Abstract
The kidney is a vital organ that carries out significant metabolic functions in our body. Due to the complexity of its role, the kidney is also susceptible to many disease conditions, such as acute kidney injury (AKI) and chronic kidney disease (CKD). Despite the prevalence and our increased understanding of the pathophysiology of both AKI and CKD as well as the transition of AKI to CKD, no well-established therapeutics have been applied clinically to these conditions, rendering an urgent need for a novel potential therapeutic target to be developed. In this article, we reviewed the function of ketone bodies in some common kidney conditions, such as drug-induced nephrotoxicity, ischemia and reperfusion injury, fibrosis development, diabetic kidney disease, kidney aging, hypertension, and CKD progression. All the selected studies reviewed were performed in animal models by primarily utilizing rodents, which also provide invaluable sources for future clinical applications. Ketone bodies have shown significant renal protective properties via attenuation of oxidative stress, increased expression of anti-inflammatory proteins, gene regulation, and a reduction of apoptosis of renal cells. A physiological level of ketone bodies could be achieved by fasting, a ketogenic diet, and an exogenous ketone supplement. Finally, the limitations of the long-term ketogenic diet were also discussed.
1. Kidney Functions and Diseases
The kidney is a vital endocrine organ involved in our body’s metabolic system [1]. For instance, the kidney participates in regulating the blood pressure system through the renin–angiotensin–aldosterone system (RAAS) by releasing renin stored in the juxtaglomerular cell to initiate RAAS in response to a decrease in blood pressure [2]. The kidney also assists in blood oxygen level monitoring by erythropoietin secretion, which also has a protective function in many organs and multiple conditions, such as protection against ischemia and reperfusion injury, hypoxia conditions, and oxidative damage induced by reactive oxygen species [3,4]. Due to the variety and complexity of duties that the kidney bears, the kidney is also susceptible to multiple disease conditions, which could harm the body as a whole. Kidney disease is generally classified into two categories: acute kidney injury (AKI) and chronic kidney disease (CKD) [5,6,7].
AKI is characterized by a sudden loss of kidney glomerular filtration rate within a short period of time, ranging from a few hours to a few days [8]. AKI could cause an accumulation of waste products in the blood and disrupts the electrolyte balance of the system, which could cause fluid retention, leading to oliguria and an increase in serum creatinine, which are important markers in AKI patients [9]. According to Kidney Disease: Improving Global Outcomes, AKI can be diagnosed if serum creatinine increases more than 0.3 mg/dL or 1.5 times baseline within 7 days, as well as less than 0.5 mL/Kg/h urine output in 6 h [9]. Typically, AKI is classified into three categories: prerenal, intrinsic, and postrenal [8,10,11]. Prerenal AKI is due to decreased renal perfusion and insufficient blood flow into the kidney, which could be caused by renal artery stenosis, heart failure, and some common drugs, such as nonsteroid anti-inflammatory drugs (NSAIDs) and ACE inhibitors [9]. Intrinsic AKI is due to a process within the kidney, which includes acute tubular necrosis (ATN) and acute interstitial nephritis (AIN) [12]. ATN normally is localized in proximal and distal tubules in the loop of Henle, which is caused by nephrotoxin or ischemia of tubules [9]. AIN is a type of delayed hypersensitivity reaction induced by drugs or streptococcus infections in the tubular and interstitial space. Postrenal AKI can be due to an inadequate amount of urine leaving the kidney caused by benign prostatic hyperplasia or kidney stones [9,13].
CKD is characterized by a persistent decrease in the glomerular filtration rate (GFR) of less than 60 mL/min/1.73 m2 [14,15]. Some significant clinical presentations of CKD include albuminuria, urine sediment abnormalities, electrolyte imbalance, other abnormalities detected by histology and imaging, and a history of kidney transplant [16,17]. There are multiple complications associated with CKD that could impact an individual’s quality of life, including anemia, cardiovascular disease, mineral and bone disorders, and malnutrition [18]. AKI could transit into CKD if left untreated. Although many studies illustrate that multiple types of cells are involved in the progression from AKI to CKD, the exact mechanism is still unclear [19]. In contrast to the high prevalence of AKI and CKD, no well-established therapeutics have been used to treat these disease conditions.
2. Ketone Bodies
Ketone bodies are alternative sources of energy that the brain and other organs can use when the body is in a prolonged fasting or starved state [20,21]. Generally, ketone bodies refer to three biomolecules which include β-hydroxybutyrate, acetoacetate, and acetone (Figure 1). Among these three molecules, acetone cannot be used and is exhaled via the respiratory tract [22]. The ketogenesis process starts with fatty acid being broken down in the liver to generate acetyl-CoA and ATP (Figure 2) [23], especially during the condition when there is a high glucagon-to-insulin ratio that stimulates the β oxidation of fatty acids [24]. As a result, an excessive amount of acetyl-CoA is produced and overwhelms the oxidation ability of the citric acid cycle. Consequently, acetyl-CoA is converted to ketone bodies. Following the enzymatic metabolism of thiolase, HMG-CoA synthase, and HMG-CoA lyase, ketone bodies are synthesized in the liver and released into the bloodstream [25]. However, the liver and red blood cells cannot utilize ketone bodies as energy sources due to the lack of succinyl CoA-3-oxaloacid CoA transferase (SCOT) which prevents ketone bodies from being broken down before they reach other tissues and organs [25].
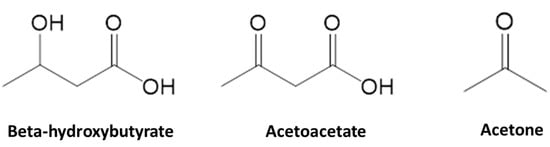
Figure 1.
The chemical structures of the three ketone bodies.
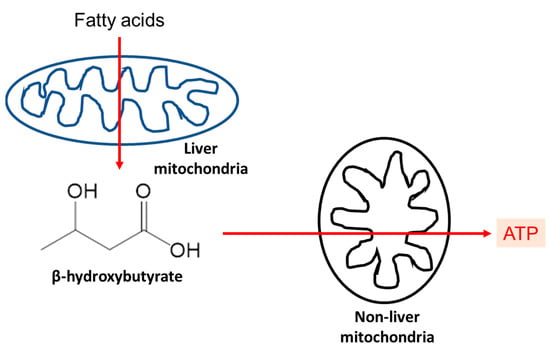
Figure 2.
Production of ketone bodies in the liver and their use as energy sources by other organs. Mainly depicted ketone body here is β-hydroxybutyrate, as this is the major ketone body species that is used for signaling and energy purpose. This figure is based on reference [20].
After β-hydroxybutyrate and acetoacetate enter the circulation, they will be reuptaken by other tissue cells via passive transport through monocarboxylate transporters 1 and 2 (MCT1, MCT2) for energy production purposes [26]. β-hydroxybutyrate is enzymatically converted back to acetoacetate by mitochondrial 3-hydroxybutyrate dehydrogenase (BDH 1) [26]. SCOT maintains the equilibrium between acetoacetate and acetoacetate-CoA that is then cleaved by acetoacetyl-CoA thiolase into two molecules of acetyl-CoA for an energy source [25,26]. A physiological level of ketone bodies has shown renal protective effects via multiple pathways in a wide range of disease conditions, including AKI and CKD as well as kidney aging in animal models [6,27,28,29,30,31,32,33,34]. With both endogenous ketone bodies during the fasting state and exogenous ketone bodies by ingesting a ketogenic diet or β-hydroxybutyrate, the patient could satisfy the concentration requirement for the renal protective effect to be activated [35,36,37].
In addition to being a nonhepatic energy source, ketone bodies, in particular, β-hydroxybutyrate, have unique signaling properties and biological functions [20,38,39]. As shown in Figure 3, β-hydroxybutyrate can serve as a direct antioxidant, can inhibit the mitochondrial generation of reactive oxygen species (ROS), and can also serve as a signaling molecule to upregulate transcriptional activities, leading to the expression of cytoprotective proteins, such as HO-1, NQO1, SOD2, catalase, glucose-6-phosphate dehydrogenase (G6PD), and γ-glutathione-cysteine ligase modifier subunit (GCLM) (Figure 3). The transcriptional factors involved in this upregulation by β-hydroxybutyrate have been demonstrated to include FOXO1, FOXO3a, and Nrf2 [20] (Figure 3).
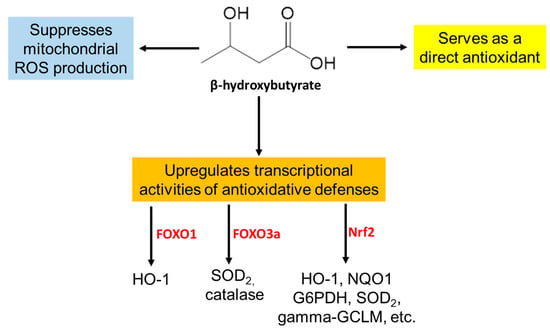
Figure 3.
General properties of β-hydroxybutyrate. Shown here is that this ketone body can promote the transcriptional activities of second defense antioxidant systems, including HO-1, NQO1, G6PD, SOD2, and catalase. This figure is based on reference [20].
In this review article, we will focus on the renal protective properties of ketone bodies. In particular, we will focus on selected animal models that are represented in the field of ketone body protection in kidney disease, including AKI and CKD. The animal models covered include sepsis-, paraquat- and cisplatin-induced AKI, ischemic kidney injury, hypertension and CKD progression, diabetic kidney disease (DKD), and kidney aging (Figure 4). All these animal models of kidney disease and aging have been widely explored for understanding the pathophysiology of kidney disease and for testing the therapeutic potentials of numerous natural and artificial compounds.
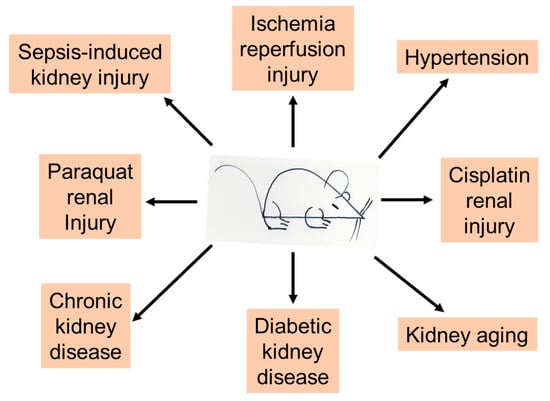
Figure 4.
Animal models of kidney injury and kidney disease covered in this article. All these animal models show renal protective effects of ketone bodies regardless of whether they are endogenous or exogenous.
2.1. Ketone Bodies in Sepsis-Induced AKI
Sepsis, as a life-threatening condition, is a major cause of AKI [40]. Animal models of sepsis-induced AKI are often created by the use of lipopolyssaccharide (LPS), an outer membrane component of gram-negative bacteria [41]. Indeed, LPS-induced septic AKI is an excellent model for studying AKI pathology and testing AKI therapeutics [42,43,44]. A recent study by Soni et al. [45] demonstrated that exogenous ketone ester administration could ameliorate LPS-induced kidney injury in a mouse model, and the underlying protective mechanisms of ketone ester is likely due to suppression of inflammatory and stress-responsive proteins, including IL-6, ccl-2, and Nos2. Additionally, renal INOS, p-JNK, and P-ERK were also decreased by ketone ester. Furthermore, ketone ester-treated mice exhibited less inflammasome activation [45]. Therefore, ketone bodies are nephroprotective in LPS-induced AKI.
It should be noted that while ketone bodies can be used to mitigate sepsis-induced kidney injury, there might be a toxic threshold for the use of ketone bodies such as β-hydroxybutyrate. Weckx et al. [46] have found that when the β-hydroxybutyrate dosage was raised from 150 mg/day to 300 mg/day, the illness severity scores of the septic animals reflected by muscle weakness doubled, and mortality of the animals increased from 30.45 to 87.5%. This study concludes that while 150 mg/day of β-hydroybutyrate is a safe dosage, dosages beyond 180 mg/day would induce toxicity and compromise the therapeutic value of β-hydroxybutyrate [46], at least in the mouse model that was used.
2.2. Ketone Bodies in Paraquat-Induced AKI
Paraquat (1,1-dimethyl-4-4-bipiridinium dichloride) is a widely used herbicide [47], especially in developing countries. It has been reported that a high death rate is associated with paraquat toxicity due to either accidental or intentional poisoning in mammals [48]. Paraquat rapidly distributes into tissues regardless of the route of administration, with the highest concentration being observed in the kidney where it produces severe nephrotoxicity. Impaired renal clearance of paraquat will result in an increased amount of plasma paraquat, thereby causing tissue damage in other vital organs. Respiratory failure in paraquat-induced AKI is responsible for most deaths triggered by paraquat [48]. Paraquat is reduced by nicotinamide adenine dinucleotide phosphate (NADPH)-cytochrome P-450 reductase to form free radicals [49]. β-hydroxybutyrate could rapidly cross the cell membrane and peripheral tissues according to a study conducted by Wei et al. Such studies indicate that β-hydroxybutyrate could be a potential therapeutic target for paraquat-induced toxicity [34]. Indeed, a significant drop in BUN and serum creatinine in the treatment group was detected compared to the paraquat-only group. Paraquat sharply decreased the levels of SOD and catalase (CAT) in rats, whereas β-hydroxybutyrate increased the concentration for both when compared to the control groups. A similar result was reported for glutathione (GSH), which is also an antioxidant molecule. When the levels of SOD, CAT, and GSH are low, the body has less capacity to fight against oxidative stress, and these decreases were attenuated by β-hydroxybutyrate [34]. The concentration of malondialdehyde, a byproduct of polyunsaturated fatty acid oxidation [50,51], was found to be decreased in the condition of oxidative stress. β-hydroxybutyrate is also involved in the regulation of the paraquat-induced apoptosis pathway by suppressing the concentration of three apoptotic proteins, including caspase-3, caspase-9, and poly-ADP ribose polymerase [34]. β-hydroxybutyrate also increased the level of antiapoptotic protein Bcl-2 and decreased the level of proapoptotic protein Bax as measured by western blot, leading to attenuated paraquat toxicity [34]. β-hydroxybutyrate could also regulate the oxidative and inflammatory response by regulating gene transcription factor NF E2-related factor 2 (Nrf2), which can further activate the expression of endogenous cytoprotective proteins, such as HO-1 and NQO-1 [52,53]. Western blot assay indicated that β-hydroxybutyrate contributed to an increase in transcription factor Nrf2, which was suppressed by the effect of paraquat [34].
It should be pointed out that paraquat-induced AKI may involve numerous mechanisms. For example, it has been reported in rats that paraquat-induced AKI may be regulated by NF-KB and death-associated protein kinase (DAPK), leading to inflammasome activation [54]. However, whether this NF-KB and DAPK regulation of inflammasome could be modulated by ketone bodies for AKI therapy has not been assessed [54].
2.3. Ketone Bodies in Cisplatin-Induced AKI
Cisplatin is one of the first metal-based drugs used to treat cancer via DNA lesion generation by purine interaction, resulting in the apoptosis of cancer cells [55]. However, cisplatin is also associated with many side effects across the body [56], in particular, renal toxicity [57,58]. Mikami et al. have reported that β-hydroxybutyrate exhibits a renal protective effect in cisplatin-induced AKI. Similar to the result in paraquat-induced AKI, β-hydroxybutyrate decreased BUN and serum creatinine, indicating an improved kidney function by β-hydroxybutyrate. The level of caspase-3 in human renal cortical epithelial cells (HRCE), an apoptotic cysteine protease, was also significantly lower in the group treated with β-hydroxybutyrate plus cisplatin compared to the group treated with cisplatin alone. Additionally, the level of antiapoptotic protein Bcl-2 was increased, and DNA damage marker p53 expression was decreased in the β-hydroxybutyrate treated group [32]. Moreover, β-hydroxybutyrate not only significantly attenuated a cisplatin-mediated decrease of HDAC 4/5/6 level and cisplatin-induced histone H3 acetylation through HDAC-associated pathway [32] but also significantly attenuated cisplatin-induced AMP-activated kinase (AMPK) phosphorylation, leading to further downregulation of HDAC5 and the apoptosis pathway [32]. Beta-hydroxybutyrate has also been shown to attenuate cisplatin-induced kidney injury by inhibiting NLRP3 inflammasome and oxidative stress [59].
2.4. Ketone Bodies in Ischemic Kidney Injury
Ischemic kidney injury is defined as a transient interruption of normal blood flow to the kidney, which could lead to an increase in oxidative stress, inflammation, and kidney dysfunction [60,61,62,63,64]. It has been established that ketone bodies are nephroprotective in ischemic kidney injury [65,66]. For example, when a ketogenic diet containing high fats, moderate proteins, and very low carbohydrates was given to Wistar rats for 3 days followed by ischemic surgery [33], less atrophied tubules and tubular degeneration, as well as hyaline casts in the rats, were observed [33]. It was further found that the ketogenic diet increased the activity of antioxidant enzyme SOD and glutathione peroxidases but not CAT after ischemic and reperfusion (IR) injury in rats. The same study also found that the ketogenic diet suppressed the concentration of proinflammation factors, such as tumor necrosis factor alpha (TNFα), interleukin 6 (IL-6), and monocyte chemoattractant protein 1 (MCP1) [33]. These changes were related to less expression of nuclear factor kappa B (NF-κB) subunit P50. Ketogenic diets also reduced the concentration of serum creatinine and BUN when compared to the control diet group [33]. Sirius red staining indicated that a ketogenic diet decreased IR-induced areas of fibrosis and western blots showed that fibrosis alpha smooth muscle actin (αSMA) and fibronectin (FN) levels were both decreased by a ketogenic diet when compared to the control diet group [33].
2.5. Ketone Bodies in Hypertension and CKD Progression
Hypertension is a well-known contributing factor to the progression of CKD [67], and treatment to lower blood pressure could slow the progression of CKD as well as preserve renal function [68]. When 20% 1,3-butanediol, a precursor of β-hydroxybutyrate [69], was given to a group of female spontaneously hypertensive rats via drinking water, there was a significant decrease in systolic blood pressure, diastolic blood pressure, and mean arterial pressure after 9 weeks of 1,3-butanediol treatment along with an increased concentration of β-hydroxybutyrate in the blood [30]. CKD progression markers were also assessed in this experiment, including serum creatinine concentration, proteinuria level as well as renal fibrosis, whereby all three markers had shown a significant decrease in the 1,3-butanediol-fed rats group when compared to the control group, while excretion of KIM-1 and NGAL remained unchanged [30]. Both proinflammation and anti-inflammation markers in the kidney were also assessed in this study. There was an increased level of anti-inflammatory cytokines, including IL-10, IL-2, and granulocyte-macrophage colony-stimulating factor (GM-CSF), in renal cortex lysates in the 1,3-butanediol-treated group. Interestingly, 1,3-butanediol also mildly increased proinflammatory factors, including IL-18, IL-1β, IL-7, TNFα, monocyte chemotactic protein-1 (MCP-1), and macrophage inflammatory protein-3α, potentially due to the long-term treatment of 1,3-butanediol in this experiment [30]. Overall, this study provides some clues that ketone bodies could be utilized to delay CKD progression by decreasing blood pressure in hypertension, lowering proteinuria and serum creatinine levels as well as contributing to anti-inflammatory cytokine production [30].
2.6. Ketone Bodies in Diabetic Kidney Disease
Diabetic kidney disease (DKD) is a complication associated with diabetes mellitus and is a leading cause of developing CKD [7,70,71]. Diabetic kidney disease is also responsible for the development of decreased glomerular filtration rates, glomerulus hyperfiltration, albuminuria, and end-stage renal failure which requires a kidney transplant for an individual to survive [72]. Ketone bodies have been shown to be beneficial for metabolic kidney disease as selectively discussed below.
2.6.1. SGLT2 Inhibitor (SGLT2i) and Ketones
Excessive activation of the mechanistic target of rapamycin complex 1 (mTORC1) has been associated with renal disease progression, whereby the suppression effect of 1,3-butanediol and empagliflozin on mTORC1 was also examined [29]. When 1,3-butanediol was given to high-fat diet-fed ApoE knockout mice, a significant increase in blood β-hydroxybutyrate was observed, which was thought to contribute to less elevation in plasma cystatin C, decreased mitochondrial morphological changes in proximal tubule epithelial cells, decreased glomerular damage and tubular vacuolar formation, fibrosis, and inflammation [29]. Additionally, similar results were also observed in the high-fat diet-fed ApoE knockout mice treated with an SGLT-2 inhibitor, empagliflozin. Such results could potentially explain how the mechanism of action of the SGLT-2 inhibitor contributed to the renal protective effect in diabetic kidney disease [29]. The renal protective effect of 1,3-butanediol and empagliflozin was also assessed in nondiabetic 5/6 nephrectomized rats; histology amelioration was detected in both podocytes and tubulointerstitial lesions through inhibition of mTORC1 signaling in both podocytes and proximal tubule epithelial cells, which agrees with the results observed in diabetic mice [29]. It should be pointed out that in addition to preventing glucose reabsorption, SGLT-2 inhibitors may act by promoting the use of ketone bodies in proximal tubular cells [73,74,75,76]. In a well-validated porcine model of heart failure [77], SGLT2i could cause a metabolic shift in fuel utilization from glucose-inefficient toward the consumption of ketone bodies [78]. In fact, this enhanced metabolic utilization of ketone bodies by SGLT2i has also been confirmed in human patients [79]. Hence, this increased ketone body utilization in the kidney could explain the benefits of SGLT2i for CKD patients [80,81] and quality of life [82]. These studies could also explain why SGLT2 inhibitors are efficacious on patients independent of CKD status [83].
2.6.2. Ketone Bodies and Glomerular Podocyte Senescence
Ketone bodies can also attenuate the senescence response of glomerular podocytes in diabetic kidney disease [84]. Fang et al. have reported that when streptozotocin (STZ)-induced diabetic mice were given β-hydroxybutyrate via I.P. injection every other day for 4 weeks, the Nrf2 response in podocytes was enhanced in the diabetic kidney, and Nrf2 signaling was required for the mitigation of podocyte senescence and the protection of podocytes against diabetes-induced oxidative stress [84,85]. The authors also found that GSK3β inhibition by β-hydroxybutyrate was also essential for podocytes senescence mitigation and antioxidation, and there is likely a physical interaction between β-hydroxybutyrate and GSK3β [84]. Regardless of the underlying mechanisms, the overall effects of β-hydroxybutyrate on Nrf2 activation and GSK3β was that STZ-induced diabetic kidney injury was much attenuated as levels of albuminuria were significantly decreased [84]. Additionally, SGLT2i’s cellular senescence mitigation in DKD by promoting ketone body-trigged Nrf2 activation has also been independently reported by others [86].
2.7. Ketone Bodies in Kidney Aging
Kidney aging results in a lesser amount of functional nephrons and kidney function [87,88], which could be affected by inflammation, autophagy, and redox stress [7,89,90]. Multiple factors could accelerate the aging process, especially diabetes, which could cause complications, such as hypertension, inflammation, oxidative stress, and advanced glycation end products [91]. A study conducted by Kim et al. indicates that β-hydroxybutyrate could potentially be a treatment plan for kidney-aging-related inflammation [31]. The β-hydroxybutyrate treated group observed an increase in both MnSOD and CAT when compared to the control group. Moreover, there was a decreased level of reactive oxygen species and peroxynitrite, decreased mRNA levels for IL-1β, IL-6, and TNFα, enhanced FoxO1 activation, and a decreased level of NF-κB phosphorylation. In the meantime, there were increased interactions between FoxO1 and proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), which may lead to attenuated aging-related inflammation [31]. Interestingly, β-hydroxybutyrate was also found to decrease the level of circulating insulin, leading to attenuated activation of PI3K and Akt [31].
Of note, given that aging and age-associated disease can be retarded by caloric restriction [92,93,94,95], it has been reported that ketone bodies can mimic the effects of caloric restriction [96,97,98,99]. This caloric restriction effect of ketone bodies has been linked to their activation of the Nrf2 signaling pathway and/or the FOXO signaling pathway [98,99], as shown in Figure 5. Therefore, it is not surprising that a ketogenic diet can mimic the effect of caloric restriction and retards both AKI and CKD by attenuating renal oxidative stress and inflammation in rats [33].
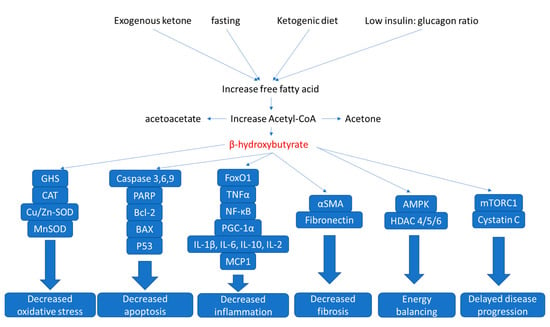
Figure 5.
Nephroprotective mechanisms of ketone bodies. Shown are sources of ketone bodies and the potential underlying mechanisms of renal protection by ketone bodies involved in kidney diseases discussed in this text.
3. Summary
AKI and CKD are global health problems affecting millions of people worldwide with an increasing prevalence in recent years [100,101]. Kidney function can be easily disrupted by drugs or toxicants, such as cisplatin [32] and paraquat [34]. Kidney function can also be impaired by comorbid conditions, such as diabetes [29,102,103,104], hypertension [105,106], thyroid hormone dysregulation [107], and epilepsy [108]. There are still a very limited number of therapies established to treat AKI and CKD besides traditional supportive care [109], electrolyte management [110], dialysis [111], and transplantation [112]. More experiments support the fact that ketone bodies have a renal protective effect through their antioxidants and anti-inflammatory properties. Ketone bodies could be a potential therapeutic regimen in clinical settings to treat a variety of kidney conditions as discussed in this paper. Ketone bodies exert these antioxidants and anti-inflammatory effects when appearing at physiological concentrations that can be achieved by fasting, a ketogenic diet, or exogenous ketone supplements, such as β-hydroxybutyrate and 1,3-butanediol [35,36,37]. Figure 5 depicts and summarizes the pertinent mechanisms and molecular players involved in ketone body renal protection discussed above.
Even though the ketogenic diet showed some distinct clinical benefits, such as ameliorating polycystic kidney disease [113], long-term treatments also raise concerns that the ketogenic diet could cause acidosis, anemia, increased cardiovascular risk, and increased oxidative stress [33,114]. A long-term ketogenic diet could also cause kidney stones, which is detrimental to kidney function [115]. Therefore, more research is needed to elucidate the comprehensive mechanisms of the ketone body renal protective effect and the potential side effects of ketone body treatments.
4. Future Directions
In this article, we have discussed the protective roles of ketone bodies in animal models of kidney disease. Most of the studies covered mainly focused on the cell signaling properties of ketone bodies in kidney injury, which include activation of second phase antioxidant defense systems involving transcriptional activation of Nrf2 and FOXO1. All these signaling events converge on the decrease in cell death and the amelioration of kidney injury. As redox imbalance between NADH and NAD+ and oxidative stress are tightly linked in the pathology of kidney injury [7], the role of NADH/NAD redox players and mitochondrial biogenesis and homeostasis in ketone body nephroprotection will need to be comprehensively investigated in the near future. These redox players would include those metabolic components involved in NADH generation and NAD+ regeneration, such as dihydrolipoamide dehydrogenase [116], mitochondrial complex I [4,117], NADPH oxidases [118], and apoptosis-inducing factor [119]. Additionally, the role of mitochondrial dynamics involving mitochondrial fusion and fission [120,121] in ketone body nephroprotection will also need to be evaluated.
Author Contributions
Conceptualization, H.L. and L.-J.Y. original draft preparation; H.L.; review and editing, L.-J.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work received no external funding.
Institutional Review Board Statement
Not applicable.
Informed consent statement: Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Acharya, V.; Olivero, J. The kidney as an endocrine organ. Methodist Debakey Cardiovasc. J. 2018, 14, 305–307. [Google Scholar] [CrossRef]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar]
- van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res. Rev. 2008, 59, 22–33. [Google Scholar] [CrossRef]
- Wu, J.; Luo, X.; Thangthaeng, N.; Sumien, N.; Chen, Z.; Rutledge, M.A.; Jing, S.; Forster, M.J.; Yan, L.J. Pancreatic mitochondrial complex i exhibits aberrant hyperactivity in diabetes. Biochem. Biophys. Rep. 2017, 11, 119–129. [Google Scholar] [CrossRef]
- Diwan, V.; Brown, L.; Gobe, G.C. Adenine-induced chronic kidney disease in rats. Nephrology 2018, 23, 5–11. [Google Scholar] [CrossRef]
- Yan, L.J. Folic acid-induced animal model of kidney disease. Animal Model. Exp. Med. 2021, 4, 329–342. [Google Scholar] [CrossRef]
- Yan, L.J. Nadh/nad(+) redox imbalance and diabetic kidney disease. Biomolecules 2021, 11, 730. [Google Scholar] [CrossRef]
- Turgut, F.; Awad, A.S.; Abdel-Rahman, E.M. Acute kidney injury: Medical causes and pathogenesis. J. Clin. Med. 2023, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; James, M.T. Acute kidney injury. Ann. Intern. Med. 2018, 168, 837. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.S.; Kaushik, R.; Ahmad, S.; Kaushik, R.M. Profile of acute kidney injury in patients with decompensated cirrhosis at a tertiary-care center in uttarakhand, india. Dig. Dis. 2020, 38, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, M.; Nayfeh, T.; Atieh, A.; AbuShamma, O.; Babaa, B.; Baniowda, M.; Hrizat, A.; Hasan, B.; Hassett, L.; Hamadah, A.; et al. Diagnostic performance of fractional excretion of sodium for the differential diagnosis of acute kidney injury: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2022, 17, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Dipiro, J.T.; Talbet, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L.M. Pharmacotherapy: A Pathophysiological Approach, 9th ed.; McGraw-Hill Education: New York, NY, USA, 2014. [Google Scholar]
- Mulay, S.R.; Evan, A.; Anders, H.J. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol. Dial. Transplant. 2014, 29, 507–514. [Google Scholar] [CrossRef]
- Prasad, R.; Jha, R.K.; Keerti, A. Chronic kidney disease: Its relationship with obesity. Cureus 2022, 14, e30535. [Google Scholar] [CrossRef]
- Yan, L.J. The nicotinamide/streptozotocin rodent model of type 2 diabetes: Renal pathophysiology and redox imbalance features. Biomolecules 2022, 12, 1225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, Y.; Zhao, X.; Cui, H.; Han, M.; Ren, X.; Gang, X.; Wang, G. Obesity and chronic kidney disease. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E24–E41. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Levin, A.; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef]
- Ammirati, A.L. Chronic kidney disease. Rev. Assoc. Med. Bras. (1992) 2020, 66 (Suppl. S1), s03–s09. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in aki to ckd progression. Am. J. Physiol. Renal. Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies, stress response, and redox homeostasis. Redox. Biol. 2020, 29, 101395. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Rohling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Lieberman, M.; Marks, A.D. Marks’ Basic Medical Biochemistry: A Clinical Approach, 4th ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Ghimire, P.; Dhamoon, A.S. Ketoacidosis. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Fukao, T.; Mitchell, G.; Sass, J.O.; Hori, T.; Orii, K.; Aoyama, Y. Ketone body metabolism and its defects. J. Inherit. Metab. Dis. 2014, 37, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies for kidney injury and disease. Adv. Redox Res. 2021, 2, 100009. [Google Scholar] [CrossRef]
- Yan, L.J.; Allen, D.C. Cadmium-induced kidney injury: Oxidative damage as a unifying mechanism. Biomolecules 2021, 11, 1575. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. Sglt2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mtorc1 inhibition. Cell Metab. 2020, 32, 404–419 e406. [Google Scholar] [CrossRef]
- Ishimwe, J.A.; Garrett, M.R.; Sasser, J.M. 1,3-butanediol attenuates hypertension and suppresses kidney injury in female rats. Am. J. Physiol. Renal. Physiol. 2020, 319, F106–F114. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, M.H.; Ha, S.; Bang, E.J.; Lee, Y.; Lee, A.K.; Lee, J.; Yu, B.P.; Chung, H.Y. Anti-inflammatory action of beta-hydroxybutyrate via modulation of pgc-1alpha and foxo1, mimicking calorie restriction. Aging 2019, 11, 1283–1304. [Google Scholar] [CrossRef]
- Mikami, D.; Kobayashi, M.; Uwada, J.; Yazawa, T.; Kamiyama, K.; Nishimori, K.; Nishikawa, Y.; Morikawa, Y.; Yokoi, S.; Takahashi, N.; et al. Beta-hydroxybutyrate, a ketone body, reduces the cytotoxic effect of cisplatin via activation of hdac5 in human renal cortical epithelial cells. Life Sci. 2019, 222, 125–132. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Leon-Contreras, J.C.; Sanchez-Tapia, M.; Silva-Palacios, A.; Cano-Martinez, A.; Gonzalez-Reyes, S.; Jimenez-Osorio, A.S.; Hernandez-Pando, R.; Osorio-Alonso, H.; Sanchez-Lozada, L.G.; et al. A ketogenic diet attenuates acute and chronic ischemic kidney injury and reduces markers of oxidative stress and inflammation. Life Sci. 2022, 289, 120227. [Google Scholar] [CrossRef]
- Wei, T.; Tian, W.; Liu, F.; Xie, G. Protective effects of exogenous beta-hydroxybutyrate on paraquat toxicity in rat kidney. Biochem. Biophys. Res. Commun. 2014, 447, 666–671. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef]
- Thai, P.N.; Miller, C.V.; King, M.T.; Schaefer, S.; Veech, R.L.; Chiamvimonvat, N.; Bers, D.M.; Dedkova, E.N. Ketone ester d-beta-hydroxybutyrate-(r)-1,3 butanediol prevents decline in cardiac function in type 2 diabetic mice. J. Am. Heart Assoc. 2021, 10, e020729. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Kumar, S.; Behl, T.; Sachdeva, M.; Sehgal, A.; Kumari, S.; Kumar, A.; Kaur, G.; Yadav, H.N.; Bungau, S. Implicating the effect of ketogenic diet as a preventive measure to obesity and diabetes mellitus. Life Sci. 2021, 264, 118661. [Google Scholar] [CrossRef]
- Li, R.J.; Liu, Y.; Liu, H.Q.; Li, J. Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 2020, 44, e13140. [Google Scholar] [CrossRef]
- Doi, K.; Leelahavanichkul, A.; Yuen, P.S.; Star, R.A. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Investig. 2009, 119, 2868–2878. [Google Scholar] [CrossRef]
- Kiyonaga, N.; Moriyama, T.; Kanmura, Y. Effects of dexmedetomidine on lipopolysaccharide-induced acute kidney injury in rats and mitochondrial function in cell culture. Biomed. Pharmacother. 2020, 125, 109912. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Cha, D.R.; Kim, B.; An, E.J.; Lee, S.R.; Cha, J.J.; Kang, Y.S.; Ghee, J.Y.; Han, J.Y.; Bae, Y.S. Lps-induced acute kidney injury is mediated by nox4-sh3yl1. Cell Rep. 2020, 33, 108245. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Song, Y.; Zhao, M.; Yi, Z.; Zeng, Q. Protective effects of edaravone, a free radical scavenger, on lipopolysaccharide-induced acute kidney injury in a rat model of sepsis. Int. Urol. Nephrol. 2015, 47, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Lu, G. Thioredoxin relieves lipopolysaccharide-induced acute kidney injury in mice by reducing inflammation, oxidative stress and apoptosis. Exp. Ther. Med. 2021, 21, 629. [Google Scholar] [CrossRef]
- Soni, S.; Martens, M.D.; Takahara, S.; Silver, H.L.; Maayah, Z.H.; Ussher, J.R.; Ferdaoussi, M.; Dyck, J.R.B. Exogenous ketone ester administration attenuates systemic inflammation and reduces organ damage in a lipopolysaccharide model of sepsis. Biochim. Biophys. Acta Mol. Basis. Dis. 2022, 1868, 166507. [Google Scholar] [CrossRef] [PubMed]
- Weckx, R.; Goossens, C.; Derde, S.; Pauwels, L.; Vander Perre, S.; Van den Bergh, G.; Langouche, L. Identification of the toxic threshold of 3-hydroxybutyrate-sodium supplementation in septic mice. BMC Pharmacol. Toxicol. 2021, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Onur, B.; Cavusoglu, K.; Yalcin, E.; Acar, A. Paraquat toxicity in different cell types of swiss albino mice. Sci. Rep. 2022, 12, 4818. [Google Scholar] [CrossRef]
- Safaei Asl, A.; Dadashzadeh, P. Acute kidney injury in patients with paraquat intoxication; a case report and review of the literature. J. Renal. Inj. Prev. 2016, 5, 203–206. [Google Scholar] [CrossRef]
- Fussell, K.C.; Udasin, R.G.; Gray, J.P.; Mishin, V.; Smith, P.J.; Heck, D.E.; Laskin, J.D. Redox cycling and increased oxygen utilization contribute to diquat-induced oxidative stress and cytotoxicity in chinese hamster ovary cells overexpressing nadph-cytochrome p450 reductase. Free Radic. Biol. Med. 2011, 50, 874–882. [Google Scholar] [CrossRef]
- Yan, L.J.; Lodge, J.K.; Traber, M.G.; Matsugo, S.; Packer, L. Comparison between copper-mediated and hypochlorite-mediated modifications of human low density lipoproteins evaluated by protein carbonyl formation. J. Lipid Res. 1997, 38, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Lodge, J.K.; Traber, M.G.; Packer, L. Apolipoprotein b carbonyl formation is enhanced by lipid peroxidation during copper-mediated oxidation of human low-density lipoproteins. Arch. Biochem. Biophys. 1997, 339, 165–171. [Google Scholar] [CrossRef]
- Wu, J.; Li, R.; Li, W.; Ren, M.; Thangthaeng, N.; Sumien, N.; Liu, R.; Yang, S.; Simpkins, J.W.; Forster, M.J.; et al. Administration of 5-methoxyindole-2-carboxylic acid that potentially targets mitochondrial dihydrolipoamide dehydrogenase confers cerebral preconditioning against ischemic stroke injury. Free Radic. Biol. Med. 2017, 113, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jin, Z.; Yang, X.; Yan, L.J. Post-ischemic administration of 5-methoxyindole-2-carboxylic acid at the onset of reperfusion affords neuroprotection against stroke injury by preserving mitochondrial function and attenuating oxidative stress. Biochem. Biophys. Res. Commun. 2018, 497, 444–450. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Wang, Y.; Zhao, M. Nlrp3 inflammasome activation regulated by nf-kappab and dapk contributed to paraquat-induced acute kidney injury. Immunol. Res. 2017, 65, 687–698. [Google Scholar] [CrossRef]
- Iskander, A.; Yan, L.J. Cisplatin-induced kidney toxicity: Potential roles of major nad(+)-dependent enzymes and plant-derived natural products. Biomolecules 2022, 12, 1078. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Sears, S.M.; Orwick, A.; Siskind, L.J. Modeling cisplatin-induced kidney injury to increase translational potential. Nephron 2022, 147, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.N.; Yan, D.Q.; Zhang, X.P.; Chen, X.; Zhang, W.H.; Jia, H.L. Kidney mesenchymal stem cells alleviate cisplatin-induced kidney injury and apoptosis in rats. Tissue Cell 2023, 80, 101998. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Yang, M.; Han, Y.; Zhao, H.; Jiang, N.; Li, L.; Chen, W.; Li, C.; Yang, J.; Liu, Y.; et al. Beta-hydroxybutyrate against cisplatin-induced acute kidney injury via inhibiting nlrp3 inflammasome and oxidative stress. Int. Immunopharmacol. 2022, 111, 109101. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal. Inj. Prev. 2015, 4, 20–27. [Google Scholar]
- Yan, Y.; Bai, J.; Zhou, X.; Tang, J.; Jiang, C.; Tolbert, E.; Bayliss, G.; Gong, R.; Zhao, T.C.; Zhuang, S. P2x7 receptor inhibition protects against ischemic acute kidney injury in mice. Am. J. Physiol. Cell Physiol. 2015, 308, C463–C472. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Min, S.J.; Xu, B.; Zhang, C.; Pei, J.; Zhang, W.; Luo, G.H. The protective effects of exogenous spermine on renal ischemia-reperfusion injury in rats. Transl. Androl. Urol. 2021, 10, 2051–2066. [Google Scholar] [CrossRef] [PubMed]
- Andrianova, N.V.; Zorov, D.B.; Plotnikov, E.Y. Targeting inflammation and oxidative stress as a therapy for ischemic kidney injury. Biochemistry 2020, 85, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jang, H.R. Role of t cells in ischemic acute kidney injury and repair. Korean J. Intern. Med. 2022, 37, 534–550. [Google Scholar] [CrossRef]
- Makievskaya, C.I.; Popkov, V.A.; Andrianova, N.V.; Liao, X.; Zorov, D.B.; Plotnikov, E.Y. Ketogenic diet and ketone bodies against ischemic injury: Targets, mechanisms, and therapeutic potential. Int. J. Mol. Sci. 2023, 24, 2576. [Google Scholar] [CrossRef]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Beta-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef] [PubMed]
- Ameer, O.Z. Hypertension in chronic kidney disease: What lies behind the scene. Front. Pharmacol. 2022, 13, 949260. [Google Scholar] [CrossRef]
- Johnson, R.J.; Segal, M.S.; Srinivas, T.; Ejaz, A.; Mu, W.; Roncal, C.; Sanchez-Lozada, L.G.; Gersch, M.; Rodriguez-Iturbe, B.; Kang, D.H.; et al. Essential hypertension, progressive renal disease, and uric acid: A pathogenetic link? J. Am. Soc. Nephrol. 2005, 16, 1909–1919. [Google Scholar] [CrossRef]
- Evans, M.; McClure, T.S.; Koutnik, A.P.; Egan, B. Exogenous ketone supplements in athletic contexts: Past, present, and future. Sports Med. 2022, 52, 25–67. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Schnellmann, R.G. Pharmacological targeting of mitochondria in diabetic kidney disease. Pharmacol. Rev. 2023, 75, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Natesan, V.; Kim, S.J. Diabetic nephropathy-a review of risk factors, progression, mechanism, and dietary management. Biomol. Ther. 2021, 29, 365–372. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Lupsa, B.C.; Kibbey, R.G.; Inzucchi, S.E. Ketones: The double-edged sword of sglt2 inhibitors? Diabetologia 2023, 66, 23–32. [Google Scholar] [CrossRef]
- Hattori, Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: Proposed role of ketone utilization. Heart Fail. Rev. 2021, 26, 947–952. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Maleki, M.; Butler, A.E.; Jamialahmadi, T.; Sahebkar, A. New insights into cellular links between sodium-glucose cotransporter-2 inhibitors and ketogenesis. J. Cell. Biochem. 2022, 123, 1879–1890. [Google Scholar] [CrossRef]
- Ito, M.; Tanaka, T. The anticipated renoprotective effects of sodium-glucose cotransporter 2 inhibitors. Intern. Med. 2018, 57, 2105–2114. [Google Scholar] [CrossRef]
- Ishikawa, K.; Aguero, J.; Tilemann, L.; Ladage, D.; Hammoudi, N.; Kawase, Y.; Santos-Gallego, C.G.; Fish, K.; Levine, R.A.; Hajjar, R.J. Characterizing preclinical models of ischemic heart failure: Differences between lad and lcx infarctions. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1478–H1486. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Mayr, M.; Badimon, J. Sglt2 inhibitors in heart failure: Targeted metabolomics and energetic metabolism. Circulation 2022, 146, 819–821. [Google Scholar] [CrossRef]
- Larmour, K.; Levin, A. Slowing progression in ckd: Dapa ckd and beyond. Clin. J. Am. Soc. Nephrol. 2021, 16, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.J.; Jardine, M.J.; Chertow, G.M.; Mahaffey, K.W. Dedicated kidney disease-focused outcome trials with sodium-glucose cotransporter-2 inhibitors: Lessons from credence and expectations from dapa-hf, dapa-ckd, and empa-kidney. Diabetes. Obes. Metab. 2020, 22 (Suppl. S1), 46–54. [Google Scholar] [CrossRef]
- Requena-Ibanez, J.A.; Santos-Gallego, C.G.; Rodriguez-Cordero, A.; Vargas-Delgado, A.P.; Badimon, J.J. Empagliflozin improves quality of life in nondiabetic hfref patients. Sub-analysis of the empatropism trial. Diabetes Metab. Syndr. 2022, 16, 102417. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Van Spall, H.G.C. In hfref, adding empagliflozin to medical therapy reduced a composite outcome, regardless of ckd status. Ann. Intern. Med. 2021, 174, JC68. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Chen, B.; Gong, A.Y.; Malhotra, D.K.; Gupta, R.; Dworkin, L.D.; Gong, R. The ketone body beta-hydroxybutyrate mitigates the senescence response of glomerular podocytes to diabetic insults. Kidney Int. 2021, 100, 1037–1053. [Google Scholar] [CrossRef]
- Kume, S. Ketone bodies: Back to a place in the sun. Kidney Int. 2021, 100, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced nrf2 activation. Diabetes Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- Uddin, M.J.; Farjana, M.; Moni, A.; Hossain, K.S.; Hannan, M.A.; Ha, H. Prospective pharmacological potential of resveratrol in delaying kidney aging. Int. J. Mol. Sci. 2021, 22, 8258. [Google Scholar] [CrossRef] [PubMed]
- Chang-Panesso, M. Acute kidney injury and aging. Pediatr. Nephrol. 2021, 36, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yuan, L.; Liu, F.; Li, L.; Liu, J.; Chen, Y.; Lu, Y.; Yuan, Y. Molecular mechanisms and physiological functions of autophagy in kidney diseases. Front. Pharmacol. 2022, 13, 974829. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.; Tian, Z.; Ma, K.; Li, Y.; Wang, L.; Nasser, M.I.; Liu, C. The diseased kidney: Aging and senescent immunology. Immun. Ageing 2022, 19, 58. [Google Scholar] [CrossRef]
- Guo, J.; Zheng, H.J.; Zhang, W.; Lou, W.; Xia, C.; Han, X.T.; Huang, W.J.; Zhang, F.; Wang, Y.; Liu, W.J. Accelerated kidney aging in diabetes mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 1234059. [Google Scholar] [CrossRef]
- Sohal, R.S.; Forster, M.J. Caloric restriction and the aging process: A critique. Free Radic. Biol. Med. 2014, 73, 366–382. [Google Scholar] [CrossRef]
- Dubey, A.; Forster, M.J.; Lal, H.; Sohal, R.S. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch. Biochem. Biophys. 1996, 333, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of age and caloric restriction on mitochondrial protein oxidative damage in mice. Mech. Ageing Dev. 2012, 133, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Mank, M.M.; Reed, L.F.; Walton, C.J.; Barup, M.L.T.; Ather, J.L.; Poynter, M.E. Therapeutic ketosis decreases methacholine hyperresponsiveness in mouse models of inherent obese asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L243–L257. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.P.; Cunningham, R.P.; Davis, R.A.H.; Deemer, S.E.; Roberts, B.M.; Plaisance, E.P.; Rector, R.S. A dietary ketone ester mitigates histological outcomes of nafld and markers of fibrosis in high-fat diet fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G564–G572. [Google Scholar] [CrossRef] [PubMed]
- Izuta, Y.; Imada, T.; Hisamura, R.; Oonishi, E.; Nakamura, S.; Inagaki, E.; Ito, M.; Soga, T.; Tsubota, K. Ketone body 3-hydroxybutyrate mimics calorie restriction via the nrf2 activator, fumarate, in the retina. Aging Cell 2018, 17, e12699. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Lan, J.; Xu, G.; Zhu, Y.; Lin, C.; Yan, Z.; Shao, S. Association of body mass index and acute kidney injury incidence and outcome: A systematic review and meta-analysis. J. Ren. Nutr. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Bao, Y.W.; Yuan, Y.; Chen, J.H.; Lin, W.Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Zool Res. 2018, 39, 72–86. [Google Scholar]
- Pillai, A.; Fulmali, D. A narrative review of new treatment options for diabetic nephropathy. Cureus 2023, 15, e33235. [Google Scholar] [CrossRef]
- Georgianos, P.I.; Vaios, V.; Eleftheriadis, T.; Papachristou, E.; Liakopoulos, V. Therapeutic advances in diabetic kidney disease. Int. J. Mol. Sci. 2023, 24, 2803. [Google Scholar] [CrossRef]
- Guo, M.; Chen, Q.; Huang, Y.; Wu, Q.; Zeng, Y.; Tan, X.; Teng, F.; Ma, X.; Pu, Y.; Huang, W.; et al. High glucose-induced kidney injury via activation of necroptosis in diabetic kidney disease. Oxid. Med. Cell. Longev. 2023, 2023, 2713864. [Google Scholar] [CrossRef]
- Kelsey, R. Metabolic changes in the kidney. Nat. Rev. Nephrol. 2020, 16, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fang, X.; Zhang, H.; Gao, W.; Hsu, H.J.; Roman, R.J.; Fan, F. Genetic susceptibility of hypertension-induced kidney disease. Physiol. Rep. 2021, 9, e14688. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P.; Bajo, M.A.; Selgas, R.; Diez, J.J. Thyroid dysfunction and kidney disease: An update. Rev. Endocr. Metab. Disord. 2017, 18, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Sazgar, M. Kidney disease and epilepsy. J. Stroke Cerebrovasc. Dis. 2021, 30, 105651. [Google Scholar] [CrossRef]
- Roy, P.J.; Weltman, M.; Dember, L.M.; Liebschutz, J.; Jhamb, M.; Consortium, H. Pain management in patients with chronic kidney disease and end-stage kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 671–680. [Google Scholar] [CrossRef]
- Langston, C. Managing fluid and electrolyte disorders in kidney disease. Vet. Clin. N. Am. Small Anim. Pract. 2017, 47, 471–490. [Google Scholar] [CrossRef]
- Collister, D.; Rigatto, C.; Tangri, N. Anemia management in chronic kidney disease and dialysis: A narrative review. Curr. Opin. Nephrol. Hypertens. 2017, 26, 214–218. [Google Scholar] [CrossRef]
- Woo, Y.M.; Pereira, B.J.; Gill, J.S. Chronic kidney disease progression in native and transplant kidneys. Curr. Opin. Nephrol. Hypertens. 2004, 13, 607–611. [Google Scholar] [CrossRef]
- Strubl, S.; Oehm, S.; Torres, J.A.; Grundmann, F.; Haratani, J.; Decker, M.; Vuong, S.; Kaur Bhandal, A.; Methot, N.; Haynie-Cion, R.; et al. Ketogenic dietary interventions in autosomal dominant polycystic kidney disease-a retrospective case series study: First insights into feasibility, safety and effects. Clin. Kidney J. 2022, 15, 1079–1092. [Google Scholar] [CrossRef]
- Kosinski, C.; Jornayvaz, F.R. Effects of ketogenic diets on cardiovascular risk factors: Evidence from animal and human studies. Nutrients 2017, 9, 517. [Google Scholar] [CrossRef]
- Sampath, A.; Kossoff, E.H.; Furth, S.L.; Pyzik, P.L.; Vining, E.P. Kidney stones and the ketogenic diet: Risk factors and prevention. J. Child Neurol. 2007, 22, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Sumien, N.; Thangthaeng, N.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic. Res. 2013, 47, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jin, Z.; Yan, L.J. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox. Biol. 2017, 11, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Dendooven, A.; Ishola, D.A., Jr.; Nguyen, T.Q.; Van der Giezen, D.M.; Kok, R.J.; Goldschmeding, R.; Joles, J.A. Oxidative stress in obstructive nephropathy. Int. J. Exp. Pathol. 2011, 92, 202–210. [Google Scholar] [CrossRef]
- Brosey, C.A.; Ho, C.; Long, W.Z.; Singh, S.; Burnett, K.; Hura, G.L.; Nix, J.C.; Bowman, G.R.; Ellenberger, T.; Tainer, J.A. Defining nadh-driven allostery regulating apoptosis-inducing factor. Structure 2016, 24, 2067–2079. [Google Scholar] [CrossRef]
- Mao, H.; Zhang, Y.; Xiong, Y.; Zhu, Z.; Wang, L.; Liu, X. Mitochondria-targeted antioxidant mitoquinone maintains mitochondrial homeostasis through the sirt3-dependent pathway to mitigate oxidative damage caused by renal ischemia/reperfusion. Oxid. Med. Cell. Longev. 2022, 2022, 2213503. [Google Scholar] [CrossRef]
- Qin, L.; Xi, S. The role of mitochondrial fission proteins in mitochondrial dynamics in kidney disease. Int. J. Mol. Sci. 2022, 23, 14725. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).