Exploring the Structural Chemistry of Pyrophosphoramides: N,N′,N″,N‴-Tetraisopropylpyrophosphoramide




Abstract
1. Introduction
2. Materials and Methods
2.1. General Considerations
2.2. Synthesis of N,N′,N″,N‴-Tetraisopropylpyrophosphoramide (O((iPrNH)2PO)2)
2.3. Purification by Column Chromatography
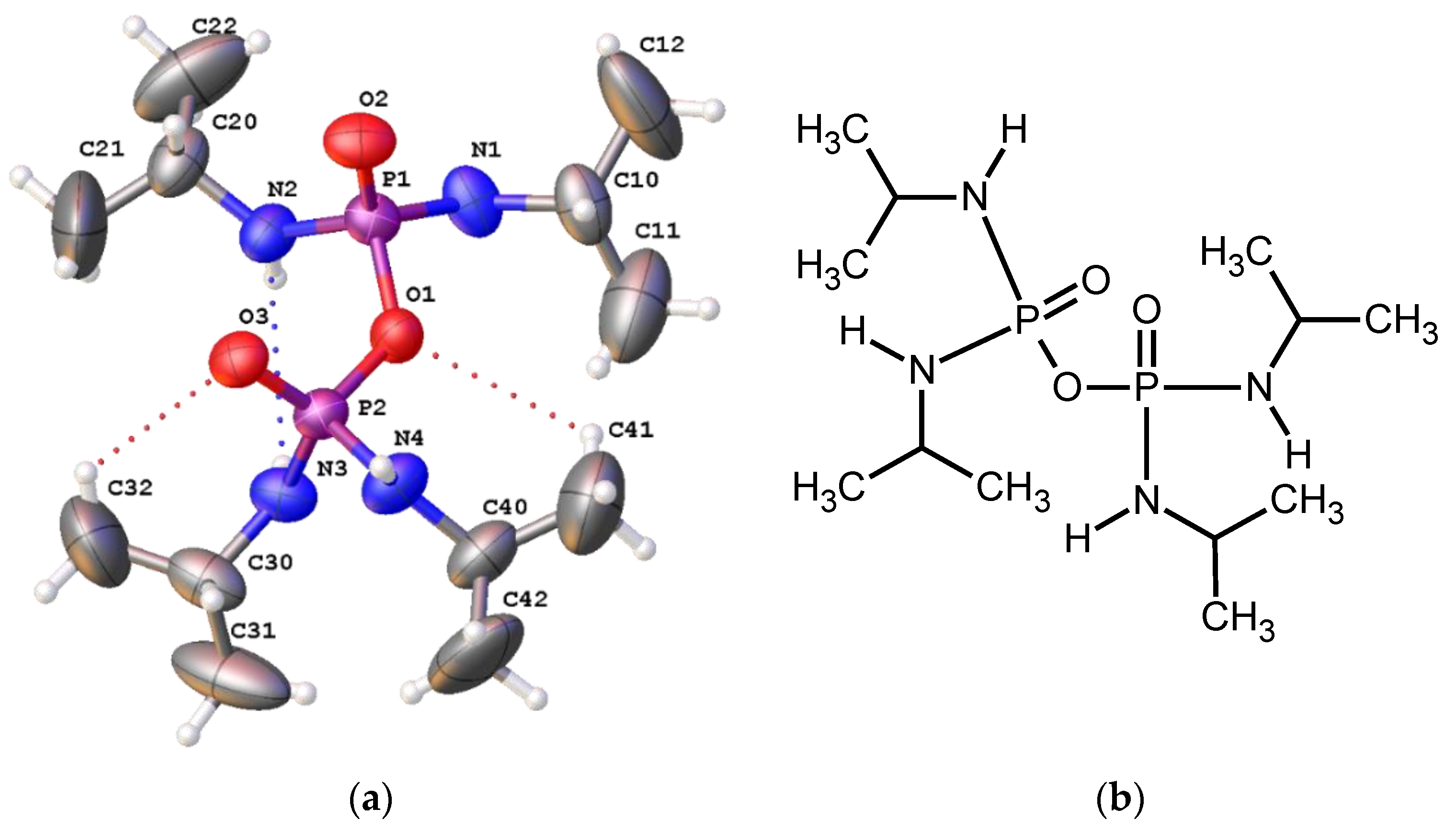
2.4. Single-Crystal X-ray Diffraction
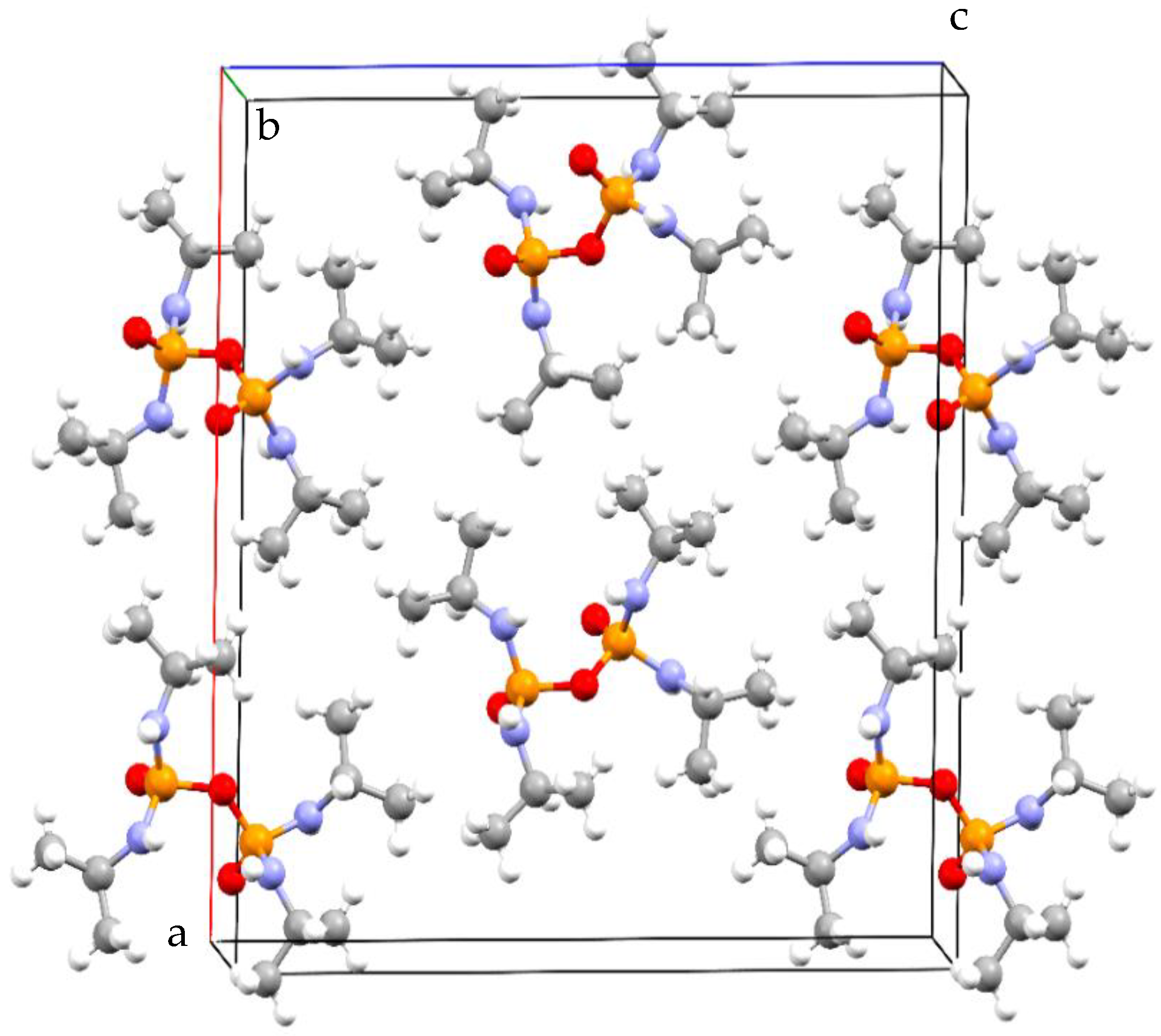
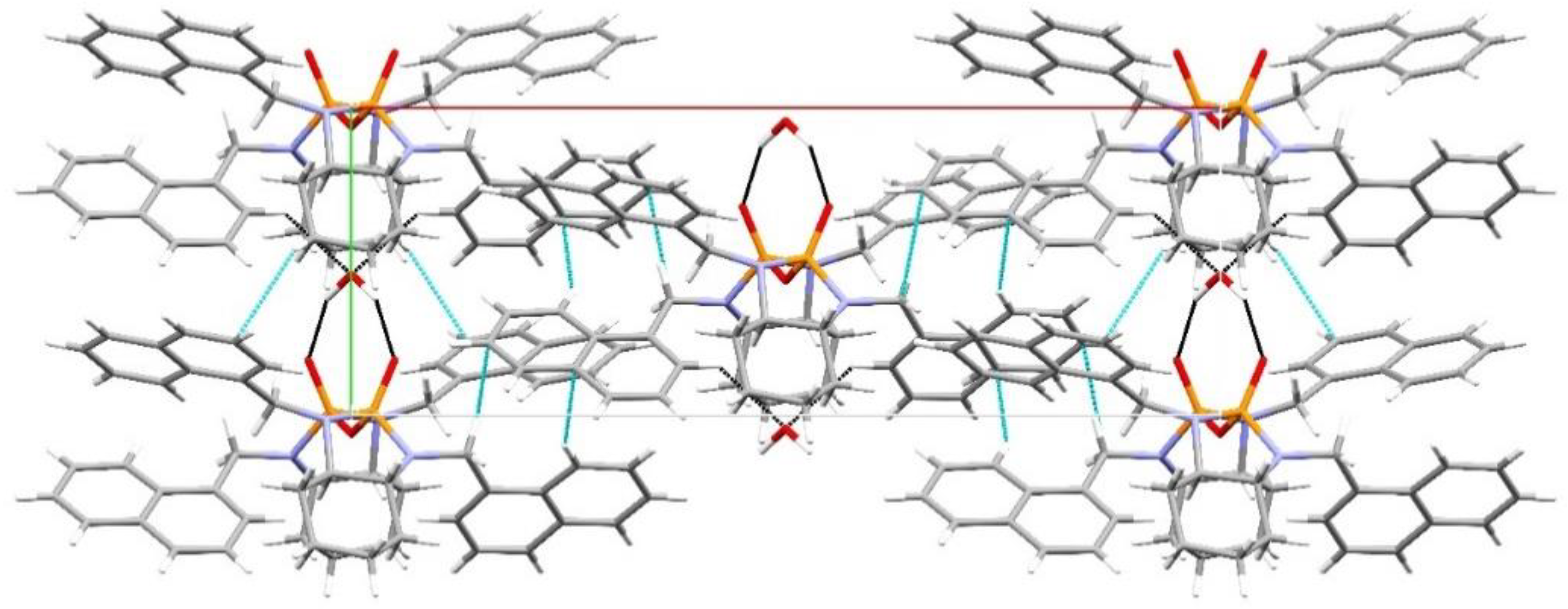
3. Results and Discussion
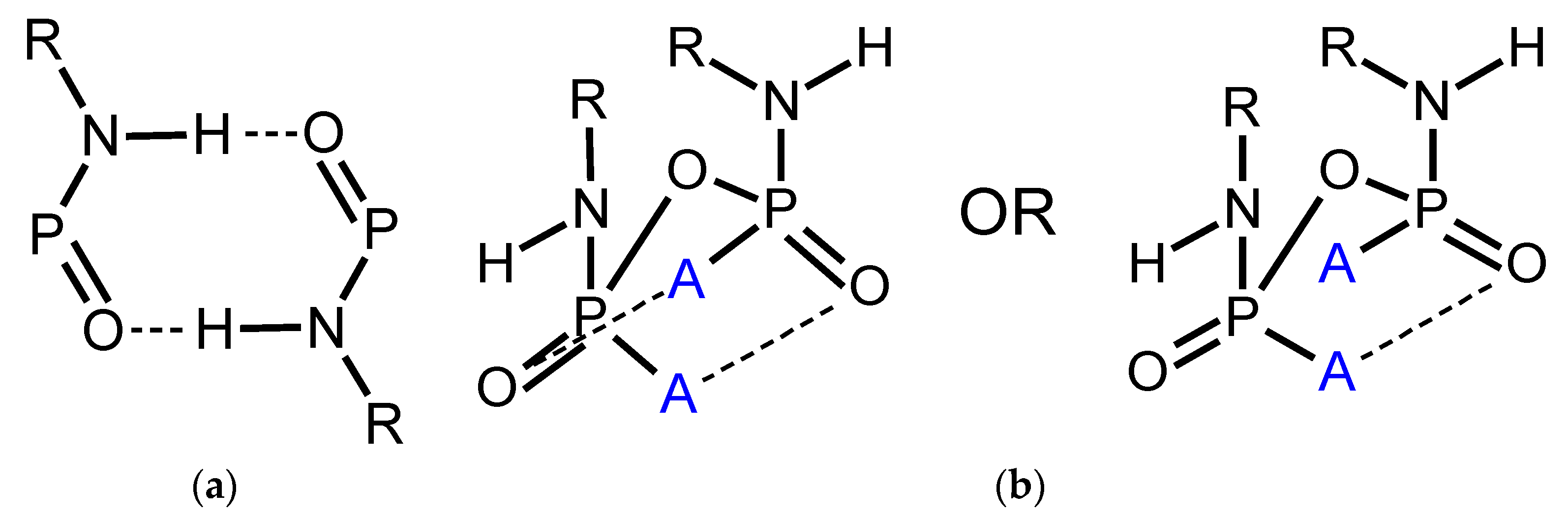
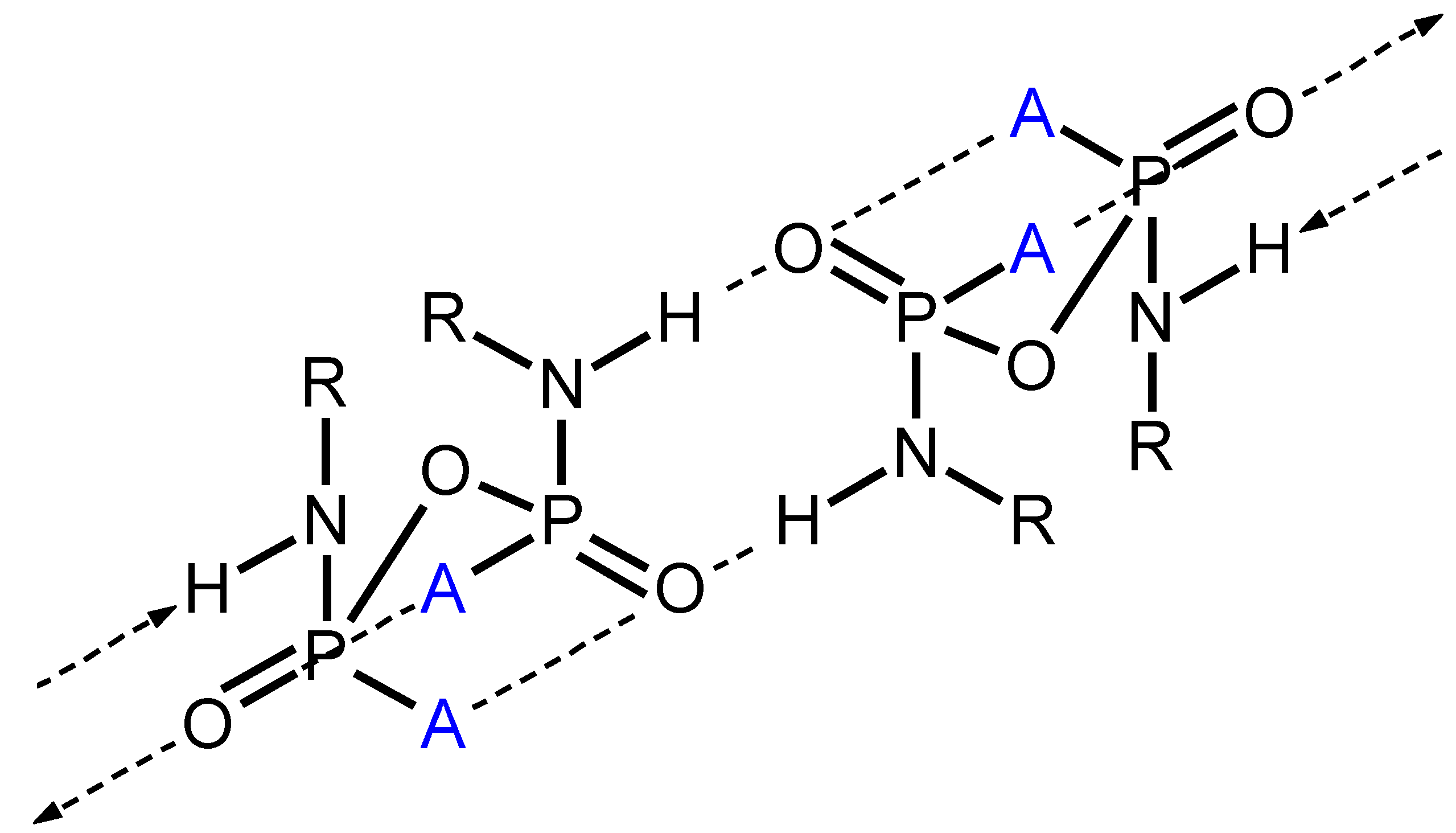
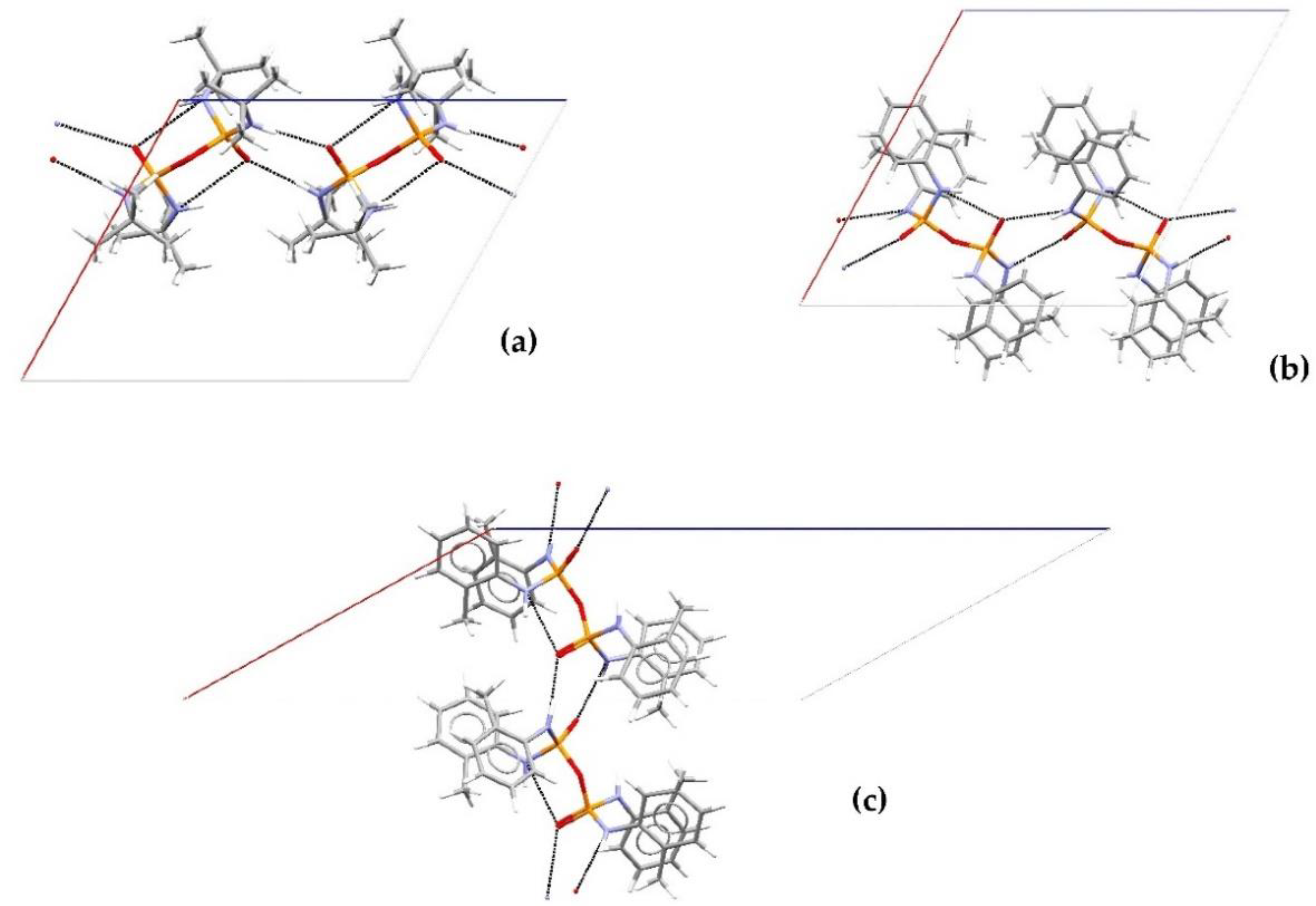
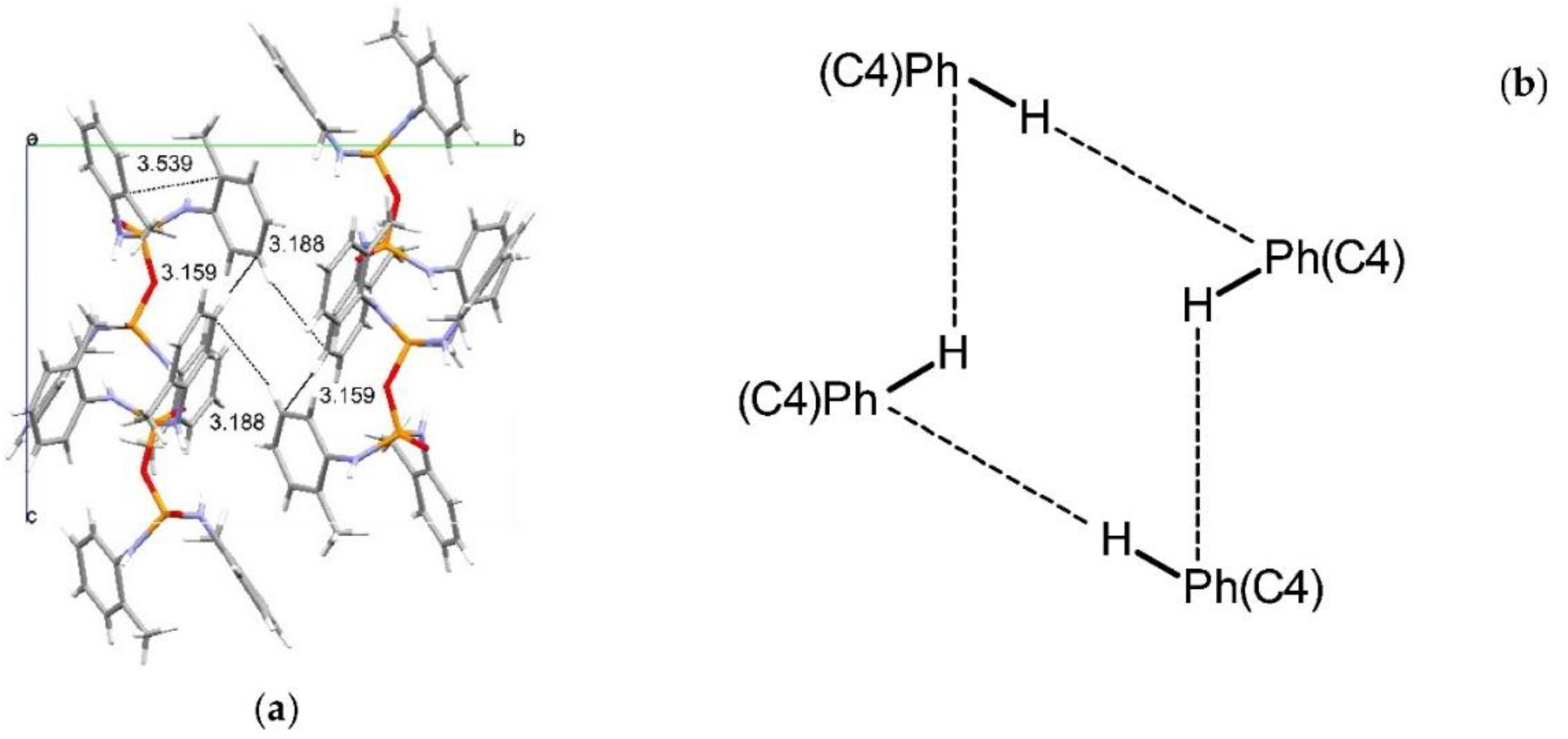
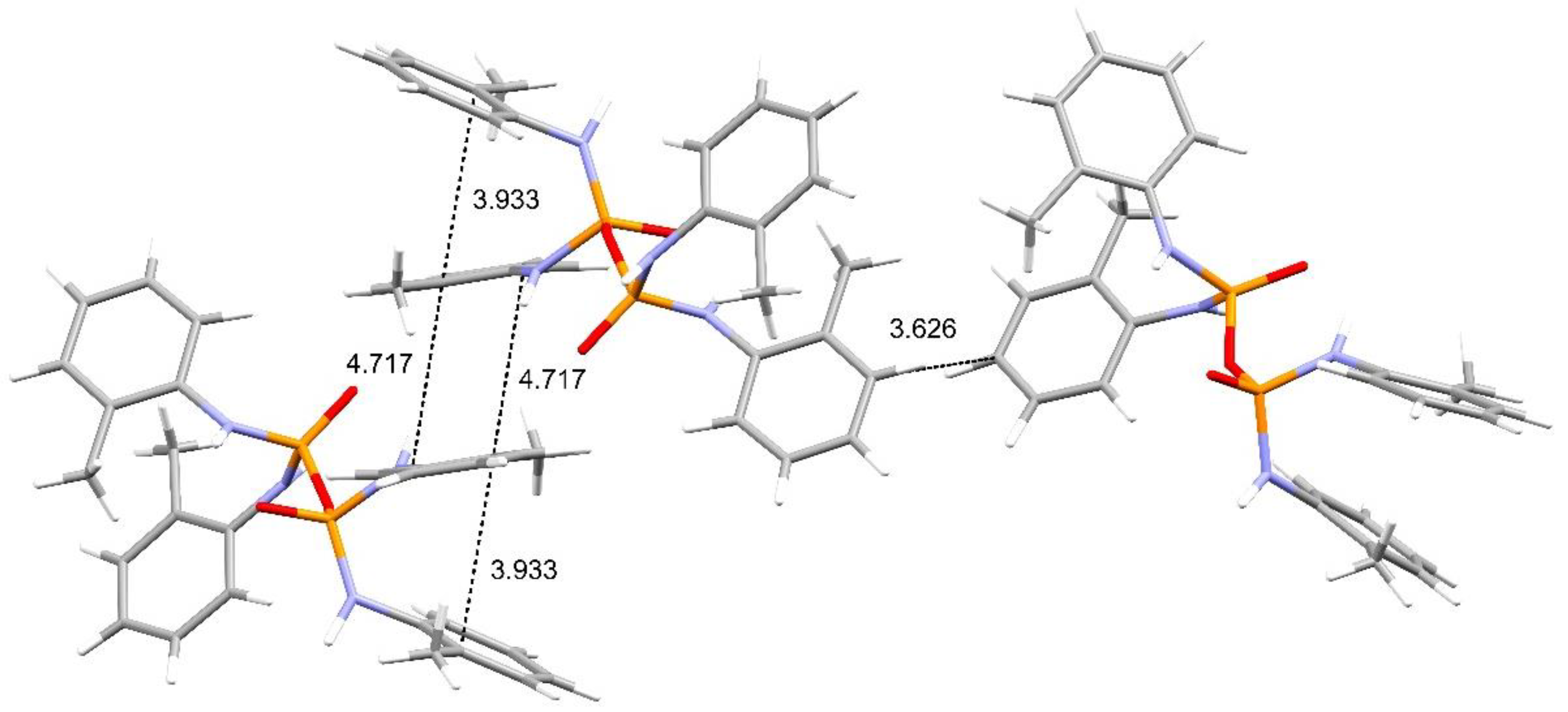
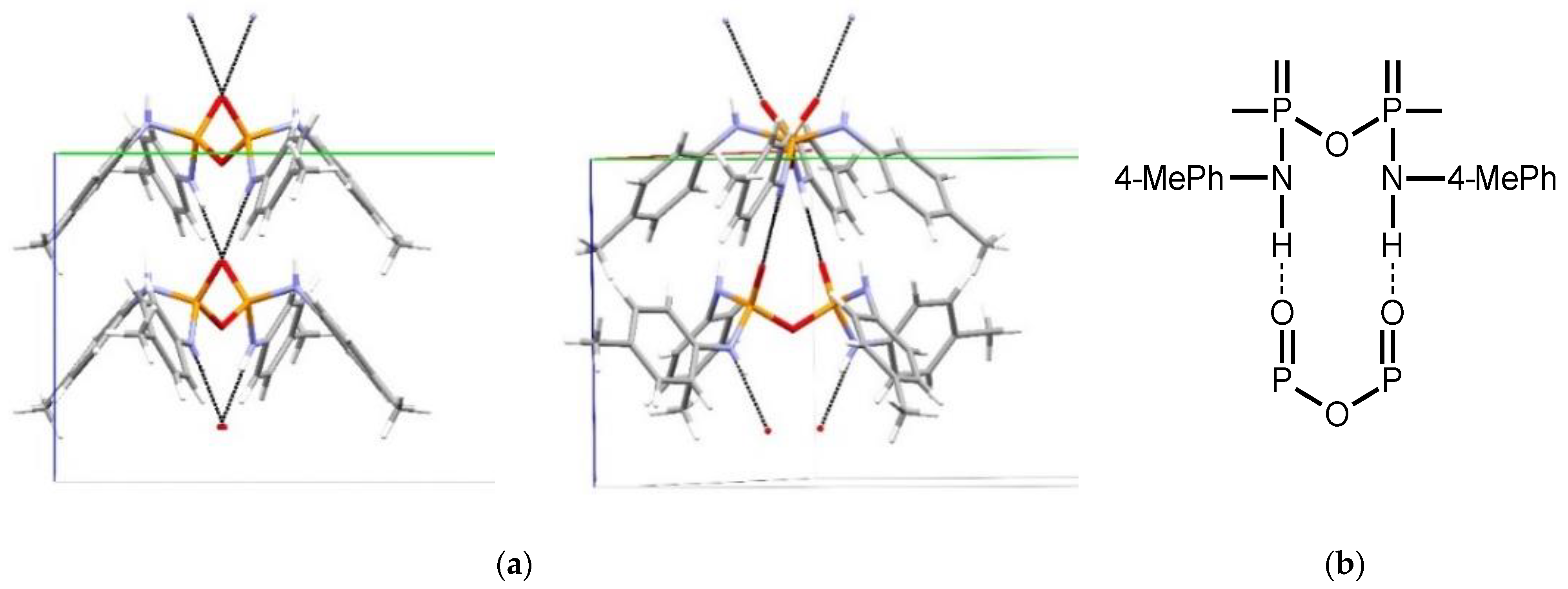
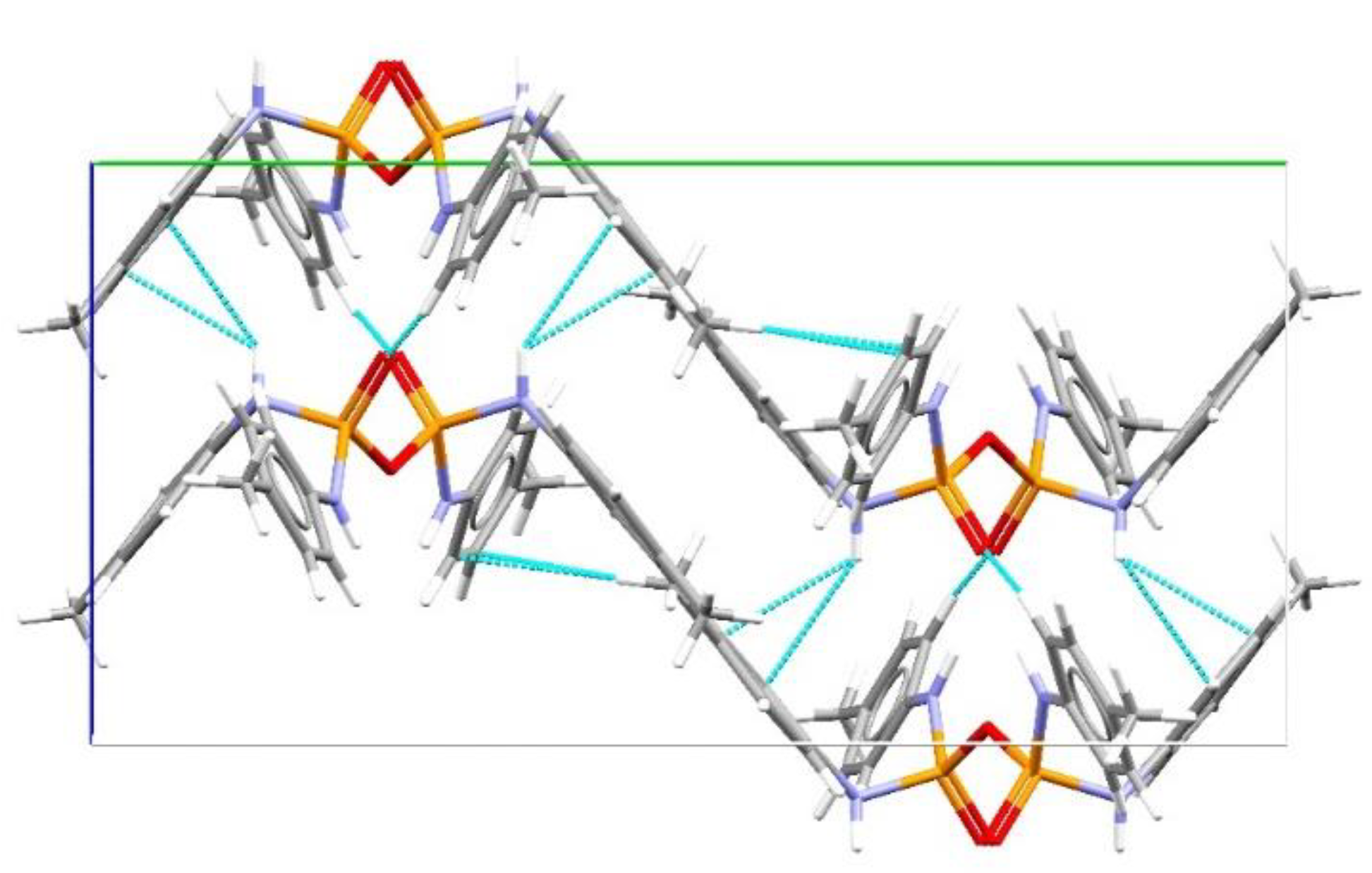
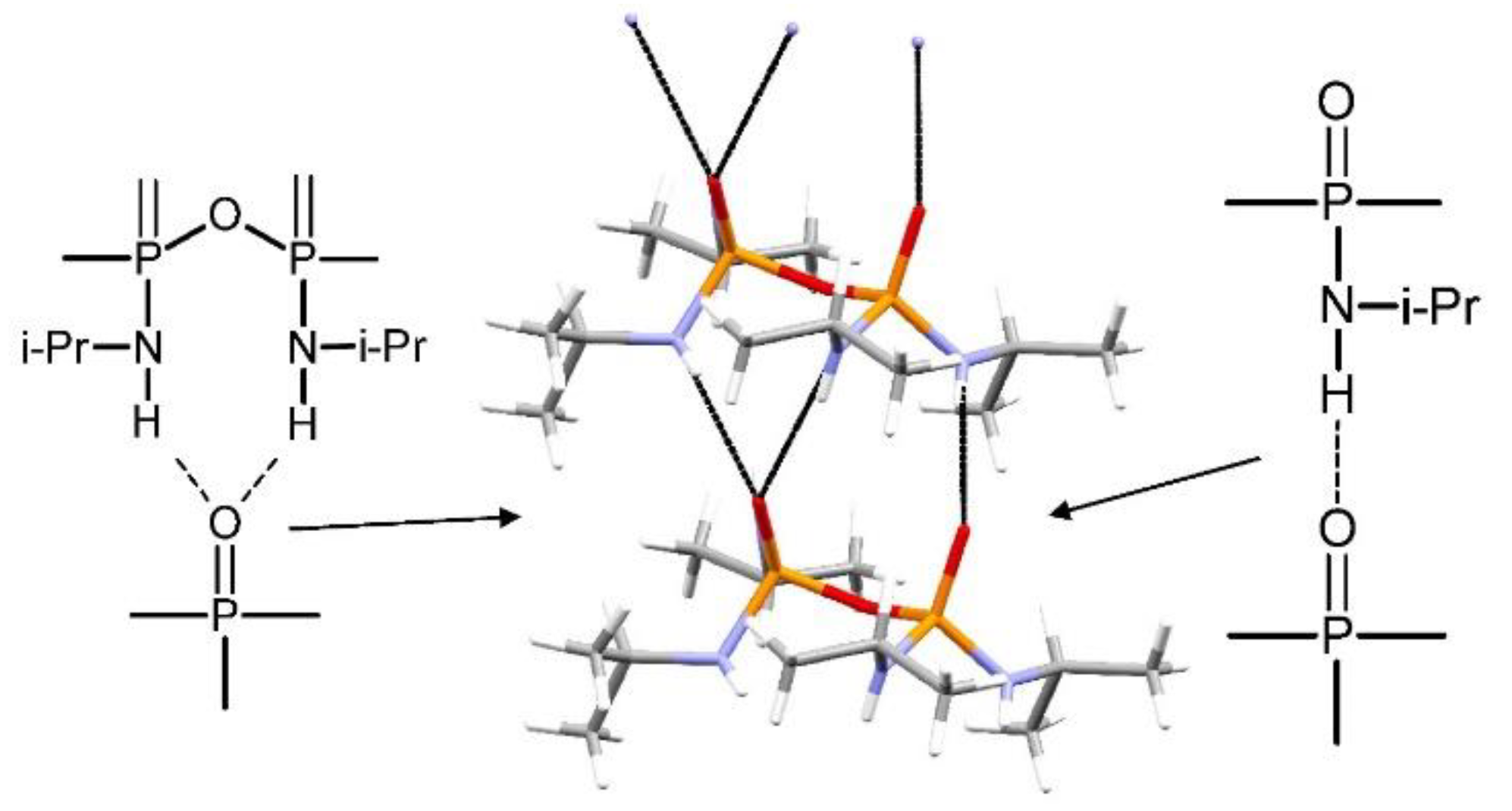
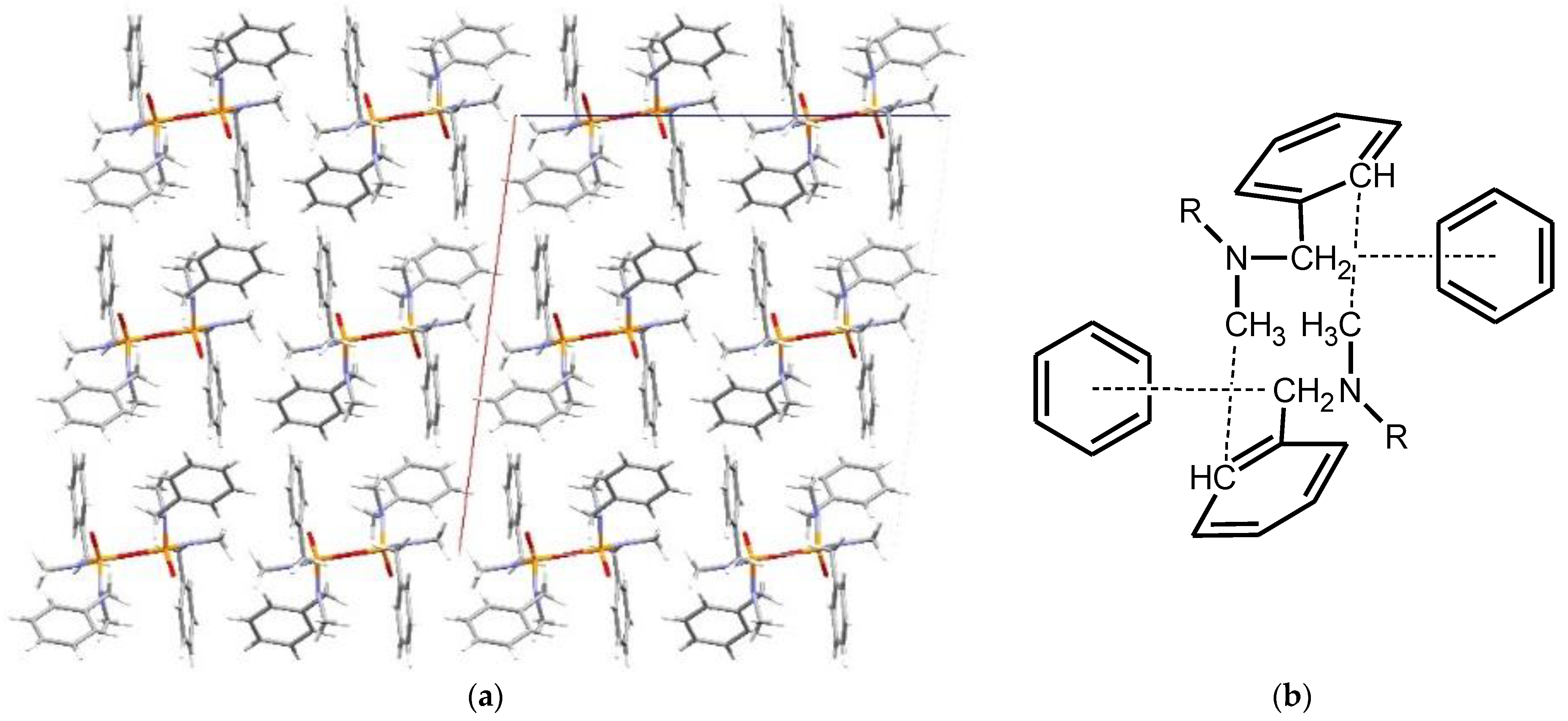
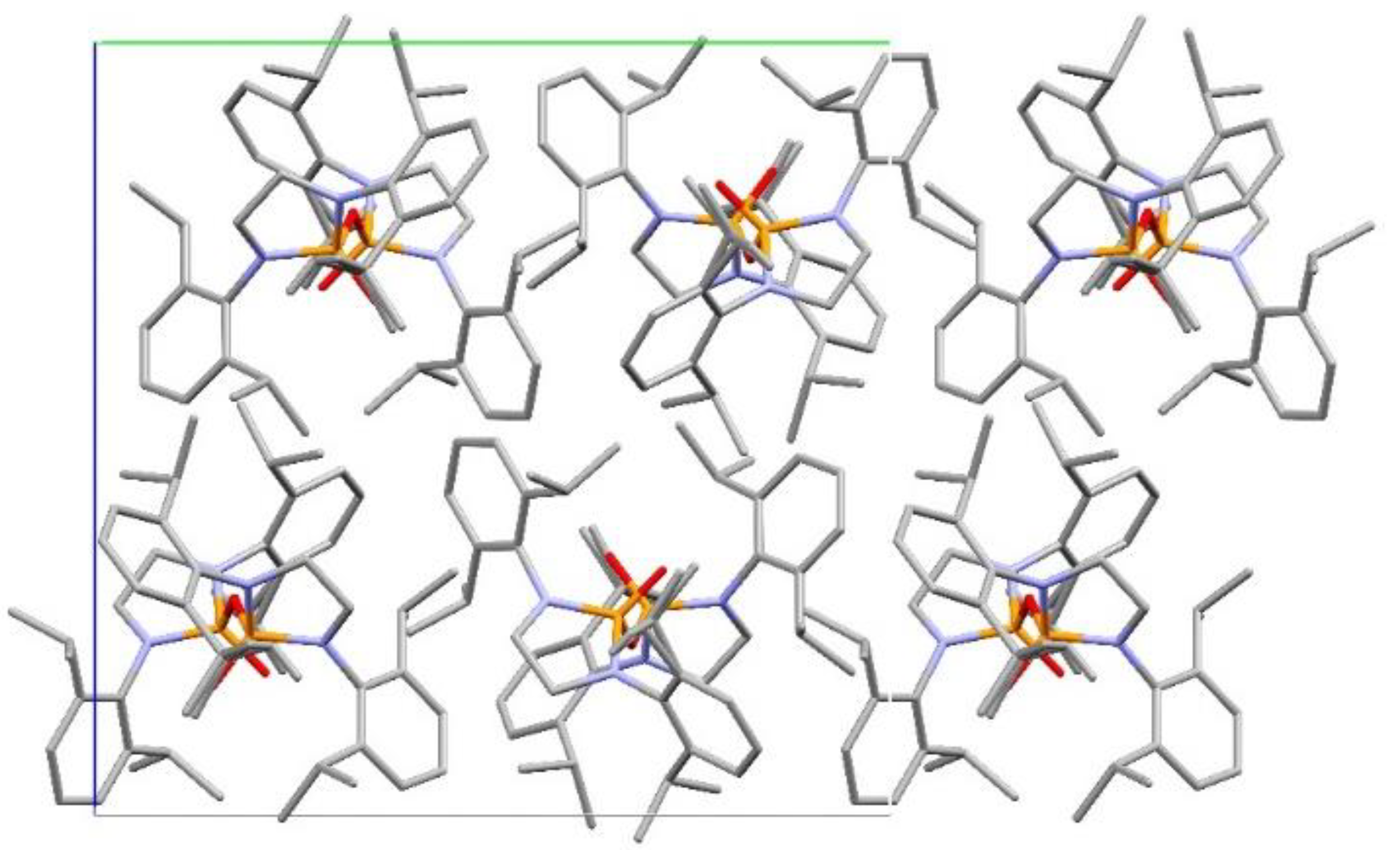
Structural Comparison with Previously Described Pyrophosphoramides
3.1.1. Structural Comparison of Mono-N-Substituted Pyrophosphoramides.
3.1.2. Structural Comparison of di-N-Substituted Pyrophosphoramides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Single Crystal X-ray Diffraction Experimental Description
- Dolomanov, O.V., Bourhis, L.J., Gildea, R.J, Howard, J.A.K. & Puschmann, H. (2009), J. Appl. Cryst. 42, 339–341.
- Sheldrick, G.M. (2015). Acta Cryst. A71, 3–8.
- Sheldrick, G.M. (2015). Acta Cryst. C71, 3–8.
Appendix A.2. Refinement Model Description
- Fixed Uiso
- At 1.2 times of all C(H) groups
- At 1.5 times of all CH3 groups
- 2a.
- Ternary CH refined with riding coordinates: C10(H10), C20(H20), C30(H30), C40(H40)
- 2b.
- Idealised Me refined as rotating group: C31(H31A,H31B,H31C), C11(H11A,H11B,H11C), C41(H41A,H41B,H41C), C42(H42A,H42B,H42C), C12(H12A,H12B,H12C), C22(H22A,H22B,H22C), C32(H32A,H32B,H32C), C21(H21A, H21B,H21C)
References
- Li, B.; Stribley, J.A.; Ticu, A.; Xie, W.; Schopfer, L.M.; Hammond, P.; Brimijoin, S.; Hinrichs, S.H.; Lockridge, O. Abundant Tissue Butyrylcholinesterase and Its Possible Function in the Acetylcholinesterase Knockout Mouse. J. Neurochem. 2002, 75, 1320–1331. [Google Scholar] [CrossRef]
- Pohanka, M. Cholinesterases, a target of pharmacology and toxicology. Biomed. Pap. 2011, 155, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Lickerish, L.A. Studies on commercial octamethylpyrophosphor- amide (schradan) V.*-Insecticidal Comparisons of the Two Main Constituents. J. Sci. Food Agric. 1953, 4, 24–28. [Google Scholar] [CrossRef]
- Rediske, J.H.; Lawrence, W.H. Octamethylpyrophosphoramide (OMPA) As a Systemic Animal Repellent for Douglas-Fir Seedlings. For. Sci. 1964, 10, 93–103. [Google Scholar]
- Joesten, M.D. The donor properties of pyrophosphate derivatives. V. Complexes of molybdenum (V) oxychloride, uranyl ion, and thorium (IV) ion with octamethylpyrophosphoramide. J. Inorg. Nucl. Chem. 1967, 6, 1598–1599. [Google Scholar] [CrossRef]
- du Preez, J.G.H.; Sadie, F.G. Hexamethylphosphoramide and octamethylpyrophosphoramide complexes of tetravalent metal chlorides. J. S. Afr. Chem. Inst. 1966, 19, 73–84. [Google Scholar]
- Joesten, M.D.; Hussain, M.S.; Lenhert, P.G. Structure studies of pyrophosphate chelate rings. I. Crystal structures of tris-octamethylpyrophosphoramide complexes of cobalt (II), magnesium (II), and copper (II)perchlorates. Inorg. Chem. 1970, 9, 151–161. [Google Scholar] [CrossRef]
- Hussain, M.S.; Joesten, M.D.; Lenhert, P.G. Structuee Studies of Pyrophosphate Chelate Rings. II. The Crystal Structuee of Bis(Perchlorato)Bis(Octamethylpyrophosphoramide)Copper(II). Inorg. Chem. 1970, 9, 162–168. [Google Scholar] [CrossRef]
- Kepert, D.L.; Patrick, J.M.; White, A.H. Structure and Stereochemistry in f-Block Complexes of High Co-Ordination Number. Part 3.* The [M(Bidentate Ligand)2(Unidentate Ligand)4] System: Crystal Structures of Tetrakis (Isothiocyanato)- Bis(Octamethylpyrophosphoramide. Dalton Trans. 1983, 0, 559–566. [Google Scholar]
- Cameron, T.S.; Cordes, R.E.; Jackman, F.A. Synthesis and Crystal Structure of μ-Oxo-bis(phosphenyl-ortho-toluidide). Z. Nat. B 1978, 33, 728–730. [Google Scholar] [CrossRef]
- Pourayoubi, M.; Padělková, Z.; Rostami Chaijan, M.; Růžička, A. N,N′,N″,N‴-Tetrakis(2-Methylphenyl)-Oxybis(Phosphonic Diamide): A Redetermination at 150 K with Mo K Radiation. Acta Crystallogr. Sect. E Crystallogr. Commun. 2011, 67, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Pourayoubi, M.; Tarahhomi, A.; Karimi Ahmadabad, F.; Fejfarová, K.; Van Der Lee, A.; Dušek, M. Two New XP(O)[NHC(CH3)3]2 Phosphor-Amidates, with X = (CH3)2N and [(CH3)3CNH]2P(O)(O). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2012, 68, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Tarahhomi, A.; Pourayoubi, M.; Golen, J.A.; Zargaran, P.; Elahi, B.; Rheingold, A.L.; Ramírez, M.A.L.; Percino, T.M. Hirshfeld surface analysis of new phosphoramidates. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 260–270. [Google Scholar] [CrossRef]
- Giffin, N.A.; Hendsbee, A.D.; Roemmele, T.L.; Lumsden, M.D.; Pye, C.C.; Masuda, J.D. Preparation of a Diphosphine with Persistent Phosphinyl Radical Character in Solution: Characterization, Reactivity with O2, S8, Se, Te, and P4, and Electronic Structure Calculations. Inorg. Chem. 2012, 51, 11837–11850. [Google Scholar] [CrossRef] [PubMed]
- Yerramsetti, N.; Unruh, D.K.; Li, G. CCDC 1559159: Experimental Crystal Structure Determination. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/ccdc.csd.cc1pbfgx&sid=DataCite (accessed on 28 June 2017).
- Tarahhomi, A.; Pourayoubi, M.; Fejfarová, K.; Dusek, M. A Novel Amido-pyrophosphate MnII Chelate Complex with the Synthetic Ligand O{P(O)[NHC(CH3)3]2}2(L): [Mn(L)2{OC(H)N(CH3)2}2]Cl2.2H2O. Acta Crystallogr. Sect. C Struct. Chem. 2013, 69, 225–229. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Desiraju, G.R. Designer Crystals: Intermolecular Interactions, Network Structures and Supramolecular Synthons. Chem. Commun. 1997, 16, 1475–1482. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: A Holistic View. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: A brief overview. J. Chem. Sci. 2010, 122, 667–675. [Google Scholar] [CrossRef]
- Goehring, M.; Niedenzu, K. Diphosphorsäure-tetrakis-dimethylamid. Angew. Chem. 1956, 68, 704. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- ACD/ChemSketch; Advanced Chemistry Development: Toronto, ON, Canada, 2013.
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.J.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Bernstein, J.; Sheva, B. Pinching Polymorphs. Nat. Mater. 2005, 4, 427–429. [Google Scholar] [CrossRef]
- Haleblian, J.; McCrone, W. Pharmaceutical Applications of Polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef]
- De Villiers, M.; Van Der Watt, J.; Lötter, A. Kinetic study of the solid-state photolytic degradation of two polymorphic forms of furosemide. Int. J. Pharm. 1992, 88, 275–283. [Google Scholar] [CrossRef]
- Foltz, M.F.; Coon, C.L.; Garcia, F.; Nicholas, A.L., III. The Thermal Stability of the Polymorphs of Hexanitrohexaazaisowurtzitane. Part I Propellants Explos. Pyrotech. 1994, 19, 19–25. [Google Scholar] [CrossRef]
- Hilfiker, R.; Blatter, F.; Von Raumer, M. Relevance of Solid-state Properties for Pharmaceutical Products. In Polymorphism; Wiley: Hoboken, NJ, USA, 2006; pp. 1–19. [Google Scholar]
- Haywood, A.; Mangan, M.; Grant, G.; Glass, B. Extemporaneous Isoniazid Mixture: Stability Implications. J. Pharm. Pract. Res. 2005, 35, 181–182. [Google Scholar] [CrossRef]
- Sarcevica, I.; Orola, L.; Veidis, M.V.; Podjava, A.; Belyakov, S. Crystal and Molecular Structure and Stability of Isoniazid Cocrystals with Selected Carboxylic Acids. Cryst. Growth Des. 2013, 13, 1082–1090. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C12H32N4O3P2 | μ/mm−1 | 2.133 |
|---|---|---|---|
| Formula weight/gmol−1 | 342.35 | F(000) | 744.0 |
| Temperature/K | 293 | Crystal size/mm3 | 0.8 × 0.6 × 0.3 |
| Crystal system | orthorhombic | Radiation | CuKα (λ = 1.54186) |
| Spacegroup | Pca21 | 2θ range for data collection/° | 8.696 to 137.268 |
| a/Å | 20.342(2) | Index ranges | −24 ≤ h ≤ 23, −3 ≤ k ≤ 6, −20 ≤ l ≤ 23 |
| b/Å | 5.0495(6) | Reflections collected | 43032 |
| c/Å | 19.103(3) | Independent reflections | 3317 [Rint = 0.0557, Rsigma = 0.0226] |
| α/° | 90 | Data/restraints/parameters | 3317/1/214 |
| β/° | 90 | Goodness-of-fit on F2 | 1.044 |
| γ/° | 90 | Final R indexes [I > = 2σ (I)] | R1 = 0.0402, wR2 = 0.1071 |
| Volume/Å3 | 1962.2(4) | Final R indexes [all data] | R1 = 0.0408, wR2 = 0.1075 |
| Z | 4 | Largest diff. peak/hole/eÅ−3 | 0.28/−0.29 |
| ρ calc/gcm−3 | 1.159 | Flack parameter | 0.01(3) |
| Pyropshophoramides | ||||
|---|---|---|---|---|
| Di-N-Substituted Pyrophosphoramides | Mono-N-Substituted Pyrophosphoramides | |||
| O((R2N)2PO)2 (Symmetric secondary amine) | O((R1R2N)2PO)2 (Asymmetric secondary amine) | O((R1(NR2)2)2PO)2 (N,N′-substituted diamine derivatives) | O((AlkylNH)2PO)2 (Primary alkyl amine derivatives) | O((ArlyNH)2PO)2 (Primary aryl amine derivatives) |
| O((Me2N)2PO)2 Liquid [3,4] | O((BzMeN)2PO)2 C2/c [13] | O((C2H4(2,5-iPrPhN)2)2PO)2 P21/n [14] | O((tBuNH)2PO)2 P21/c [12] | O((2-MePhNH)2PO)2 P21/c [10,11] |
| O((1,2-Cy(NaphN)2)2PO)2 C2 [15] | O((iPrNH)2PO)2 Pca21 (current) | O((4-MePhNH)2PO)2 Pccn [13] | ||
| D | H | A | d(D-A)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° |
|---|---|---|---|---|---|---|
| C41 | H41A | O1 | 0.96 | 2.99 | 3.601(11) | 122.9 |
| C42 | H42B | O2 1 | 0.96 | 2.58 | 3.505(8) | 161.4 |
| C32 | H32A | O3 | 0.96 | 2.85 | 3.511(10) | 126.4 |
| N3 | H3 | O3 2 | 0.75(5) | 2.34(5) | 3.063(4) | 164(5) |
| N3 | H3 | N2 | 0.75(5) | 2.98(5) | 3.491(5) | 128(4) |
| N1 | H1 | O2 2 | 0.70(4) | 2.22(4) | 2.907(4) | 170(5) |
| N4 | H4 | N3 3 | 0.92(6) | 2.79(6) | 2.979(4) | 161(5) |
| N2 | H2 | O3 2 | 0.85(6) | 2.16(6) | 2.979(4) | 161(5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micallef, D.; Vella-Zarb, L.; Baisch, U. Exploring the Structural Chemistry of Pyrophosphoramides: N,N′,N″,N‴-Tetraisopropylpyrophosphoramide. Chemistry 2021, 3, 149-163. https://doi.org/10.3390/chemistry3010013
Micallef D, Vella-Zarb L, Baisch U. Exploring the Structural Chemistry of Pyrophosphoramides: N,N′,N″,N‴-Tetraisopropylpyrophosphoramide. Chemistry. 2021; 3(1):149-163. https://doi.org/10.3390/chemistry3010013
Chicago/Turabian StyleMicallef, Duncan, Liana Vella-Zarb, and Ulrich Baisch. 2021. "Exploring the Structural Chemistry of Pyrophosphoramides: N,N′,N″,N‴-Tetraisopropylpyrophosphoramide" Chemistry 3, no. 1: 149-163. https://doi.org/10.3390/chemistry3010013
APA StyleMicallef, D., Vella-Zarb, L., & Baisch, U. (2021). Exploring the Structural Chemistry of Pyrophosphoramides: N,N′,N″,N‴-Tetraisopropylpyrophosphoramide. Chemistry, 3(1), 149-163. https://doi.org/10.3390/chemistry3010013