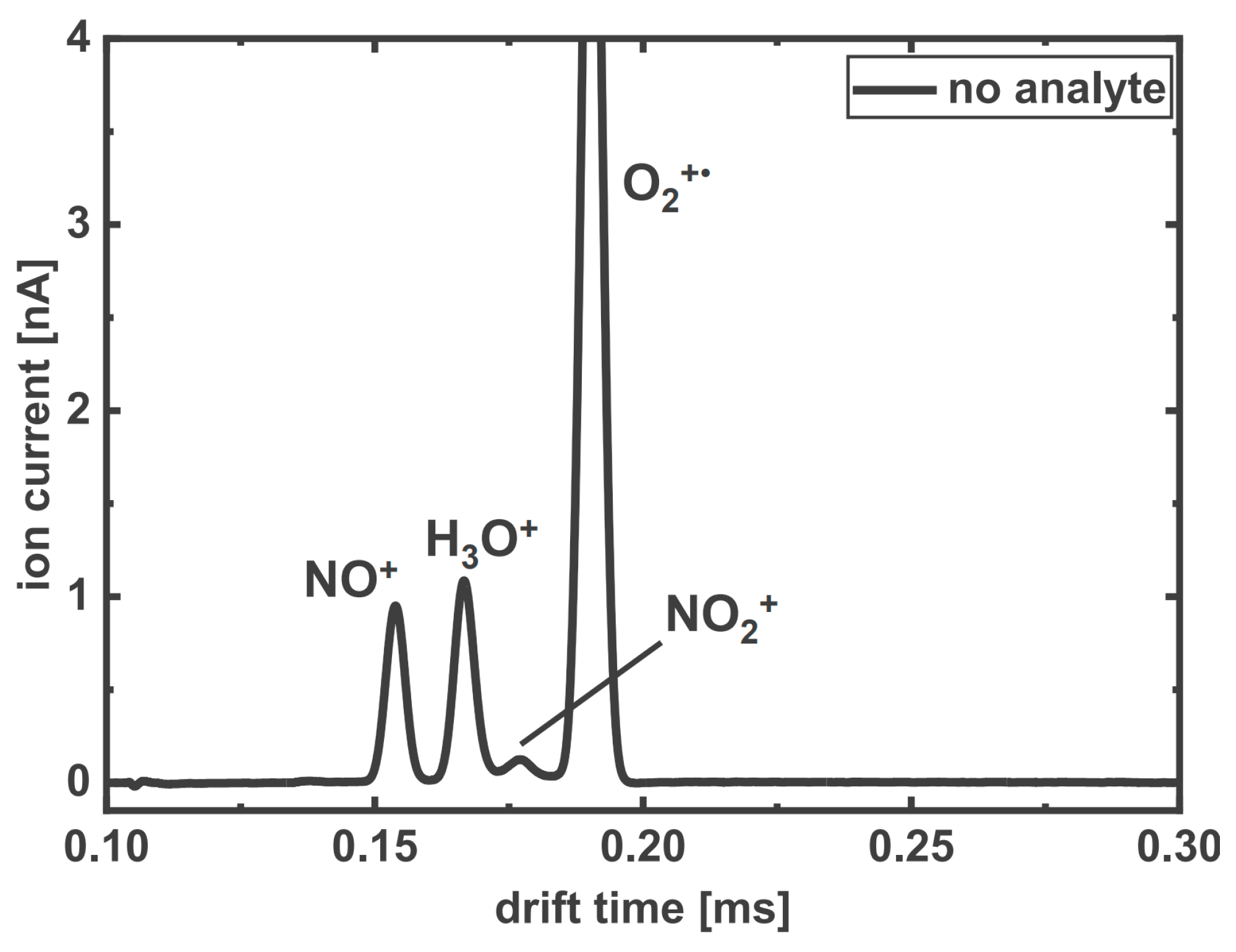
3.1.1. HiKE-IMS Reagent Ions
3.1.2. PTR/SRI-ToF-MS
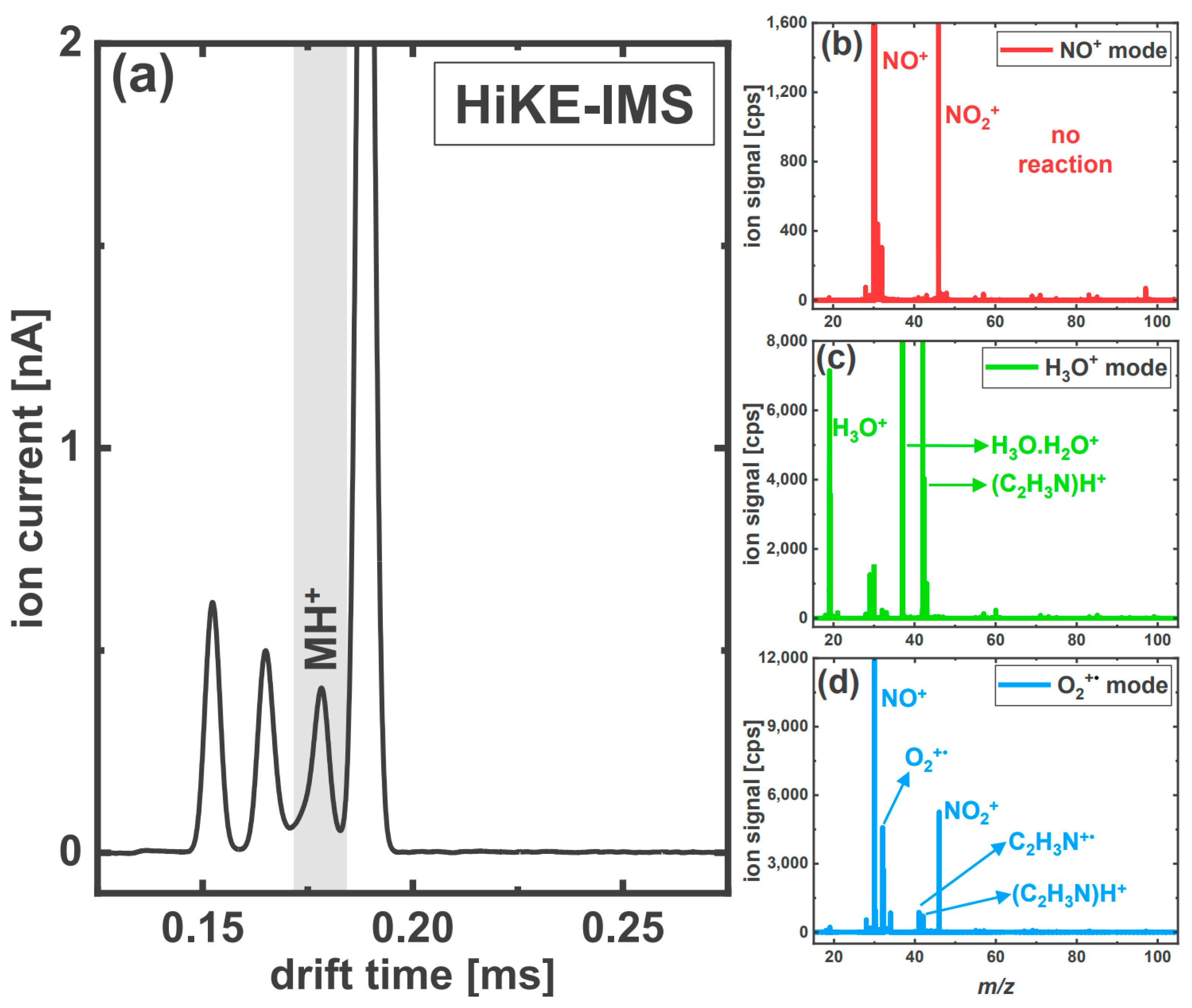
3.2. HiKE-IMS Product Ion Mobility Peaks and PTR/SRI-ToF-MS Product Ion Identification
3.2.1. Acetonitrile
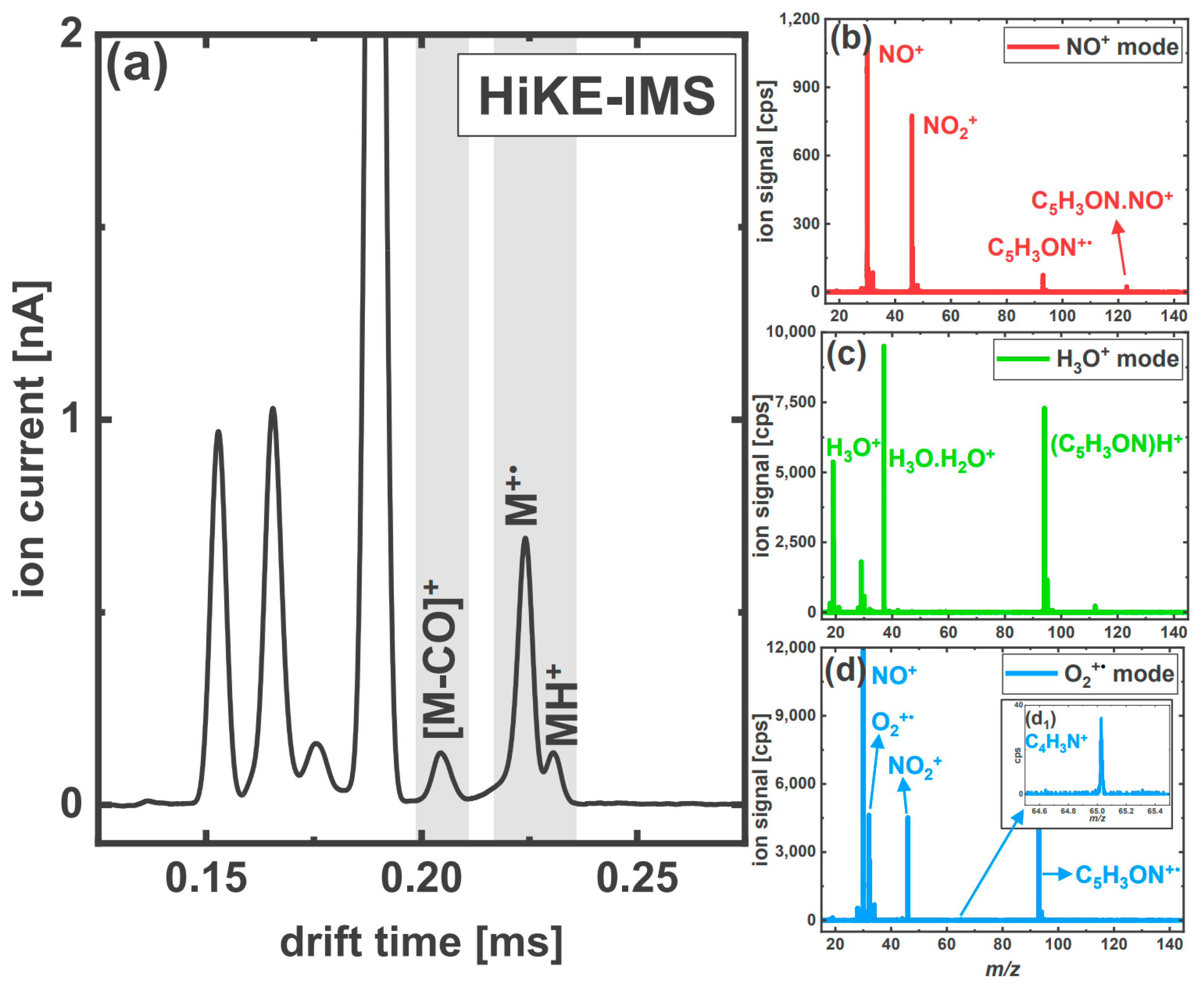
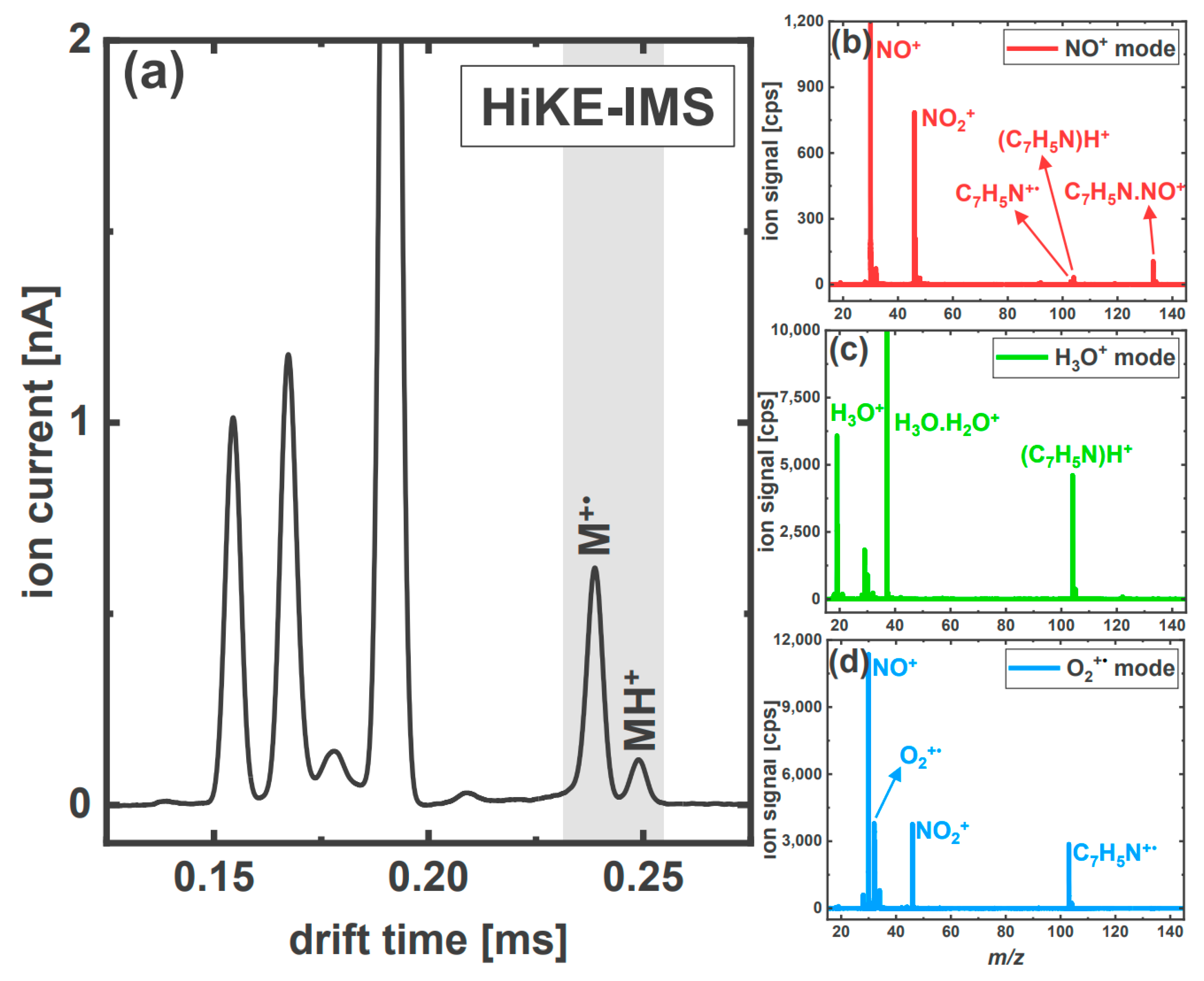
3.2.2. The Aromatic Nitriles: 2-Furonitrile and Benzonitrile
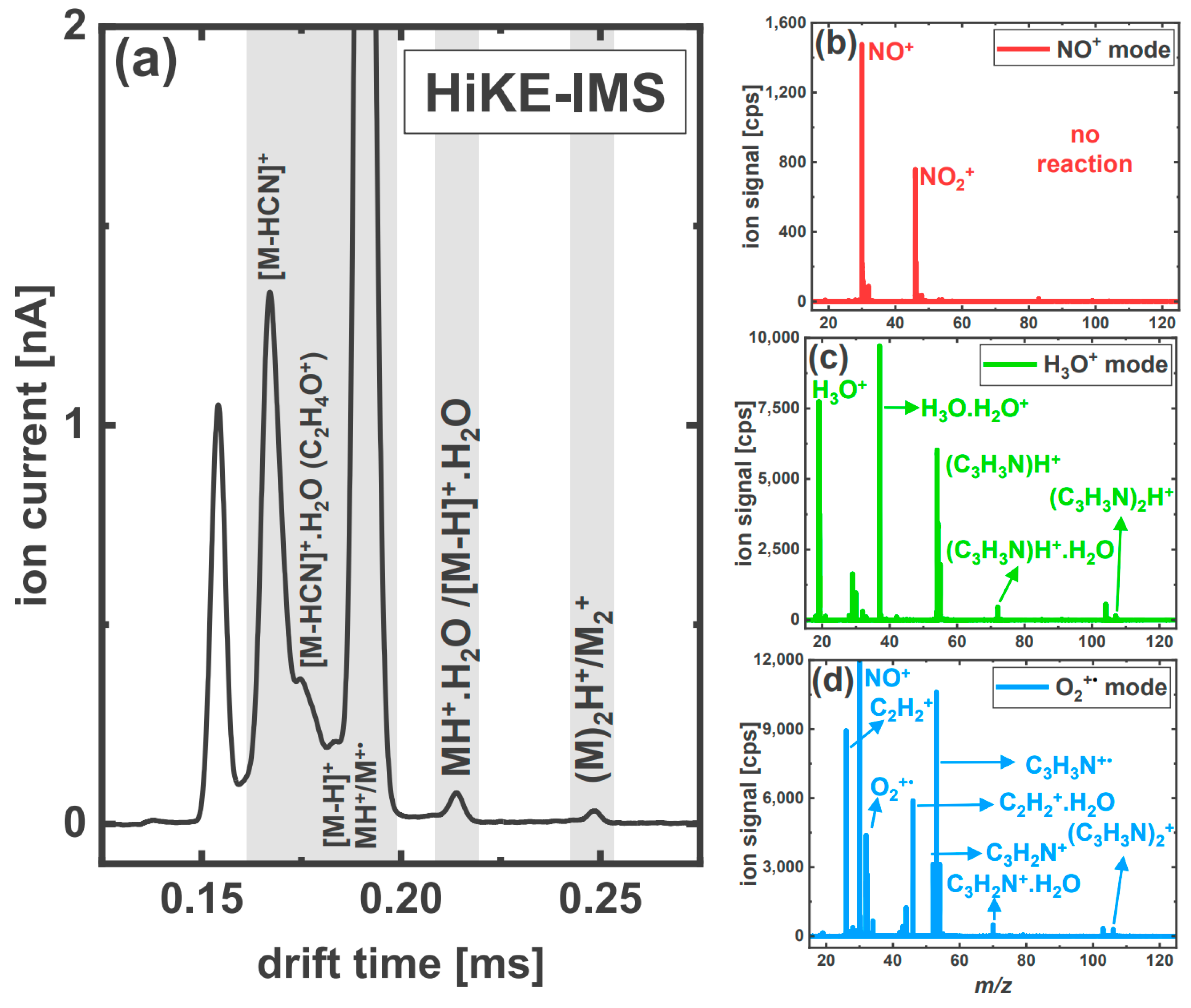
3.2.3. Acrylonitrile
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Langejuergen, J.; Allers, M.; Oermann, J.; Kirk, A.; Zimmermann, S. High Kinetic Energy Ion Mobility Spectrometer: Quantitative Analysis of Gas Mixtures with Ion Mobility Spectrometry. Anal. Chem. 2014, 86, 7023–7032. [Google Scholar] [CrossRef] [PubMed]
- Allers, M.; Kirk, A.T.; von Roßbitzky, N.; Erdogdu, D.; Hillen, R.; Wissdorf, W.; Benter, T.; Zimmermann, S. Analyzing Positive Reactant Ions in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS) by HiKE-IMS-MS. J. Am. Soc. Mass Spectrom. 2020, 31, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Allers, M.; Kirk, A.T.; Eckermann, M.; Schaefer, C.; Erdogdu, D.; Wissdorf, W.; Benter, T.; Zimmermann, S. Positive Reactant Ion Formation in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS). J. Am. Soc. Mass Spectrom. 2020, 31, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.; Schaefer, C.; Ruzsanyi, V.; Märk, T.D.; Eiceman, G.; Mayhew, C.A.; Zimmermann, S. High kinetic energy Ion Mobility Spectrometry—Mass spectrometry investigations of four inhalation anaesthetics: Isoflurane, enflurane, sevoflurane and desflurane. Int. J. Mass Spectrom. 2022, 475, 116831. [Google Scholar] [CrossRef]
- Hegan, O.; Gómez, J.I.S.; Schlögl, R. The potential of NO+ and O2+• in switchable reagent ion proton transfer reaction time-of-flight mass spectrometry. Mass Spec. Rev. 2022. [CrossRef]
- Available online: https://www.ionicon.com/products/details/ptr-tof-10k (accessed on 13 April 2023).
- Allers, M.; Kirk, A.T.; Schaefer, C.; Schlottmann, F.; Zimmermann, S. Formation of positive product ions from substances with low proton affinity in high kinetic energy ion mobility spectrometry. Rapid Commun. Mass Spectrom. 2021, 35, e8998. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.; Eiceman, G.; Märk, T.D.; Mayhew, C.A.; Ruzsanyi, V.; Schaefer, C.; Zimmermann, S. High kinetic energy-ion mobility spectrometry-mass spectrometry investigations of several volatiles and their fully deuterated analogues. Eur. Phys. J. D 2022, 76, 181. [Google Scholar] [CrossRef]
- Schlottmann, F.; Kirk, A.T.; Allers, M.; Bohnhorst, A.; Zimmermann, S. High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS) at 40 mbar. J. Am. Soc. Mass Spectrom. 2020, 31, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.M.; Mayhew, C.A. Proton Transfer Reaction Mass Spectrometry: Principles and Applications; Wiley: West Sussex, UK, 2014; ISBN 978-1-4051-7668-2. [Google Scholar]
- Weiss, F.; Mayhew, C.A.; Ruzsanyi, V.; Lederer, W.; Märk, T.D. A selective reagent ion-time-of-flight-mass spectrometric study of the reactions of O2+• with several volatile halogenated inhalation anaesthetics: Potential for breath analysis. Eur. Phys. J. D 2022, 76, 193. [Google Scholar] [CrossRef]
- Malásková, M.; Olivenza-León, D.; Chellayah, P.D.; Martini, J.; Lederer, W.; Ruzsanyi, V.; Unterkofler, K.; Mochalski, P.; Märk, T.D.; Watts, P.; et al. Studies pertaining to the monitoring of volatile halogenated anaesthetics in breath by proton transfer reaction mass spectrometry. J. Breath Res. 2020, 14, 026004. [Google Scholar] [CrossRef] [PubMed]
- Gochel-Dupuis, M.; Delwiche, J.; Hubin-Franskin, M.-J.; Collin, J.E. High resolution HeI photoelectron spectrum of acetonitrile. Chem. Phys. Lett. 1992, 193, 41–48. [Google Scholar] [CrossRef]
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]
- Bohme, D.K.; Mackay, G.I.; Schiff, H.I. Determination of proton affinities from the kinetics of proton transfer reactions. VII. The proton affinities of O2, H2, Kr, O, N2, Xe, CO2, CH4, N2O, and CO. J. Chem. Phys. 1980, 73, 4976–4986. [Google Scholar] [CrossRef]
- Klapstein, D.; MacPherson, C.D.; O’Brien, R.T. The photoelectron spectra and electronic structure of 2-carbonyl furans. Can. J. Chem. 1990, 68, 747–754. [Google Scholar] [CrossRef]
- Reiser, G.; Habenicht, W.; Müller-Dethlefs, K.; Schlag, E.W. The ionization energy of nitric oxide. Chem. Phys. Lett. 1988, 152, 119–123. [Google Scholar] [CrossRef]
- Simbizi, R.; Nduwimana, D.; Niyoncuti, J.; Cishahayo, P.; Gahungu, G. On the formation of 2- and 3-cyanofurans and their protonated forms in interstellar medium conditions: Quantum chemical evidence. RSC Adv. 2022, 12, 25332. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Linstrom, P. NIST Chemistry WebBook; NIST Standard Reference Database 69; NIST: Gaithersburg, MD, USA, 1997. [Google Scholar]
- Araki, M.; Sato, S.; Kimura, K. Two-color zero kinetic energy photoelectron spectra of benzonitrile and its van der Waals complexes with argon. Adiabatic ionization potentials and cation vibrational frequencies. J. Phys. Chem. 1996, 100, 10542–10546. [Google Scholar] [CrossRef]
- Ohno, K.; Matumoto, S.; Imai, K.; Harada, Y. Penning ionization electron spectroscopy of nitriles. J. Phys. Chem. 1984, 88, 206–209. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| reaction region length | 49 mm |
| drift region length | 149 mm |
| corona voltage | 1200 V |
| E/N—reaction region | 120 Td |
| E/N—drift region | 120 Td |
| IMS pressure | 17 mbar |
| IMS temperature | 60 °C |
| drift gas flow | 12 mLs/min |
| analyte gas flow | 18 mLs/min |
| injection time | 3 µs |
| temperature of inlet capillary | 100 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, F.; Schaefer, C.; Zimmermann, S.; Märk, T.D.; Mayhew, C.A. Revealing the Ion Chemistry Occurring in High Kinetic Energy-Ion Mobility Spectrometry: A Proof of Principle Study. Analytica 2023, 4, 113-125. https://doi.org/10.3390/analytica4020010
Weiss F, Schaefer C, Zimmermann S, Märk TD, Mayhew CA. Revealing the Ion Chemistry Occurring in High Kinetic Energy-Ion Mobility Spectrometry: A Proof of Principle Study. Analytica. 2023; 4(2):113-125. https://doi.org/10.3390/analytica4020010
Chicago/Turabian StyleWeiss, Florentin, Christoph Schaefer, Stefan Zimmermann, Tilmann D. Märk, and Chris A. Mayhew. 2023. "Revealing the Ion Chemistry Occurring in High Kinetic Energy-Ion Mobility Spectrometry: A Proof of Principle Study" Analytica 4, no. 2: 113-125. https://doi.org/10.3390/analytica4020010
APA StyleWeiss, F., Schaefer, C., Zimmermann, S., Märk, T. D., & Mayhew, C. A. (2023). Revealing the Ion Chemistry Occurring in High Kinetic Energy-Ion Mobility Spectrometry: A Proof of Principle Study. Analytica, 4(2), 113-125. https://doi.org/10.3390/analytica4020010