
Insight into Tyrosine-Containing Pharmaceuticals as Potential Inhibitors of SARS-CoV-2 3CLpro and NSP16: Structural Analysis, Docking Studies, Molecular Dynamics Simulations, and Density Functional Theory Investigations

Abstract
1. Introduction
2. Materials and Methods
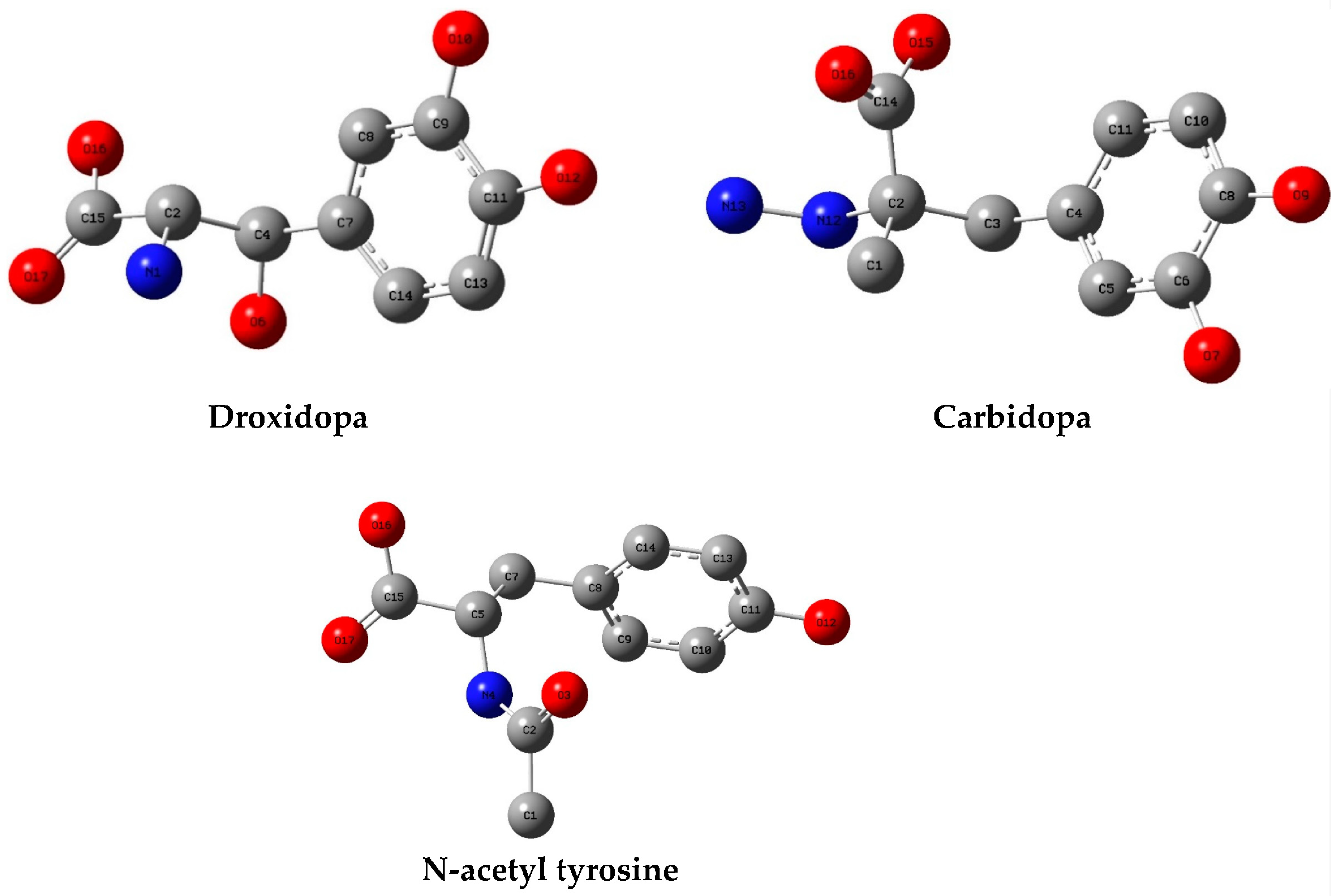
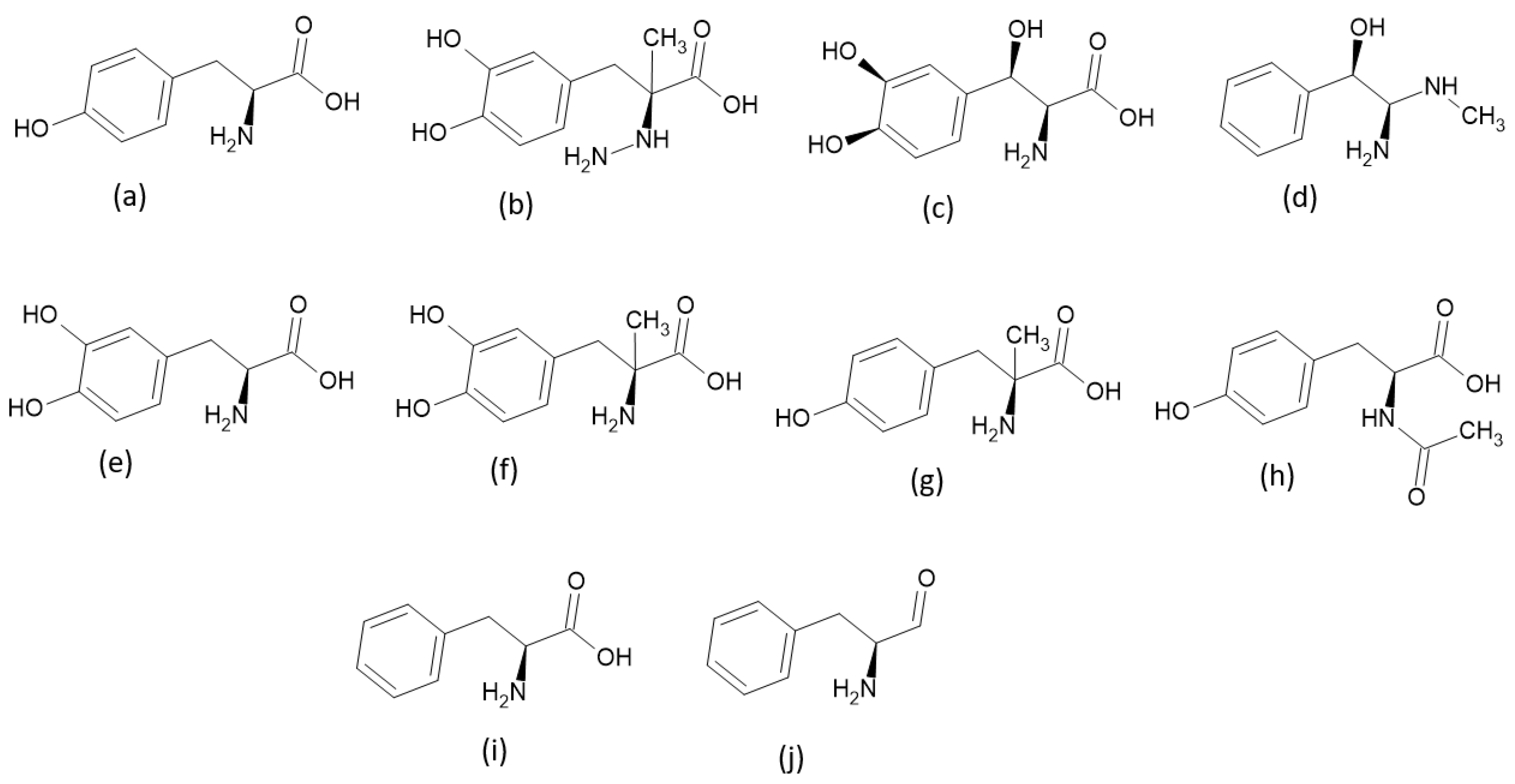
2.1. Criteria Used to Select TPh
2.2. Generation and Energy Minimization of the TPh, 3CLpro, and NSP16
2.3. Molecular Docking
2.4. Molecular Dynamics Simulations
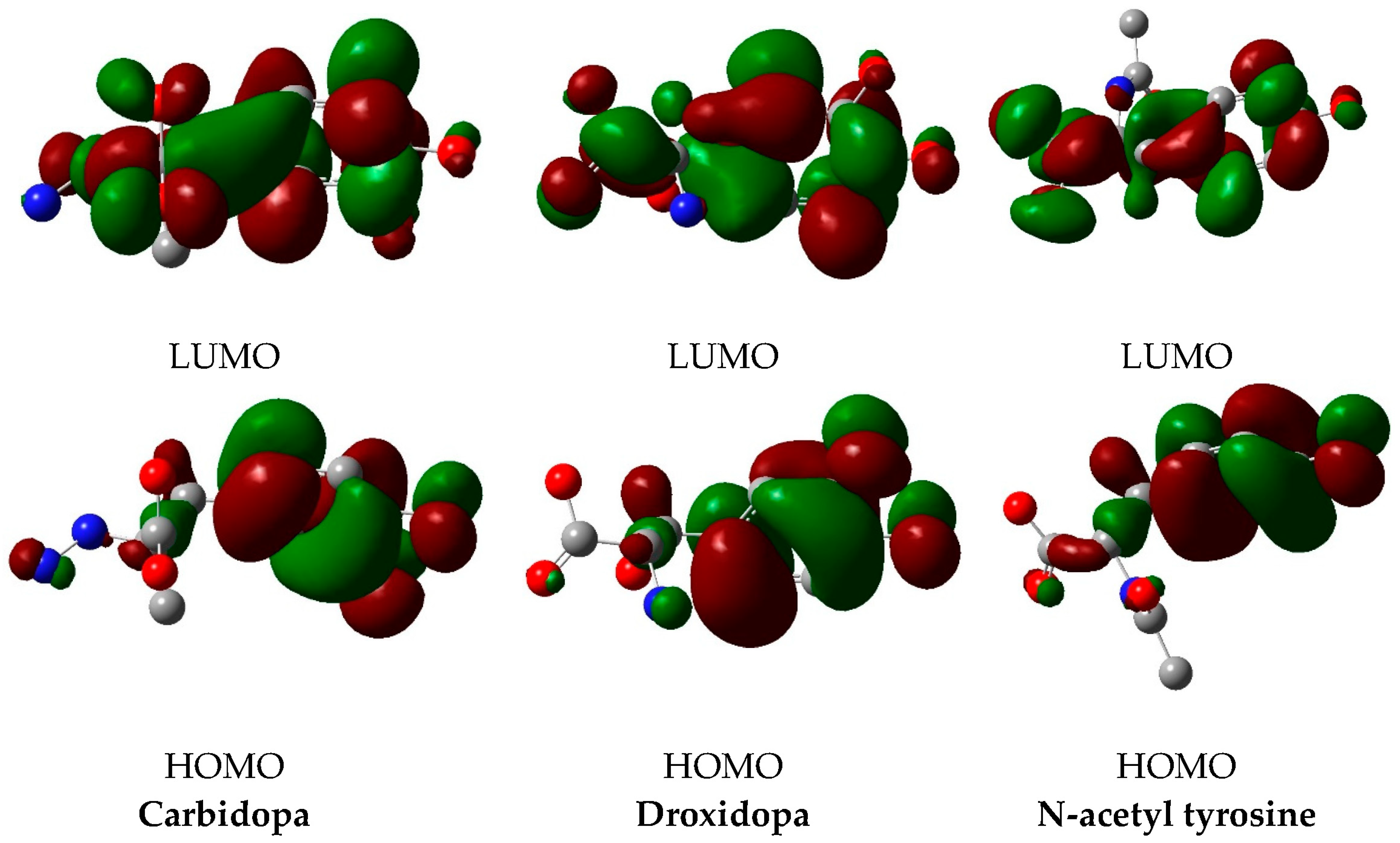
2.5. DFT Calculations and Chemical Reactivity Descriptors
3. Results and Discussion
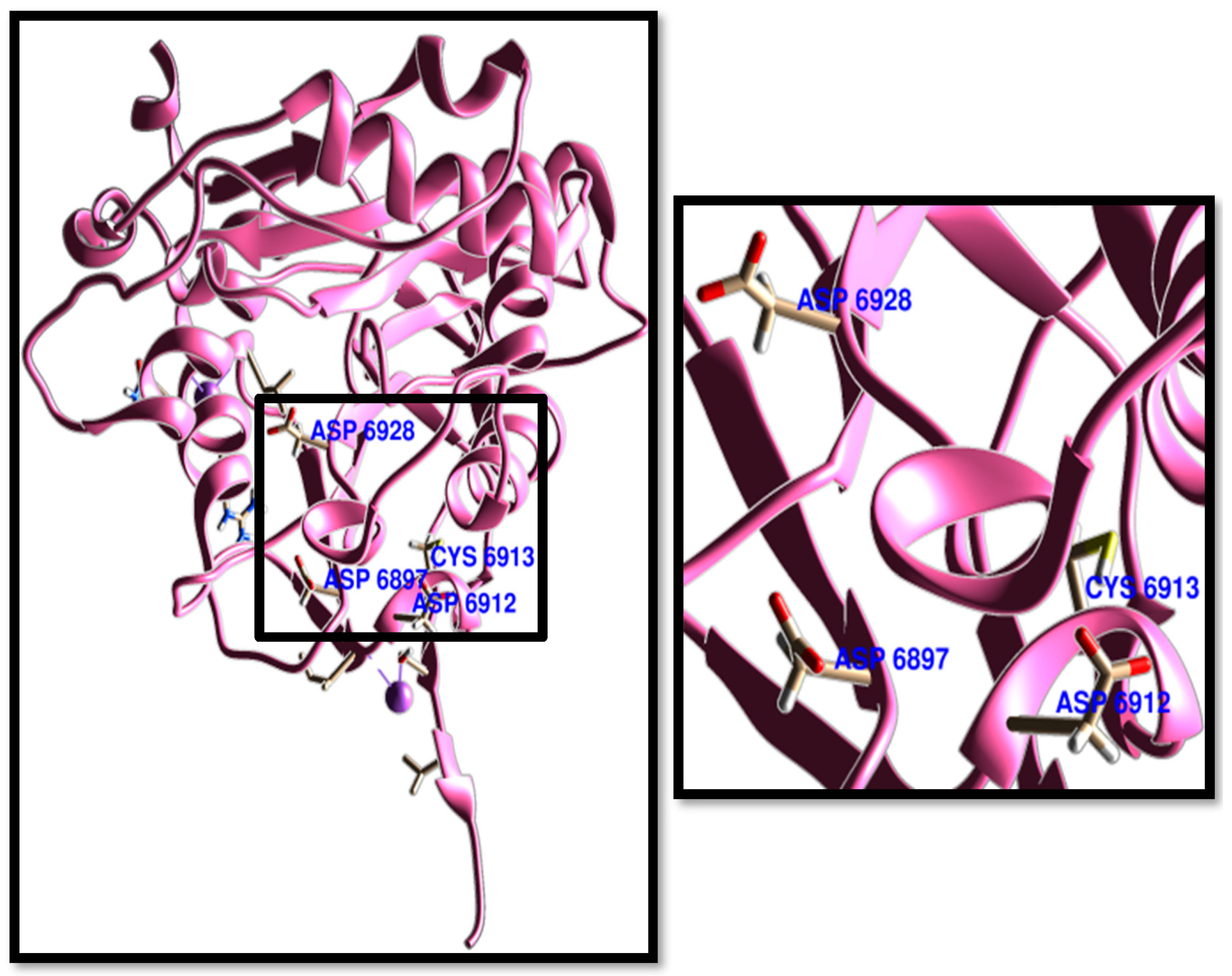
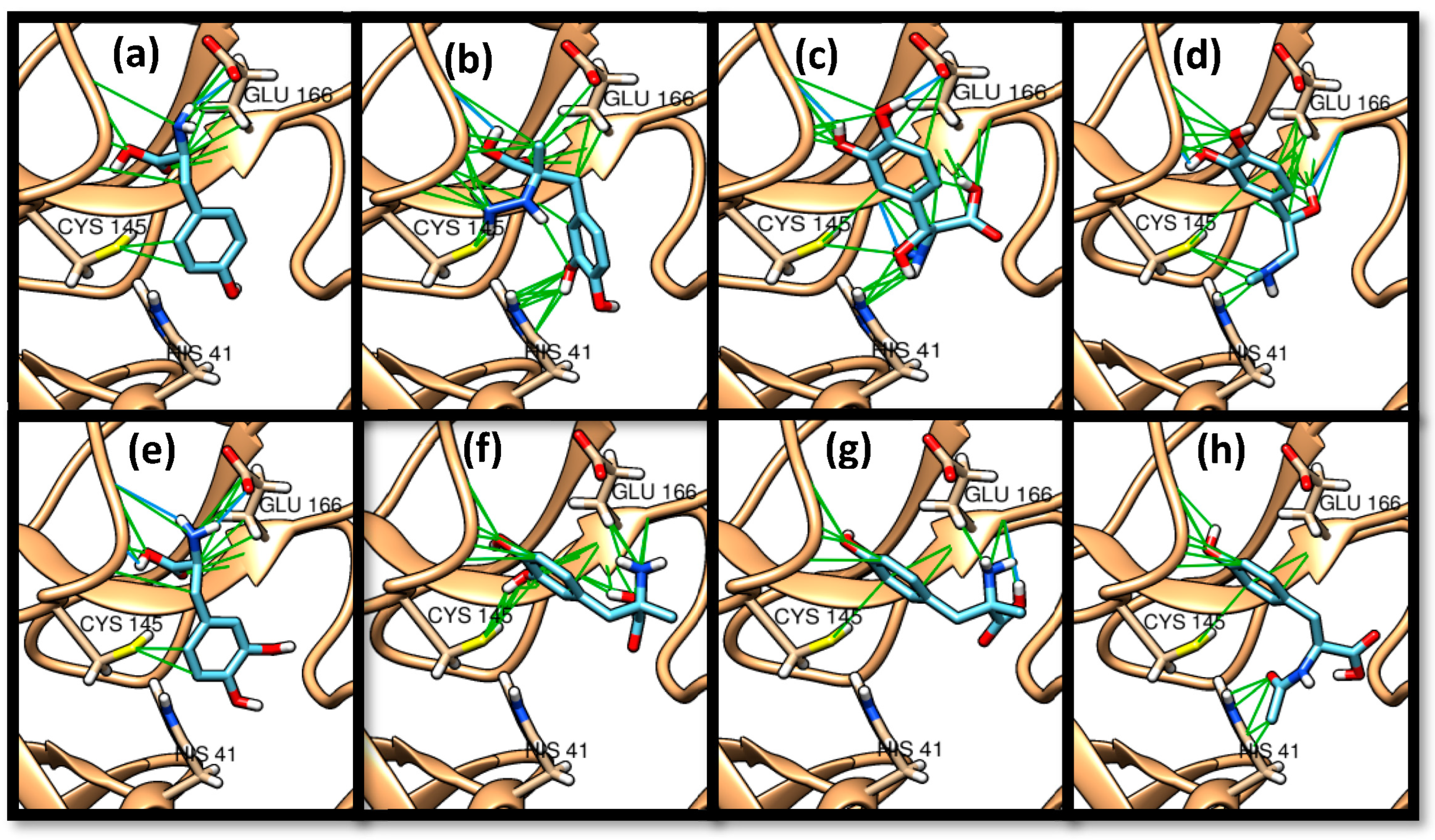
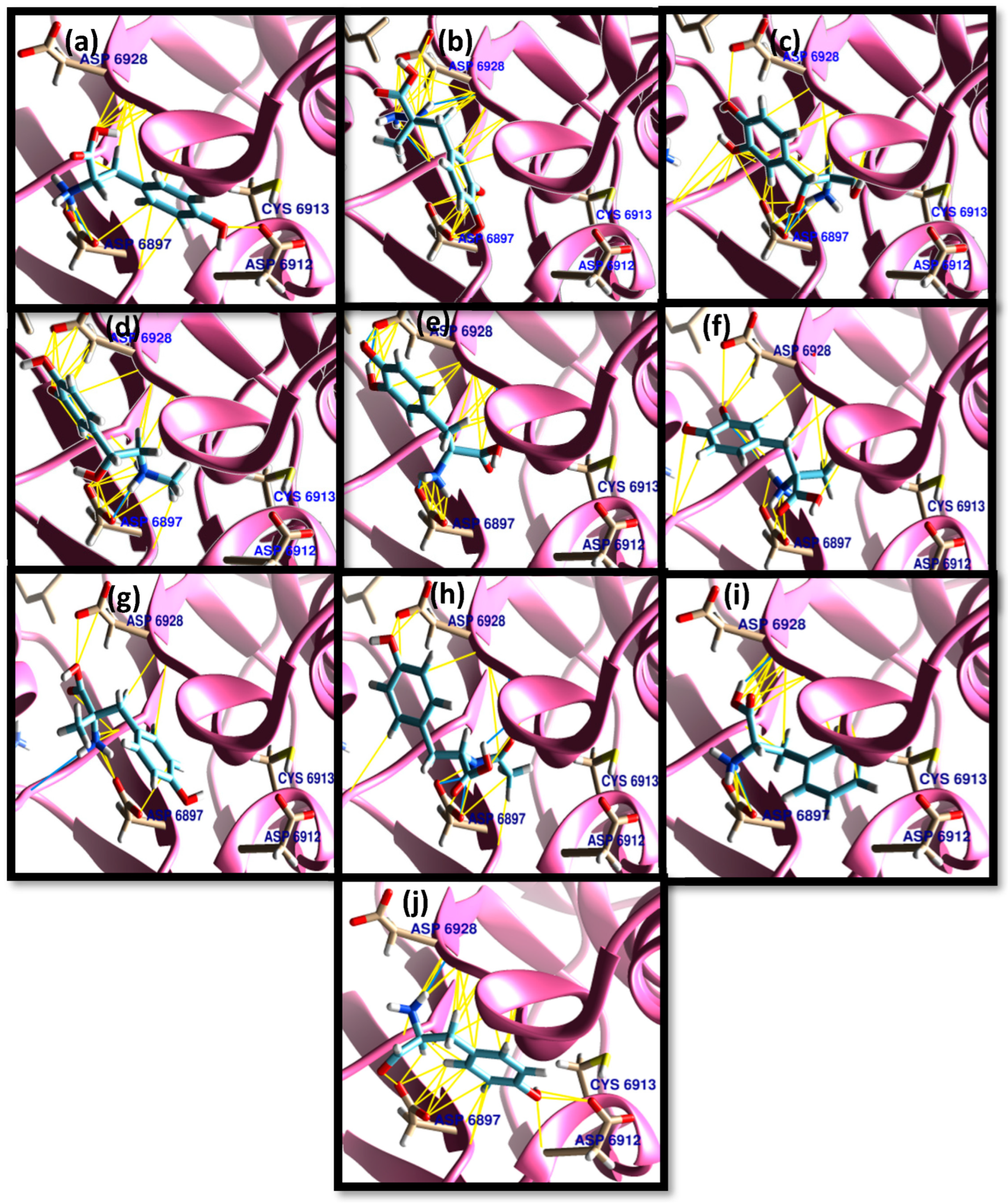
3.1. Molecular Docking
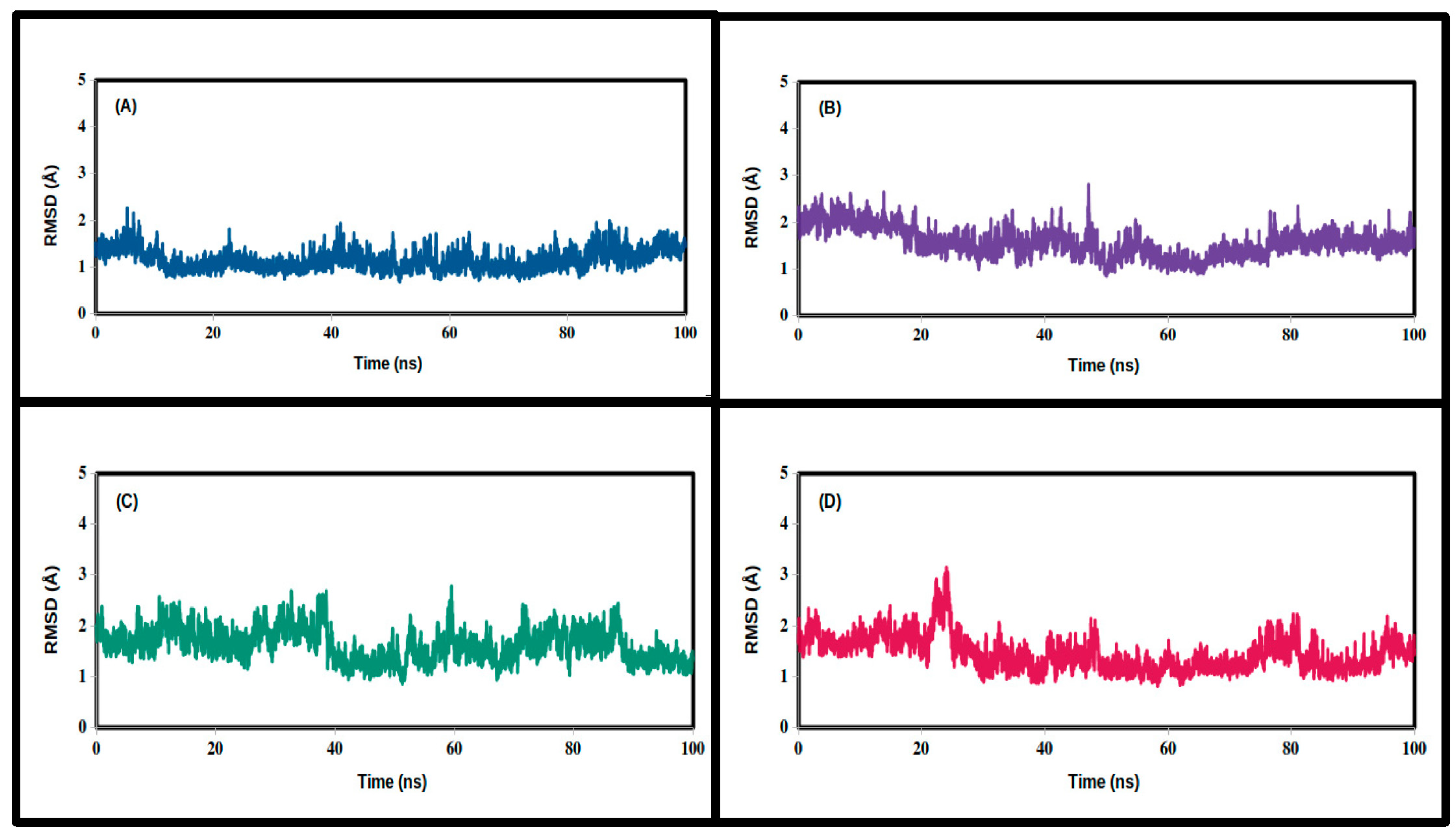
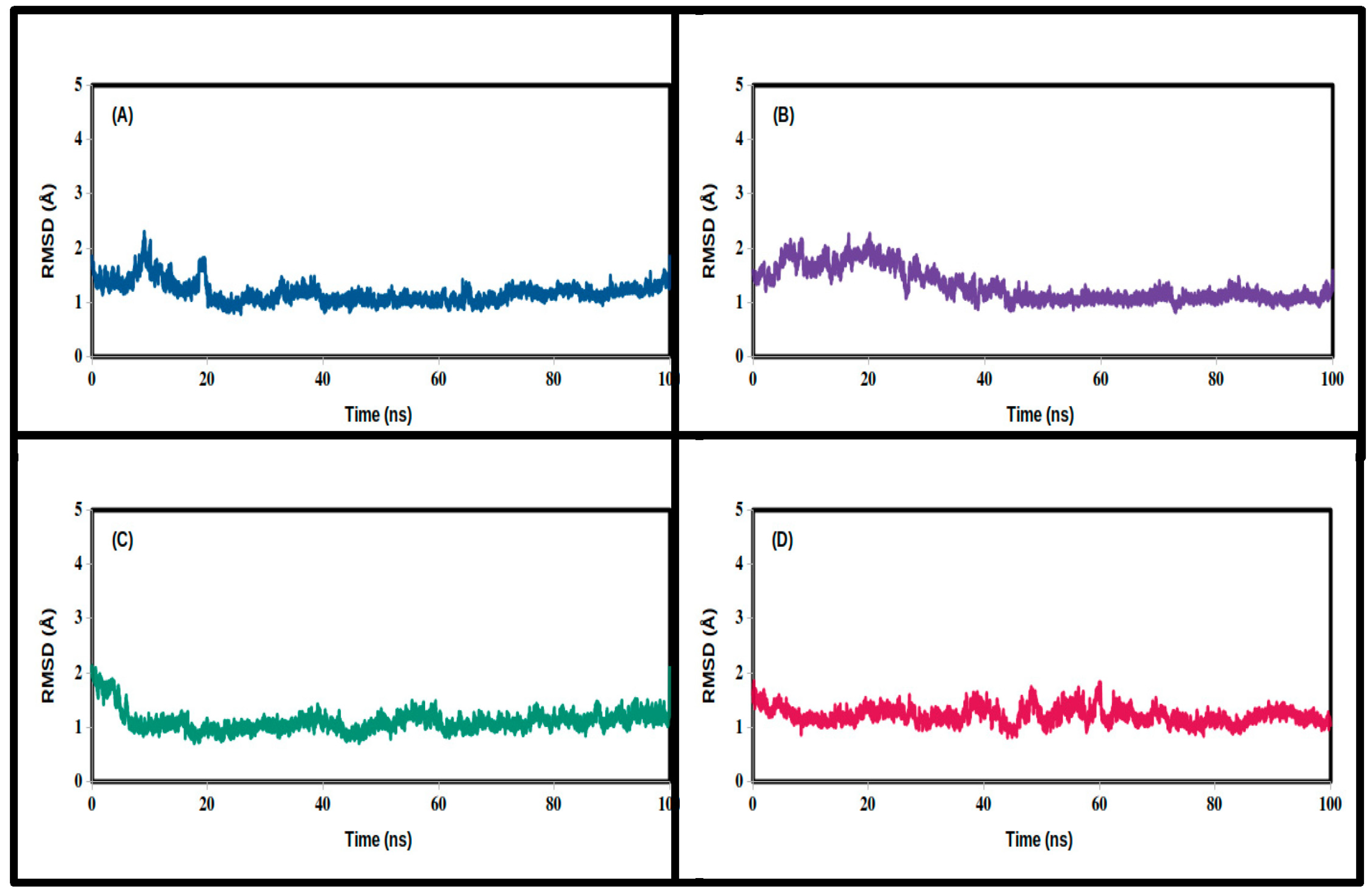
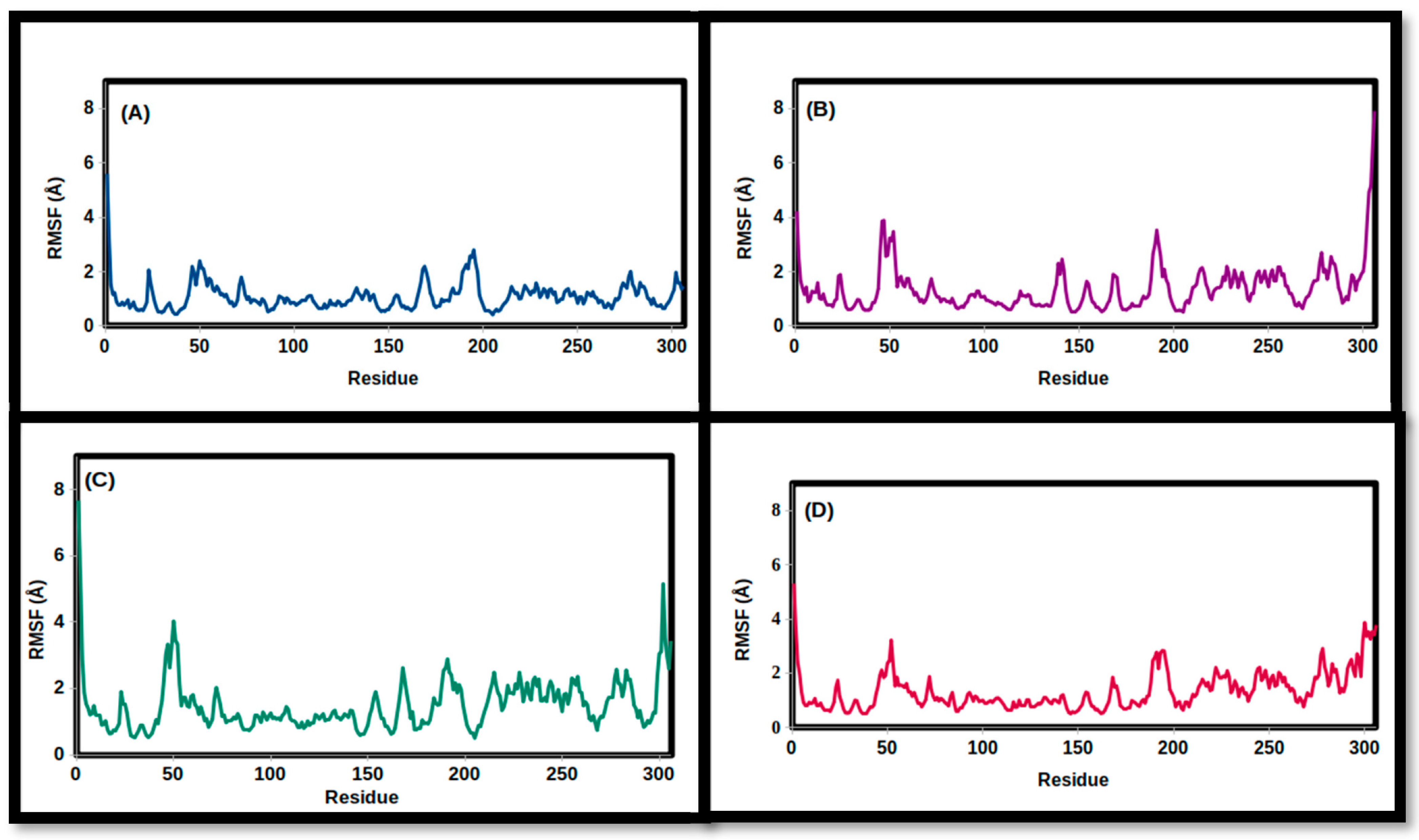
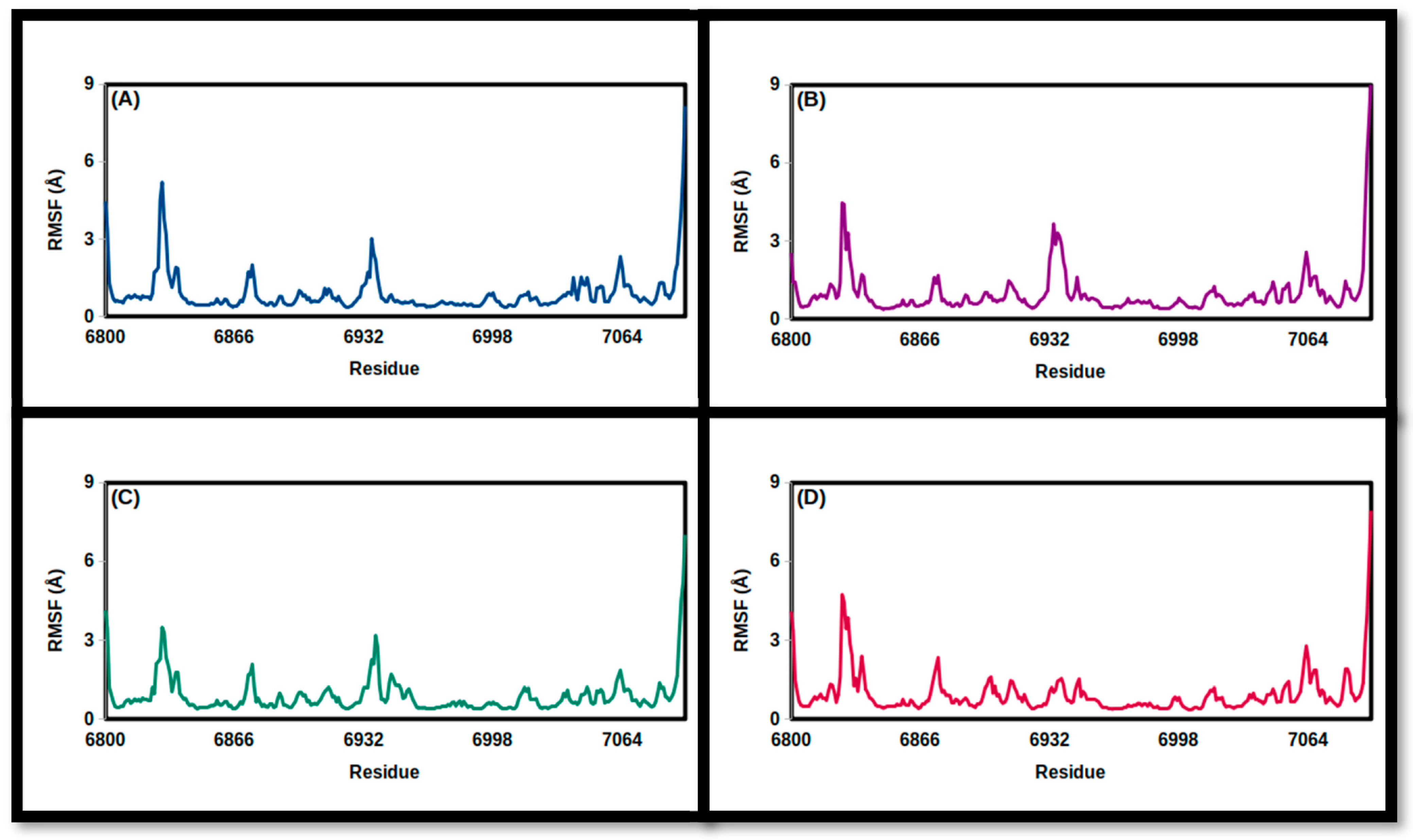
3.2. Molecular Dynamics Simulations
3.3. DFT Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lvov, D.K.; Alkhovsky, S.V. Source of the COVID-19 pandemic: Ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus). Vopr. Virusol. 2020, 65, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Mishra, A.; Joshi, R.; Jha, S.; Sharma, A.R.; Upadhyay, S.; Sarma, P.; Prakash, A.; Medhi, B. Human animal interface of SARS-CoV-2 (COVID-19) transmission: A critical appraisal of scientific evidence. Vet. Res. Commun. 2020, 44, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Maurin, M.; Fenollar, F.; Mediannikov, O.; Davoust, B.; Devaux, C.; Raoult, D. Current status of putative animal sources of sars-cov-2 infection in humans: Wildlife, domestic animals and pets. Microorganisms 2021, 9, 868. [Google Scholar] [CrossRef]
- Hedman, H.D.; Krawczyk, E.; Helmy, Y.A.; Zhang, L.; Varga, C. Host Diversity and Potential Transmission Pathways of SARS-CoV-2 at the Human-Animal Interface. Pathogens 2021, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Sardanelli, A.M.; Isgrò, C.; Palese, L.L. SARS-CoV-2 Main Protease Active Site Ligands in the Human Metabolome. Molecules 2021, 26, 1409. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Tam, N.M.; Pham, M.Q.; Ha, N.X.; Nam, P.C.; Phung, H.T.T. Computational estimation of potential inhibitors from known drugs against the main protease of SARS-CoV-2. RSC Adv. 2021, 11, 17478–17486. [Google Scholar] [CrossRef]
- Thissera, B.; Sayed, A.M.; Hassan, M.H.; Abdelwahab, S.F.; Amaeze, N.; Semler, V.T.; Alenezi, F.N.; Yaseen, M.; Alhadrami, H.A.; Belbahri, L.; et al. Bioguided Isolation of Cyclopenin Analogues as Potential SARS-CoV-2 Mpro Inhibitors from Penicillium citrinum TDPEF34. Biomolecules 2021, 11, 1366. [Google Scholar] [CrossRef] [PubMed]
- Tassakka, A.C.M.A.; Sumule, O.; Massi, M.N.; Manggau, M.; Iskandar, I.W.; Alam, J.F.; Permana, A.D.; Liao, L.M. Potential bioactive compounds as SARS-COV-2 inhibitors from extracts of the marine red alga Halymenia durvillei (Rhodophyta)-A computational study. Arab. J. Chem. 2021, 14, 103393. [Google Scholar] [CrossRef]
- Al-Janabi, A.S.; Elzupir, A.O.; Abou-Krisha, M.M.; Yousef, T.A. New Dual Inhibitors of SARS-CoV-2 Based on Metal Complexes with Schiff-Base 4-Chloro-3-Methyl Phenyl Hydrazine: Synthesis, DFT, Antibacterial Properties and Molecular Docking Studies. Inorganics 2023, 11, 63. [Google Scholar] [CrossRef]
- Omer, E.A.; Abdelfatah, S.; Riedl, M.; Meesters, C.; Hildebrandt, A.; Efferth, T. Coronavirus Inhibitors Targeting nsp16. Molecules 2023, 28, 988. [Google Scholar] [CrossRef]
- Mule, S.; Singh, A.; Greish, K.; Sahebkar, A.; Kesharwani, P.; Shukla, R. Drug repurposing strategies and key challenges for COVID-19 management. J. Drug Target. 2022, 30, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Fadlalla, M.; Ahmed, M.; Ali, M.; Elshiekh, A.A.; Yousef, B.A. Molecular docking as a potential approach in repurposing drugs against COVID-19: A systematic review and novel pharmacophore models. Curr. Pharmacol. Rep. 2022, 8, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Pongrácz, J.E.; Mátyus, P. Recent advances in influenza, HIV and SARS-CoV-2 infection prevention and drug treatment—The need for precision medicine. Chemistry 2022, 4, 216–258. [Google Scholar] [CrossRef]
- Richman, D.D. COVID-19 vaccines: Implementation, limitations and opportunities. Glob. health Med. 2021, 3, 1–5. [Google Scholar] [CrossRef]
- Vesga, L.C.; Ruiz-Hernández, C.A.; Alvarez-Jacome, J.J.; Duque, J.E.; Rincon-Orozco, B.; Mendez-Sanchez, S.C. Repurposing of four drugs as anti-SARS-CoV-2 agents and their interactions with protein targets. Sci. Pharm. 2022, 90, 24. [Google Scholar] [CrossRef]
- Elliott, P.; Eales, O.; Steyn, N.; Tang, D.; Bodinier, B.; Wang, H.; Elliott, J.; Whitaker, M.; Atchison, C.; Diggle, P.J.; et al. Twin peaks: The omicron SARS-CoV-2 BA. 1 and BA. 2 epidemics in England. Science 2022, 376, eabq4411. [Google Scholar] [CrossRef]
- Shi, J.; Xiao, Y.; Zhang, Y.; Geng, D.; Cong, D.; Shi, K.X.; Knapp, R.J. Challenges of drug development during the COVID-19 pandemic: Key considerations for clinical trial designs. Br. J. Clin. Pharmacol. 2021, 87, 2170–2185. [Google Scholar] [CrossRef]
- Wang, X.; Guan, Y. COVID-19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Med. Res. Rev. 2021, 41, 5–28. [Google Scholar] [CrossRef]
- Sorbera, C.; Brigandì, A.; Cimino, V.; Bonanno, L.; Ciurleo, R.; Bramanti, P.; Di Lorenzo, G.; Marino, S. The impact of SARS-COV2 infection on people in residential care with Parkinson Disease or parkinsonisms: Clinical case series study. PLoS ONE 2021, 16, e0251313. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- de Leeuw, S.W.; Perram, J.; Smith, E.R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. Lond. A. Math. Phys. Sci. 1980, 373, 27–56. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Humphrey, W.; ADalke Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Song, Y.; Li, J.; Wu, Q.; Yi, C.; Wu, H.; Chen, Z.; Ou-Yang, W. Study of film thickness effect on carbon nanotube based field emission devices. J. Alloys Compd. 2020, 816, 152648. [Google Scholar] [CrossRef]
- Lim, L.; Shi, J.; Mu, Y.; Song, J. Dynamically-driven enhancement of the catalytic machinery of the SARS 3C-like protease by the S284-T285-I286/A mutations on the extra domain. PLoS ONE 2014, 9, e101941. [Google Scholar] [CrossRef]
- Amin, S.A.; Banerjee, S.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem. 2021, 215, 113294. [Google Scholar] [CrossRef]
- Yousef, T. Structural, optical, morphology characterization and DFT studies of nano sized Cu (II) complexes containing schiff base using green synthesis. J. Mol. Struct. 2020, 1215, 128180. [Google Scholar] [CrossRef]
- Yousef, T.; Alduaij, O.; El-Reash, G.A.; El Morshedy, R. Semiempirical studies, spectral analysis, in vitro antibacterial and DNA degradation studies of heterocyclic thiosemicarbazone ligand and its metal complexes. J. Mol. Liq. 2016, 222, 762–776. [Google Scholar] [CrossRef]
- Yousef, T.; El-Reash, G.A.; El Morshedy, R. Quantum chemical calculations, experimental investigations and DNA studies on (E)-2-((3-hydroxynaphthalen-2-yl) methylene)-N-(pyridin-2-yl) hydrazinecarbothioamide and its Mn (II), Ni (II), Cu (II), Zn (II) and Cd (II) complexes. Polyhedron 2012, 45, 71–85. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmaceutical Name | Binding Percentage * | Score ± SD (kcal/mol) | RMSD | Hydrogen Bond (Interactions/Conformers) | Van der Waals (Interactions/Conformers) |
|---|---|---|---|---|---|
| Tyrosine | 22 | −4.9–−5.3 | 45.45–47.09 | HIS 163 (3/2), GLU 166 (1/1), PHE 140 (2/1) | HIS 163 (13/2), MET 165 (6/2), GLU 166 (20/2), PHE 140 (11/2), SER 144 (6/2), LEU 141 (4/2), GLN 189 (2/1), CYS 145 (2/1) |
| Carbidopa | 56 | −5.6–−5.1 | 0.0–5.49 | HIS 163 (3/2), PHE 140 (3/3), GLU 166 (2/2), LEU 141 (1/1) | ASN 142 (29/4), HIS 163 (27/5), HIS 41 (18/4), MET 49 (23/4), MET 165 (12/5), PHE 140 (31/5), CYS 145 (14/5), GLU 166 (42/5), SER 144 (21/5), GLY 143 (2/1), LEU 141 (24/5), GLN 189 (6/4) |
| Droxidopa | 44 | −5.8–−5.2 | 0.0–5.46 | HIS 163 (1/1), GLU 166 (3/2), PHE 140 (3/3), LEU 141 (1/1), HIS 164 (1/1) | GLU 166 (48/4), LEU 166 (18/4), PHE 140 (22/4), HIS 41 (11/3), HIS 163 (17/4), HIS 164 (7/3), MET 165 (21/4), SER 144 (10/4), MET 49 (3/2), ASN 142 (17/3), CYS 145 (7/3), GLN 189 (4/1) |
| Epinephrine | 33 | −5.2–−4.9 | 45.19–47.54 | HIS 163 (3/3), HIS 164 (1/1), GLU 166 (2/2), LEU 141 (2/2) | GLU 166 (35/3), LEU 141 (14/3), PHE 140 (17/3), PHE 140 (11/3), HIS 163 (12/3), SER 144 (12/3), MET 165 (11/3), ASN 142 (6/2), ASN 142 (9/3), HIS 41 (6/2), CYS 145 (9/3), HIS 164 (5/2) |
| Levodopa | 33 | −5.3–−5.2 | 45.49–47.71 | HIS 163 (4/2), HIS 41 (1/1), PHE 140 (3/3), GLU 166 (1/1), LEU 141 (1/1) | HIS 163 (15/2), HIS 164 (1/1), HIS 41 (11/2), PHE 140 (17/3), SER 144 (12/3), MET 49 (9/3), GLU 166 (27/3), LEU 141 (10/3), CYS 145 (7/3), MET 165 (5/3), ASN 145 (5/2) |
| Methyldopa | 22 | −5.9–−5.3 | 0.0–2.44 | - | HIS 163 (12/2), HIS 164 (5/2), GLU 166 (17/2), CYS 145 (7/2), PHE 140 (6/2), SER 144 (7/2), LEU 141 (6/2), MET 165 (8/2), MET 49 (3/1), GLN 189 (3/1), HIS 41 (7/1), ASN 142 (2/1) |
| Metyrosine | 11 | −4.9 | 23.93–26.24 | GLU 166 (1/1) | HIS 163 (8/1), GLU 166 (10/1), SER 144 (2/1), MET 165 (3/1), PHE 140 (5/1), LEU 141 (2/1), CYS 145 (1/1), ASN 142 (1/1) |
| N-acetyl tyrosine | 33 | −5.4–−5.4 | 26.78–30.63 | GLU 166 (1/1), CYS 145 (1/1) | HIS 163 (9/2), HIS 41 (14/3), HIS 164 (3/2), SER 144 (2/1), SER 46 (1/1), PHE 140 (9/2), GLU 166 (21/3), GLN 189 (6/2), LEU 141 (3/2), CYS 145 (1/1), MET 165 (8/2), MET 49 (12/2), CYS 44 (4/1), THR 25 (1/1), THR 45 (4/1), THR 26 (1/1), ASN 142 (2/1) |
| Pharmaceutical Name | Binding Percentage * | Score ± SD (kcal/mol) | RMSD | Hydrogen Bond (Interactions/Conformers) | Van der Waals (Interactions/Conformers) |
|---|---|---|---|---|---|
| Tyrosine | 56 | −5.5–−5.1 | 0.0–3.76 | ASN 6899 (1/1), TYR 6930 (1/1), ASP 6897 (5/4), GLY 6871 (1/1) | ASP 6897 (30/5), TYR 6930 (24/5), PHE 6947 (6/1), GLY 6871 (5/1), ASN 6841 (9/1), ASP 6912 (9/4), MET 6929 (37/5), GLY 6869 (14/5), ASP 6931 (4/1), LEU 6898 (21/4), ASN 6899 (16/5), ASP 6928 (9/3) |
| Carbidopa | 56 | −6.3–−5.6 | 0.0–6.04 | LYS 6844 (2/2), ASN 6899 (1/1), TYR 6930 (2/2), ASP 6928 (1/1), (GLY 6869 (1/1), ASP 6897 (1/1) | ASN 6841 (22/4), LYS 6844 (27/4), LYS 6968 (12/3), GLY 6869 (19/2), GLY 6871 (10/1), SER 6872 (5/1), ASP 6897 (27/5), ASN 6899 (34/5), ASP 6928 (34/4), MET 6929 (20/5), TYR 6930 (58/5), PRO 6932 (10/2), GLU 7001 (2/2) |
| Droxidopa | 67 | −6.1–−5.7 | 0.0–7.21 | ASP 6897 (4/3), LYS 6844 (2/2), TYR 6930 (1/1), GLY 6869 (1/1), GLY 6871 (1/1) | LYS 6844 (13/3), GLY 6869 (35/6), GLY 6871 (25/5), ASP 6897 (46/6), ASP 6928 (20/5), MET 6929 (37/4), TYR 6930 (72/6), ASN 6899 (29/6), ASN 6841 (27/5) |
| Epinephrine | 44 | −5.8–−5.2 | 0.0–6.22 | TYR 6930 (4/3), ASP 6897 (2/2), GLY 6869 (1/1), ASN 6841 (1/1) | TYR 6930 (33/4), ASP 6897 (33/4), LEU 6898 (6/2), LYS 6844 (6/2), GLY 6869 (31/3), ASP 6928 (20/3), MET 6929 (20/2), ASP 6912(3/1), ASN 6841 (29/3), ASN 6899 (8/2), GLY 6871 (6/2), PRO 6932 (3/1) |
| Levodopa | 67 | −5.9–−5.4 | 0.0–6.49 | ASP 6897 (4/4), ASP 6928 (2/1), GLY 6871(1/1), GLY 6869 (1/1), TYR 6930 (4/3), ASN 6899 (1/1) | ASP 6897(49/7), MET 6929(50/6), ASN 6841(30/3), ASP 6928(37/5), TYR 6930(49/6), GLY 6869(30/5), ASN 6899 (23/6), LEU 6898 (37/4), LYS 6844 (4/1), GLY 6871 (9/2), ASP 6912 (10/3), PHE 6947 (3/1), PRO 6932 (7/1) |
| Methyldopa | 67 | −5.8–−5.5 | 0.0–5.61 | ASN 6841 (1/1), LYS 6844 (3/3), ASN 6899 (2/2), TYR 6930 (4/3), GLY 6869 (1/1), ASP 6928 (3/2) | TYR 6930 (37/7), ASN 6899 (37/6), ASN 6841 (37/6), ASP 6928 (43/6), GLY 6869 (13/4), GLY 6871 (17/3), LYS 6844 (17/4), PRO 6932 (5/2), GLU 7001 (2/2), LYS 6968 (14/2). ALA 6870 (6/2), SER 6872 (1/1), ASP 6897 (28/6) |
| Metyrosine | 78 | −5.6–−5.0 | 0.0–7.20 | LYS 6844 (1/1), TYR 6930 (2/2), ASP 6897 (2/2), ASP 6928 (4/3), GLY 6871 (2/2) | ASP 6897 (23/8), ASP 6928 (31/7), GLY 6869 (17/5), GLY 6871 (18/4), LYS 6844 (16/3), MET 6929 (23/5), ASN 6841 (31/6), ASN 6899 (19/5), TYR 6930 (57/6), LYS 6968 (14/3), PRO 6932 (4/1), ALA 6870 (5/1), LEU 6898 (8/2), GLU 7001 (2/1), LYS 6935 (3/1), SER 6872 (1/1) |
| N-acetyl tyrosine | 67 | −6.3–−5.5 | 0.0–9.04 | LYS 6844 (2/2), LYS 6968 (1/1), ASN 6899 (2/2), TYR 6930 (3/2), ASP 6897 (4/2), ASN 6841 (1/1), GLY 6871 (1/1) | TYR 6930 (47/4), MET 6929 (43/4), GLY 6871 (14/3), GLY 6869 (30/6), ASP 6897 (40/6), ASP 6928 (20/6), ASP 6912 (7/2), ASN 6899 (26/5), ASN 6841 (40/4), LYS 6844 (4/2), LYS 6968 (6/1), LYS 6935 (2/1), LYS 6874 (1/1), LEU 6898 (13/4), SER 6872 (7/2), PRO 6878 (5/1), ALA 6870 (2/1), PHE 6947 (4/1) |
| Phenylalanine | 67 | −5.5–−4.9 | 0.0–5.05 | ASN 6899 (1/1), TYR 6930 (1/1), ASP 6897 (4/3), ASP 6928 (1/1), GLY 6871 (1/1), GLY 6869 (1/1) | TYR 6930 (49/5), MET 6929 (76/6), ASP 6912. (19/4), ASP 6897 (36/6), ASP 6928 (21/5), ASP 6931 (6/2), LEU 6898 (40/5), GLY 6869 (24/4, GLY 6871 (5/1), ASN 6899 (21/5), ASN 6841 (6/1), PRO 6932 (10/3), PHE 6947 (15/5), CYS 6913 (2/1) |
| Tyrosinal | 56 | −5.2–−4.8 | 0.0–5.77 | ASP 6928 (1/1), ASP 6897 (1/1), ASP 6912 (1/1), GLY 6871 (1/1), ASP 6931(1/1) | ASP 6912 (15/4), TYR 6930 (36/4), MET 6929 (55/5), GLY 6869 (31/4), ASP 6897 (37/5), LEU 6898 (29/5), ASN 6899 (10/3), ASP 6931 (21/2), PHE 6947 (13/3), GLY 6871 (6/1), PRO 6932 (10/2), SER 6872 (1/1) |
| 3CLpro Complex | Apo | Carbidopa | Droxidopa | N-Acetyl Tyrosine |
|---|---|---|---|---|
| RMSD (Å) | ||||
| Mean | 1.455 | 1.573 | 1.629 | 1.158 |
| SD | 0.337 | 0.309 | 0.322 | 0.219 |
| Minimum | 0.812 | 0.884 | 0.861 | 0.680 |
| Maximum | 3.151 | 2.813 | 2.781 | 2.267 |
| RMSF (Å) | ||||
| Mean | 1.345 | 1.375 | 1.489 | 1.088 |
| SD | 0.705 | 0.873 | 0.792 | 0.513 |
| Minimum | 0.505 | 0.501 | 0.501 | 0.414 |
| Maximum | 5.309 | 7.917 | 7.669 | 5.601 |
| NSP16 Complex | Apo | Carbidopa | Droxidopa | N-Acetyl Tyrosine |
|---|---|---|---|---|
| RMSD (Å) | ||||
| Mean | 1.224 | 1.301 | 1.115 | 1.188 |
| SD | 0.154 | 0.305 | 0.196 | 0.197 |
| Minimum | 0.815 | 0.821 | 0.710 | 0.789 |
| Maximum | 1.855 | 2.272 | 2.126 | 2.307 |
| FMSF (Å) | ||||
| Mean | 0.90 | 0.99 | 0.90 | 0.96 |
| SD | 0.84 | 0.94 | 0.74 | 0.83 |
| Minimum | 0.36 | 0.38 | 0.39 | 0.38 |
| Maximum | 8.16 | 9.24 | 7.02 | 7.93 |
| Parameter | Carbidopa | Droxidopa | N-Acetyl Tyrosine |
|---|---|---|---|
| Electronic Energy | −800.11 | −780.69 | −782.92 |
| Total Dipole Moment | 3.35 | 3.36 | 1.79 |
| RMS Gradient Norm | 0.000014 | 0.000009 | 0.000003 |
| Electronic Spatial Extent | 4341.77 | 3966.31 | 4270.48 |
| ID | EH/eV | EL/eV | (EL–EH) /Ev | χ/eV | μ/eV | η/eV | S/eV−1 | ω/eV | σ/eV−1 |
|---|---|---|---|---|---|---|---|---|---|
| Carbidopa | −6.13 | −0.70 | 5.43 | 3.42 | −3.42 | 2.72 | 0.37 | 2.15 | 1.36 |
| Droxidopa | −6.32 | −0.89 | 5.43 | 3.61 | −3.61 | 2.72 | 0.37 | 2.39 | 1.36 |
| N-acetyl tyrosine | −6.29 | −0.80 | 5.49 | 3.55 | −3.55 | 2.75 | 0.36 | 2.29 | 1.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elamin, M.R.; Yousef, T.A.; Elzupir, A.O. Insight into Tyrosine-Containing Pharmaceuticals as Potential Inhibitors of SARS-CoV-2 3CLpro and NSP16: Structural Analysis, Docking Studies, Molecular Dynamics Simulations, and Density Functional Theory Investigations. Chemistry 2023, 5, 762-777. https://doi.org/10.3390/chemistry5020054
Elamin MR, Yousef TA, Elzupir AO. Insight into Tyrosine-Containing Pharmaceuticals as Potential Inhibitors of SARS-CoV-2 3CLpro and NSP16: Structural Analysis, Docking Studies, Molecular Dynamics Simulations, and Density Functional Theory Investigations. Chemistry. 2023; 5(2):762-777. https://doi.org/10.3390/chemistry5020054
Chicago/Turabian StyleElamin, Mohamed R., Tarek A. Yousef, and Amin O. Elzupir. 2023. "Insight into Tyrosine-Containing Pharmaceuticals as Potential Inhibitors of SARS-CoV-2 3CLpro and NSP16: Structural Analysis, Docking Studies, Molecular Dynamics Simulations, and Density Functional Theory Investigations" Chemistry 5, no. 2: 762-777. https://doi.org/10.3390/chemistry5020054
APA StyleElamin, M. R., Yousef, T. A., & Elzupir, A. O. (2023). Insight into Tyrosine-Containing Pharmaceuticals as Potential Inhibitors of SARS-CoV-2 3CLpro and NSP16: Structural Analysis, Docking Studies, Molecular Dynamics Simulations, and Density Functional Theory Investigations. Chemistry, 5(2), 762-777. https://doi.org/10.3390/chemistry5020054