Abstract
Tyrosine-containing pharmaceuticals’ (TPh) potential to inhibit SARS CoV-2 3-chymotrypsin-like proteases (3CLpro) and nonstructural protein 16 (NSP16) has been explored using docking studies, molecular dynamics simulations, and density functional theory. The TPh with FDA approval showed excellent contact with the active site pockets of 3CLpro and NSP16. Their binding affinity scores ranged from −5.8 to −4.9 kcal/mol and −6.3 to −4.8 for 3CLpro and NSP16, respectively. A 100-ns molecular dynamics simulation confirmed the stability of the carbidopa/NSP16 complex and N-acetyl tyrosine with both target enzymes. Further, the HOMO-LUMO transitions, molecular orbitals, and dipole moments of carbidopa, droxidopa, and N-acetyl tyrosine were computed using density functional theory (DFT). Considering N-acetyl tyrosine and carbidopa’s substantial inhibitory activity, it is recommended to investigate them further in order to explore their application for the treatment of COVID-19 or any other coronaviruses in the future.
1. Introduction
Coronavirus disease (COVID-19) continues to affect the global population. SARS-CoV-2 is the causative agent of the global COVID-19 pandemic, which has currently affected 679,863,569 individuals and accounted for 6,799,565 deaths worldwide by 22 February 2023 (https://www.worldometers.info/coronavirus/) (accessed on 22 February 2023). SARS-CoV-2 is an RNA virus that belongs to the Coronaviridae family and infects humans and animals [1,2,3,4]. SARS-CoV-2 contains two open reading frames, ORF1a and ORF1b, translated by host ribosomes as two multi-proteins, pp1a and pp1ab. ORF1a encodes the papain-like protease (PLpro) and the main protease (Mpro) or the 3-chymotrypsin-like protease of SARS-CoV-2 (3CLpro), the essential cysteine proteases for viral replication and transcription. The 3CLpro cleaves 11 different sites at the C-terminus and is involved in forming 15 nonstructural proteins. The 3CLpro is unlike the human host-cell protease [5,6,7]. Therefore, it has become the main target for drug repurposing and development programs to fight the COVID-19 pandemic [8,9,10]. Further, the nonstructural protein 16 (NSP16) is methyltransferase- and S-adenosylmethionine-dependent; it forms a heterodimer with the NSP10 cofactor. They are involved in the RNA methylation of the first nucleotide transcribed by activating the action of 2′-O-methyltransferase. This is a key step in evading viral RNA-triggered immune responses by preventing host recognition [11,12].
As compared to de novo drug acquisition, drug repurposing is considered the most efficient method of identifying new uses for approved therapeutic drugs, as it involves less time and fewer expenses regarding treatment. An important benefit of repurposing drugs is the availability of information about their safety and pharmacokinetic profiles, predicting potential side effects and drug interactions [13,14]. There are several vaccines that are approved now, and millions have been vaccinated, which decreases the incidence rates. There is, however, a possibility that new variants, such as omicron and delta, may emerge that are resistant to the vaccines, and that a new treatment protocol may need to be employed [15,16,17,18]. Further, despite all efforts, scientists still cannot develop a satisfactory therapy for COVID-19. In addition, very few or limited repurposed pharmaceuticals have been approved, such as remdesivir [19,20]. Therefore, there is still a need to discover new drugs to combat COVID-19.
In several studies, pharmaceuticals, synthetic drugs, and natural products were reported to be effective in treating COVID-19 [14,15,16,17,18,19,20]. A recent study has shown that some Parkinson’s disease patients do not show severe symptoms related to infection with COVID-19 compared to control patients with no Parkinson’s disease. These Parkinson’s disease patients were given a daily dose of levodopa [21]. The levodopa structure includes tyrosine hydroxylated in the meta position. Herein, we investigated FDA-approved tyrosine-containing pharmaceuticals (TPh) as 3CLpro and NSP16 inhibitors using molecular docking and molecular dynamics simulation. The structural and physical characteristics were studied using density functional theory (DFT).
2. Materials and Methods
2.1. Criteria Used to Select TPh
The TPh were selected using the search engine of the Drug Bank database. The results revealed that eleven TPh fulfilled the criterion of having a tyrosine or tyrosine-like moiety, as shown in Scheme 1.
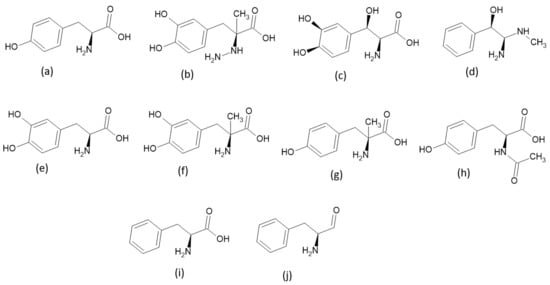
Scheme 1.
Chemical structures of TPh: (a) tyrosine, (b) carbidopa, (c) droxidopa, (d), epinephrine, (e) levodopa, (f) methyldopa, (g) metyrosine, (h) N-acetyl tyrosine, (i) phenylalanine, and (j) tyrosinal.
2.2. Generation and Energy Minimization of the TPh, 3CLpro, and NSP16
The 3D structures of the selected TPh were generated as PDB files using the build structure function in UCSF Chimera [22,23], utilizing SMILES strings offered by the Drug Bank online database. Their energy was minimized for 16,000 steepest descent steps at 8000 conjugate gradient steps. The crystal structures of SARS-CoV-2 3CLpro and NSP16 were downloaded from the Protein Data Bank database (PDB IDs: 6Y2E and 6WKQ, respectively). Then, the 3CLpro structure was minimized for 1000 steepest descent steps at 20 conjugate gradient steps after removing the water residues.
2.3. Molecular Docking
Molecular docking was achieved using the AutoDock Vina tool implemented in UCSF Chimera. A grid box (35.0, 65.0, 65.0) Å, centered at (−16 × −24.5 × 16) Å, and (43.9, 42.8, 45.7) Å, centered at (91.7 × 18.82 × −2.96) Å for 3CLpro and NSP16, respectively, were used, retaining the default parameter values. The predicted affinity score was obtained through the View Dock tool. The hydrogen bonds, van der Waals results, and images were processed and visualized using UCSF Chimera [22,23,24,25,26].
2.4. Molecular Dynamics Simulations
Carbidopa, droxidopa, and N-acetyl tyrosine docked were separated from the 3CLpro via UCSF Chimera. The missed hydrogen atoms were added to the ligands and saved as PDB files. The Amber force field of GAFF2 [27] and ff14SB [28] assigned the inhibitors and the 3CLpro and NSP16 structures, respectively. Each system was solvated using TIP3P water molecules [29] and neutralized by sodium chloride. Molecular dynamics (MD) simulations were achieved employing the Nanoscale Molecular Dynamics (NAMD) Simulation 2.6 program [30]. The systems tested were minimized at 273.15 K for 1 ps using the NVE ensemble. The temperature was gradually increased to 310 K in a protocol consisting of 2100 minimization steps. Next, they were minimized at 310 K for 10 ps, followed by 20 ns MD simulations run at 310 K and a time step of 2 fs. The periodic boundary conditions and particle mesh Ewald process were employed to calculate electrostatic interactions [31,32]. The trajectory was analyzed with the VMD 1.8 program to obtain the root-mean-square deviation (RMSD) and the root-mean-square fluctuation (RMSF) [33].
2.5. DFT Calculations and Chemical Reactivity Descriptors
The Gaussian 09 software was used for all DFT calculations, https://gaussian.com/glossary/g09/ (accessed on 29 April 2013). Theoretical DFT computations were performed using the DFT method in the gas phase with the B3LYP 6-311G (d, p) basis set. Calculations, such as the energy of the highest occupied molecular orbital (EHOMO) and energy of the lowest unoccupied molecular orbital (ELUMO), were performed to obtain the quantum chemical parameters of the organic compounds. Additional parameters were calculated; these included the separation energies (ΔE), chemical potentials (Pi), absolute electro-negativities (v), absolute hardness (g), global electrophilicity (x), absolute softness (r), and global softness (S) [34,35,36].
3. Results and Discussion
3.1. Molecular Docking
The crystal structures of SARS-CoV-2 3CLpro and NSP16 and their active sites are depicted in Figure 1 and Figure 2. The 3CLpro was active in a homodimer form. The GLU 166 in one protomer and the N-terminal amino acid residues in the other protomer were involved in forming the S1 subsite binding pocket. The catalytic dyad of HIS 41 and CYS 145 was involved in S1 site formation. Thus, the inhibitors interacting with these residues are of great interest as potential 3CLpro [37,38,39].
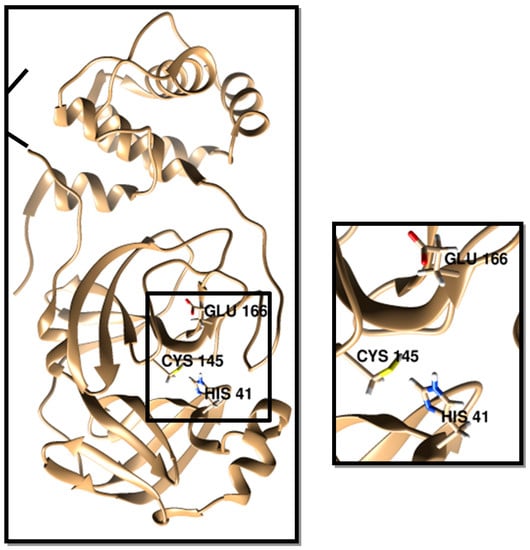
Figure 1.
SARS-CoV-2 chymotrypsin-like protease crystal structure (PDB ID: 6Y2E). The catalytic dyad of HIS 41 and CYS 145 and the active residue of GLU 166 are visualized for identification.
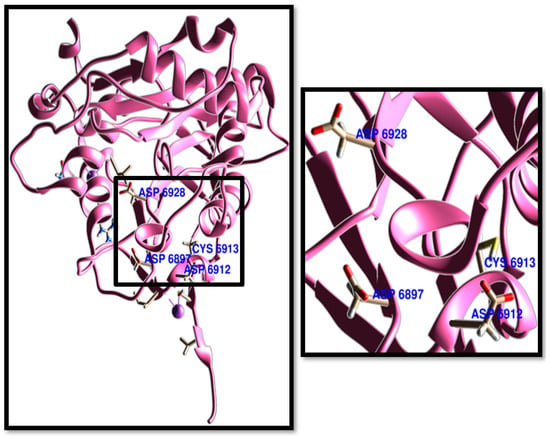
Figure 2.
SARS-CoV-2 NSP16 crystal structure (PDB ID: 6WKQ). The active residues of ASP 6928, ASP 6897, ASP 6912, and ASP 6913 are visualized for identification.
TPh docked to the 3CLpro’s active site pocket, except for tyrosinal, phenylalanine, and hydroxyamphetamine (Table 1). Carbidopa showed a higher binding affinity, followed by droxidopa. In Figure 3, hydrogen bonds and van der Waals interactions are depicted between the TPh and 3CLpro active sites. Phenyl groups in TPh have shown excellent interactions with the thiolate of the CYS 145 residue. The amino group was attracted to GLU 16’s acid, with some ability to form hydrogen bonds, as with tyrosine and levodopa. As a result of the functionalization of the phenol ring, the binding affinity of levodopa, the hydroxylated form of tyrosine, was slightly enhanced compared to its parent tyrosine. Moreover, the functionalization of the chain containing amino and acid groups with electron-donating groups, such as amino and hydroxyl groups, enhanced further the binding affinity, as in carbidopa and droxidopa (Table 1).

Table 1.
The binding affinity of the pharmaceuticals containing a tyrosine (TPh) with 3-chymotrypsin-like protease (3CLpro).
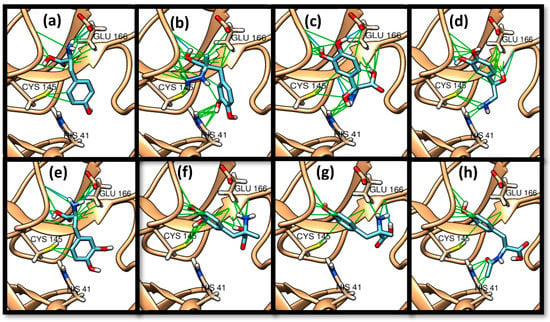
Figure 3.
The TPh docked with 3CLpro with a particular focus on contact with HIS 41, CYS 145, and GLU 166. (a) Tyrosine, (b) carbidopa, (c) droxidopa, (d) epinephrine, (e) levodopa, (f) methyldopa, (g) metyrosine, (h) N-acetyl tyrosine. Hydrocarbon skeletons of TPh are shown in cyan, nitrogen atoms in blue, and oxygens in red. Hydrogen bonds are shown in blue, and van der Waals in green.
On the other hand, the residues of ASP 6928, ASP 6897, ASP 6912, and ASP 6913 were determined as the active sites in NSP16 [27]. Interestingly, all TPh were docked successfully to the active site of NSP16, with high binding affinity for conformers to each PCT tested. Among TPh, N-acetyl tyrosine, carbidopa, and droxidopa exhibited the highest binding energies to the active sites of 3CLpro, respectively (Table 2). The acid group of the ASP 6897 residue exhibited a strong tendency to form hydrogen bonds with the amino groups of TPh. Meanwhile, ASP 6928 preferred to interact with electron-rich phenyl groups, but form hydrogen bonds, as in levodopa (Figure 4).
Table 2.
The binding affinity of the pharmaceuticals containing a tyrosine (TPh) with NSP16.

Figure 4.
The TPh docked with NSP16 with a particular focus on contact with ASP 6928, ASP 6897, ASP 6912, and ASP 6913. (a) Tyrosine, (b) carbidopa, (c) droxidopa, (d) epinephrine, (e) levodopa, (f) methyldopa, (g) metyrosine, (h) N-acetyl tyrosine, (i) phenylalanine, and (j) tyrosinal. Hydrocarbon skeletons of TPh are shown in cyan, nitrogen atoms in blue, and oxygens in red. Hydrogen bonds are shown in blue, and van der Waals in yellow.
3.2. Molecular Dynamics Simulations
RMSDs relative to the coordinate-averaged model were calculated for backbone atoms.
In order to compute the RMSDs, we used the coordinate-averaged model as a reference to calculate the trajectories lengthwise (Figure 5 and Figure 6). N-acetyl tyrosine significantly exaggerated the equilibration states of 3CLpro and NSP16 at 30 ns, with lower RMSDs throughout the production runs, confirming their stability. It appears that droxidopa and carbidopa form fewer stable complexes with 3CLpro, exhibiting higher RMSDs and fluctuations compared to apo (Table 3 and Table 4). However, carbidopa reached equilibration with NSP16 at around 50 ns.
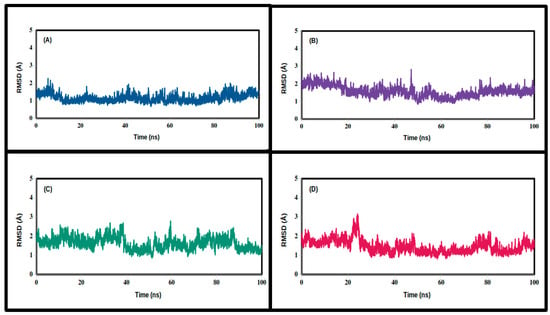
Figure 5.
Root-mean-square deviation values of the backbone carbon atoms of the simulated TPh and 3CLpro complexes throughout the production runs: (A) N-acetyl tyrosine, (B) carbidopa, (C) droxidopa, and (D) apo.
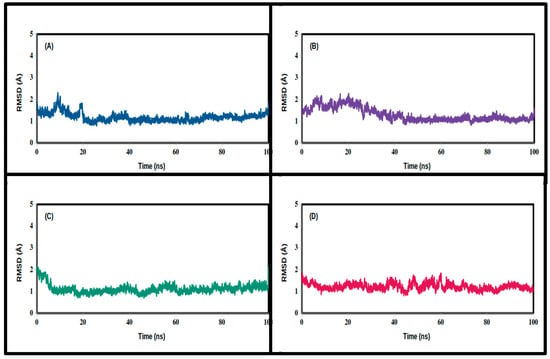
Figure 6.
Root-mean-square deviation values of the backbone carbon atoms of the simulated TPh and NSP16 complexes throughout the production runs: (A) N-acetyl tyrosine, (B) carbidopa, (C) droxidopa, and (D) apo.
Table 3.
RMSD and RMSF data of the simulated TPh and 3CLpro complexes during production runs of 100 ns.
Table 4.
RMSD and RMSF data of the simulated TPh and NSP16 complexes during production runs of 100 ns.
The fluctuations in the backbone residues of the simulated TPh/3CLpro were examined utilizing RMSF (Figure 7 and Figure 8). Carbidopa and droxidopa showed fluctuations similar to that of apo protein, whereas N-acetyl tyrosine showed a lower RMSF value, indicating a reduced degree of flexibility and a greater level of stability for 3CLpro. For Tph/NSP16 complexes, the average backbone residues’ fluctuations were reduced by droxidopa and N-acetyl tyrosine (Table 4). It is noteworthy that NSP16’s flexibility movement at residue 6932 is increased as a result of the TPh compared to the apo protein (Figure 8). This behavior suggests that the protein has undergone a conformational change, which impacts its activity.
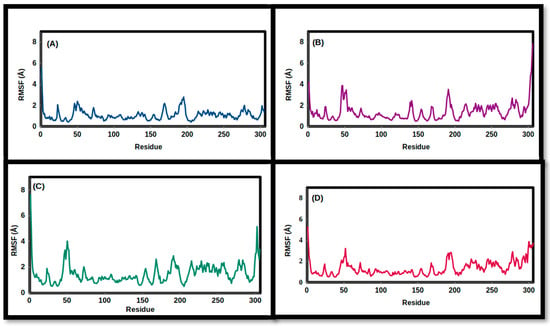
Figure 7.
Root-mean-square fluctuation values of the simulated TPh and 3CLpro complexes throughout the 100 ns trajectory: (A) N-acetyl tyrosine, (B) carbidopa, (C) droxidopa, and (D) apo.
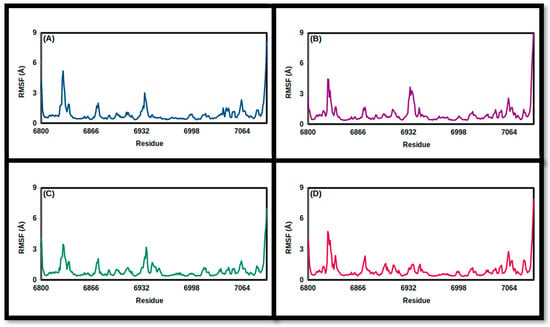
Figure 8.
Root-mean-square fluctuation values of the simulated TPh and NSP16 complexes throughout the 100 ns trajectory: (A) N-acetyl tyrosine, (B) carbidopa, (C) droxidopa, and (D) apo.
3.3. DFT Calculations
All geometrical structures of the pharmaceuticals under investigation were optimized (Figure 9). Table 5 shows a summary of the approximate DFT calculations, including the electronic energy, RMS gradient norm, electronic spatial extent, and dipole moment. The dipole moment values are in the order of N-acetyl tyrosine < droxidopa ≈ carbidopa. A lower dipole moment may demonstrate better biological availability due to higher solubility in water. Thus, N-acetyl tyrosine is the best TPh studied for research on therapy and bioactivity.
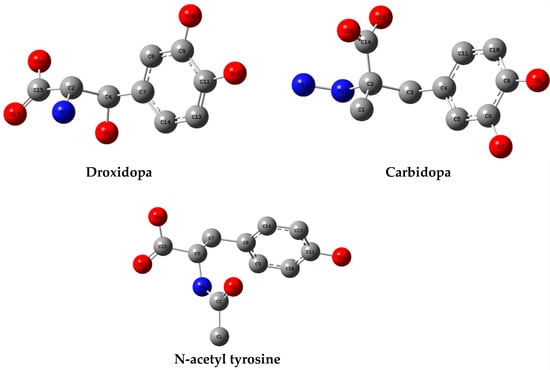
Figure 9.
Optimized geometrical structures of TPh with atomic numbering.
Table 5.
Electronic energy (Hartree/Particle), RMS gradient norm (Hartree/Bohr), electronic spatial extent (a.u.), and dipole moment (Debye) of TPh.
The frontier molecular orbitals (FMO) can provide objective qualitative information about the HOMO electrons being susceptible to transfer to the LUMO. In addition, HOMO and LUMO are very useful quantum chemical parameters to assess molecules’ reactivity and are used to measure other parameters, such as the descriptors for chemical reactivity. The values of the HOMO and LUMO for the studied TPh are listed in Table 6.
Table 6.
Calculated EHOMO (EH), ELUMO (EL), energy band gap (EH–EL), chemical potential (μ), electronegativity (χ), global hardness (η), global softness (S), global electrophilicity index (ω), and softness (σ) for TPh.
The isodensity surface plots of HOMO and LUMO for TPh are shown in Figure 10. The energy of HOMO of droxidopa is higher compared with carbidopa and N-acetyl tyrosine. However, the destabilization of the LUMO level is found to be higher in droxidopa than the others as well. Consequently, the energy gap is in the order of carbidopa ≈ droxidopa < N-acetyl tyrosine. According to FMO theory, the HOMO and LUMO energy levels influence the bioactivity of small structural drugs the most. Obviously, each TPh studied has a different level of energy for HOMO. As a result, droxidopa displayed the most HOMO lying, making it an excellent electron donor. An interesting observation is that N-acetyl tyrosine at the largest energy gap had hydrogen bonds with the active residues of the GLU 166 and CYS 145 residues. Interactions such as these could facilitate N-acetyl tyrosine’s binding to 3CLpro and NSP16. According to these results, N-acetyl tyrosine is more stable than the other TPh studied, supporting previous findings from molecular dynamics and dipole moment measurements.
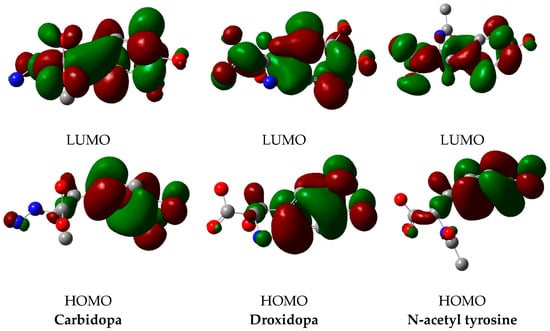
Figure 10.
HOMO and LUMO plots of the TPh.
4. Conclusions
To summarize, the inhibitory effect of the SARS-CoV-2 3CLpro and NSP16 by TPh was investigated utilizing molecular docking and MD simulations. The candidates revealed a high affinity to bind with the active sites of the target enzymes, particularly the NSP16, with high stability and a shallow binding energy affinity. The interactions of TPh with the active site pockets were thoroughly discussed. RMSF and RMSD data verified that N-acetyl tyrosine was able to form stable complexes with 3CLpro and NSP16, and carbidopa could form a stable complex with NSP16. Furthermore, the HOMO–LUMO transitions, atomic orbitals, and dipole moments of N-acetyl tyrosine, carbidopa, and droxidopa were determined using density functional theory (DFT). Of the TPh, N-acetyl tyrosine and carbidopa showed substantial inhibitory activity against SARS-CoV-2. This in silico study highlighted the crucial role of the tyrosine moiety in the inhibition of 3CLpro and NSP16 of SARS-CoV-2 for further investigations to explore their possible application for the treatment of COVID-19 or other coronaviruses in the future.
Author Contributions
M.R.E. and A.O.E.: conceptualization; A.O.E. and T.A.Y.: software; M.R.E., A.O.E. and T.A.Y.: validation, formal analysis, writing—original draft; M.R.E.: project administration, funding acquisition; A.O.E.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, Grant No. (21-13-18-046).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data will be available upon suitable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lvov, D.K.; Alkhovsky, S.V. Source of the COVID-19 pandemic: Ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus). Vopr. Virusol. 2020, 65, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Mishra, A.; Joshi, R.; Jha, S.; Sharma, A.R.; Upadhyay, S.; Sarma, P.; Prakash, A.; Medhi, B. Human animal interface of SARS-CoV-2 (COVID-19) transmission: A critical appraisal of scientific evidence. Vet. Res. Commun. 2020, 44, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Maurin, M.; Fenollar, F.; Mediannikov, O.; Davoust, B.; Devaux, C.; Raoult, D. Current status of putative animal sources of sars-cov-2 infection in humans: Wildlife, domestic animals and pets. Microorganisms 2021, 9, 868. [Google Scholar] [CrossRef]
- Hedman, H.D.; Krawczyk, E.; Helmy, Y.A.; Zhang, L.; Varga, C. Host Diversity and Potential Transmission Pathways of SARS-CoV-2 at the Human-Animal Interface. Pathogens 2021, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Sardanelli, A.M.; Isgrò, C.; Palese, L.L. SARS-CoV-2 Main Protease Active Site Ligands in the Human Metabolome. Molecules 2021, 26, 1409. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Tam, N.M.; Pham, M.Q.; Ha, N.X.; Nam, P.C.; Phung, H.T.T. Computational estimation of potential inhibitors from known drugs against the main protease of SARS-CoV-2. RSC Adv. 2021, 11, 17478–17486. [Google Scholar] [CrossRef]
- Thissera, B.; Sayed, A.M.; Hassan, M.H.; Abdelwahab, S.F.; Amaeze, N.; Semler, V.T.; Alenezi, F.N.; Yaseen, M.; Alhadrami, H.A.; Belbahri, L.; et al. Bioguided Isolation of Cyclopenin Analogues as Potential SARS-CoV-2 Mpro Inhibitors from Penicillium citrinum TDPEF34. Biomolecules 2021, 11, 1366. [Google Scholar] [CrossRef] [PubMed]
- Tassakka, A.C.M.A.; Sumule, O.; Massi, M.N.; Manggau, M.; Iskandar, I.W.; Alam, J.F.; Permana, A.D.; Liao, L.M. Potential bioactive compounds as SARS-COV-2 inhibitors from extracts of the marine red alga Halymenia durvillei (Rhodophyta)-A computational study. Arab. J. Chem. 2021, 14, 103393. [Google Scholar] [CrossRef]
- Al-Janabi, A.S.; Elzupir, A.O.; Abou-Krisha, M.M.; Yousef, T.A. New Dual Inhibitors of SARS-CoV-2 Based on Metal Complexes with Schiff-Base 4-Chloro-3-Methyl Phenyl Hydrazine: Synthesis, DFT, Antibacterial Properties and Molecular Docking Studies. Inorganics 2023, 11, 63. [Google Scholar] [CrossRef]
- Omer, E.A.; Abdelfatah, S.; Riedl, M.; Meesters, C.; Hildebrandt, A.; Efferth, T. Coronavirus Inhibitors Targeting nsp16. Molecules 2023, 28, 988. [Google Scholar] [CrossRef]
- Mule, S.; Singh, A.; Greish, K.; Sahebkar, A.; Kesharwani, P.; Shukla, R. Drug repurposing strategies and key challenges for COVID-19 management. J. Drug Target. 2022, 30, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Fadlalla, M.; Ahmed, M.; Ali, M.; Elshiekh, A.A.; Yousef, B.A. Molecular docking as a potential approach in repurposing drugs against COVID-19: A systematic review and novel pharmacophore models. Curr. Pharmacol. Rep. 2022, 8, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Pongrácz, J.E.; Mátyus, P. Recent advances in influenza, HIV and SARS-CoV-2 infection prevention and drug treatment—The need for precision medicine. Chemistry 2022, 4, 216–258. [Google Scholar] [CrossRef]
- Richman, D.D. COVID-19 vaccines: Implementation, limitations and opportunities. Glob. health Med. 2021, 3, 1–5. [Google Scholar] [CrossRef]
- Vesga, L.C.; Ruiz-Hernández, C.A.; Alvarez-Jacome, J.J.; Duque, J.E.; Rincon-Orozco, B.; Mendez-Sanchez, S.C. Repurposing of four drugs as anti-SARS-CoV-2 agents and their interactions with protein targets. Sci. Pharm. 2022, 90, 24. [Google Scholar] [CrossRef]
- Elliott, P.; Eales, O.; Steyn, N.; Tang, D.; Bodinier, B.; Wang, H.; Elliott, J.; Whitaker, M.; Atchison, C.; Diggle, P.J.; et al. Twin peaks: The omicron SARS-CoV-2 BA. 1 and BA. 2 epidemics in England. Science 2022, 376, eabq4411. [Google Scholar] [CrossRef]
- Shi, J.; Xiao, Y.; Zhang, Y.; Geng, D.; Cong, D.; Shi, K.X.; Knapp, R.J. Challenges of drug development during the COVID-19 pandemic: Key considerations for clinical trial designs. Br. J. Clin. Pharmacol. 2021, 87, 2170–2185. [Google Scholar] [CrossRef]
- Wang, X.; Guan, Y. COVID-19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Med. Res. Rev. 2021, 41, 5–28. [Google Scholar] [CrossRef]
- Sorbera, C.; Brigandì, A.; Cimino, V.; Bonanno, L.; Ciurleo, R.; Bramanti, P.; Di Lorenzo, G.; Marino, S. The impact of SARS-COV2 infection on people in residential care with Parkinson Disease or parkinsonisms: Clinical case series study. PLoS ONE 2021, 16, e0251313. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- de Leeuw, S.W.; Perram, J.; Smith, E.R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. Lond. A. Math. Phys. Sci. 1980, 373, 27–56. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Humphrey, W.; ADalke Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Song, Y.; Li, J.; Wu, Q.; Yi, C.; Wu, H.; Chen, Z.; Ou-Yang, W. Study of film thickness effect on carbon nanotube based field emission devices. J. Alloys Compd. 2020, 816, 152648. [Google Scholar] [CrossRef]
- Lim, L.; Shi, J.; Mu, Y.; Song, J. Dynamically-driven enhancement of the catalytic machinery of the SARS 3C-like protease by the S284-T285-I286/A mutations on the extra domain. PLoS ONE 2014, 9, e101941. [Google Scholar] [CrossRef]
- Amin, S.A.; Banerjee, S.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem. 2021, 215, 113294. [Google Scholar] [CrossRef]
- Yousef, T. Structural, optical, morphology characterization and DFT studies of nano sized Cu (II) complexes containing schiff base using green synthesis. J. Mol. Struct. 2020, 1215, 128180. [Google Scholar] [CrossRef]
- Yousef, T.; Alduaij, O.; El-Reash, G.A.; El Morshedy, R. Semiempirical studies, spectral analysis, in vitro antibacterial and DNA degradation studies of heterocyclic thiosemicarbazone ligand and its metal complexes. J. Mol. Liq. 2016, 222, 762–776. [Google Scholar] [CrossRef]
- Yousef, T.; El-Reash, G.A.; El Morshedy, R. Quantum chemical calculations, experimental investigations and DNA studies on (E)-2-((3-hydroxynaphthalen-2-yl) methylene)-N-(pyridin-2-yl) hydrazinecarbothioamide and its Mn (II), Ni (II), Cu (II), Zn (II) and Cd (II) complexes. Polyhedron 2012, 45, 71–85. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).