Abstract
Globally, hepatocellular carcinoma (HCC) is a significant cause of mortality and morbidity among chronically infected HCV patients. It is established that HCV is a primary risk factor for HCC progression. The treatment of HCV infection has been transformed by the introduction of DAAs with high rates of virological clearance. The reduction in cirrhosis-related consequences, particularly HCC, is the long-term objective of DAAs therapy for HCV. Although the risk of developing HCC is decreased in HCV patients who achieve a disease-sustaining virological response, these patients are nevertheless at risk, especially those with severe fibrosis and cirrhosis. Previous studies have shown that HCV induce several mechanisms of hepatocarcinogenesis in the host’s hepatic micro- and macro-environment, which leads to HCC progression. In an HCV-altered environment, compensatory liver regeneration favors chromosomal instability and irreversible alterations, which encourage hepatocyte neoplastic transformation and the development of malignant clones. These mechanisms involve a series of genetic and epigenetic modifications including host genetic factors, dysregulation of several signaling pathways, histone, and DNA modifications including methylation and acetylation. This review highlights the genetic and epigenetic factors that lead to the development of HCC in chronic HCV-infected individuals and can be targeted for earlier HCC diagnosis and prevention.
1. Introduction
The analysis of global cancer burden signifies that hepatic cancer is one of the most fatal tumors [1]. It is reported that hepatic cancer is the second leading cause of mortality in men and the sixth leading cause of mortality in women due to cancer. Within the subtypes of hepatic cancer, 70–85% cases are of hepatocellular carcinoma (HCC) [2]. According to the International Agency for Research on Cancer Monographs Program, the most significant risk factors for HCC are infectious agents, especially hepatitis C virus (HCV) [3]. The estimated 5-year survival rate of patients with an advanced stage of HCC still remains very low. This is mainly because of late HCC diagnoses, high recurrence rates, and the frequent resistance to HCC therapies [4,5].
The prevalence of HCC induced by HCV varies by both ethnicity and region. Hepatitis B virus (HBV) is the primary cause of HCC in the bulk of Asia and Africa, while HCV is the leading cause in the USA, Europe, Japan, and South America. Viral genotype, diabetes mellitus, lifestyle variables, obesity, and concurrent liver disease are the main risk factors for the development of HCC in chronic HCV infection [6].
HCV is an enveloped virus belonging to the family Flaviviridae. It contains a positive-sense, RNA genome of 9.6kb that is translated into a single, large polyprotein. This polyprotein is cleaved into several structural (E1, E2, and core protein) and non-structural (NS2, p7, NS3, NS4A, NS4B, NS5A, and NS5B) proteins via the activity of several host and viral proteases [7]. Apart from playing a role in viral life cycle, these HCV proteins also interfere in various host cell processes including the regulation of cell signaling pathways, apoptosis, transcriptional activation, angiogenic mechanisms, and the transformation of cells. HCV regulates these processes by modulating several genetic and epigenetic factors of host cellular machinery and plays a role in HCC carcinogenesis and oncogenesis [8].
The development of HCV-induced HCC is a multi-factorial process involving several viral, host, and environmental factors. Multiple host responses initiated by host–virus interactions lead to hepatic inflammation, then fibrosis, and eventually cirrhosis. A hepatic oncogenic environment is created by HCV through the combination of chronic hepatic inflammation, viral protein expression, chronically deranged cellular signaling events, and oxidative stress that ultimately leads to host genetic and epigenetic instability. Hepatocarcinogenesis is also partially associated with injury to hepatocytes by molecules accumulated during hepatic inflammation, including the reactive oxygen species (ROS) [9].
The frequency of HCV-related HCC is reduced after achieving a sustained virologic response (SVR) after interferon-based or direct-acting antiviral (DAA) treatments [10], but the risk of HCC development still exists and remains relatively high, as reviewed recently [11]. This review focuses on the genetic and epigenetic modifications induced by HCV which lead to the development of HCC in HCV-infected patients (Figure 1).
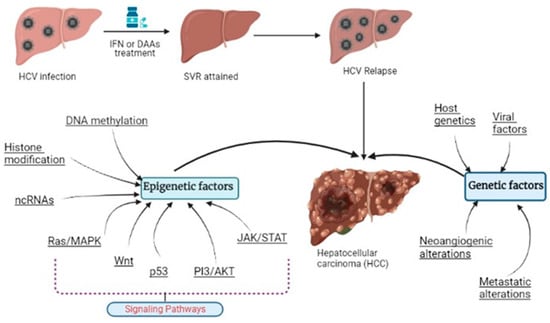
Figure 1.
HCV-induced HCC development along with the modified genetic and epigenetic factors.
In contrast to genetic alterations, epigenetic modifications are reversible and have a greater impact on gene expression than genetic changes. Epigenetics refers to heritable states of gene expression without altering the DNA sequence itself. Understanding the mechanisms behind the emergence and heterogeneity of HCC is thus considerably aided by investigations of epigenetic control and the related molecular machinery. Additionally, this information may be used to find biomarkers for HCC diagnosis and prognosis as well as potential new targets for more effective therapy strategies in the future (epigenetic remodeling in human hepatocellular carcinoma).
2. Host and Virus Genetic Factors
Host and viral genetic factors of patients with HCV infection play a significant role in HCC development. Numerous host genetic variables have been discovered by genome-wide association studies (GWAS), which affect the effectiveness of antiviral medication, side effects, or the development of HCV infection naturally. Single nucleotide polymorphisms (SNPs) in or around the IFNL3 and IFNL4 loci, originally reported as IL28B loci6, were found to be significantly associated with both the responsiveness to IFN-based therapy for chronic hepatitis C (CHC) and the spontaneous clearance of HCV. Several GWAS have found genetic variants connected to hepatic fibrogenesis and carcinogenesis in HCV patients, which are relevant to HCC development. Recently, a TLL1 variation linked to the emergence of HCC was discovered in HCV patients following SVR. Interestingly, using a candidate gene approach, genetic variants of many cytokine and chemokine genes, including IL6, CCL2, CCL8, TNF, and TGFB1, have demonstrated substantial relationships with the advancement of hepatic fibrosis [12].
HCV is classified into more than 80 subtypes and six main genotypes based on nucleotide variation. Every known HCV genotype is hepatotropic and harmful. However, it has been proposed that the infectivity and pathogenicity of the genotype may change depending on the rate of development of cirrhosis and the chance of HCC. The viral load and the HCV genotype should be taken into consideration when determining the period of therapy. This is contrary to a previous meta-analysis, which linked genotype 1 with a 78% increased risk of HCC relative to all other genotypes and a 60% increased risk among patients with cirrhosis [13]. Certain HCV genotypes appear to be associated with a higher risk of HCC, particularly genotype 3 which is associated with an 80% higher risk of HCC compared to genotype 1 [14].
3. Neoangiogenic and Metastatic Alterations
HCV-induced hepatic neoangiogenesis and metastasis contribute to the progression and invasion of HCC in patients by providing additional sources of nutrients and oxygen to cancer cells for their growth, proliferation, and dissemination to secondary sites primarily through the portal vein. An increased density of microvasculature has been reported in HCV patients. The HCV core protein transcriptionally upregulates hypoxia inducible factor 1α [15] which increases the transcription of vascular endothelial growth factor (VEGF) and modulates the expression of matrix metalloproteases (MMP-9 and MMP-2) and cyclooxygenase 2 [16]. The HCV core protein also interacts with Smad3 resulting in the upregulation of the TGF-β signaling pathway via NF-Kβ (nuclear factor kappa-light-chain-enhancer of activated B cells) [17]. Moreover, it has been reported that NS5A, E1, and E2 proteins activate metastatic EMT (endothelial mesenchymal transformation) process via Twist2 receptor activation [18] and VEGF and TGF-β signaling, respectively [19]. These changes induce a proangiogenic state in host cells and promote cancer cells’ permeability and migration.
HCV regulates the transcription of the MT1-MMP gene via the activation of the NS3/4A-TC-PTP axis and kinase signaling. This, in turn, activates the EGFR gene along with its ligands including BTC and HB-EGF, which causes the degradation of the extracellular matrix and the activation of the genes necessary for invadopodia formation. These events contribute to the invasiveness and metastasis of cancer cells for the development of HCC and extra-hepatic manifestations [20]. It has also been established that the HCV core protein modulates the Nm23-H1 protein in the host cell and inhibits its tumor metastasis suppressor ability by increasing Nm23-H1 SUMOylation with SUMO2 and SUMO3 [21].
4. Regulation of Noncoding RNAs (ncRNAs)
HCV infection dysregulates noncoding RNAs’ (ncRNAs) transcriptome of host cells and contributes to HCC development [22]. Recent studies have reported the dysregulation of several miRNAs (micro RNAs) and long noncoding RNAs (lncRNAs) via HCV infection, resulting in impaired innate and adaptive immune responses that lead to hepatic fibrosis and cirrhosis [23,24]. HCV modulates the activity of these miRNAs and lncRNAs by different pathways including cell-cycle, lipid metabolism, and immune-response pathways [25].
It has also been reported that during HCV infection the expression of miR-122 is epigenetically regulated by the binding of RXRA to the DR1 and DR2 motifs at the promoter region of miR-122. The upregulation of miR-122 contributes to the development of HCC in HCV-infected patients [26]. Other studies have also reported that HCV infection causes the upregulation of several additional miRNAs, including miR-371-5p, miR-24, miR-25, miR-200, miR-381, miR-34a, miR-130a/b, miR-146a, miR-200a, and miR-23b. These miRNAs contribute to HCC development mainly by upregulating the activity of CREB1 and SCD (stearoyl-CoA desaturase) genes and downregulating the activity of FN1 (fibronectin 1) and PPARG genes [27]. Similarly, the HCV core protein epigenetically downregulates miR-124 by upregulating DNMT1 promoting HCC progression and invasion in the liver [28]. Additionally, miR-484, miR-524, miR-615, and miR-628 are also found to be dysregulated in HCV-induced HCC [29].
Most lncRNAs with a pro-viral effect are upregulated by HCV infection. The HCV core protein upregulates the expression of HOTAIR which alters the lipid metabolic pathway and silences the sirtuin 1 (SIRT1) promoter which aids in the efficient release of viral particles from the cell [30]. It has been reported that the expression of lncRNA-8 is increased in Huh-7.5 hepatoma cells after HCV infection. LncRNA-8 increases HCV replication by promoting GPR55 (G-protein coupled receptor 55) expression and downregulating the expression of interferon-stimulated genes (ISGs) including IFITM1, Mx1, and ISG15 [31]. HCV also induces the upregulation of HULC (highly upregulated in liver cancer) via the transcription of retinoid X receptor-α (RXRA) [32], IGF2-AS, 7SK [33], lncRNA-IFI6 (AL445490.1) [34], and lncATV (AL391832.2) [35], promoting viral release. Additionally, PAR5 and aHIF lncRNAs are also reported to be dysregulated in HCV-induced HCC [36]. However, the detailed mechanisms of HCC development by up- or downregulation of various ncRNAs induced by HCV infection still need to be explored.
5. DNA Methylation Patterns
DNA hypermethylation, hypomethylation, and imprinting loss are methylation-related changes which are closely tied to a number of human illnesses, including cancer. According to the RNA sequence study, tumor-suppressive genes’ promoter hypermethylation lowers their protein levels, while oncogenic genes’ promoter hypomethylation raises their expression levels. The expression of several tumor suppressor genes is specifically suppressed by DNA hypermethylation at their promoter regions, which directly contributes to the carcinogenic processes of HCC caused by HCV infection. Importantly, the primary mechanism for the inactivation of these genes is hypermethylation of the promoter CpG island. In addition to the tumor suppressor gene p53, several other tumor suppressor genes have also been found to be hypermethylated in earlier studies. The development of HCC caused by HCV involves 11 tumor suppressor genes, including RASAL1, EGLN3, CSMD1, CDKN2A, BCORL1, SFRP1, ZNF382, RUNX3, LOX, RB1, and p73. All of these tumor suppressor genes are downregulated in HCC cells with an HCV connection because the hypermethylation of these genes typically transcriptionally suppresses protein production [37,38,39].
6. Histone Modifications
Histone methylation, acetylation, phosphorylation, sumoylation, and ubiquitination have received the most attention in a variety of cancer model studies. Histone acetylation and methylation have been extensively studied during HCC development [40].
6.1. Histone Methylation
It has been shown that the overexpression of histone demethylase could increase HCC cell proliferation through the regulation of its downstream genes E2F1 and E2F2 [41], regarding the involvement of histone methylation in specific HCV-induced HCC. Another histone lysine-specific demethylase (LSD1), whose expression has been connected to HCV infection in HCC, is similarly elevated compared to normal cells; LSD1 knockout cells exhibit greater levels of H3K4me1/2 and H3K9me1/2. H3K4/2 and H3K9me1/2 are expressed at greater levels in LSD1 knockout cells [42].
Histone lysine or arginine methylases are strongly associated with the development of the prognosis for HCC. G9a, a histone methyltransferase, is primarily responsible for the epigenetic silencing of the tumor suppressor gene RARRES3, which leads to the development of HCC [43]. Ribavirin (RBV), a synthetic triazole derivative of guanosine used in anti-HCV therapy, works by reducing the mRNA levels of a number of interferon (IFN)-stimulated genes (ISGs) that are preactivated in HCC patients. These chosen ISGs are changed with histone H3 lysine 9 dimethylation/trimethylation (H3K9me2/H3K9me3) for transcriptional suppression by attracting additional G9a. ISG downregulation is impaired by G9a defect, and RBV is prevented from activating IFN for anti-HCV activity [44]. The polycomb-related gene EZH2 is a histone methyltransferase for histone 3’s lysine 27. Through its interaction with other transcription factors, EZH2 is essential for gene silencing and chromatin remodeling [45]. In China, the overexpression of EZH2 is thought to be a promising biomarker for people with HCC [46].
6.2. Histone Acetylation
Histone acetylation, which is dynamically regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), is also frequently observed in the development of HCC in HCV-infected individuals. Examples include histone H3 acetylated on lysine 9 (H3K9Ac) and histone 3 acetylated on lysine 27 (H3K27Ac). In mice, histone deacetylase 3 (HDAC3) knockout results in hyperacetylated H3K9 and decreased H3K9 trimethylation (H3K9me3), which impairs DSB (double-strand DNA breaks) repair and encourages the growth of HCC tumors. H3K27Ac stimulates universal cell-type-specific gene expression in cancer cells, where it is primarily located in core euchromatin areas. Whether it is caused by HCV infection or HBV infection, HCC tissues exhibit higher H3K27Ac levels than the normal liver [47].
Multiple biological mechanisms are often involved in HCC-related histone acetylation. For instance, iron excess is a risk factor for the progression of HCV to HCC in the liver, which is typically controlled by a peptide called human hepcidin. HDAC activation on histone acetylation lowers the expression level of hepcidin during HCV infection. A potential mechanism for the anti-HCV defense is provided by the involvement of hepcidin in inhibiting HCV replication by activating the signal transducer and activating transcription 3 (STAT3) signaling [29,45,48].
Histone deacetylase (HDAC) has also been linked positively to HCC and HCV, according to reports. Because the HDAC3 inhibitor was discovered to reduce HCV replication by boosting the liver-expressed antimicrobial peptide (LEAP1) and lowering apolipoprotein A1, it has consequently been evaluated for clinical HCC treatment (ApoA1). Hypoxia-inducible factor 1 (HIF1) and STAT3 are transcription factors that can change the histone acetylation of these gene promoter areas, which could account for the antiviral action of the HDAC3 inhibitor [49].
7. mRNA Modifications
RNA stability and translation are regulated by m6A modification, which takes place in RNAs during co-transcription. Viruses with RNA genomes have been found to have m6A methylation. These m6A modifications govern numerous pathogeneses and components of the viral life cycle that are connected to particular viral diseases [6]. The most prevalent mRNA modification in mammals, N6-methyladenosine (m6A) [50], has contributed to the development of various diseases, including cancers [51]. There are three classes of enzymes that are involved in the regulation of m6A modifications which include “erasers” (demethylases, such as FTO and ALKBH5), “readers” (YTH domain-containing heterogeneous nuclear ribonucleoprotein and RNA binding proteins), and “Writers” (methyltransferases, such as KIAA1429, METTL14, METTL3, and WTAP), whereby gene expression regulation is mediated through reversible m6A RNA methylation [52].
An essential methyltransferase involved in m6A modification is KIAA1429 [53]. KIAA1429 contributes to the emergence of HCC by regulating the mRNA methylation levels of ID2 in cell lines. Significantly reducing KIAA1429 at the cellular level prevented HCC cells from migrating, proliferating, and invading. The processes mediated by KIAA1429 in HCC were at least largely carried out by ID2 through methylation, according to the mechanisms [54]. More specifically, in HCC, KIAA1429 increased the m6A level of ID2 mRNA, which in turn decreased ID2 expression and aided cell invasion and migration. ID2 was a basic helix–loop–helix transcription factor antagonist and retinoblastoma (Rb) family protein antagonist. (Id2 is a retinoblastoma protein target and mediates signaling by Myc oncoproteins.) The ID proteins, master regulators of cancer stem cells and tumor aggressiveness, reported that the development of numerous disorders including cancer has been linked to ID2 [55].
The metastatic potential of HCC is suppressed by METTL14 and METTL14’s forced expression prevents tumor invasion and metastasis in HCC. PRI-miR126 is regulated by the METTL14-m6A module, and this has functional effects on tumor metastasis. Our findings imply that m6A alteration is important for tumor biology and that the association between METTL14 and miRNA signaling may be a potential target for HCC intervention [56]. The overexpression of YTHDF1 is linked to a poor prognosis for the disease. According to Kaplan–Meier analysis, the reduced YTHDF1 expression level was associated with improved survival for HCC patients. Additionally, it was examined that the co-expressed genes for YTHDF1 were using GO and KEGG pathways, and it was discovered that YTHDF1 was crucial for controlling HCC cell cycle progression and metabolism. This study may provide a brand-new therapeutic and prognostic target for HCC [51].
8. Modifications in Different Signaling Pathways
8.1. PI3K/AKT Pathway
The phosphoinositol-3-kinase (PI3K) pathway contributes in HCC carcinogenesis and metastasis by signaling cell proliferation and the inhibition of apoptosis. The activation of PI3K by various growth factors including EGF and IGF results in the activation of AKT via PIP3 (phosphoinositol triphosphate 3) [57]. AKT functions in the phosphorylation of many downstream cytoplasmic protein substrates for the regulation of apoptosis, RNA processing, cell division, and cell proliferation. HCV activates the PI3K/AKT pathway and, as a consequence, enhances HCV entry and cellular proliferation [58]. The activation of this pathway stimulates another downstream effector, SREBP, that facilitates the translocation of HCV via NS5A. Additionally, it has been found that NS5A inhibits apoptosis by downregulating the activity of a tumor suppressor gene PTEN (phosphatase and tensin homolog) which further upregulates the PI3K pathway, leading to the accumulation of AKT in cells and contributing to HCC carcinogenesis [59].
Similarly, HCV induces MDSC (myeloid-derived suppressor cell)-like monocytes which maintain persistent HCV infection by upregulating the PI3K signaling pathway [60]. Moreover, the activation of this pathway is also necessary for the stabilization of HIF-1α which leads to the stimulation of VEGF, triggering angiogenesis for HCC progression [61].
8.2. Wnt Pathway
The Wnt signaling pathway regulates tissue homeostasis, embryonic development, and several cellular processes including cell migration, polarity, and mobility. The Wnt pathway is activated by the binding of the FZD (Frizzled) receptor (7-transmembrane protein with cystine-rich extracellular domain for ligand binding) and Wnt ligands. The activated FZD receptor inhibits β-catenin degradation. β-catenin is degraded by a degradation complex of GSK3β (glycogen synthase kinase 3β), casein kinase I, APC (adenomatous polyposis coli), Axin, and Diversin. If not degraded, the resultant accumulation of β-catenin in the nucleus activates several cell proliferation regulators including c-MYC, CCND1, and WISP-1 [62].
Mutations in the Wnt signaling pathway—such as protein mutations and abnormal expressions—have been observed in more than 50% of HCC patients with 30% mutations caused by the accumulation of β-catenin [63]. HCV infection downregulates Auxin2 or APC, leading to β-catenin accumulation in the cell which stimulates cancerous mutations within hepatocytes by rendering the cells chemo-resistant and anti-apoptotic. It increases the expression of the p53 repressor, Deltanp73 [64]. HCV dysregulates the pathway by its structural and non-structural proteins. It has been reported that the NS5A protein can directly activate the Wnt signaling pathway by interacting with PI3K, inhibiting the activity of GSK3β, and preventing the degradation of β-catenin [65]. HCV also epigenetically downregulates PPARγ which causes myo-fibroblastic trans-differentiation (MTD) in the liver and leads to fibrosis by impairing the Wnt signaling pathway [66]. Moreover, the HCV core protein induces the Wnt signaling pathway via increasing cellular proliferation by upregulating Wnt1 expression and WISP2, its target gene [67]. In cell cultures, HCV core protein and NS5A have been shown to upregulate β-catenin via indirect mechanisms, including its phosphorylation and stabilization [64].
8.3. JAK/STAT Pathway
Differentiation, transcription activation, and other biological processes are regulated by the Janus kinase-signal transducer and activator of the transcription (JAK/STAT) pathway. JAK/STAT signaling has also been reported to regulate liver regeneration and gluconeogenesis. The JAK/STAT pathway is activated by various cytokines and growth factors, such as ILs, IFNs, and EGF family members. This signaling system is essential in HCC caused by HCV. HCV stimulates STAT3 and STAT3-mediated transcription. STAT3 is regarded as an oncogene and may accelerate the progression of HCC by affecting cell differentiation, proliferation, and death [68]. HCV promotes STAT3 both directly through contact with the HCV core protein and indirectly by NS5A, which stimulates STAT3 via ROS generation [69]. IFN-γ activation causes phosphorylation of JAK1/2 and transcription driven by STAT3. According to mechanistic research, HCV prohibits the IFN impact on the JAK/STAT signaling pathway deteriorating elements along this journey [70].
Additionally, STAT regulation enhances viral replication and inhibits the production of ISGs, such as PKR, ISG15, MxB, and OAS2. The signaling-related protein STAT1 expression is notably downregulated in the tissues of HCC tumors. Moreover, STAT1 is evidently necessary for HCV replication, even though it is diminished in HCV-infected cells as a result of the HCV core, E1, E2, and NS3-proteasome-dependent 4A degradation. This is because STAT1 prevents HCC growth by encouraging apoptotic and p53-related processes and causes cell death. A recent study showed that STAT1 overexpression promotes G0 and G1 cell differentiation cycle arrest via cyclin A and cyclin D1, downregulates expressions of the CDK2 protein and cyclin E, and also encourages apoptosis by reducing the expression of Hes-1 and NF-kB p65 and boosting p53 and Fbxw7 expression [71,72].
8.4. p53 Pathway
Based on different cohorts, p53 can be mutated in 13–39% of HCV-related HCC cases [62,63]. The activation of the p53 pathway is a central event in DNA damage response, chemosensitivity, and the prognosis of HCC. p53-pathway-mediated apoptosis is affected at multiple levels in HCC. The p53 family acts as a target for novel therapeutic strategies in HCC. A crucial goal in the development of anticancer strategies in HCC must therefore be the restoration of physiologic apoptosis in response to cellular stress signals by reconstitution of the tumor suppressor function of p53 family members [73].
Interestingly, a recent study revealed that the mutations in TP53 are more frequent in tumors in patients that achieved a sustained virological response (SVR) after the treatment with direct-acting antivirals (DAAs) than in HCV-SVR-IFN tumors [74]. As the number of patients was low, the possible impact of HCV treatment on the p53 mutation rate should be further investigated.
8.5. Ras/MAPK Pathway
The development and occurrence of HCC has found to be associated with abnormal activation of the Ras/MAPK signaling pathway. The Ras/MAPK signaling pathway has been shown to stabilize the effect of hypoxia caused by HIF-1a and the inactivate tumor suppressor p53 gene in cells, thereby promoting angiogenesis via amplification [75]. It has been demonstrated that the HCV core protein can incorporate the Ras oncogene within rodent fibroblasts under specific conditions and can transcriptionally repress the p53 promoter [9].
Once activated by extracellular growth factor, the GTP-Ras complex phosphorylates ERK1/2 and MAP2Ks and activates two transcription factors, c-Fos and c-Jun, to upregulate the expression of genes involved in cell division and differentiation [76]. The core protein of HCV upregulates the Ras/MAPK signaling pathway by inducing the expression of histone methyltransferase SET and MYND domain containing 3 (SMYD3) which promotes the methylation of a negative regulator of this pathway, namely, the RASSF1A promoter [77,78]. Moreover, HCV infection downregulates the activation of RASALI and DUSP1 (dual-specificity phosphatase 1) via DNA methylation, thereby facilitating the activation of Ras in GTP-form. It has also been reported that the cellular kinase MEKI phosphorylates NS5A and regulates HCV replication in HCV-replicon systems. In this way, the Ras/MAPK signaling pathway is modulated and the anti-HCV activity of IFN-γ is inhibited by indirect or direct regulation of the phosphorylation of NS5A [69].
9. Conclusions
In account of the extensive genetic and epigenetic network that underlies the molecular cause of HCC induced by HCV, it can be concluded that HCV dysregulates various cellular processes and signaling pathways and facilitates in the pathogenesis of HCC. These HCV regulated pathways and processes can be used for developing more effective therapies for delaying HCC progression. Combined therapy may be required to target several proteins in light of the fact that HCV-induced HCC transcriptionally results in the activation of oncogenic genes and the suppression of tumor suppressor genes.
Author Contributions
Conceptualization, S.A.; writing—original draft preparation, A.S. & H.R.A.; writing—review and editing, Z.M.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the HEC Pakistan, MEAE and the MESRI as part of Franco-Pakistani Hubert Curien Partnership.
Institutional Review Board Statement
Not applicable as it is a review article.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.; Piacentini, M. Molecular mechanisms of hepatitis C virus–induced hepatocellular carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Axley, P.; Ahmed, Z.; Ravi, S.; Singal, A.K. Hepatitis C Virus and Hepatocellular Carcinoma: A Narrative Review. J. Clin. Transl. Hepatol. 2017, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, R.; Deng, Y.; Zhao, L. Conditional survival of patients with hepatocellular carcinoma: Results from the Surveillance, Epidemiology, and End Results registry. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; Henley, S.J.; Cucinelli, J.E.; McGlynn, K.A. Changing Hepatocellular Carcinoma Incidence and Liver Cancer Mortality Rates in the United States. Am. J. Gastroenterol. 2014, 109, 542–553. [Google Scholar] [CrossRef]
- Kim, G.-W.; Siddiqui, A. N6-methyladenosine modification of HCV RNA genome regulates cap-independent IRES-mediated translation via YTHDC2 recognition. Proc. Natl. Acad. Sci. USA 2021, 118, e2022024118. [Google Scholar] [CrossRef]
- Dubuisson, J.; Cosset, F.-L. Virology and cell biology of the hepatitis C virus life cycle—An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef]
- Bhattacharjee, C.; Singh, M.; Das, D.; Chaudhuri, S.; Mukhopadhyay, A. Current therapeutics against HCV. Virusdisease 2021, 32, 228–243. [Google Scholar] [CrossRef]
- Virzì, A.; Suarez, A.A.R.; Baumert, T.F.; Lupberger, J. Oncogenic Signaling Induced by HCV Infection. Viruses 2018, 10, 538. [Google Scholar] [CrossRef]
- Janjua, N.Z.; Wong, S.; Darvishian, M.; Butt, Z.A.; Yu, A.; Binka, M.; Alvarez, M.; Woods, R.; Yoshida, E.M.; Ramji, A.; et al. The impact of SVR from direct-acting antiviral- and interferon-based treatments for HCV on hepatocellular carcinoma risk. J. Viral Hepat. 2020, 27, 781–793. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Saleem, K.; Afzal, S.; Decaens, T. Predictive Factors for Hepatocellular Carcinoma Development after Direct-Acting Antiviral Treatment of HCV. Livers 2021, 1, 313–321. [Google Scholar] [CrossRef]
- Matsuura, K.; Tanaka, Y. Host genetic variations associated with disease progression in chronic hepatitis C virus infection. Hepatol. Res. 2018, 48, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Bruno, S.; Mondelli, M.U.; Maisonneuve, P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: A meta-analysis. J. Hepatol. 2009, 50, 1142–1154. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Koga, H.; Yoshida, T.; Masuda, H.; Iwamoto, H.; Sakata, M.; Sata, M. Hepatitis C virus core protein upregulates the expression of vascular endothelial growth factor via the nuclear factor-κB/hypoxia-inducible factor-1α axis under hypoxic conditions. Hepatol. Res. 2012, 42, 591–600. [Google Scholar] [CrossRef]
- Zhu, C.; Liu, X.; Wang, S.; Yan, X.; Tang, Z.; Wu, K.; Li, Y.; Liu, F. Hepatitis C virus core protein induces hypoxia-inducible factor 1α-mediated vascular endothelial growth factor expression in Huh7.5.1 cells. Mol. Med. Rep. 2014, 9, 2010–2014. [Google Scholar] [CrossRef]
- Mahmoudvand, S.; Shokri, S.; Taherkhani, R.; Farshadpour, F. Hepatitis C virus core protein modulates several signaling pathways involved in hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 42–58. [Google Scholar] [CrossRef]
- Akkari, L.; Grégoire, D.; Floc’H, N.; Moreau, M.; Hernandez, C.; Simonin, Y.; Rosenberg, A.R.; Lassus, P.; Hibner, U. Hepatitis C viral protein NS5A induces EMT and participates in oncogenic transformation of primary hepatocyte precursors. J. Hepatol. 2012, 57, 1021–1028. [Google Scholar] [CrossRef]
- Wilson, G.K.; Brimacombe, C.L.; Rowe, I.; Reynolds, G.M.; Fletcher, N.; Stamataki, Z.; Bhogal, R.H.; Simões, M.L.; Ashcroft, M.; Afford, S.C.; et al. A dual role for hypoxia inducible factor-1α in the hepatitis C virus lifecycle and hepatoma migration. J. Hepatol. 2012, 56, 803–809. [Google Scholar] [CrossRef]
- Ninio, L.; Nissani, A.; Meirson, T.; Domovitz, T.; Genna, A.; Twafra, S.; Srikanth, K.D.; Dabour, R.; Avraham, E.; Davidovich, A.; et al. Hepatitis C Virus Enhances the Invasiveness of Hepatocellular Carcinoma via EGFR-Mediated Invadopodia Formation and Activation. Cells 2019, 8, 1395. [Google Scholar] [CrossRef]
- Paul, C.; Khera, L.; Kaul, R. Hepatitis C virus core protein interacts with cellular metastasis suppressor Nm23-H1 and promotes cell migration and invasion. Arch. Virol. 2019, 164, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Unfried, J.P.; Fortes, P. LncRNAs in HCV Infection and HCV-Related Liver Disease Juan. Int. J. Mol. Sci. 2020, 21, 2255. [Google Scholar] [CrossRef] [PubMed]
- Kerr, T.A.; Korenblat, K.M.; Davidson, N.O. MicroRNAs and liver disease. Transl. Res. 2011, 157, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Plissonnier, M.-L.; Herzog, K.; Levrero, M.; Zeisel, M.B. Non-Coding RNAs and Hepatitis C Virus-Induced Hepatocellular Carcinoma. Viruses 2018, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Bonkovsky, H.L. Non-coding RNAs in hepatitis C-induced hepatocellular carcinoma: Dysregulation and implications for early detection, diagnosis and therapy. World J. Gastroenterol. 2013, 19, 7836–7845. [Google Scholar] [CrossRef]
- Song, K.; Han, C.; Wu, T. Epigenetic regulation of miR-122 by PPARgammar and hepatitis B virus X protein in hepatocellular carcinoma cells. FASEB J. 2013, 27, 872.10. [Google Scholar] [CrossRef]
- Steuerwald, N.M.; Parsons, J.C.; Bennett, K.; Bates, T.C.; Bonkovsky, H.L. Parallel microRNA and mRNA expression profiling of (genotype 1b) human hepatoma cells expressing hepatitis C virus. Liver Int. 2010, 30, 1490–1504. [Google Scholar] [CrossRef]
- Zeng, B.; Li, Z.; Chen, R.; Guo, N.; Zhou, J.; Zhou, Q.; Lin, Q.; Cheng, D.; Liao, Q.; Zheng, L.; et al. Epigenetic regulation of miR-124 by Hepatitis C Virus core protein promotes migration and invasion of intrahepatic cholangiocarcinoma cells by targeting SMYD3. FEBS Lett. 2012, 586, 3271–3278. [Google Scholar] [CrossRef]
- El-Maraghy, S.A.; Adel, O.; Zayed, N.; Yosry, A.; El-Nahaas, S.M.; Gibriel, A.A. Circulatory miRNA-484, 524, 615 and 628 expression profiling in HCV mediated HCC among Egyptian patients; implications for diagnosis and staging of hepatic cirrhosis and fibrosis. J. Adv. Res. 2019, 22, 57–66. [Google Scholar] [CrossRef]
- Li, Z.-Q.; Gu, X.-Y.; Hu, J.-X.; Ping, Y.; Li, H.; Yan, J.-Y.; Li, J.; Sun, R.; Yu, Z.-J.; Zhang, Y. Hepatitis C virus core protein impairs metabolic disorder of liver cell via HOTAIR-Sirt1 signalling. Biosci. Rep. 2016, 36, e00336. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Wilhelm, J.; Gerresheim, G.K.; Shalamova, L.A.; Niepmann, M. Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus. Viruses 2019, 11, 549. [Google Scholar] [CrossRef]
- Sharma, G.; Tripathi, S.K.; Das, S. lncRNA HULC facilitates efficient loading of HCV-core protein onto lipid droplets and subsequent virus-particle release. Cell. Microbiol. 2019, 21, e13086. [Google Scholar] [CrossRef]
- Xiong, Y.; Jia, M.; Yuan, J.; Zhang, C.; Zhu, Y.; Kuang, X.; Lan, L.; Wang, X. STAT3-regulated long non-coding RNAs lnc-7SK and lnc-IGF2-AS promote hepatitis C virus replication. Mol. Med. Rep. 2015, 12, 6738–6744. [Google Scholar] [CrossRef]
- Liu, X.; Duan, X.; Holmes, J.A.; Li, W.; Lee, S.H.; Tu, Z.; Chung, R.T. A novel lncRNA regulates HCV infection through IFI6. Hepatology 2019, 69, 1004–1019. [Google Scholar] [CrossRef]
- Fan, J.; Cheng, M.; Chi, X.; Liu, X.; Yang, W. A Human Long Non-coding RNA LncATV Promotes Virus Replication Through Restricting RIG-I–Mediated Innate Immunity. Front. Immunol. 2019, 10, 1711. [Google Scholar] [CrossRef]
- Zhang, Q.; Matsuura, K.; Kleiner, D.E.; Zamboni, F.; Alter, H.J.; Farci, P. Analysis of long noncoding RNA expression in hepatocellular carcinoma of different viral etiology. J. Transl. Med. 2016, 14, 328. [Google Scholar] [CrossRef]
- Lim, J.S.; Park, S.-H.; Jang, K.L. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012, 321, 154–161. [Google Scholar] [CrossRef]
- Zekri, A.E.-R.N.; Nassar, A.A.-M.; El-Rouby, M.N.E.-D.; Shousha, H.I.; Barakat, A.B.; El-Desouky, E.D.; Zayed, N.A.; Ahmed, O.S.; Youssef, A.S.E.-D.; Kaseb, A.O.; et al. Disease Progression from Chronic Hepatitis C to Cirrhosis and Hepatocellular Carcinoma is Associated with Increasing DNA Promoter Methylation. Asian Pac. J. Cancer Prev. 2013, 14, 6721–6726. [Google Scholar] [CrossRef]
- Sun, G.; Zhang, C.; Feng, M.; Liu, W.; Xie, H.; Qin, Q.; Zhao, E.; Wan, L. Methylation analysis of p16, SLIT2, SCARA5, and Runx3 genes in hepatocellular carcinoma. Medicine 2017, 96, e8279. [Google Scholar] [CrossRef]
- Braghini, M.R.; Re, O.L.; Romito, I.; Fernandez-Barrena, M.G.; Barbaro, B.; Pomella, S.; Rota, R.; Vinciguerra, M.; Avila, M.A.; Alisi, A. Epigenetic remodelling in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 1–21. [Google Scholar] [CrossRef]
- Zhu, L.; Huang, F.; Wan, T.; Xu, H.; Zhao, Q. Overexpression of long noncoding RNA LINC00882 is associated with poor prognosis in hepatocellular carcinoma. OncoTargets Ther. 2018, 11, 5209–5217. [Google Scholar] [CrossRef]
- Kim, S.; Bolatkan, A.; Kaneko, S.; Ikawa, N.; Asada, K.; Komatsu, M.; Hayami, S.; Ojima, H.; Abe, N.; Yamaue, H.; et al. Deregulation of the Histone Lysine-Specific Demethylase 1 Is Involved in Human Hepatocellular Carcinoma. Biomolecules 2019, 9, 810. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chiu, D.K.-C.; Tsang, F.H.-C.; Law, C.-T.; Cheng, C.L.-H.; Au, S.L.-K.; Lee, J.M.-F.; Wong, C.C.L.; Ng, I.O.-L.; Wong, C.-M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Golden-mason, L.; Rosen, H.R. Revisiting the Paradox of ISG expression as a predictor of HCV treatment response, a decade later. Hepatology 2018, 68, 2053–2055. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Yu, Z.; Tian, Y.; Lee, Y.-Y.; Li, M.S.; Go, M.Y.; Cheung, Y.-S.; Lai, P.B.; Chan, A.M.; To, K.-F.; et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J. Hepatol. 2014, 62, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.-Y.; Tong, Z.-T.; Zheng, F.; Liao, Y.-J.; Wang, Y.; Rao, H.-L.; Chen, Y.-C.; Wu, Q.-L.; Liu, Y.-H.; Guan, X.-Y.; et al. EZH2 protein: A promising immunomarker for the detection of hepatocellular carcinomas in liver needle biopsies. Gut 2011, 60, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xia, J.; Zhou, Y.-J.; Wan, J.; Li, L.; Bao, J.; Shi, Y.-J.; Bu, H. Proportions of acetyl-histone-positive hepatocytes indicate the functional status and prognosis of cirrhotic patients. World J. Gastroenterol. 2015, 21, 6665–6674. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yang, X.; Shen, Y.; Wang, Y.; Xia, X.; Zhang, A. STAT3 signaling pathway plays importantly genetic and functional roles in HCV infection. Mol. Genet. Genom. Med. 2019, 7, e821. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Q.; Yang, Q.; Tang, J.; Xu, C.; Gai, D.; Chen, X.; Chen, J. Histone Deacetylase 3 Inhibitor Suppresses Hepatitis C Virus Replication by Regulating Apo-A1 and LEAP-1 Expression. Virol. Sin. 2018, 33, 418–428. [Google Scholar] [CrossRef]
- Batista, P.J. The RNA Modification N 6 -methyladenosine and Its Implications in Human Disease. Genom. Proteom. Bioinform. 2017, 15, 154–163. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, Y.; Mao, Q.; Jiang, X.; Jiang, W.; Chen, J.; Xu, W.; Zhong, L.; Sun, X. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomarkers 2018, 21, 859–868. [Google Scholar] [CrossRef]
- Huo, F.-C.; Zhu, Z.-M.; Pei, D.-S. N(6)-methyladenosine (m(6)A) RNA modification in human cancer. Cell Prolif. 2020, 53, e12921. [Google Scholar] [CrossRef]
- Cheng, X.; Li, M.; Rao, X.; Zhang, W.; Li, X.; Wang, L.; Huang, G. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. OncoTargets Ther. 2019, 12, 3421–3428. [Google Scholar] [CrossRef]
- Han, Q.; Yang, J.; Yang, H.; Li, C.; Li, J.; Cao, Y. RETRACTED ARTICLE: KIAA1429 promotes osteosarcoma progression by promoting stem cell properties and is regulated by miR-143-3p. Cell Cycle 2020, 19, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Roschger, C.; Cabrele, C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun. Signal. 2017, 15, 1–26. [Google Scholar] [CrossRef]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hoffman, B.; Liu, Q. PI3K-Akt signaling pathway upregulates hepatitis C virus RNA translation through the activation of SREBPs. Virology 2016, 490, 99–108. [Google Scholar] [CrossRef]
- Liu, Z.; Tian, Y.; Machida, K.; Lai, M.M.C.; Luo, G.; Foung, S.K.H.; Ou, J.H.J. Transient activation of the PI3K-AKT pathway by hepatitis C virusto enhance viral entry. J. Biol. Chem. 2012, 287, 41922–41930. [Google Scholar] [CrossRef]
- Cheng, D.; Zhang, L.; Yang, G.; Zhao, L.; Peng, F.; Tian, Y.; Xiao, X.; Chung, R.T.; Gong, G. Hepatitis C virus NS 5A drives a PTEN - PI 3K/Akt feedback loop to support cell survival. Liver Int. 2014, 35, 1682–1691. [Google Scholar] [CrossRef]
- Zhai, N.; Li, H.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Tu, Z. Hepatitis C virus induces MDSCs-like monocytes through TLR2/PI3K/AKT/STAT3 signaling. PLoS ONE 2017, 12, e0170516. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, Z.; Yang, L.; Gao, Y.; Zhu, Q.; Hu, L.; Huang, D.; Xu, Q. Hypoxia-inducible factors in hepatocellular carcinoma (Review). Oncol. Rep. 2019, 43, 3–15. [Google Scholar] [CrossRef]
- Gurbuz, I.; Chiquet-Ehrismann, R. CCN4/WISP1 (WNT1 inducible signaling pathway protein 1): A focus on its role in cancer. Int. J. Biochem. Cell Biol. 2015, 62, 142–146. [Google Scholar] [CrossRef]
- Khalaf, A.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef]
- Liu, J.; Ding, X.; Tang, J.; Cao, Y.; Hu, P.; Zhou, F.; Shan, X.; Cai, X.; Chen, Q.; Ling, N.; et al. Enhancement of Canonical Wnt/β-Catenin Signaling Activity by HCV Core Protein Promotes Cell Growth of Hepatocellular Carcinoma Cells. PLoS ONE 2011, 6, e27496. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2016, 52, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.-G.; Yang, Y.-Y.; He, X.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.-W.; Jin, Y.; Li, J. Wnt signaling in liver fibrosis: Progress, challenges and potential directions. Biochimie 2013, 95, 2326–2335. [Google Scholar] [CrossRef]
- Jiang, X.-H.; Xie, Y.-T.; Cai, Y.-P.; Ren, J.; Ma, T. Effects of hepatitis C virus core protein and nonstructural protein 4B on the Wnt/β-catenin pathway. BMC Microbiol. 2017, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Wonganan, O.; He, Y.J.; Shen, X.F.; Wongkrajang, K.; Suksamrarn, A.; Zhang, G.L.; Wang, F. 6-Hydroxy-3-O-methyl-kaempferol 6-O-glucopyranoside potentiates the anti-proliferative effect of interferon α/β by promoting activation of the JAK/STAT signaling by inhibiting SOCS3 in hepatocellular carcinoma cells. Toxicol. Appl. Pharmacol. 2017, 336, 31–39. [Google Scholar] [CrossRef]
- Roca Suarez, A.A.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef]
- Zhao, L.-J.; He, S.-F.; Liu, Y.; Zhao, P.; Bian, Z.-Q.; Qi, Z.-T. Inhibition of STAT Pathway Impairs Anti-Hepatitis C Virus Effect of Interferon Alpha. Cell. Physiol. Biochem. 2016, 40, 77–90. [Google Scholar] [CrossRef]
- Stevenson, N.J.; Bourke, N.; Ryan, E.; Binder, M.; Fanning, L.; Johnston, J.A.; Hegarty, J.E.; Long, A.; O’Farrelly, C. Hepatitis C virus targets the interferon-α JAK/STAT pathway by promoting proteasomal degradation in immune cells and hepatocytes. FEBS Lett. 2013, 587, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Aftab, A.; Afzal, S.; Idrees, M.; Shahid, A.A. p53 and rb promoter methylation in hepatitis C virus-related chronic hepatitis and hepatocellular carcinoma. Futur. Virol. 2021, 16, 15–25. [Google Scholar] [CrossRef]
- Imamura, T.; Okamura, Y.; Ohshima, K.; Uesaka, K.; Sugiura, T.; Ito, T.; Yamamoto, Y.; Ashida, R.; Ohgi, K.; Otsuka, S.; et al. Hepatocellular carcinoma after a sustained virological response by direct-acting antivirals harbors TP53 inactivation. Cancer Med. 2022, 11, 1769–1786. [Google Scholar] [CrossRef]
- Markert, E.K.; Levine, A.J.; Vazquez, A. Proliferation and tissue remodeling in cancer: The hallmarks revisited. Cell Death Dis. 2012, 3, e397. [Google Scholar] [CrossRef]
- Zhang, Q.; Gong, R.; Qu, J.; Zhou, Y.; Liu, W.; Chen, M.; Liu, Y.; Zhu, Y.; Wu, J. Activation of the Ras/Raf/MEK Pathway Facilitates Hepatitis C Virus Replication via Attenuation of the Interferon-JAK-STAT Pathway. J. Virol. 2012, 86, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Chen, R.; Li, Z.; Liu, Y.; Cheng, D.; Zhou, Q.; Zhou, J.; Lin, Q. Hepatitis C virus core upregulates the methylation status of the RASSF1A promoter through regulation of SMYD3 in hilar cholangiocarcinoma cells. Acta Biochim. Biophys. Sin. 2011, 43, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Huynh, V.T.; Lim, Y.-S.; Tran, S.C.; Pham, T.M.; Nguyen, L.N.; Hwang, S.B. Hepatitis C Virus Nonstructural 5A Protein Interacts with Abelson Interactor 1 and Modulates Epidermal Growth Factor-mediated MEK/ERK Signaling Pathway. J. Biol. Chem. 2016, 291, 22607–22617. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).