
4,4-Bis(isopropylthio)-1,1-diphenyl-2-azabuta-1,3-diene Adducts with Cadmium(II), Mercury(II) and Copper(I) Iodides: Crystal, Molecular and Electronic Structures of d10 Transition Metal Chelate Complexes

 , and
, and
Abstract
:1. Introduction
2. Experimental Section
2.1. Apparatus
2.2. X-ray Diffraction
2.3. Computational Studies
3. Results and Discussion
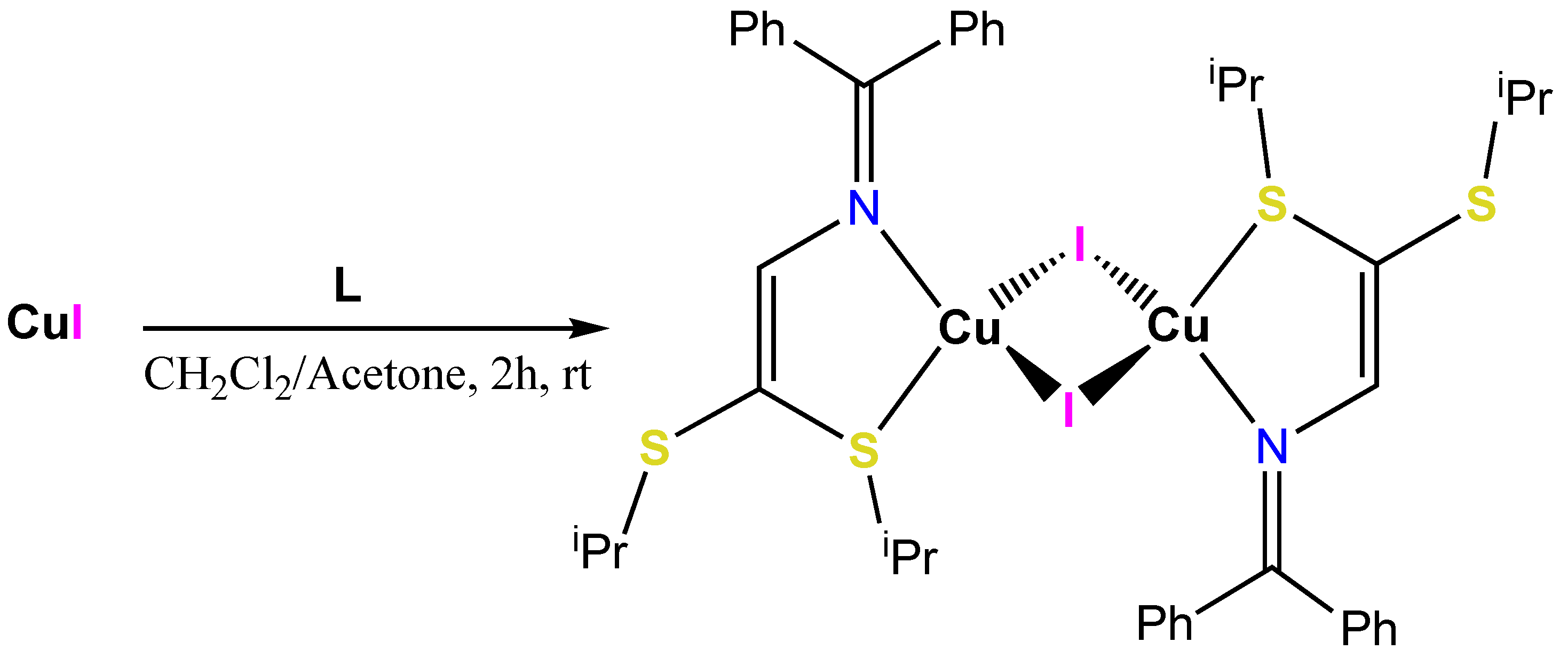
3.1. Synthesis of the CdI2, HgI2 and CuI Adduct Complexes
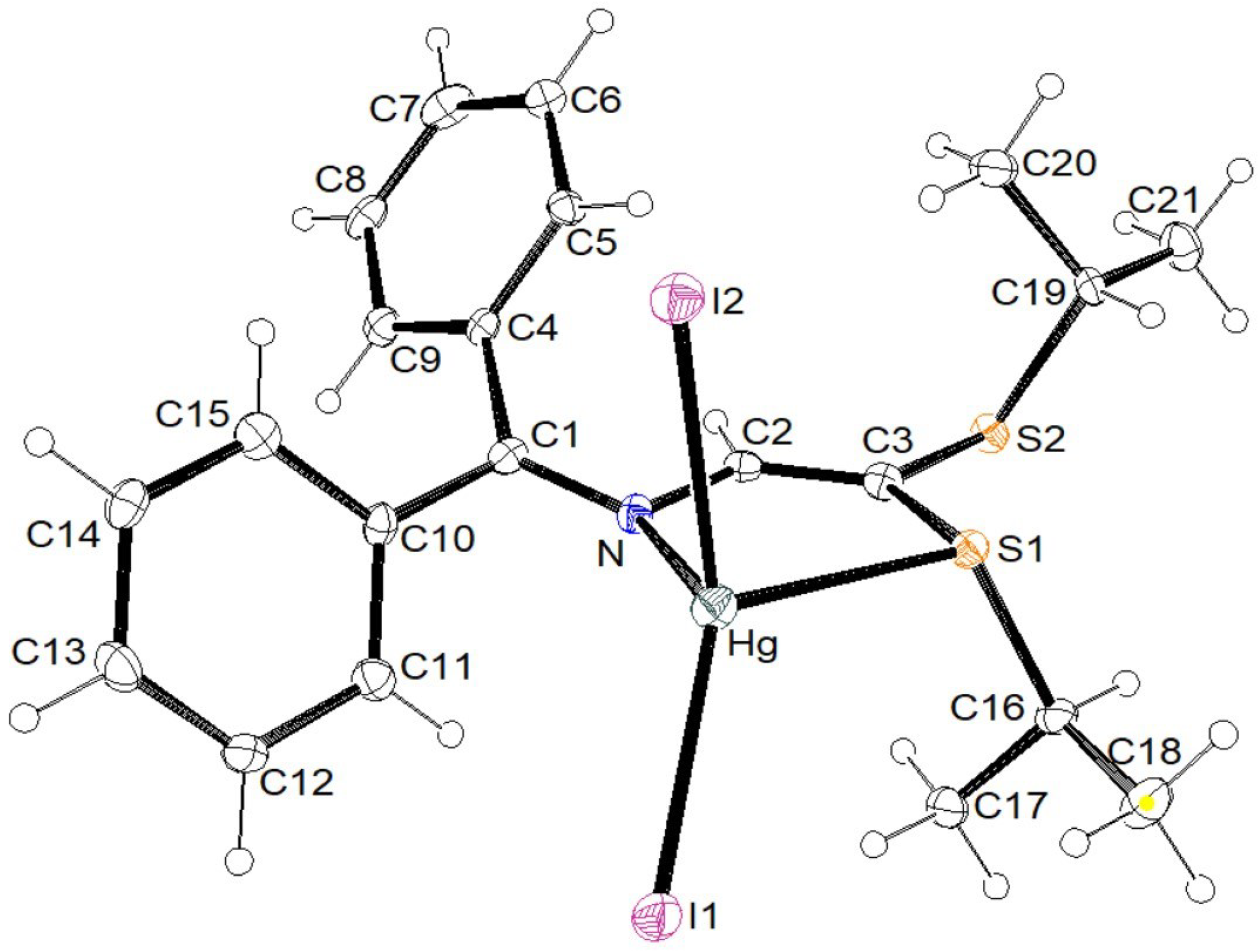
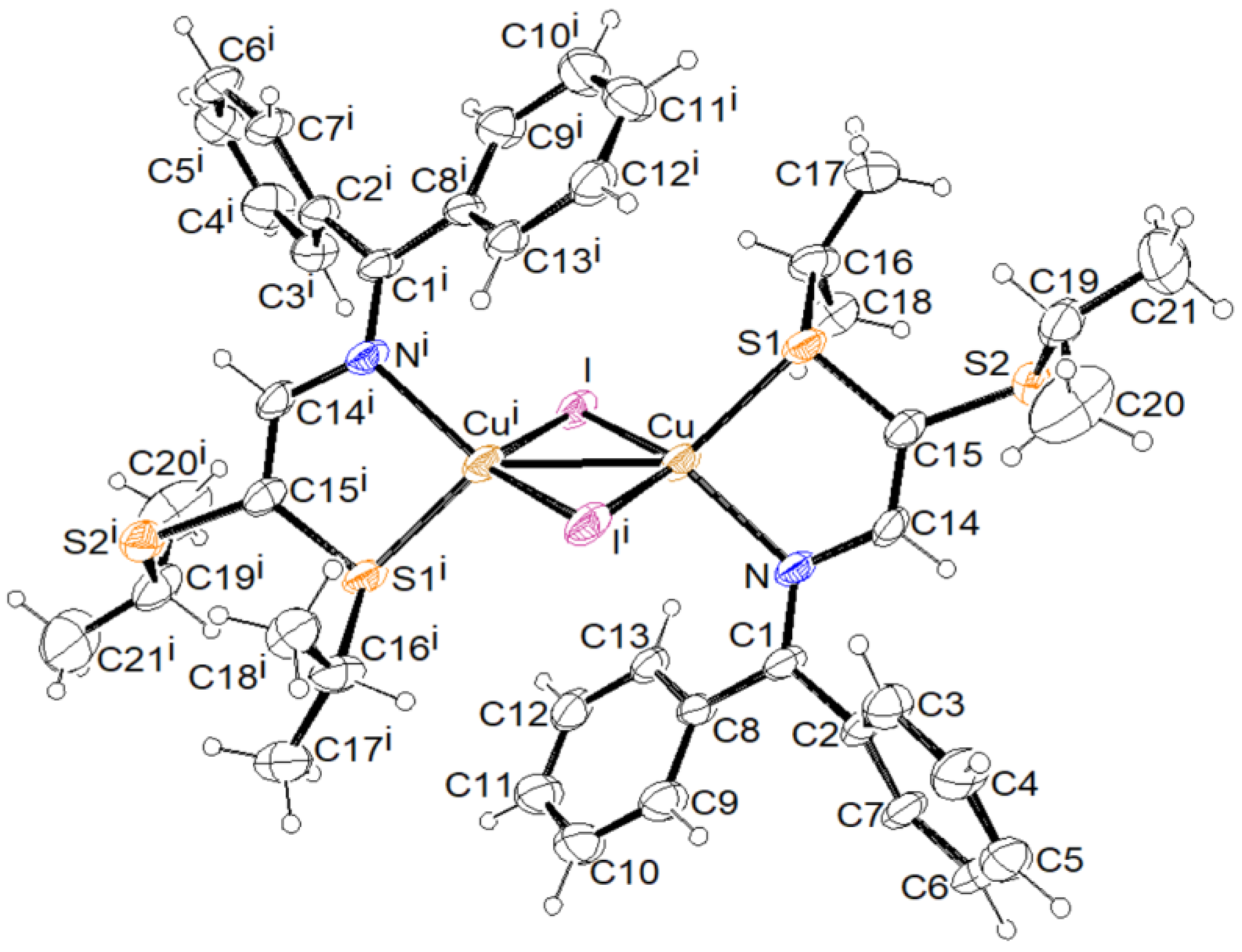
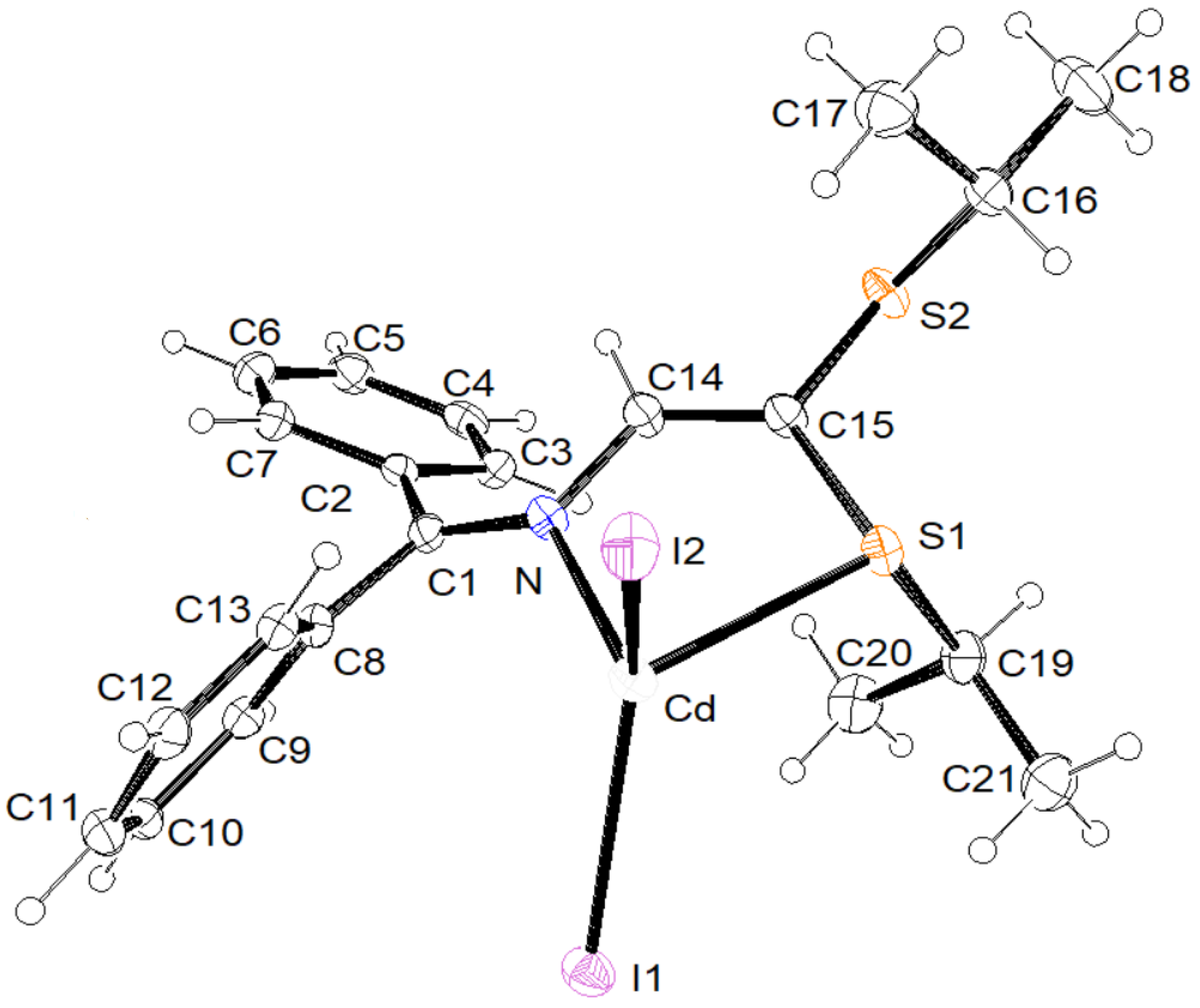
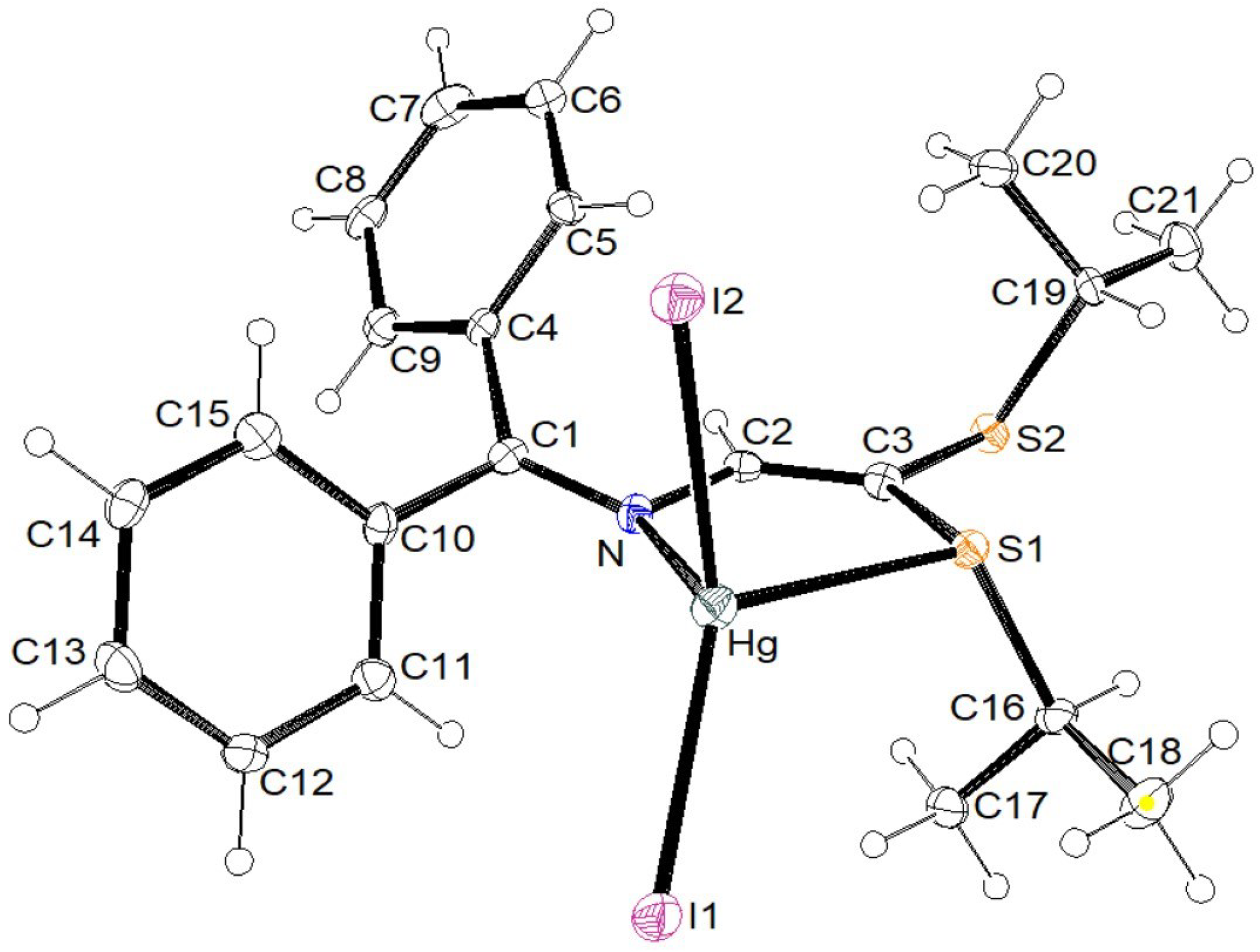
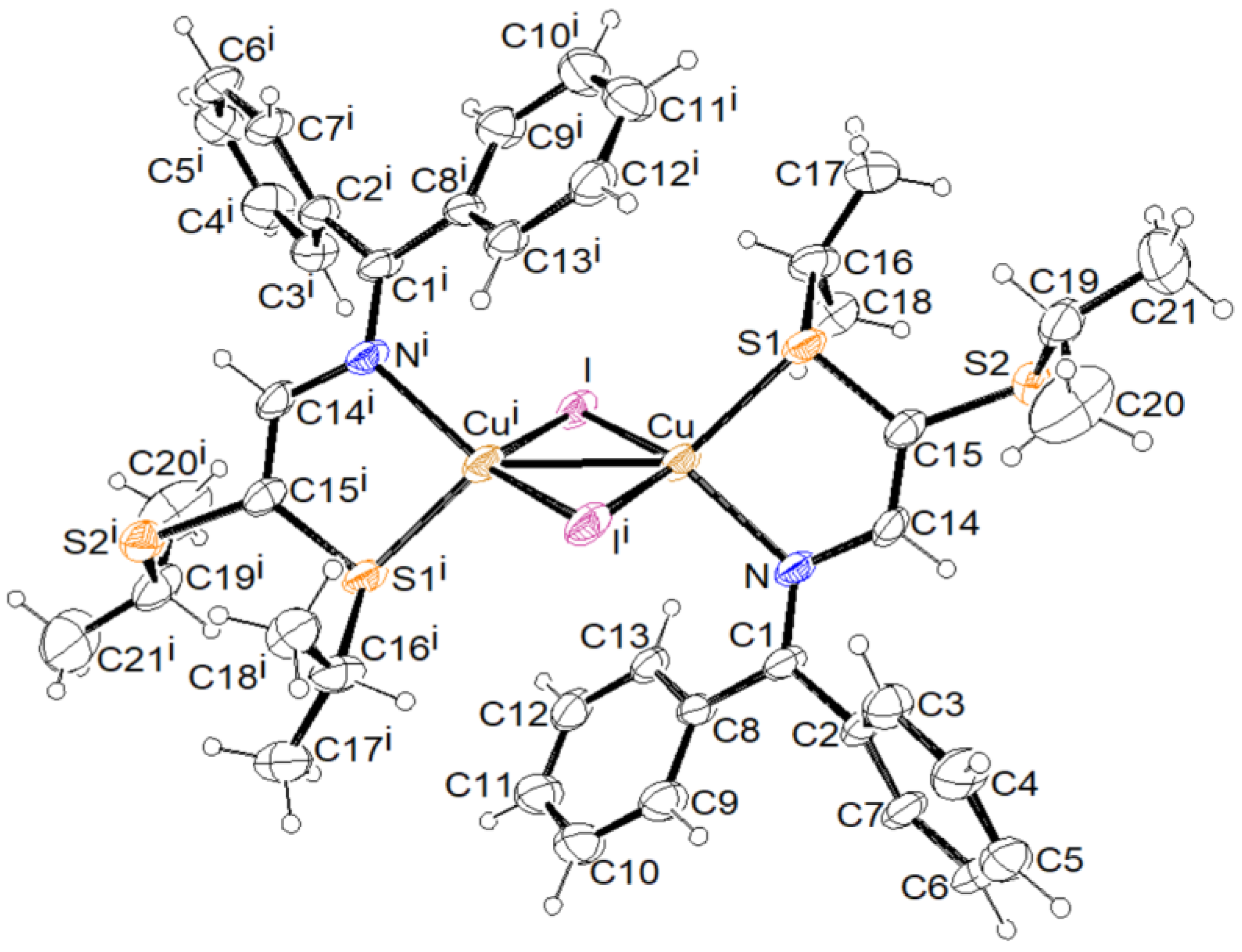
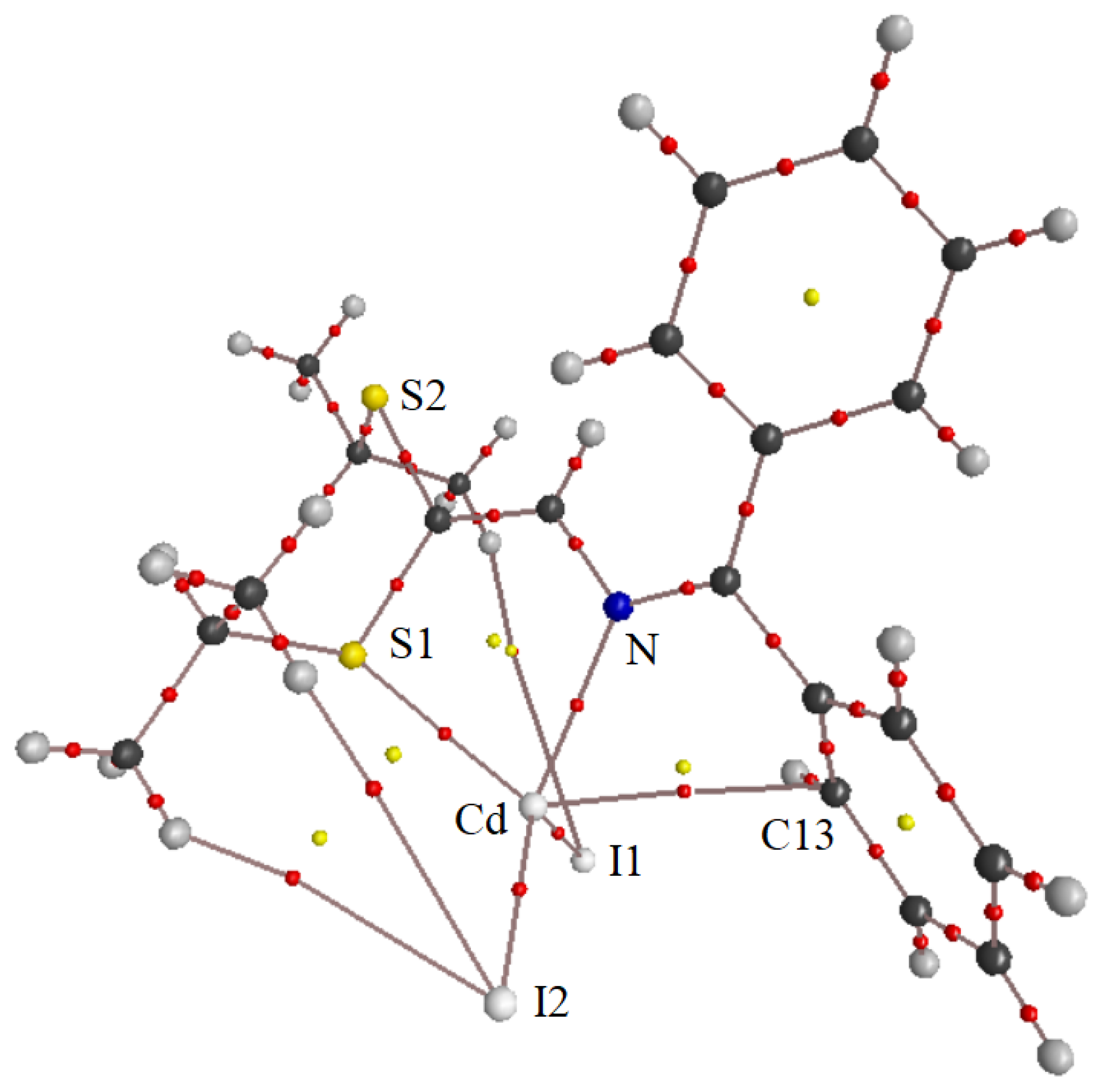
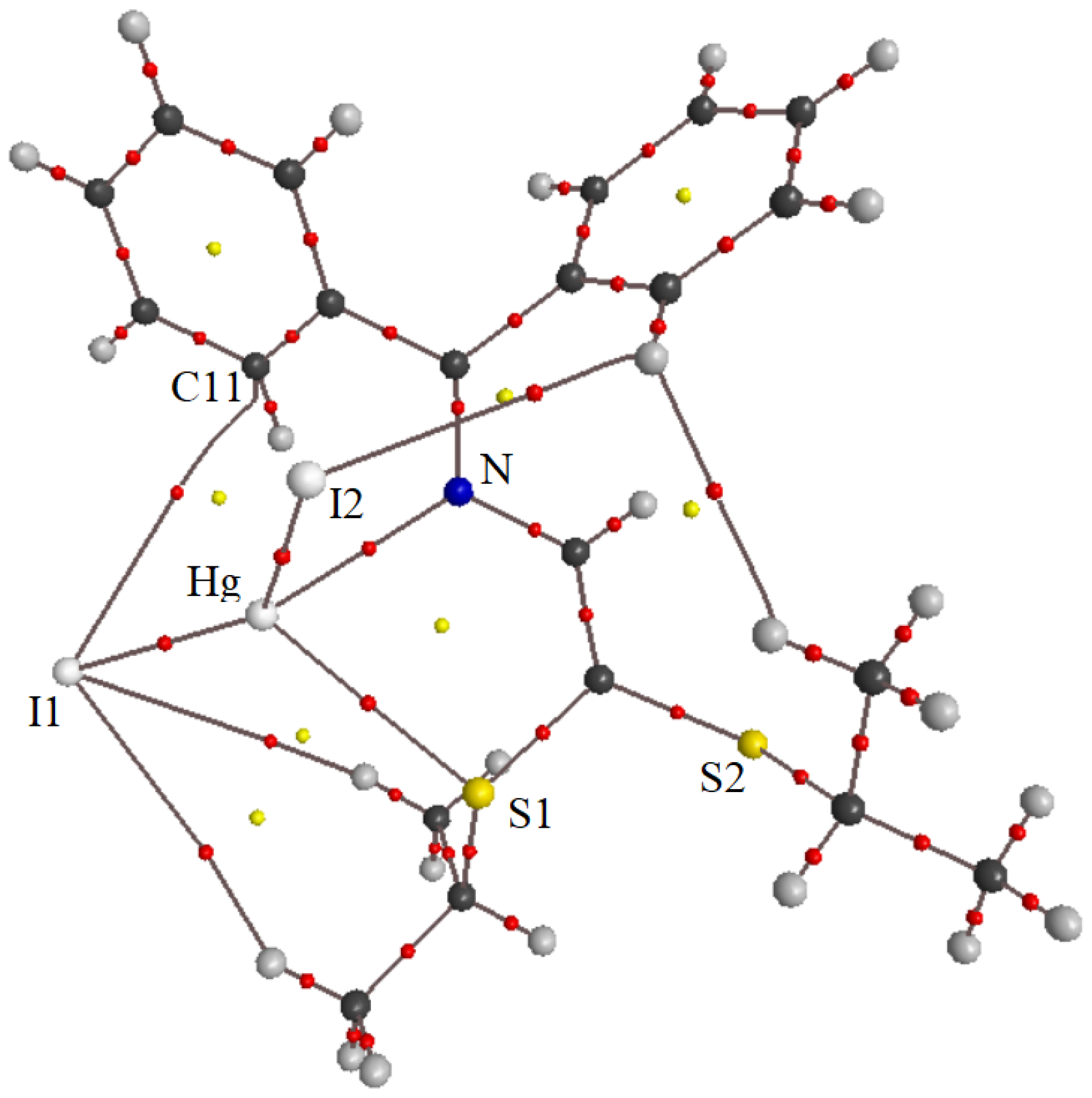
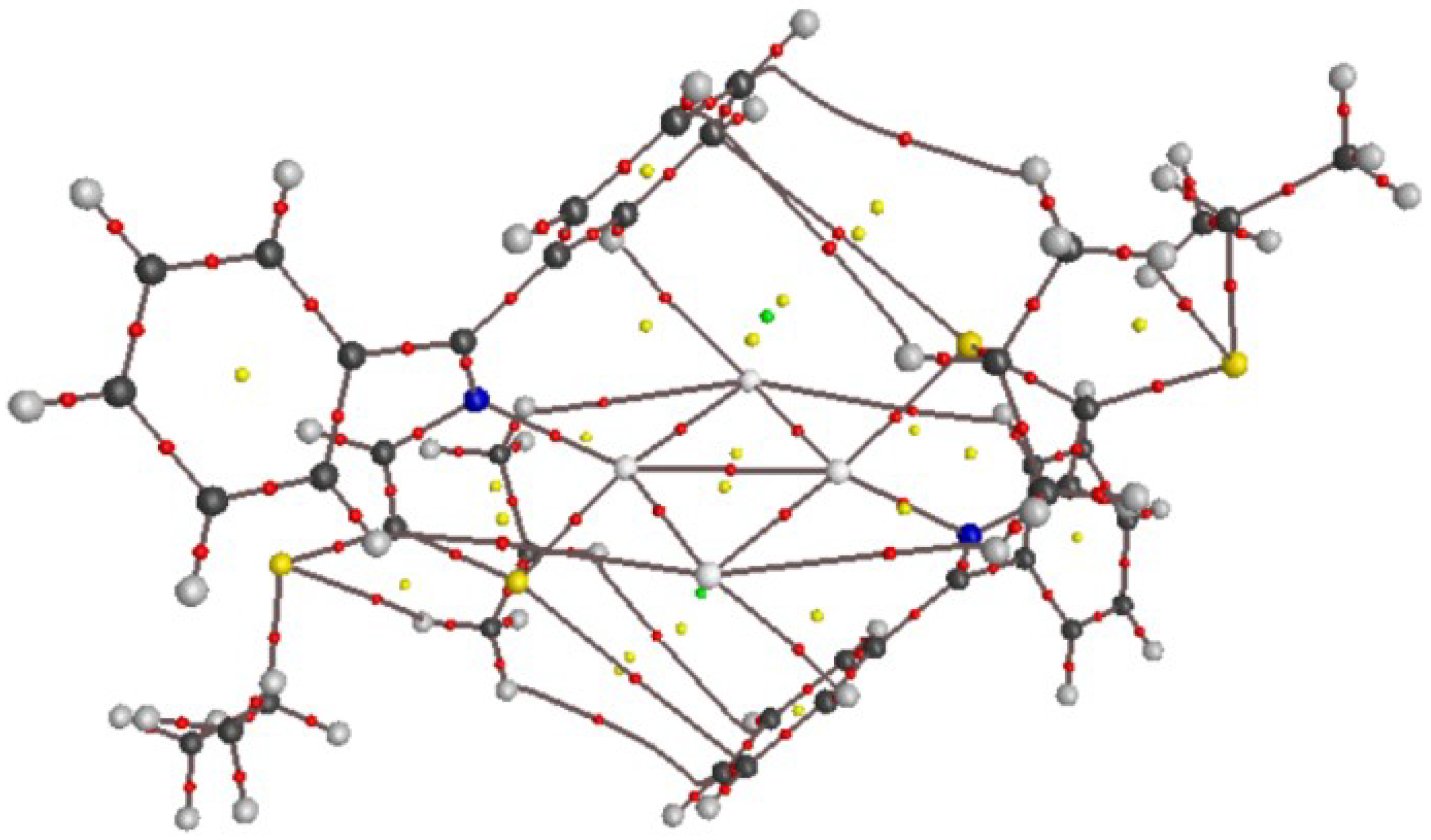
3.2. X-ray Structural Studies
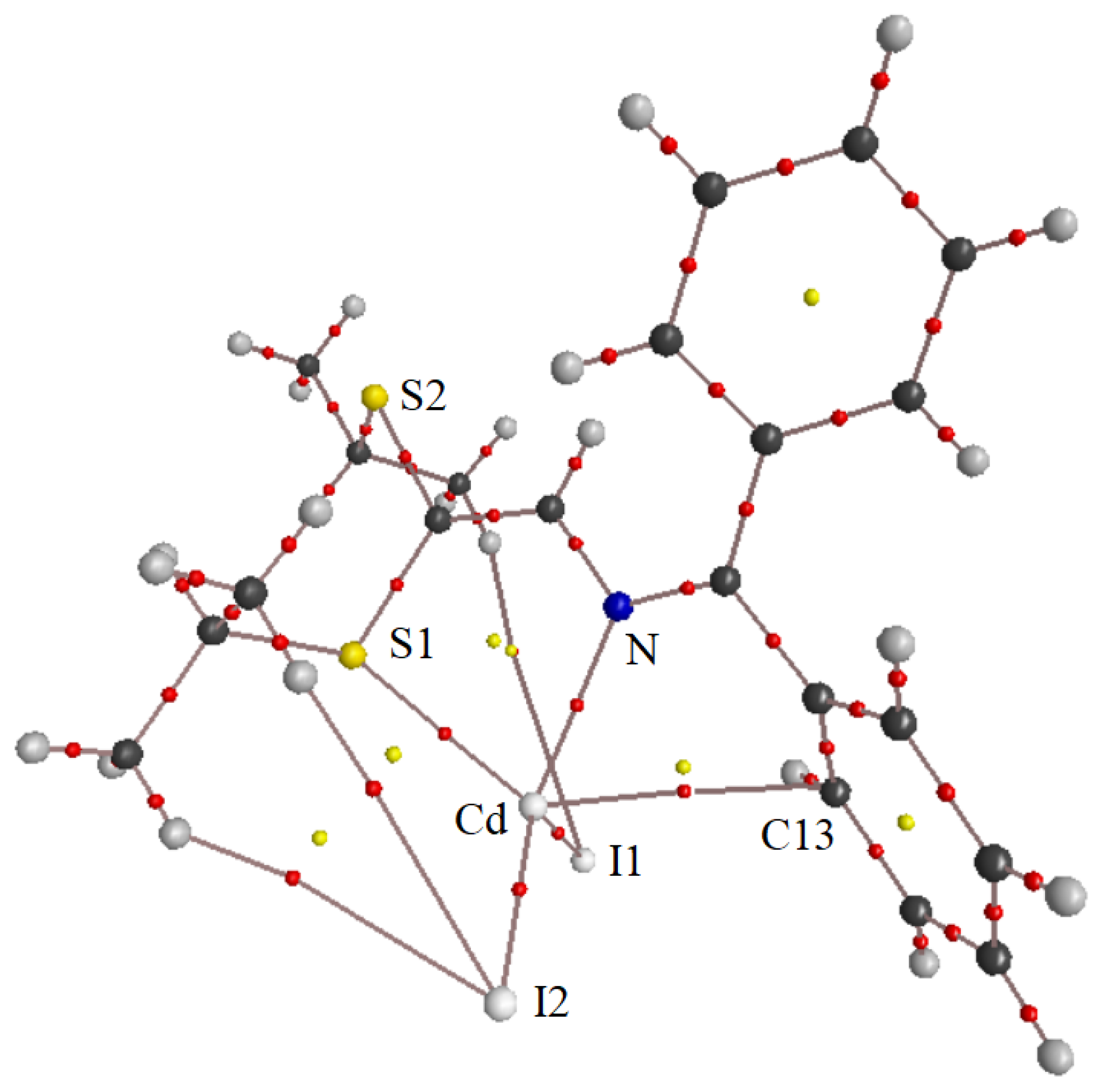
3.2.1. Molecular Structures
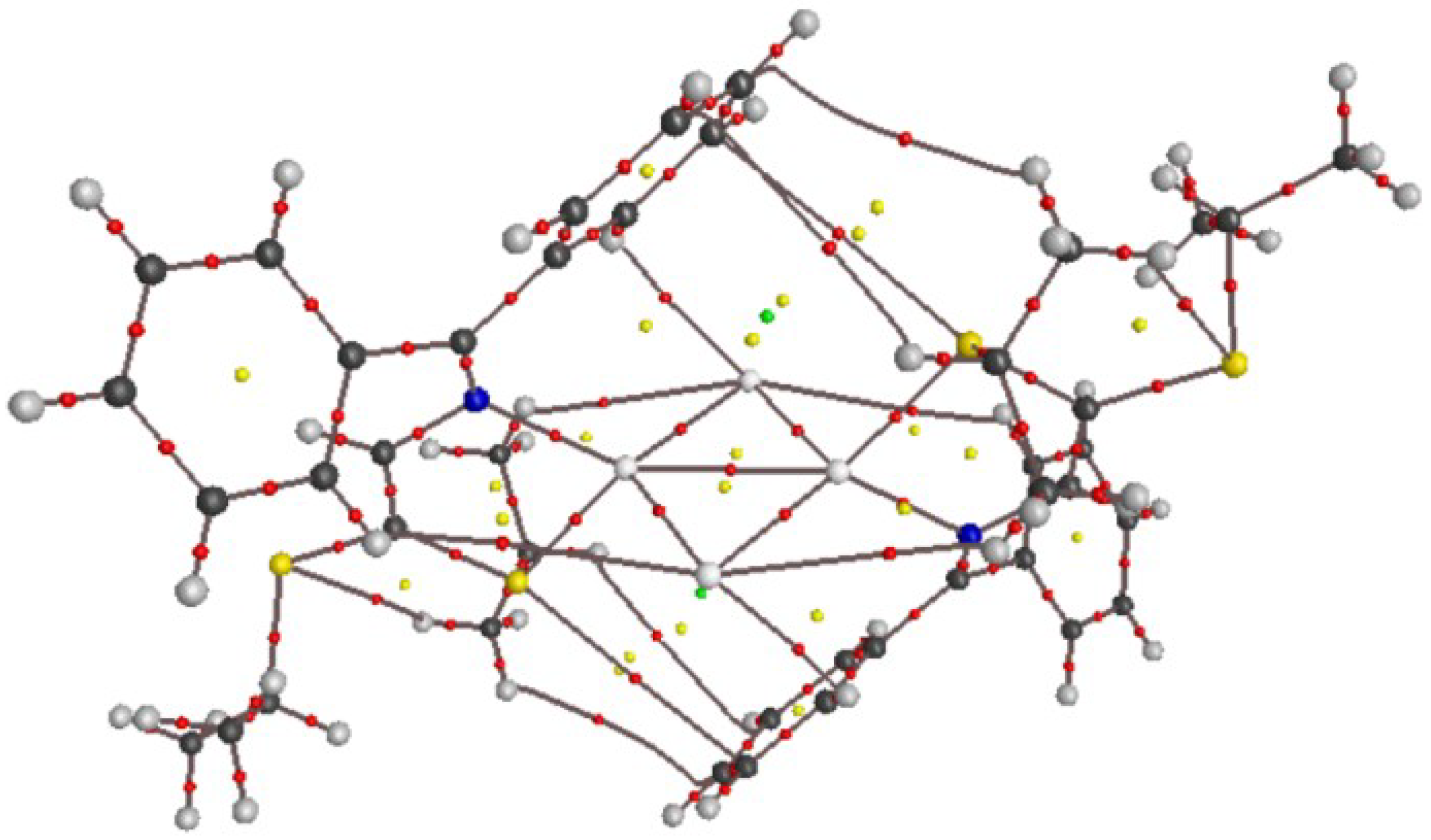
3.2.2. Crystal Structures and Supramolecular Features
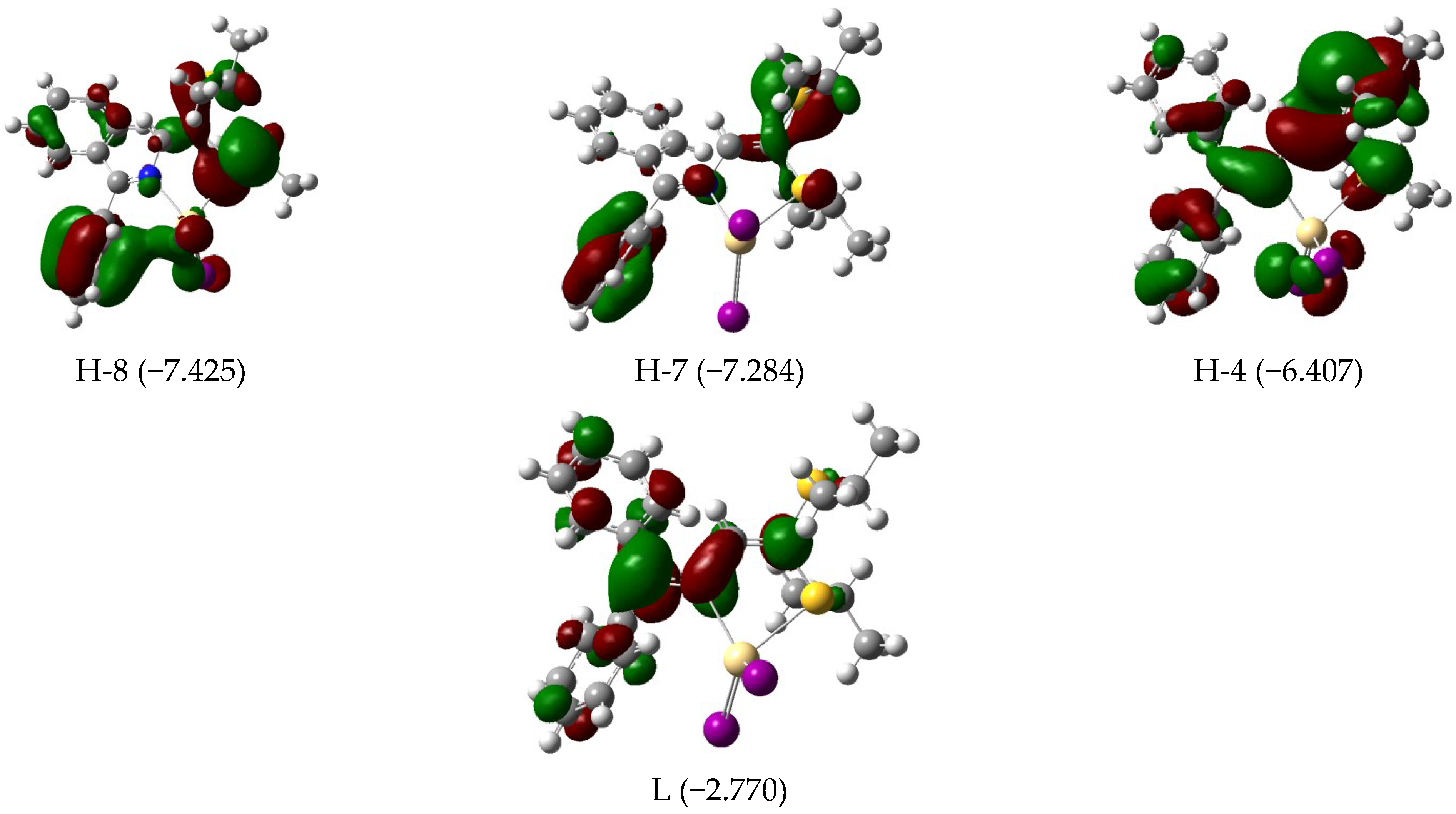
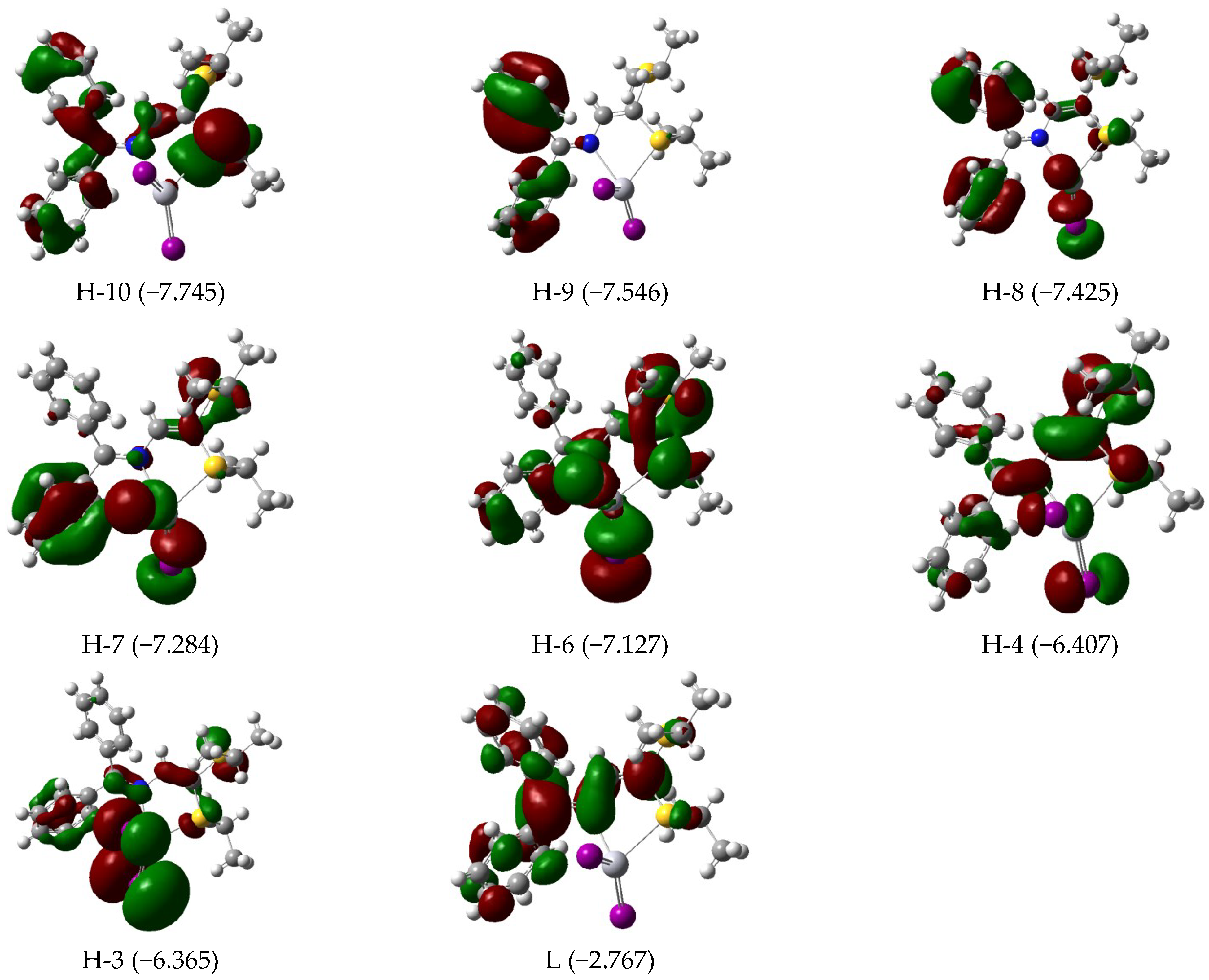
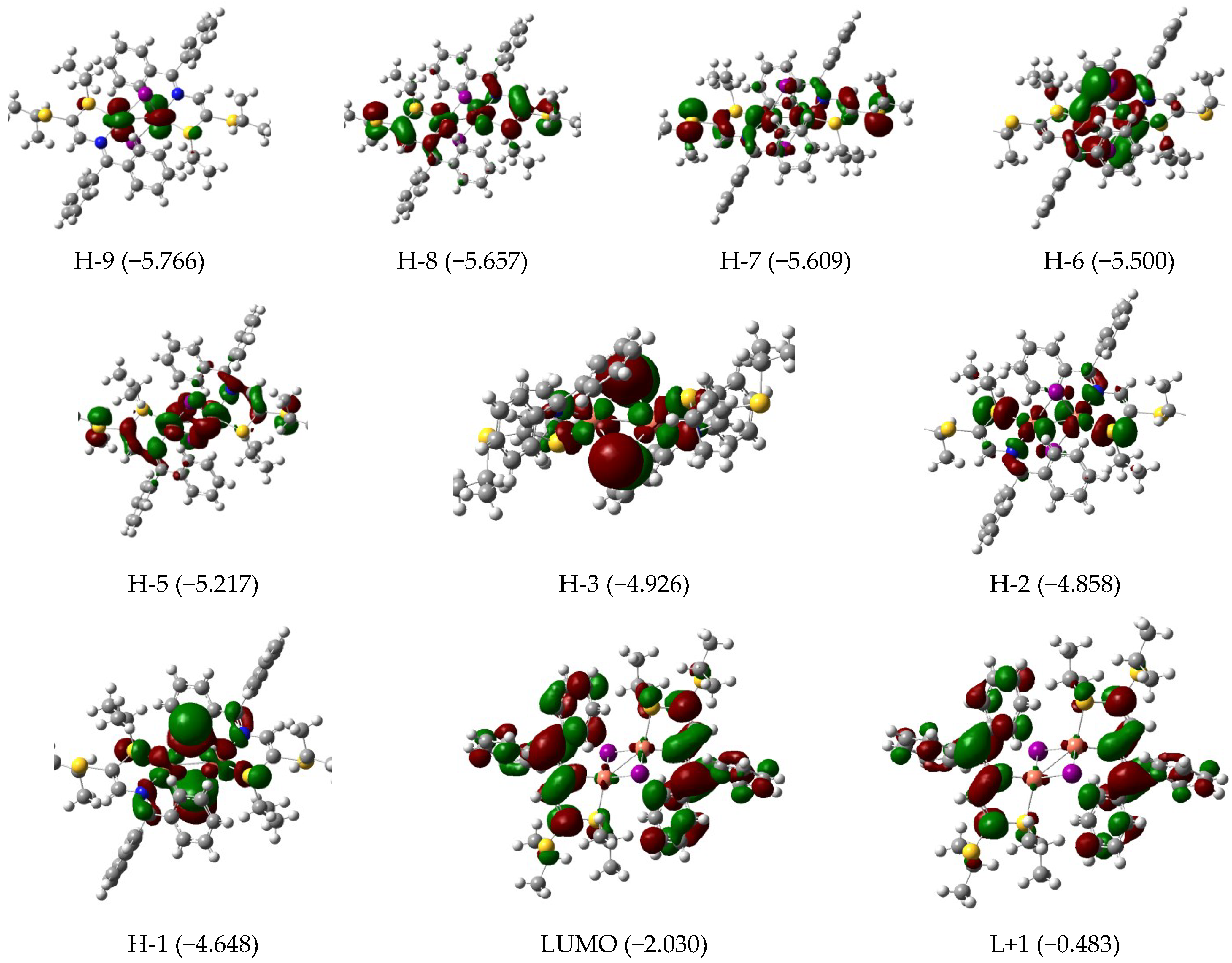
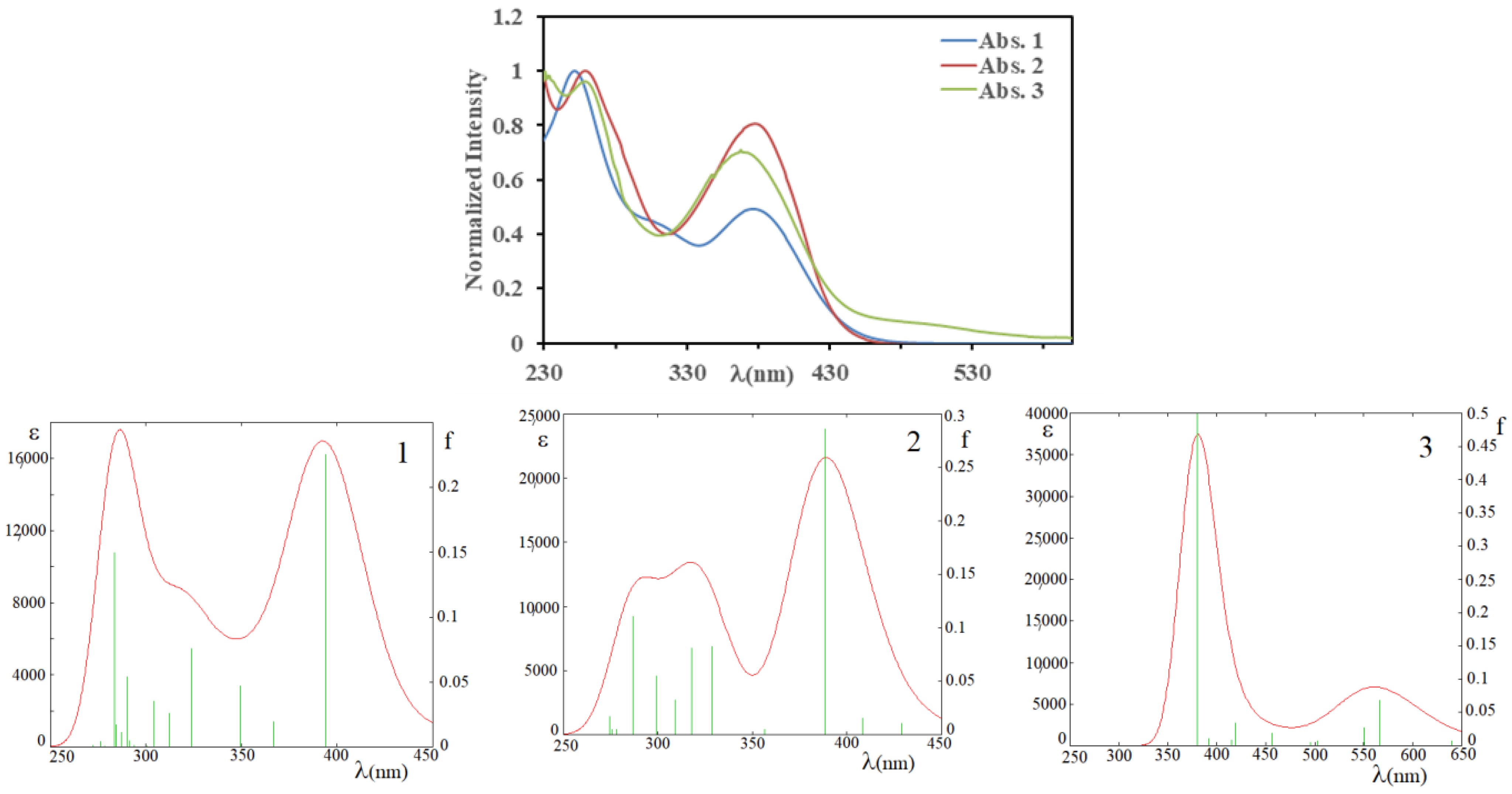
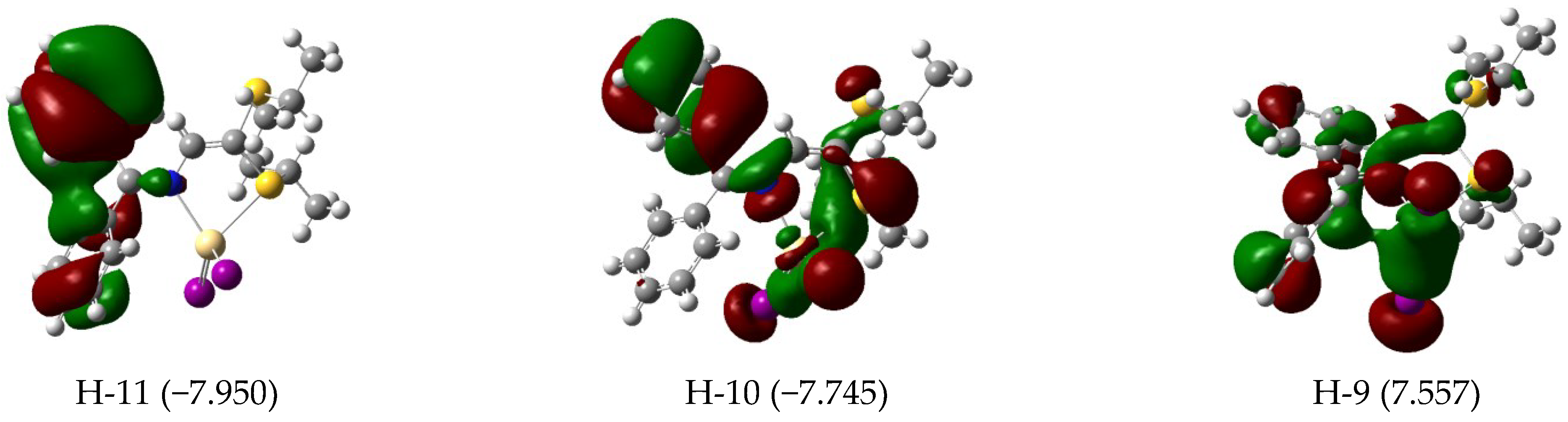
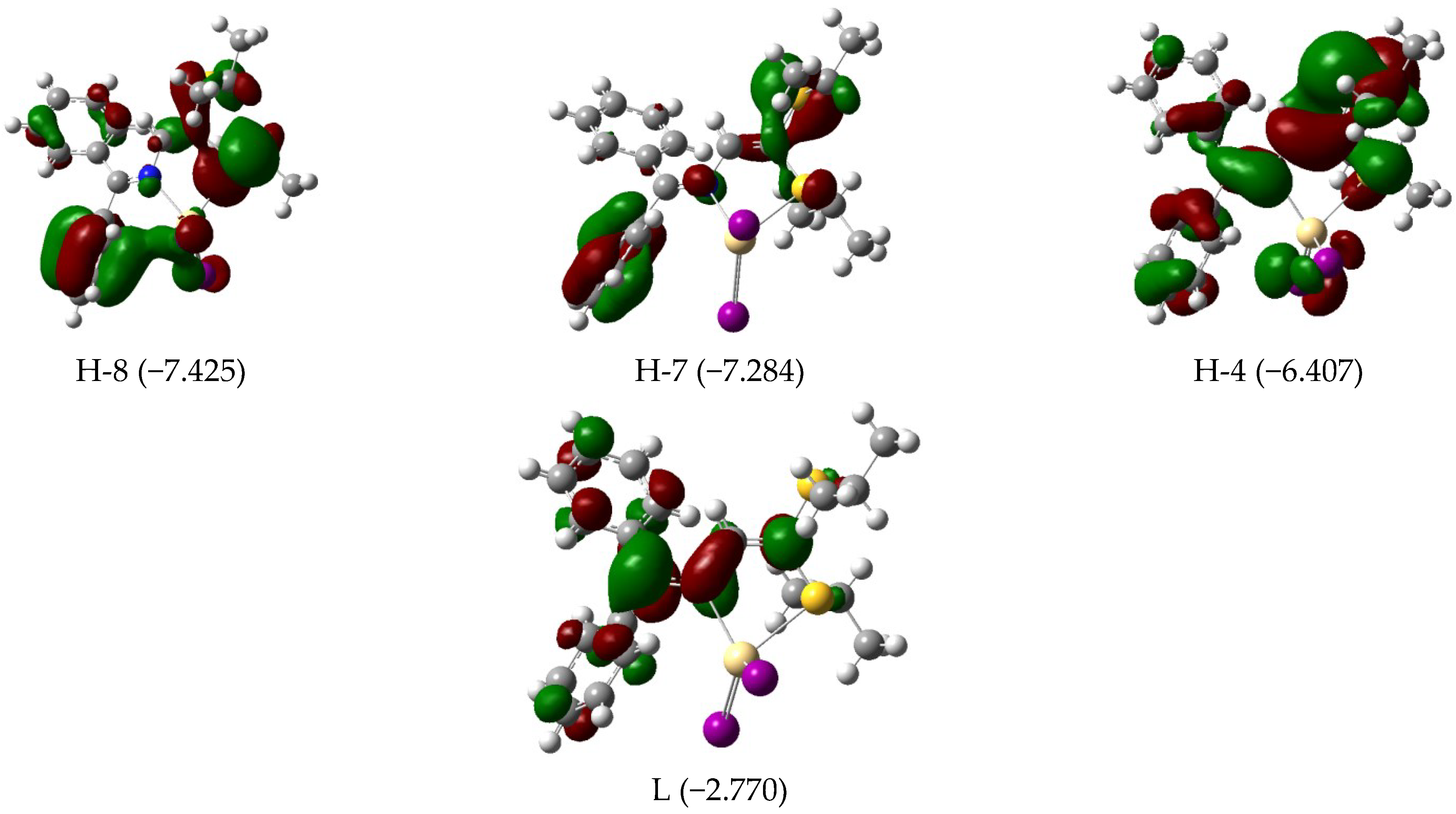
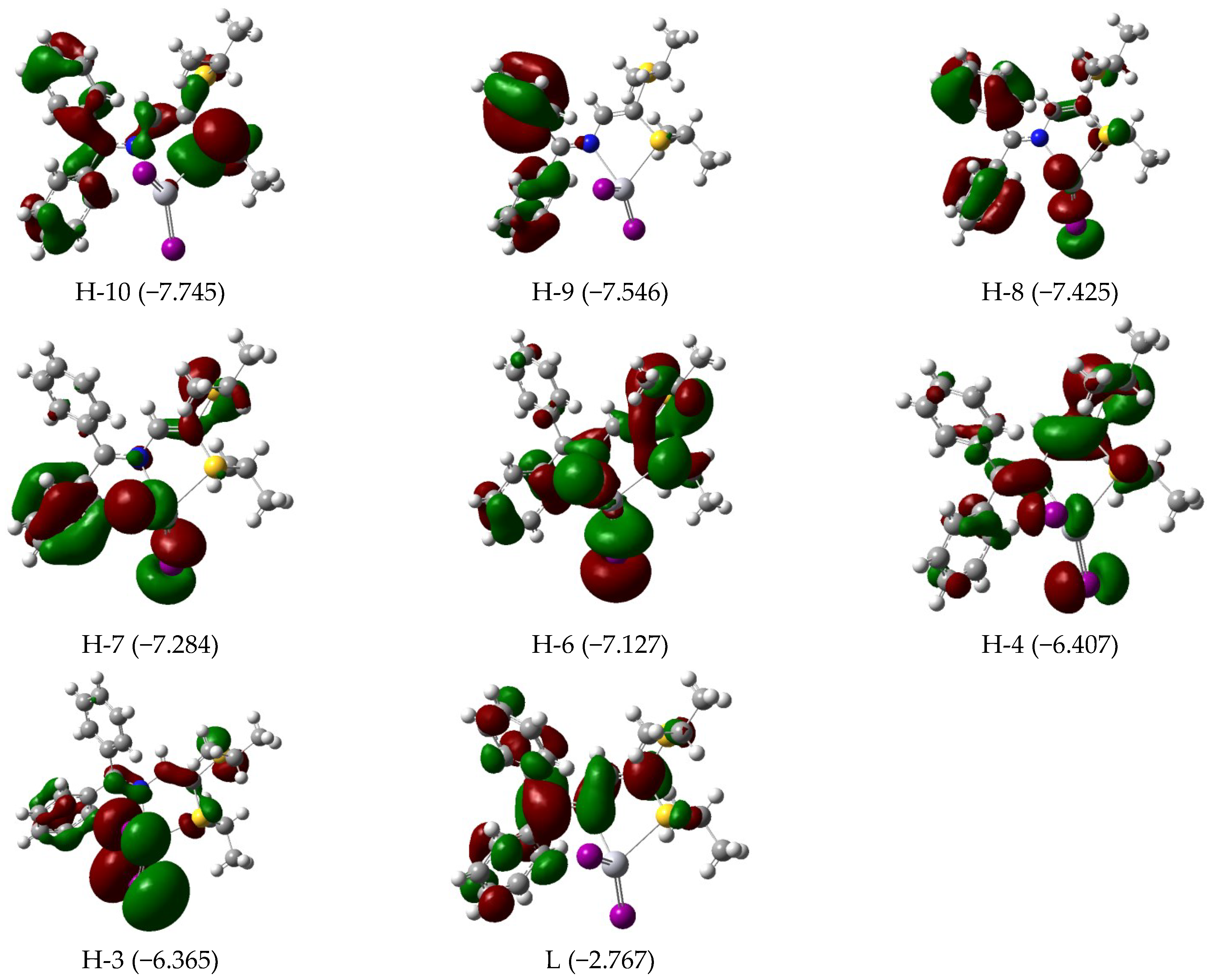
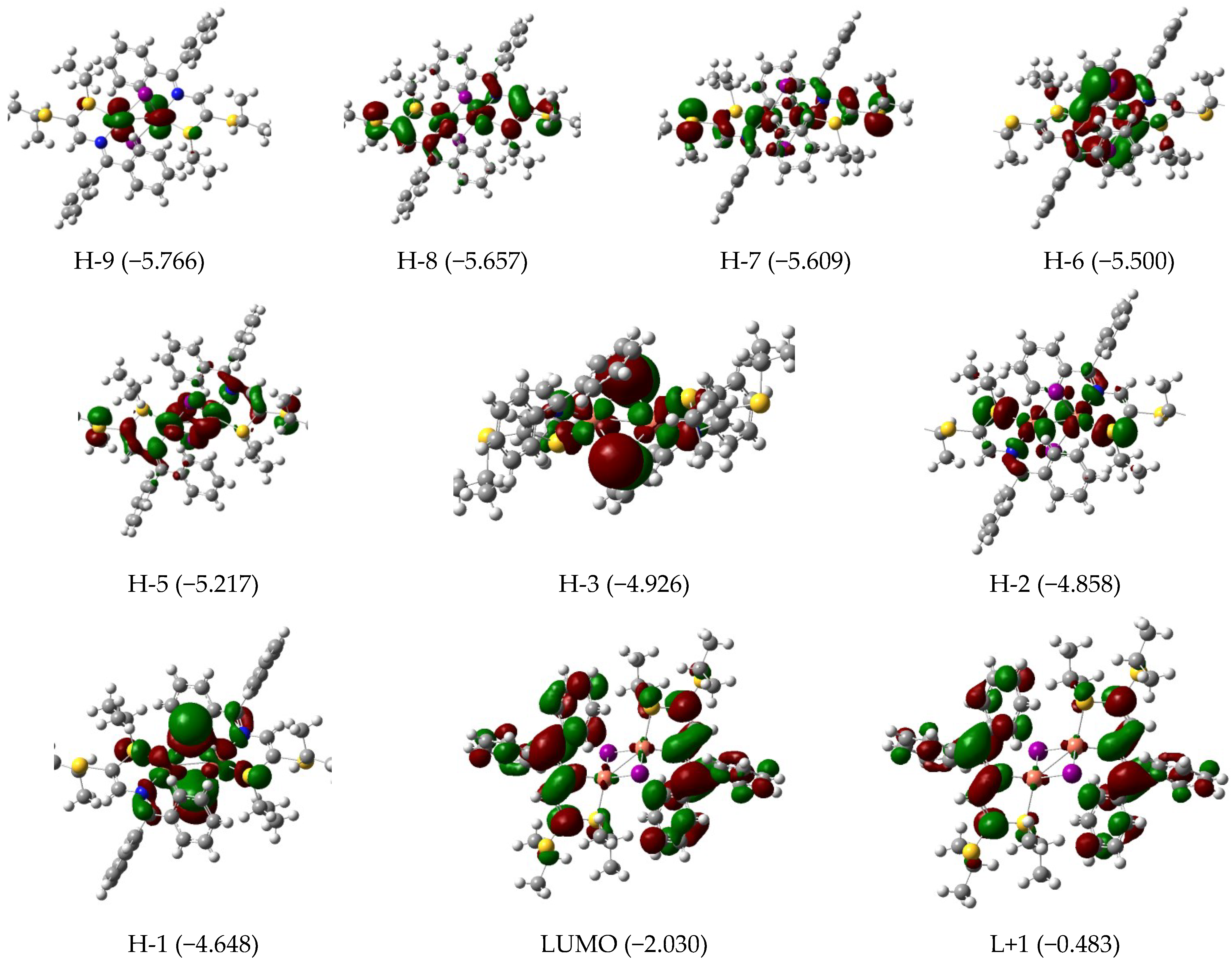
3.3. Electronic Features and AIM Approach
- 1—shared–shared (SS), covalent, (∇2ρ < 0, H < 0 and |V|/G > 2;
- 2—shared–closed (SC), transit or intermediate, (∇2ρ > 0, H < 0 and 1< |V|/G < 2;
- 3—closed–closed (CC), ionic, van der Waals…, (∇2ρ> 0, H > 0 and |V|/G < 1.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coleman, A.; Brennan, C.; Vos, J.G.; Pryce, M.T. Photophysical properties and applications of Re(I) and Re(I)–Ru(II) carbonylpolypyridyl complexes. Coord. Chem. Rev. 2008, 252, 2585–2595. [Google Scholar] [CrossRef]
- Raghavana, A.; Venugopal, A. Review: Structurally characterized α-diimine complexes of s- and p-block elements. J. Coord. Chem. 2014, 67, 2530–2549. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Zhao, Y.; Redshaw, C.; Fedushkin, I.L.; Wu, B.; Yang, X.-J. Main-group metal complexes of α-diimine ligands: Structure, bonding and reactivity. Dalton Trans. 2021, 50, 13634–13650. [Google Scholar] [CrossRef] [PubMed]
- Knorr, M.; Schmitt, G.; Kubicki, M.M.; Vigier, E. Formation of (σ-alkenyl)- and (μ-vinylidene)palladium and -platinum complexes by oxidative addition of 4,4-dichloro-1,1-diphenyl-2-azabuta-1,3-diene—The molecular structure of an unusual asymmetric (μ-vinylidene)Pd-Pd complex. Eur. J. Inorg. Chem. 2003, 2003, 514–517. [Google Scholar] [CrossRef]
- Jacquot, S.; Belaissaoui, A.; Schmitt, G.; Laude, B.; Kubicki, M.M.; Blacque, O. Reaction of diphenyldiazomethane with N-methyloxy- and N-ethyloxycarbonyl-N-(2,2,2-trichloroethylidene)amines. Eur. J. Org. Chem. 1999, 1999, 1541–1544. [Google Scholar] [CrossRef]
- Jacquot, S.; Schmitt, G.; Laude, B.; Kubicki, M.M.; Blacque, O. Nucleophilic additions of sodium alkoxides to 4,4-dichloro-1,1-diphenyl- 2-azabuta-1,3-diene. Eur. J. Org. Chem. 2000, 2000, 1235–1239. [Google Scholar] [CrossRef]
- Jacquot-Rousseau, S.; Schmitt, G.; Khatyr, A.; Knorr, M.; Kubicki, M.M.; Vigier, E.; Blacque, O. Reactivity of 4,4-Dichloro-1,1-diphenyl-2-azabutadiene Towards Alkoxides and Thiolates: Synthesis of Functionalised π-Conjugated 2-Azabutadienes and Unexpected 1,4-Thiazine Formation. Eur. J. Org. Chem. 2006, 2006, 1555–1562. [Google Scholar] [CrossRef]
- Kinghat, R.; Boudiba, H.; Khatyr, A.; Knorr, M.; Kubicki, M.M. 4,4-Bis(4-methylphenylsulfanyl)-1,1-diphenyl-2-azabuta-1,3-diene. Acta Cryst. 2008, E64, o370. [Google Scholar] [CrossRef]
- Kinghat, R.; Schmitt, G.; Ciamala, K.; Khatyr, A.; Knorr, M.; Jacquot-Rousseau, S.; Rousselin, Y.; Kubicki, M.M. 1,3-Dipolar cycloaddition of diaryldiazomethanes across N-ethoxy-carbonyl-N-(2,2,2-trichloroethylidene)amine and reactivity of the resulting 2-azabutadienes towards thiolates and cyclic amides. Comptes Rendus Chim. 2016, 19, 320–332. [Google Scholar] [CrossRef]
- Jacquot-Rousseau, S.; Khatyr, A.; Schmitt, G.; Knorr, M.; Kubicki, M.M.; Blacque, O. Synthesis and reactivity of an 2-azabutadiene-based π-conjugated dithioether: Formation of a N,S-ligated molybdenum chelate complex and C,N,S-pincer complexes of palladium and platinum. Inorg. Chem. Commun. 2005, 8, 610–613. [Google Scholar] [CrossRef]
- Schlachter, A.; Juvenal, F.; Kinghat Tangou, R.; Khatyr, A.; Guyon, F.; Karsenti, P.L.; Strohmann, C.; Kubicki, M.M.; Rousselin, Y.; Harvey, P.D.; et al. 2-Azabutadiene complexes of rhenium(I): S,N-chelated species with photophysical properties heavily governed by the ligand hidden traits. Dalton Trans. 2021, 50, 2945–2963. [Google Scholar] [CrossRef] [PubMed]
- Schramm, V. Crystal and molecular structure of tetrameric copper(I) iodide-piperidine, a complex with a tetrahedral tetrakis[copper(I) iodide] core. Inorg. Chem. 1978, 17, 714–718. [Google Scholar] [CrossRef]
- Jasinski, J.P.; Rath, N.P.; Holt, E.M. Structural comparison of (CuICH3CN4·dibenzo-18-crown-6 (I) and (CuICH3CN)x (II) fluorescent copper(I) materials. Inorg. Chim. Acta 1985, 7, 91–97. [Google Scholar] [CrossRef]
- Vega, A.; Saillard, J.-Y. Bonding in Tetrahedral Cu4(μ3-X)4L4 Copper(I) Clusters: A DFT Investigation. Inorg. Chem. 2004, 43, 4012–4018. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.D.; Knorr, M. Luminescent Coordination Polymers Built Upon Cu4X4 (X = Br,I) Clusters and Mono- and Dithioethers. Macromol. Rapid Commun. 2010, 31, 808–826. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Ozawa, Y.; Toriumi, K. Flexibility of cubane-like Cu4I4 framework: Temperature dependence of molecular structure and luminescence thermochromism of [Cu4I4(PPh3)4] in two polymorphic crystalline states. Chem. Commun. 2010, 46, 6302–6304. [Google Scholar] [CrossRef] [PubMed]
- Benito, Q.; Le Goff, X.F.; Nocton, G.; Fargues, A.; Garcia, A.; Berhault, A.; Kahlal, S.; Saillard, J.-Y.; Martineau, C.; Trébosc, J.; et al. Geometry Flexibility of Copper Iodide Clusters: Variability in Luminescence Thermochromism. Inorg. Chem. 2015, 54, 4483–4494. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bian, G.-Q.; Dai, J.; Zhang, Y.; Zhu, Q.-Y.; Lu, W. Luminescent 2-D Double-layered Polymer, [(CuI)4(CH3SCH3)3]∞, Containing Helical Chains Constructed by Flower-Basket-Shaped Cu4I4 Clusters. Inorg. Chem. 2006, 45, 8486–8488. [Google Scholar] [CrossRef]
- Knorr, M.; Bonnot, A.; Lapprand, A.; Khatyr, A.; Strohmann, C.; Kubicki, M.M.; Rousselin, Y.; Harvey, P.D. Reactivity of CuI and CuBr toward Dialkyl Sulfides RSR: From Discrete Molecular Cu4I4S4 and Cu8I8S6 Clusters to Luminescent Copper(I) Coordination Polymers. Inorg. Chem. 2015, 54, 4076–4409. [Google Scholar] [CrossRef]
- Cran, G.A.; Gibson, C.L.; Handa, S.; Kennedy, A.R. Scalemic β-Amino Sulfide Ligands: Use in Enantioselective Conjugate Additions and X-ray Analysis of a Dimeric Copper(I) Complex. Tetrahedron Asymmetry 1996, 7, 2511–2514. [Google Scholar] [CrossRef]
- Zhang, S.-M.; Hu, T.-L.; Du, J.-L.; Bu, X.-H. Tuning the formation of copper(I) coordination architectures with quinoxaline-based N,S-donor ligands by varying terminal groups of ligands and reaction temperature. Inorg. Chim. Acta. 2009, 362, 3915–3924. [Google Scholar] [CrossRef]
- Cheon, S.; Kim, T.H.; Jeon, Y.; Kim, J.; Park, K.-M. Novel heptanuclear copper(I) iodide cluster with a pinwheel shape. CrystEngComm 2013, 15, 451–454. [Google Scholar] [CrossRef]
- Lee, H.-H.; Park, I.-H.; Kim, S.; Lee, E.; Ju, H.; Jung, J.H.; Ikeda, M.; Habatac, Y.; Lee, S.S. Anion exchange coupled with the reduction and dimerisation of a copper(II) nitrate complex of tripyridyl dithioether via a single-crystal-to-single-crystal transformation. Chem. Sci. 2017, 8, 2592–2596. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.; Pankhurst, J.; Sproules, R.S.; Mignard, D.; Love, J.B. Triggering Redox Activity in a Thiophene Compound: Radical Stabilization and Coordination Chemistry. Angew. Chem. Int. Ed. 2017, 56, 7939–7943. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97 and SHELXL-97, Program for Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Cryst. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Parkin, S.; Moezzi, B.; Hope, H. XABS2: An empirical absorption correction program. J. Appl. Crystal. 1995, 28, 53–56. [Google Scholar] [CrossRef]
- Walker, N.; Stuart, D. An Empirical Method for Correcting Diffractometer Data for Absorption Effects. Acta Cryst. 1983, A39, 158–166. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; C.01, revision; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Barros, C.L.; de Oliveira, P.J.P.; Jörge, F.E.; Canal Neto, A.; Campos, M. Gaussian basis set of double zeta quality for atoms Rb through Xe: Application in non-relativistic and relativistic calculations of atomic and molecular properties. Mol. Phys. 2010, 108, 1965–1972. [Google Scholar] [CrossRef]
- Canal Neto, A.; Jörge, F.E. All-electron double zeta basis sets for the most fifth-row atoms: Application in DFT spectroscopic constant calculations. Chem. Phys. Lett. 2013, 582, 158–162. [Google Scholar] [CrossRef]
- Biegler-König, F.; Schönbohm, J.; Bayles, D. AIM2000—A Program to Analyze and Visualize Atoms in Molecules. J. Comp. Chem. 2001, 22, 545–559. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, version 17.11.14; TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Castineiras, A.; Diaz, G.; Florenzio, F.; Garcia-Blanco, S.; Martinez-Carrera, S. Crystal and molecular structure of tetraiodo (1,8-di-2-pyridyl-3,6-dithiaoctane-N,S,S′,N′)dimercury(II). J. Crystallogr. Spectrosc. Res. 1988, 18, 395–401. [Google Scholar] [CrossRef]
- Lee, E.; Lee, I.S.-G.; Park, I.-H.; Kim, S.; Ju, H.; Jung, J.H.; Ikeda, M.; Habata, Y.; Lee, S.S. Endo- and Exocyclic Coordination of a 20-Membered N2O2S2-Macrocycle and Cascade Complexation of a 40-Membered N4O4S4-Macrocycle. Inorg. Chem. 2018, 57, 6289–6299. [Google Scholar] [CrossRef]
- Zhang, J.-A.; Pan, M.; Zhang, J.-Y.; Kang, B.-S.; Su, C.-Y. Syntheses, structures and bioactivities of cadmium(II) complexes with a tridentate heterocyclic N- and S-ligand. Inorg. Chim. Acta 2009, 362, 3519–3525. [Google Scholar] [CrossRef]
- Kim, S.; Huiyeong, J.; Park, K.-M.; Jung, J.H.; Lee, S.S.; Lee, E. Influence of the Reaction Sequence on the Complexation of an NS4-Macrocycle with CdII and CuI Salts Leading to the Formation of Supramolecular Isomers and an Endo/Exocyclic CuI Complex. Inorg. Chem. 2021, 60, 13637–13645. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. J. Chem. Soc. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Okuniewski, A.; Rosiak, D.; Chojnacki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Hostettler, M.; Birkedal, H.; Schwarzenbach, D. Polymorphs and structures of mercuric iodide. Chimia 2001, 55, 541–545. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Vlasse, M. On the Crystal Structures of the Red, Yellow, and Orange Forms of Mercuric Iodide. Inorg. Chem. 1967, 6, 396–399. [Google Scholar] [CrossRef]
- Auvray, P.; Genet, F. Affinement de la structure cristalline du cinabre α–HgS. Bull. Soc. Fr. Minéral. 1973, 96, 218–219. [Google Scholar]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part I: Fundamental principles. J. Chem. Educ. 1968, 45, 581–586. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part II: Underlying theories. J. Chem. Educ. 1968, 45, 643–648. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Effective Ionic Radii on Oxides and Fluorides. Acta Cryst. 1969, B25, 925–945. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Davies, J.E.; Galloy, J.J.; Johnson, O.; Kennard, O.; Macrae, C.F.; Mitchell, E.M.; Mitchell, G.F.; Smith, J.M.; Watson, D.G. The Development of Versions 3 and 4 of the Cambridge Structural Database System. J. Chem. Inf. Comput. Sci. 1991, 31, 187–204. [Google Scholar] [CrossRef]
- Caradoc-Davies, P.L.; Hanton, L.R. Molecular rectangles from metallomacrocycles: Development of dibenzofuranligands. Dalton Trans. 2003, 9, 1754–1758. [Google Scholar] [CrossRef]
- Noren, B.; Oskarsson, A. Bond Length Variations in Tetrahydrothiophene Solvates of the Coinage Metals. The Crystal Structure of Di-mu-iodo-bis[bis(tetrahydrothiophene)copper(I)]. Acta Chem. Scand. 1987, A41, 12–17. [Google Scholar] [CrossRef]
- Lu, W.; Yan, Z.-M.; Dai, J.; Zhang, Y.; Zhu, Q.-Y.; Jia, D.-X.; Guo, W.-J. Coordination Assembly of TTF Derivatives through CuI Bridges. Eur. J. Inorg. Chem. 2005, 2005, 2339–2345. [Google Scholar] [CrossRef]
- Munakata, M.; Wu, L.P.; Kuroda-Sowa, T.; Maekawa, M.; Suenaga, Y.; Nakagawa, S.J. One-, two- and three-dimensional copper(I) and silver(I) complexes of 2,11-dithia[3.3]paracyclophane. J. Chem. Soc. Dalton Trans. 1996, 25, 1525–1530. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Zhou, H.; Lin, P.; Li, Z.-H.; Du, S.-W. Four new coordination polymers based on multinuclear Cu(I) halide clusters and 1,3-bis(imidazol-1-yl-methyl)-benzene. J. Mol. Struct. 2008, 881, 21–27. [Google Scholar] [CrossRef]
- Popelier, P. Atoms in Molecules, an Introduction; Prentice Hall: Essex, UK, 2000. [Google Scholar]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H⋯F-Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Alkorta, I.; Blanco, F.; Elguero, J. The use of a molecular balance derived from 5,5′-bipyrazole to calculate π−π stacking interactions. Tetrahedron Lett. 2008, 49, 7246–7249. [Google Scholar] [CrossRef]
- Biswas, C.; Drew, M.G.B.; Escudero, D.; Frontera, A.; Ghosh, A. Anion–π, lone-pair–π, π–π and hydrogen-bonding interactions in a CuII complex of 2-picolinate and protonated 4,4′-bipyridine: Crystal structure and theoretical studies. Eur. J. Inorg. Chem. 2009, 15, 2238–2246. [Google Scholar] [CrossRef]
- Estarellas, C.; Frontera, A.; Quinonero, D.; Deya, P.M. Theoretical ab initio study of substituted benzene trimer: Interplay between hydrogen bonding and π-π interactions. Comput. Theor. Chem. 2011, 975, 106–110. [Google Scholar] [CrossRef]
- Dinda, S.; Samuelson, A.G. The nature of bond critical points in dinuclear copper(I) complexes. Chem. Eur. J. 2012, 18, 3032–3042. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental electron density analysis of Mn2(CO)10: Metal-metal and metal-ligand bond characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J.; Senn, H.M. Metal-metal and metal-ligand bonding at a QTAIM catastrophe: A combined experimental and theoretical charge density study on the alkylidyne cluster Fe3(μ-H)(μ-COMe)(CO)10. J. Phys. Chem. A 2010, 114, 13418–13433. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Marques, H.M. The physical chemistry of coordinated aqua-, ammine-, and mixed-ligand Co2+ complexes: DFT studies on the structure, energetics, and topological properties of the electron density. Phys. Chem. Chem. Phys. 2010, 12, 2126–2138. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor—Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
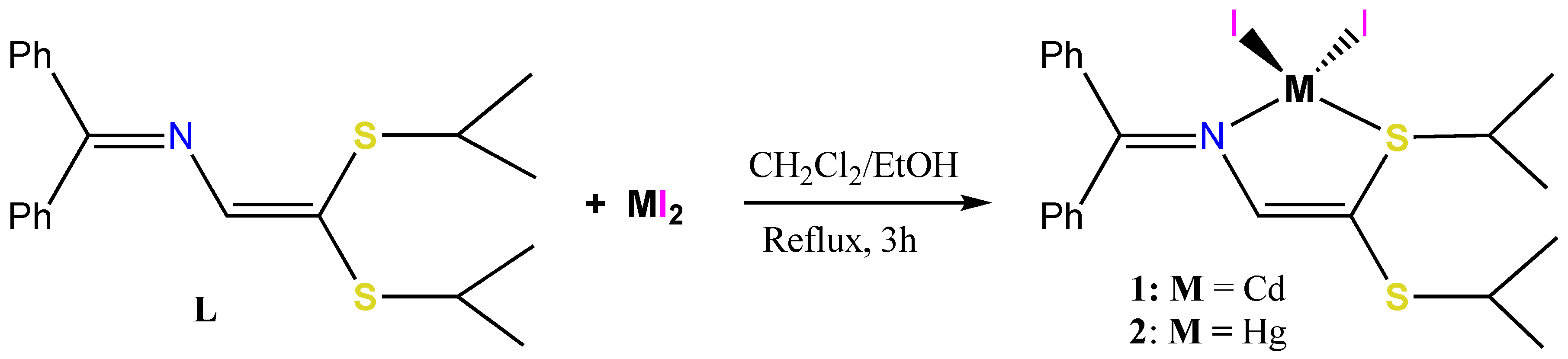






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
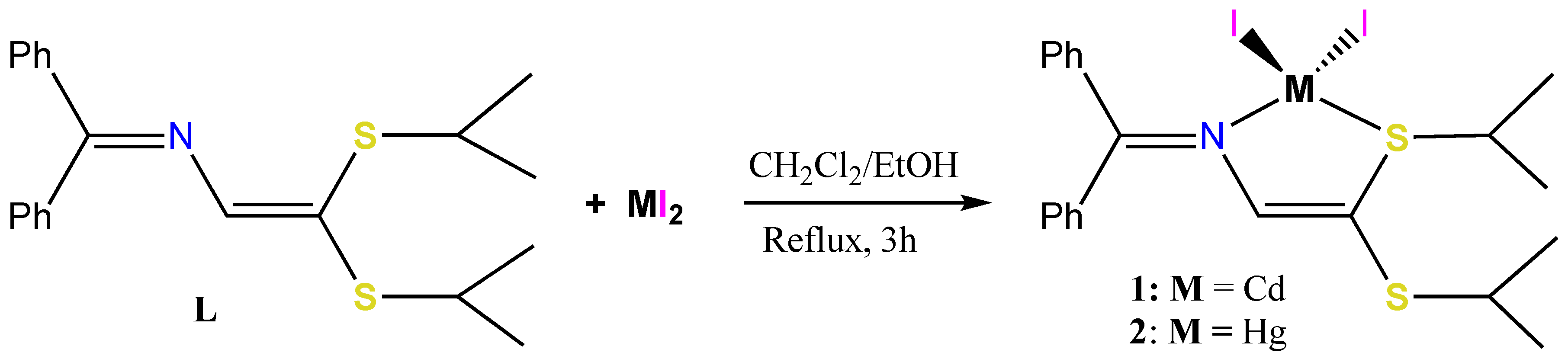{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Formula | C21H25CdI2NS2 | C22H27Cl2HgI2NS2 | C22H25CuI2NS2 |
| Formula weight | 721.74 | 894.85 | 545.98 |
| Temperature/K | 173 | 115 | 193.15 |
| Wavelength/Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | orthorhombic | triclinic |
| Space group | P21 | P212121 | P |
| a/Å | 9.006(3) | 9.7582(2) | 9.3935(19) |
| b/Å | 9.454(3) | 15.7719(3) | 9.6540(19) |
| c/Å | 15.081(4) | 17.9490(4) | 13.689(3) |
| a/° | 90.0 | 90 | 101.92(3) |
| β/° | 101.587(6) | 90 | 96.52(3) |
| γ/° | 90.0 | 90 | 90.30(3) |
| Volume/Å3 | 1257.9(6) | 2762.45(10) | 1206.3(4) |
| Z | 2 | 4 | 2 |
| ρ(calc.) g/cm3 | 1.906 | 2.152 | 1.503 |
| μ/mm−1 | 3.495 | 8.162 | 2.363 |
| F(000) | 688 | 1672 | 544 |
| Crystal size/mm | 0.16 × 0.15 × 0.12 | 0.10 × 0.10 × 0.08 | 0.20 × 0.20 × 0.10 |
| θ range for data collection/° | 2.308 to 26.997 | 2.611 to 27.476 | 4.366 to 49.998 |
| Index ranges | −11 ≤ h ≤ 11, −11 ≤ k ≤ 12, −19 ≤ l ≤ 19 | −12 ≤ h ≤ 12, −20 ≤ k ≤ 20, −23 ≤ l ≤ 23 | −11 ≤ h ≤ 11, −00 ≤ k ≤ 10, −0 ≤ l ≤ 16 |
| Reflections collected | 20,409 | 6299 | 3965 |
| Independent reflections | 5388 [R(int) = 0.0273] | 6299 | 3965 [R(int) = 0.0651] |
| Refl. greater [I > 2σ(I)] | 5325 | 5445 | 2805 |
| Absorption correction Transmission max Transmission min Refinement method | CrysAlis PRO 0.999 0.704 Full-matrix least squares on F2 | DIFABS 0.4419 0.3386 Full-matrix least squares on F2 | FACEIT in IPDS 0.7980 0.6494 Full-matrix least squares on F2 |
| Data/restraints/parameters | 5388/1/248 | 6299/0/277 | 3695/6/249 |
| Goodness-of-fit on F2 Flack parameter | 1.124 0.02(2) | 1.044 0.014(7) | 1.012 - |
| Final R indexes [I > 2σ(I)] | R1 = 0.0292, wR2 = 0.0774 | R1 = 0.0368, wR2 = 0.0601 | R1 = 0.0894, wR2 = 0.1456 |
| R indexes (all data) | R1 = 0.0297, wR2 = 0.0787 | R1 = 0.0484, wR2 = 0.0633 | R1 = 0.0805, wR2 = 0.1549 |
| Largest diff. peak and hole/e.Å−3 | 0.95 and −0.37 | 1.08 and −0.80 | 0.67 and −0.75 |
| 1 Cd | 2 Hg | 3 Cu | 1 Cd | 2 Hg | 3 Cu | ||
|---|---|---|---|---|---|---|---|
| M–I1 (a) | 2.6787(8) | 2.6412(7) | 2.576(2) | I1-M-I2 | 124.80(2) | 138.56(2) | 119.09(5) |
| M–I2 (a) | 2.7140(9) | 2.6941(7) | 2.692(2) | I1-M-S | 118.25(4) | 117.93(5) | 112.4(1) |
| M–S | 2.662(2) | 2.689(2) | 2.342(2) | I1-M-N | 114.5(1) | 121.1(2) | 127.4(2) |
| M–N | 2.321(5) | 2.496(7) | 2.143(5) | I2-M-S | 97.93(4) | 96.04(5) | 108.6(1) |
| I2-M-N | 112.7(1) | 88.9(2) | 98.0(2) | ||||
| Cu–Cu | 2.672(2) | S-M-N | 78.1(1) | 74.5(2) | 86.0(2) |
| Complex 1 | Complex 2 | Complex 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| λ (nm) | Osc. Strength | MO | λ (nm) | Osc. Strength | MO | λ (nm) | Osc. Strength | MO |
| 393 | 0.23 | H-4 (95) | 388 | 0.29 | H-4 (80) H-3 (19) | 567 | 0.07 | H-3/L+1 (10) H-2/L (73) H-1/L+1 (12) |
| 323 | 0.08 | H-7 (87) | 328 | 0.08 | H-6 (95) | 550 | 0.03 | H-1/L+1 (84) |
| 311 | 0.03 | H-8 (82) | 318 | 0.08 | H-7 (92) | 457 | 0.02 | H-7/L+1 (14) H-5/L (63) |
| 304 | 0.04 | H-9 (80) | 309 | 0.03 | H-8 (93) | 419 | 0.04 | H-9/L (14) H-6/L (76) |
| 290 | 0.05 | H-11 (21) H-10 (66) | 299 | 0.05 | H-9 (87) | 380 | 0.50 | H-9/L (33) H-8/L+1 (21) H-7/L+1 (24) |
| 283 | 0.15 | H-11 (61) H-10 (19) | 287 | 0.11 | H-10 (91) | |||
| Bond Property | M—I1 Cd Hg Cu | M—I2 Cd Hg Cu | M—S Cd Hg Cu | M—N Cd Hg Cu | Cd–C13 | Cu–Cu |
|---|---|---|---|---|---|---|
| d, Å | 2.679 2.641 2.576 | 2.714 2.694 2.692 | 2.662 2.689 2.342 | 2.321 2.496 2.143 | 3.157(5) | 2.672 |
| ρ, e−/a03 | 0.060 0.064 0.054 | 0.056 0.059 0.046 | 0.044 0.045 0.064 | 0.057 0.043 0.061 | 0.010 | 0.031 |
| ∇2ρ, e−/a05 | 0.127 0.180 0.109 | 0.119 0.165 0.085 | 0.149 0.150 0.214 | 0.301 0.184 0.295 | 0.031 | 0.021 |
| −V, Ha/a03 | 0.048 0.079 0.051 | 0.044 0.070 0.041 | 0.037 0.051 0.073 | 0.059 0.052 0.0733 | −0.0054 | 0.0288 |
| G, Ha/a03 | 0.040 0.062 0.039 | 0.037 0.055 0.031 | 0.037 0.044 0.061 | 0.067 0.049 0.0736 | 0.0066 | 0.0177 |
| −H, Ha/a03 | 0.008 0.017 0.012 | 0.007 0.014 0.010 | −0.0003 0.006 0.010 | −0.008 0.003 −0.0003 | −0.0012 | 0.0105 |
| −H/ρ, Ha/e− | 0.136 0.266 0.219 | 0.126 0.244 0.218 | −0.048 0.144 0.149 | −0.143 0.065 −0.0043 | −0.039 | 0.341 |
| |V|/G | 1.203 1.276 1.303 | 1.192 1.258 1.319 | 0.992 1.150 1.189 | 0.879 1.058 0.996 | 0.818 | 1.605 |
| DIcalc | 0.778 0.589 0.587 | 0.728 0.523 0.479 | 0.367 0.287 0.483 | 0.372 0.221 0.379 | 0.038 | 0.149 |
| DInorm | 0.682 0.727 0.565 | 0.638 0.646 0.461 | 0.322 0.354 0.465 | 0.326 0.273 0.365 | 0.018 | 0.143 |
| Wbxcalc | 0.785 0.563 0.230 | 0.756 0.471 0.192 | 0.306 0.168 0.141 | 0.216 0.101 0.100 | 0.026 | 0.097 |
| Wbxnorm | 0.752 0.809 | 0.724 0.758 | 0.293 0.270 | 0.207 0.163 | 0.025 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinghat, R.; Khatyr, A.; Knorr, M.; Strohmann, C.; Kubicki, M.M. 4,4-Bis(isopropylthio)-1,1-diphenyl-2-azabuta-1,3-diene Adducts with Cadmium(II), Mercury(II) and Copper(I) Iodides: Crystal, Molecular and Electronic Structures of d10 Transition Metal Chelate Complexes. Chemistry 2024, 6, 62-80. https://doi.org/10.3390/chemistry6010004
Kinghat R, Khatyr A, Knorr M, Strohmann C, Kubicki MM. 4,4-Bis(isopropylthio)-1,1-diphenyl-2-azabuta-1,3-diene Adducts with Cadmium(II), Mercury(II) and Copper(I) Iodides: Crystal, Molecular and Electronic Structures of d10 Transition Metal Chelate Complexes. Chemistry. 2024; 6(1):62-80. https://doi.org/10.3390/chemistry6010004
Chicago/Turabian StyleKinghat, Rodolphe, Abderrahim Khatyr, Michael Knorr, Carsten Strohmann, and Marek M. Kubicki. 2024. "4,4-Bis(isopropylthio)-1,1-diphenyl-2-azabuta-1,3-diene Adducts with Cadmium(II), Mercury(II) and Copper(I) Iodides: Crystal, Molecular and Electronic Structures of d10 Transition Metal Chelate Complexes" Chemistry 6, no. 1: 62-80. https://doi.org/10.3390/chemistry6010004
APA StyleKinghat, R., Khatyr, A., Knorr, M., Strohmann, C., & Kubicki, M. M. (2024). 4,4-Bis(isopropylthio)-1,1-diphenyl-2-azabuta-1,3-diene Adducts with Cadmium(II), Mercury(II) and Copper(I) Iodides: Crystal, Molecular and Electronic Structures of d10 Transition Metal Chelate Complexes. Chemistry, 6(1), 62-80. https://doi.org/10.3390/chemistry6010004