Abstract
Electrically driven adsorption, electroadsorption, is at the core of technologies for water desalination, energy production, and energy storage using electrolytic capacitors. Modeling can be crucial for understanding and optimizing these devices, and hence different approaches have been taken to develop multiple models, which have been applied to explain capacitive deionization (CDI) device performances for water desalination. Herein, we first discuss the underlying physics of electroadsorption and explain the fundamental similarities between the suggested models. Three CDI models, namely, the more widely used modified Donnan (mD) model, the Randles circuit model, and the recently proposed dynamic Langmuir (DL) model, are compared in terms of modeling approaches. Crucially, the common physical foundation of the models allows them to be improved by incorporating elements and simulation tools from the other models. As a proof of concept, the performance of the Randles circuit is significantly improved by incorporating a modeling element from the mD model and an implementation tool from the DL model (charge-dependent capacitance and system identification, respectively). These principles are accurately validated using data from reports in the literature showing significant prospects in combining modeling elements and tools to properly describe the results obtained in these experiments.
1. Introduction
Because the global demand for potable water, energy, and energy storage is expanding [1], it is imperative to develop current technologies and innovate new ones for wider accessibility. Supercapacitors [2,3,4] (electrolytic capacitors) have received a lot of attention because of their high capacity for storing energy by adsorbing ions into highly porous electrodes [2]. Because supercapacitors are based on ion adsorption in porous electrodes, electrically driven adsorption (electroadsorption) is at the core of electrolytic capacitor technologies.
When a highly porous material with a large surface area is immersed in a liquid containing high concentration of ions, applying an external voltage (inducing an electric field between the solution and material) accelerates the adsorption of ions on the electrode surface [5,6]. Models are built to understand electroadsorption processes as well as ion transport through pores [7], thus generally leading to the prediction of ion adsorption and thereby assisting in improving the performance of electroadsorption devices.
In capacitive deionization (CDI) [8,9,10,11], the electroadsorption process (from saline water) takes place in a cell comprising two electrodes separated by a spacer (Figure 1), and the ion adsorption is utilized to remove ions from the water [7]. Modeling of CDI processes has evolved in different directions [9,12,13,14,15], wherein models consider the same underlying physics but with widely varying approaches for its implementation. Limited crossover in formulation and implementation limits their potential usefulness, meaning they could be profitably and integrally developed by incorporating ideas and tools from the other modeling approaches.

Figure 1.
A schematic representation of capacitive deionization (CDI) cell typically comprising two porous electrodes separated by a spacer. When a potential is applied across the electrodes, the ions in the water flowing through such device adsorb on the electrodes.
Herein, we discuss the underlying model-independent physics governing electroadsorption. Primarily, this is used as a starting point for comparing the approaches and tools used in three different CDI models: the modified Donnan (mD) model [7,8,9,16,17,18], the Randles circuit [12], and the dynamic Langmuir (DL) model [13,19,20]. Crucially, we add value by discussing how tools used in the respective models could be implemented to enhance the other models. As a proof of concept, a system identification method from the DL model is implemented to significantly improve simulation performance of the Randles circuit.
2. Materials and Methods
2.1. Theory
This section discusses the fundamental physics involved in electroadsorption processes. Mainly, the principles behind the Randles circuit, mD, and DL modeling approaches are discussed and compared. Finally, we discuss the differences in how these models are implemented, including how fitting parameters are extracted.
2.1.1. The Physics Behind the Models
In CDI devices, highly porous electrodes with high surface area are used for maximum adsorption sites for the ions. Let us consider a piece of porous material thoroughly soaked with saltwater [21,22,23] (Figure 2). Now, activated carbon cloth (ACC) is a popular choice of electrode material [23,24,25]. To clarify the porous structure, note that ACC, for instance, has been shown to comprise ~70% pores [7], which can be classified according to size (diameter ) as micropores ( < 2 nm), mesopores (2 nm < < 50 nm), or macropores ( > 50 nm) [26]. Thus, the ACC cloth can be seen as an amalgamation of fibers with tiny holes (micro- and mesopores) and open spaces between them (macropores).
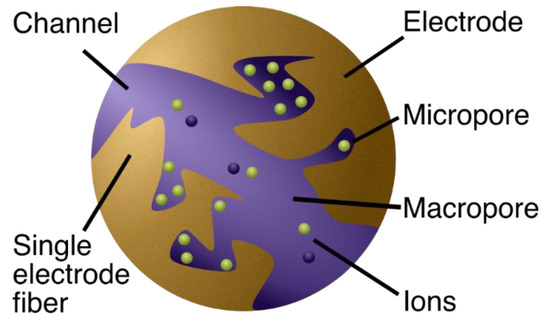
Figure 2.
A schematic illustration showing a zoomed-in view (two carbon fibers from the knitted carbon cloth) of an electrode. The macropores in the electrode are the primary pathways for the water flow. Most of the ion adsorption occurs in the micropores because they carry most of the electrode area.
Water naturally passes through the open space, making the macropores the primary pathways for water flow, while the micropores intuitively hold most of the surface area because of their high area-to-volume ratio, rendering them suitable as the primary location for salt adsorption [7]. Most of the adsorption is thus thought to occur in pores smaller than 2 nm, and around half of the micropore volume constitutes of pores smaller than 1 nm (see Figure S3 in Ref. [27]). Incidentally, the micropores and hydrated sizes of atoms are quite comparable (hydrated diameter of sodium is 0.72 nm) [28], suggesting that few atoms can fit neck-to-neck across the diameter of a single micropore (for a more thorough discussion regarding ion and pore sizes, see [2]).
When a potential difference is abruptly induced between the pore material and the solution, ions will rapidly begin to electroadsorb on the electrodes by migrating to the micropores because of the induced electric field, cations adsorbing on the anodes, and vice versa. The exact performance varies between electrodes; however, such porous electrodes hold around mg salt per gram of electrode [25,29,30,31], while the effective specific surface area is around m2/g [22,32,33]. To put this in perspective, let us consider the fraction of the projected area of adsorbed ions to the total electrode area, denoted by (Equation (1)). In this equation, is the NaCl molar weight, is the Avogadro number, and is the hydrated radius of species . To clarify, is thus the projected area per atom, scaled to mole through , scaled to weight through . This area per weight is compared to the mass adsorption, scaled by the electrode weight per area A. Using hydrated radii from [28], the hypothetical fractional coverage if all adsorbed ions are stacked neck-to-neck is thus computed to 8%.
Ideally, electroadsorption is the only process occurring in the pores. However, Faradaic reactions (carbon oxidation [34]) can lead to a transfer of charge from the electrode through the solution [16]. In addition, it has been generally found that micropores can carry a net charge, which is balanced by ions from the solution even before an external potential is applied, altering the point of zero charge (PZC) [8,35]. Some of these balancing ions present in the micropores (well-known Langmuir adsorption) are expelled upon the application of electric field [7]. This means not all charges induced on the electrodes will have corresponding counterions adsorbed from the solution [33,36,37,38].
2.1.2. From Physics to Model
It has been said that “all models are wrong but some are useful” [39]. Generally, a model is a simplification representing some aspects of reality, and the value of a model is logically judged not purely by how closely it matches reality but rather how well it completes the task for which it was constructed. Thus, models can be constructed from different starting points in terms of modeling tools.
Several models have been constructed considering an electric circuit with a capacitive element [40,41,42] as the electrode together with the ions constitutes an electrolytic (double layer) capacitor. Thus, a Randles circuit [12] accounts for storage through a capacitive element , the time scales of adsorption through a resistive element , and charge leakages through a second resistive element (Figure 3). This basic principle can also be specified further through a transmission line model where more elements are added [43,44,45]. Interestingly, such a model might be able to calculate charge storage over time [12], even though the physics at a pore-scale level is ignored and the impact of changing parameters, such as the ion concentration, cannot be predicted.
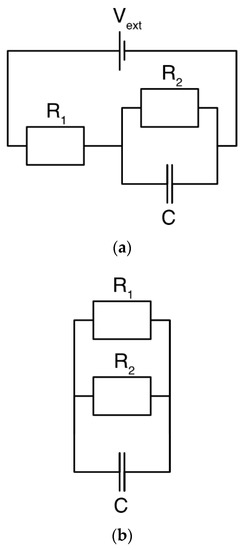
Figure 3.
A simple circuit for describing the dynamics of a charging electrode. (a) During charging, the capacitive element relates to how much charge can be stored, the series resistance governs the rate of the current, and the resistance determines the charge leakage. is the applied voltage. (b) When the voltage is removed, the capacitive element acts as a battery, and the stored charge is released. The charge can be released either through the external circuit () or as leakage current through the resistive element ().
Models starting from a pore-scale level often consider an electric double layer (EDL) being formed near the electrode surface [46]. The EDL theory has developed over time. The early Helmholtz model viewed ions as being able to form a monolayer on the electrode surface [47]. Gouy and Chapman argued that diffusive forces should create a concentration gradient out from the electrode surface [48], and Stern introduced the notion of a thicker layer, the Stern layer, close to the surface followed by a less concentrated region, the diffuse layer [47]. Finally, in the modified Donnan model [7,8,9,16,17,18], it is recognized that the micropores are so small that the diffuse layers overlap, meaning it is enough to only consider a Stern layer [49] (Figure 4).
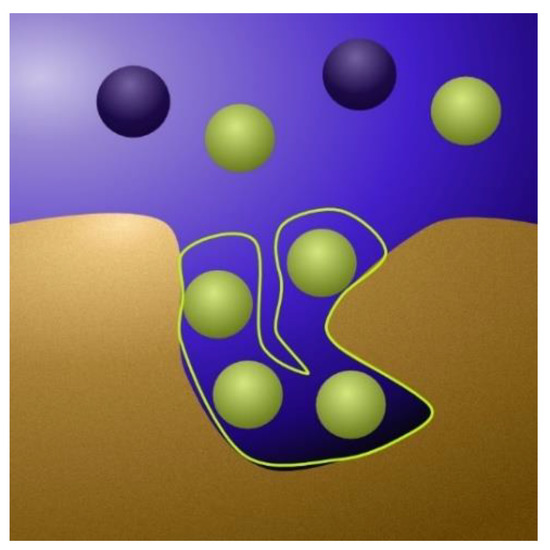
Figure 4.
A detailed view of the interface between a micropore and the surrounding macropore. In the Donnan model, the water flows through the macropores, which are net uncharged. The ion adsorption occurs mainly in the micropores (the cavity in the image) and is enhanced when voltage is applied. Mathematically, the ions are considered to form a small Stern layer against the pore walls (circled green in the image).
Mathematically (see full model including a derivation in [7]), it is thus argued that the difference in potential between the pore matrix, , and the solution in the macropores, , can be split into two components (Equation (2)). The potential drop from the pore matrix to the micropore center, , determines the charge in the micropore based on the Stern layer capacitance. In addition, the potential drop from the micropore center to the macropore, , determines the charge and salt concentration in the micropore relative to the macropore. Finally, the driving force enhancing adsorption is the potential distribution across macropores from different regions in the cell.
In CDI devices, the mD theory can describe the performance under various structural conditions, such as varying electrode sizes [50], spacer thicknesses [50], and electrolyte ion compositions [51,52] as well as operational conditions, such as flow rates [53] and ion concentrations [49]. It has also been further developed to describe not only the outlet ion concentrations but also the concentration variation within the CDI cell in 1D [18] and 2D [7,27] models.
Rather than considering the circuit or EDL approaches, some other models have approached electroadsorption of ions from an adsorption kinetics point of view. The voltage is the driving attractive force and the rate at which ions can reach the electrode surface depending on the ion concentration in the water. Moreover, the more ions already adsorbed in the tiny micropores, the more difficult it is for new ions to reach the surface in such a way that they can get adsorbed (Figure 5).

Figure 5.
The same situation as in Figure 4 but from a Langmuir adsorption perspective. Depending on the material, applied voltage, etc., there are only a fixed number of sites onto which the ions can be adsorbed.
The Langmuir isotherm was created to describe monolayer adsorption of gases on a surface [54], which has traditionally been adopted for liquids as well [55]. Equation (3) shows the rate at which the fractional surface coverage increases with time. Here, is the ion concentration in a liquid, and and are constants. Equation (4) shows the Langmuir isotherm, that is, the equilibrium fractional surface coverage , depending on the constant and the equilibrium concentration . Crucially, the Langmuir isotherm thus models the variation in adsorption based on the ion concentration.
Because of the above similarities between traditional Langmuir adsorption and electroadsorption, it is interesting to note that several studies have successfully implemented the Langmuir isotherm to accurately describe variations in electroadsorption based on the ion concentration in the liquid [56,57,58,59,60,61,62,63,64,65]. Note also that the Langmuir model (monolayer adsorption) was shown to be more accurate than the Freundlich model (multilayer adsorption) [57,58], which might be related to the limited size of the micropores in which the ions adsorb.
The Langmuir isotherm has mainly been implemented to describe the equilibrium state. However, there are fundamental similarities between the Langmuir model and Lagergren pseudo-first-order kinetics [65], as the Lagergren first-order model has been implemented to describe reaction kinetics in terms of how the adsorption varies with time in CDI [57,63,66,67,68]. Equation (5) (or, equivalently, Equation (6)) demonstrates how the adsorbed quantity (unit mg/g) varies with time, depending on the rate constant and the equilibrium adsorption [65]. Notably, this model has been very accurate in modeling electroadsorption, with R2 regression values as high as 0.99 [67].
However, while these formulations have been able to describe some aspects of electroadsorption, including concentration dependence and performance over time, several key elements are not included, such as co-ion expulsion.
2.1.3. Dynamic Langmuir Model
While traditional adsorption and electroadsorption processes are quite similar, there are several key features of an electroadsorption system that are not included in the classic Langmuir model. To address this, note that the fundamental mechanism of electroadsorption consists of the charged species, (unit mM), being adsorbed on the electrode surface due to the application of an electrical potential (Equation (7)) [13]. At equilibrium, the time derivative is zero and in the solution ( is the ion valency, subscript “e” denotes equilibrium quantity), and Equation (7) thus becomes Equation (8) for the specific charge storage . Here, denotes the effective number of surface sites, is the electrode mass per cell volume, and A, B, A’, B’ are constants.
Not all charge storage contributes to net ion adsorption (there is unideal charge efficiency). Thus, it has been shown experimentally that a pure Langmuir isotherm cannot accurately describe variation in adsorption over a wide concentration range [9]. One major effect that contributes to this discrepancy is co-ion expulsion. If some of the capacity for storing charged species is used for pushing away co-ions rather than adsorbing counterions, the net ion adsorption is reduced.
Co-ions can be present close to the electrode to neutralize native charge, that is, charged groups that are situated on the electrode surface. In addition, the ions can be passively present close to the electrode surface due to the ion concentration in the water. With and being constants, the effective number of sites is thus reduced proportionately to the number of ions that are pushed out, including a constant factor () and one that is linear with the concentration (), resulting in Equation (9) for the ion adsorption over time. At equilibrium, this can be simplified to Equation (10) for the specific ion adsorption. Here, is the salt molar weight, and are constants.
In classical Langmuir adsorption, the process is driven by an affinity for the molecules to reach the adsorption sites. Because these sites ultimately determine the total adsorption, they will change depending on experimental conditions and specifically the material used for adsorption of the ions. In the DL model, the sites S are expected to change depending on the electrode material, electrode surface modifications, cell structure, etc. Because the magnitude of the applied voltage determines the total possible charge storage, the sites will ultimately be dependent on the applied voltage, that is, denotes “voltage-induced” sites. Specifically, because the charge on a capacitor increases proportionately with the applied voltage, so too does . In Equations (8) and (10), this corresponds to the constant (and therefore also and ) being proportional to the voltage.
These fundamental equations have been shown to accurately model the charge storage and ion adsorption over varying concentrations and voltages. The full DL model, as derived elsewhere [13,19,20], has also been able to predict performance for multi-ion systems [20] and adsorption over time [19].
2.1.4. Ion Transport
A desalination device based on the porous electrodes discussed in previous sections typically comprises two electrodes separated by a spacer. Depending on the device, the water can flow either between the electrodes or directly through them during operation. Either way, the flow rate is important as it influences how quickly cleaned water is collected at the output and how quickly new ions are brought to the electrodes. Thus, the desalination efficiency of the devices is affected by not only ion adsorption but also the flow rate.
Principally, the change in concentration at a given time and location in the cell depends on the adsorption at that location and the net influx of ions. Therefore, it can be described by the mass transport equation, where is the total ion content (sum of adsorbed and free ions) and is the ion flux (Equation (11)) [7]. In some studies, this has been implemented directly to create partial differential equations (PDEs) describing the system [7].
However, such a formulation can be computationally heavy and difficult to compute. Note, therefore, that the equivalent principle for a region of any size that is “well-stirred” (the ion concentration is uniform) is that the concentration changes at a rate determined by the adsorption rate and the ion influx. The ion influx, in turn, is derived from the flow and the influent concentration (Equations (12) and (13)) [27]. Thus, the mass transport calculation can be simplified by treating the entire cell as one volume (influent concentration , the cell inlet ion concentration) [19]. Alternatively, the cell volume could be divided into some number of subcells along the flow axis, where the influent concentration to subcell is , and the concentration flowing out of the cell is [27].
2.1.5. Implementing a Model
All the presented models contain fitting parameters that can only be deduced by comparing the model formulation to experimental data. Doing so will reveal whether the model can describe the data, that is, if there is any set of parameter values for which the model matches the experimental data. Ideally, the model should then be able to predict the performance under new conditions using the already extracted parameter values.
Traditionally, experimental measurements are done to extract specific parameters. In the circuit model shown in Figure 3, for instance, the current and voltage over time are determined. When the voltage is just applied, the voltage across the capacitor is zero, so the entire external voltage works towards pushing current through , meaning . Here, is the externally applied voltage to the cell (see Figure 3), and is the current through shortly after the voltage is applied. Similarly, no current goes through the capacitor at equilibrium, so from Ohms law, Normally in a capacitor, , where is the total charge built up in the capacitor (integral of current). Because of charge leakages, the voltage here should be the voltage across the capacitor. At equilibrium, that voltage is . However, the charge cannot be deduced without knowing how much of the passed current has leaked instead of passing through the capacitor. Because generally (there is a high resistance against charge leakages), an approximate value of can be recorded by measuring how much charge is released (externally through ) upon removal of the electrical potential.
Another example of fitting can be seen in a full 2D modeling study of a flow-between CDI cell using the mD model [7]. The total passed current and the total salt adsorption at equilibrium were measured. These were used to extract parameters for the capacitance of the device and the baseline (nonelectrostatic) force attracting the ions to the micropores. Then, different values of the fractional volume of micropores, , in the electrode material were manually inserted until the simulation matched the experimental data for the effluent ion concentration over time.
Based on the above examples, at least three issues can arise in the classic view on parameter fitting. Firstly, the extracted values may not be accurate because of the utilization of a limited set of data. Secondly, it may not be possible to make a measurement such that the parameter value can be automatically calculated because there is a complex relationship between parameter values and what can be measured. Thirdly, as will be shown in the result section, extracting values for each parameter separately can result in small errors that accumulate over several cycles of operation.
A more effective way of extracting parameter values would be to implement a system identification method. If a problem can be formulated in a system identification form, programs such as MATLAB can be used because they contain toolboxes to automatically find parameter values for linear and nonlinear systems [68]. These toolboxes match the experimental data over time to the model formulation to automatically extract the best parameter values. This method brings forward several advantages. Firstly, all data, from the beginning when the voltage is applied until saturation is reached, can be used for the fitting, which means that mode reliable values can be found. Secondly, the parameter values are automatically extracted. Thirdly, all parameters are extracted at the same time and multiple cycles can be considered, meaning that the stability of the simulated model can be confirmed over multiple cycles of operation.
In setting up a model using the mentioned toolboxes [68], a set of internal states are defined that can correspond to different properties (such as adsorption and charge storage) and/or different points in space. It is required that the time derivative of each state can be expressed in terms of the current state and the input (e.g., voltage, influent ion concentration) at a given time. In addition, it should be possible to express the measured output data (e.g., the ion concentration of the effluent water) in terms of the current state and the input . Thus, the simulated values of will be related to the experimental data to find the best parameter values.
As an example, the internal states for the circuit model would be the charge passing through , and , while the output state would be the current through . Thus, the parameter values are directly and automatically extracted based on the voltage and current over time. Because these toolboxes require computation at a discrete number of points, they are not directly compatible with some of the more detailed models available, including the 2D PDE-based mD model [7]. However, the toolboxes are not entirely new in describing CDI processes as they have previously been implemented with the DL model [19].
2.2. Data for Model Validation
To validate claims from the theory section and to illustrate some aspects of the modeling and implementation of models, experimental data were extracted from published reports in the literature. The data extraction was carried out using the WebPlotDigitizer software [69].
For the Randles circuit model, the report was chosen based on displaying passed charge over time for an operation where the voltage was applied for a long time so the leakages were accumulated so as to be clearly distinguished in a graph.
3. Results
3.1. DL Model and the Randles Circuit
Here, we illustrate some differences between classical parameter extraction and the system identification used in the DL model in the context of CDI modeling. Because the Randles circuit model contains few components, it is chosen to transparently begin to highlight these differences.
Bouhadana et al. studied the charge leakages in CDI and measured how much charge was supplied to their CDI cell over time [70]. Thus, in Figure 6, the charge can be seen as increasing when the voltage is applied (adsorption phase “Ads.”), decreasing when the voltage is removed (desorption phase “Des.”), and the leakages constitute the differences between the supplied and released charged ions over a full cycle of applying and removing the voltage.
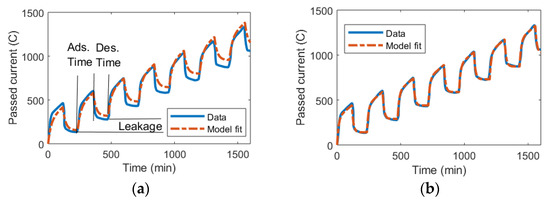
Figure 6.
The total accumulated current passed through a CDI device over multiple cycles of adsorption at 0.9 V and desorption with no applied voltage. Data from [70]. Note that the difference between the adsorbed and released charge constitutes the leakage. (a) The circuit model was implemented using classic parameter extraction as derived in the theory section. (b) The circuit model was implemented using a nlgreyest system identification method in MATLAB for parameter extraction (program code in Supplementary S1).
Using the classic parameter extraction method as described in the theory section, the Randles circuit model can be implemented for this data set. Figure 6a indicates that there is quite a reasonable fit between the model and experiment for the extracted values. However, the errors that are present tend to accumulate over multiple cycles of operation, meaning the data for the experiment and results from the model for charge accumulation diverge over time.
If the same underlying model is implemented with the nlgreyest system identification method in MATLAB, a perfect fit between the model and experiment is obtained. Crucially, this fit endures over many cycles of operation (Figure 6b). The code for automatically implementing this method has been supplied in the Supplementary Materials.
An interesting point will emerge when looking specifically at how random experimental error transfers to the model fit. Figure 7 shows an experiment for the same device as in Figure 6 but with an applied voltage of 1.1 V. Using the classic parameter extraction method, there is a significantly worse model fit even in the first cycle of operation compared to Figure 6a. Crucially, this quickly accumulates between cycles and automatically makes the transferred charge described by the model diverge from what is obtained from experimental data.
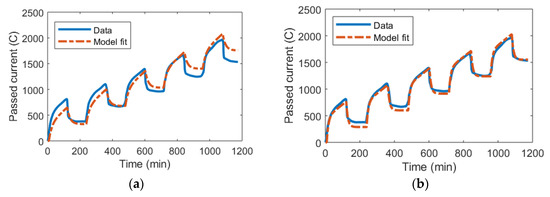
Figure 7.
The total accumulated current passed through a CDI device over multiple cycles of adsorption at 1.1 V and desorption with no applied voltage. Data from [70]. Note that the difference between the adsorbed and released charge constitutes the leakage. (a) The circuit model was implemented using classic parameter extraction as derived in the theory section. (b) The circuit model was implemented using a nlgreyest system identification method in MATLAB for parameter extraction.
When the system identification method is implemented, there is still an error in the first cycle of operation, suggesting that the model formulation could not accurately describe what happened at the beginning of the operation. However, the cumulative charge as described by the model converges with the experimental data over time, making the errors smaller. This suggests that the system identification method can reach accurate results even if there are random variations at the beginning of the experiment data.
This finding is especially relevant to CDI because it typically takes time before a CDI device reaches a stable operation, meaning the quality of the experimental data can be poor for the first few adsorption/desorption cycles before stability is reached. With a system identification method, the sensitivity to such variations is low.
The crucial point to note is that the difference between the fitting methods shown above means the fitting method fundamentally impacts the model performance even when the underlying model structure is the same. Ultimately, this means it could be valuable to investigate how to make models compatible with advanced parameter extraction methods.
3.2. mD Model and the Randles Circuit
Now, there are also prospects from incorporating elements from the mD model into the Randles circuit. As an example of this, it can be noted that the charge accumulation on electrolytic capacitors is not directly proportional to the applied voltage. This means that the capacitance varies depending on the charge state of the device. Empirically, it has been noted that the capacitance increases with the square of the charge as in Equation (14), where is the baseline capacitance and is a correction factor [7].
Bouhadana et al. studied, in the same report, the charge state of the device for four voltages. Using the classical parameter extraction method as described in the theory section, the experimental capacitances can be deduced as shown in Figure 8a. From this graph, the trend that the capacitance increases with the voltage can be clearly seen. Thus, the model without the correction factor provides an averaged approximation of the capacitance, while the model with the added correction factor (Equation (14)) yields an excellent fit over the entire voltage range. Thus, the Randles model with the correction factor is also able to more accurately describe the charge in the device as a function of the applied voltage (Figure 8b).
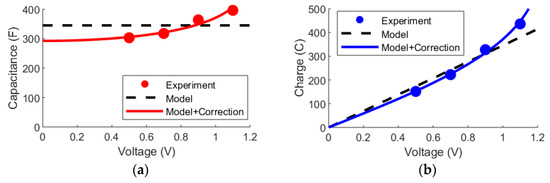
Figure 8.
The data is from [70]. Dots show experimental data, while the dashed line corresponds to the basic Randles circuit and the full line corresponds to adding the charge-dependent correction factor in Equation (14) to express the capacitance. (a) The capacitance of the cell depending on the applied voltage. (b) The accumulated charge in the cell depending on the applied voltage.
A key point to note here is that the empirical charge-dependent capacitance increase is naturally applicable to more model formulations than the mD model, which means that models could generally obtain performance gains by directly incorporating elements from other models.
3.3. DL and mD Models
Starting from the underlying physics, the DL and mD models have taken different approaches for implementing a model that can describe and predict electroadsorption in porous electrodes. Still, both can predict charge storage and ion adsorption under varying operating conditions. Below, some of the fundamental differences between the models are discussed.
Presently, the widely used mD model includes some elements that are not present in the newly proposed DL model, mainly related to the detailed spatial resolution of what happens inside the cell during the desalination processes. For instance, 2D [7] and 1D [18] coupled adsorption and transport models for flow-between and flow-through cell structures have been formulated. Such models could indicate, for instance, if there is a concentration shock during the desalination process, that is, the salt is rapidly removed locally in the cell [7]. This would mean that the cell works below maximum capacity because there is no more salt to adsorb until new ions can move by diffusion or advection to those adsorption sites. In addition, the mD model is more accurate than the DL model for describing highly nonlinear behavior. When looking at the impact of increasing the voltage, for instance, the mD model provides a more detailed description of the performance at very low voltages [13,49].
In addition, presently, the DL model shows advantages in being straightforward to apply. Because it is not constructed around solving coupled partial differential equations, different properties can be modeled independently of each other. For instance, internal states, such as ion adsorption and charge storage, as well as operational conditions, such as voltage and ion concentration, can be modeled with separate “isotherms” [13]. While the isotherms describe only equilibrium properties, the ability to independently model different properties holds also for the performance variation with time during the adsorption process [19]. Crucially, this structure makes the model well adapted for both system identification and control.
3.4. Outlook
Based on the present state of the models for electroadsorption, there is potential for further developing them in different directions based on considering both the model structure and parameter-fitting aspects.
To date, no system identification method has been implemented with the mD model. Successfully implementing this would naturally begin to increase the accuracy of the model further while automating the parameter extraction process because system identification methods remove the need to manually calculate or test different parameter values. However, for this to be possible for 2D systems, a system identification method would be required that can solve algebraic PDEs. Optionally, the mD model would have to be manually discretized and relaxed in such a way that the existing toolboxes can complete the parameter extraction.
Presently, the basic DL model does not account for electrode redox reactions inside the cell, which limits its accurately workable voltages to the non-Faradaic regime. In contrast, Figure 6 has shown that calculating leakages is at the core of the Randles circuit. This means successfully incorporating circuit-based leakages into the DL model could allow it to incorporate redox reaction as well.
In addition, to date, the DL model has not been implemented with spatial resolution inside the cell. Doing so might further increase the accuracy of the model, including the impact of varying the flow rate. In principle, computationally dividing a flow-between cell along the flow axis (Equation (13) for multiple compartments) could be directly implemented with the existing modeling framework. Further research might also make the model more detailed, for instance, to predict the difference in performances when using different electrode structures or membranes. Still, full 2D modeling would be more difficult to implement with the premade MATLAB system identification toolbox in this case because the transport equations for diffusion and advection would have to be discretized to make it possible to compute a solution.
4. Conclusions
This work has focused on expanding the understanding of how different modeling approaches can be used to predict the performance of ion electroadsorption processes in porous electrodes for capacitive deionization systems. Because there are widely different approaches to electroadsorption modeling, the underlying physics was first discussed to illuminate the common starting point for all models, namely, that ions get adsorbed into tiny micropores because of a driving voltage.
Then, three different modeling approaches were compared in terms of fundamental modeling principles. The Randles circuit implements a circuit representation, the mD model considers how charged species are distributed in electric double layers, and the DL model considers adsorption–desorption kinetics onto voltage-induced sites on the electrode.
Lastly, the quality of the parameter extraction process is found to have a strong impact on modeling accuracy even if the underlying model is the same. Thus, it is concluded that the widely used mD model might be significantly enhanced if a system identification method could be implemented for parameter extraction. However, for 1D or 2D coupled transport and adsorption inside a capacitive deionization device, this would require a system identification method that could handle spatially continuous algebraic PDEs, which is not possible using standard MATLAB toolboxes, or it would require that the model is manually discretized. The DL model is inherently well suited for implementation with system identification methods, and future research could focus on making the structure of the DL model more detailed to enable it to be integrated into 1D and 2D computations.
Supplementary Materials
The following are available online at https://www.mdpi.com/2624-8174/2/2/16/s1, MATLAB Code S1: System identification model implementing the Randles circuit, including the data used in this work.
Author Contributions
Conceptualization, J.N. and J.D.; methodology, J.N.; software, J.N.; validation, J.N.; formal analysis, J.N.; investigation, J.N.; resources, J.D.; data curation, J.N.; writing—original draft preparation, J.N.; writing—review and editing, J.D.; visualization, J.N.; supervision, J.D.; project administration, J.D.; funding acquisition, J.D. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to thank the Swedish Research Council (Diary No. 2018-05387) for funding the work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- The Sustainable Development Goals Report; UN: New York. NY, USA, 2018.
- González, A.; Goikolea, E.; Barrena, J.A.; Mysyk, R. Review on Supercapacitors: Technologies and Materials. Renew. Sustain. Energy Rev. 2016, 58, 1189–1206. [Google Scholar] [CrossRef]
- Zhong, C.; Deng, Y.; Hu, W.; Qiao, J.; Zhang, L.; Zhang, J. A Review of Electrolyte Materials and Compositions for Electrochemical Supercapacitors. Chem. Soc. Rev. 2015, 44, 7484–7539. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, L.; Zhang, J. A Review of Electrode Materials for Electrochemical Supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef]
- Gan, L.; Wu, Y.; Song, H.; Zhang, S.; Lu, C.; Yang, S.; Wang, Z.; Jiang, B.; Wang, C.; Li, A. Selective Removal of Nitrate Ion Using a Novel Activated Carbon Composite Carbon Electrode in Capacitive Deionization. Sep. Purif. Technol. 2019, 212, 728–736. [Google Scholar] [CrossRef]
- Qu, Y.; Campbell, P.G.; Hemmatifar, A.; Knipe, J.M.; Loeb, C.K.; Reidy, J.J.; Hubert, M.A.; Stadermann, M.; Santiago, J.G. Charging and Transport Dynamics of a Flow-Through Electrode Capacitive Deionization System. J. Phys. Chem. B 2018, 122, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Hemmatifar, A.; Stadermann, M.; Santiago, J.G. Two-Dimensional Porous Electrode Model for Capacitive Deionization. J. Phys. Chem. C 2015, 119, 24681–24694. [Google Scholar] [CrossRef]
- Porada, S.; Zhao, R.; Van Der Wal, A.; Presser, V.; Biesheuvel, P.M. Review on the Science and Technology of Water Desalination by Capacitive Deionization. Prog. Mater. Sci. 2013, 58, 1388–1442. [Google Scholar] [CrossRef]
- Suss, M.E.; Porada, S.; Sun, X.; Biesheuvel, P.M.; Yoon, J.; Presser, V. Water Desalination via Capacitive Deionization: What Is It and What Can We Expect from It? Energy Environ. Sci. 2015, 8, 2296–2319. [Google Scholar] [CrossRef]
- Ahualli, S.; Iglesias, G.R. Principles and Theoretical Models of CDI: Experimental Approaches. Interface Sci. Technol. 2018, 24, 169–192. [Google Scholar] [CrossRef]
- Anderson, M.A.; Cudero, A.L.; Palma, J. Capacitive Deionization as an Electrochemical Means of Saving Energy and Delivering Clean Water. Comparison to Present Desalination Practices: Will It Compete? Electrochim. Acta 2010, 55, 3845–3856. [Google Scholar] [CrossRef]
- Qin, M.; Deshmukh, A.; Epsztein, R.; Patel, S.K. Comparison of Energy Consumption in Desalination by Capacitive Deionization and Reverse Osmosis. Desalination 2019, 455, 100–114. [Google Scholar] [CrossRef]
- Nordstrand, J.; Dutta, J. Dynamic Langmuir Model: A Simpler Approach to Modeling Capacitive Deionization. J. Phys. Chem. C 2019, 123, 16479–16485. [Google Scholar] [CrossRef]
- Dykstra, J.E.; Zhao, R.; Biesheuvel, P.M.; Wal, A.V.D. Resistance Identification and Rational Process Design in Capacitive Deionization. Water Res. 2016, 88, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Jande, Y.A.C.; Kim, W.S. Predicting the Lowest Effluent Concentration in Capacitive Deionization. Sep. Purif. Technol. 2013, 224–230. [Google Scholar] [CrossRef]
- Biesheuvel, P.M.; Fu, Y.; Bazant, M.Z. Diffuse Charge and Faradaic Reactions in Porous Electrodes. Phys. Rev. E 2011, 83. [Google Scholar] [CrossRef]
- Suss, M.E.; Biesheuvel, P.M.; Baumann, T.F.; Stadermann, M.; Santiago, J.G. In Situ Spatially and Temporally Resolved Measurements of Salt Concentration between Charging Porous Electrodes for Desalination by Capacitive Deionization. Environ. Sci. Technol. 2014, 48, 2008–2015. [Google Scholar] [CrossRef]
- Guyes, E.N.; Shocron, A.N.; Simanovski, A.; Biesheuvel, P.M.; Suss, M.E. A One-Dimensional Model for Water Desalination by Flow-through Electrode Capacitive Deionization. Desalination 2017, 415, 8–13. [Google Scholar] [CrossRef]
- Nordstrand, J.; Laxman, K.; Myint, M.T.Z.; Dutta, J. An Easy-to-Use Tool for Modeling the Dynamics of Capacitive Deionization. J. Phys. Chem. A 2019, 123, 6628–6634. [Google Scholar] [CrossRef]
- Nordstrand, J.; Dutta, J. Simplified Prediction of Ion Removal in Capacitive Deionization of Multi-Ion Solutions. Langmuir 2020. [Google Scholar] [CrossRef]
- Laxman, K.; Husain, A.; Nasser, A.; Al, M.; Dutta, J. Tailoring the Pressure Drop and Fluid Distribution of a Capacitive Deionization Device. Desalination 2019, 449, 111–117. [Google Scholar] [CrossRef]
- Laxman, K.; Myint, M.T.Z.; Khan, R.; Pervez, T.; Dutta, J. Improved Desalination by Zinc Oxide Nanorod Induced Electric Field Enhancement in Capacitive Deionization of Brackish Water. Desalination 2015, 359, 64–70. [Google Scholar] [CrossRef]
- Myint, M.T.Z.; Al-Harthi, S.H.; Dutta, J. Brackish Water Desalination by Capacitive Deionization Using Zinc Oxide Micro/Nanostructures Grafted on Activated Carbon Cloth Electrodes. Desalination 2014, 344, 236–242. [Google Scholar] [CrossRef]
- Maddah, H.A.; Shihon, M.A. Activated Carbon Cloth for Desalination of Brackish Water Using Capacitive Deionization. In Desalination and Water Treatment; BoD—Books on Demand: Norderstedt, Germany, 2018. [Google Scholar] [CrossRef]
- Myint, M.T.Z.; Dutta, J. Fabrication of Zinc Oxide Nanorods Modified Activated Carbon Cloth Electrode for Desalination of Brackish Water Using Capacitive Deionization Approach. Desalination 2012, 305, 24–30. [Google Scholar] [CrossRef]
- Sefara, M. Pore Classification in the Characterization of Porous Materials: A Perspective. Cent. Eur. J. Chem. 2007, 5, 385–395. [Google Scholar] [CrossRef]
- Porada, S.; Borchardt, L.; Oschatz, M.; Bryjak, M.; Atchison, J.S.; Keesman, K.J.; Kaskel, S.; Biesheuvel, P.M.; Presser, V. Direct Prediction of the Desalination Performance of Porous Carbon Electrodes for Capacitive Deionization. Energy Environ. Sci. 2013, 6, 3700–3712. [Google Scholar] [CrossRef]
- Railsback, B. Variation in Hydrated Radius of Ions. 2019. Available online: http://www.gly.uga.edu/railsback/Fundamentals/815HydratedCationRadii04LS.pdf (accessed on 11 March 2019).
- Laxman, K.; Al Gharibi, L.; Dutta, J. Capacitive Deionization with Asymmetric Electrodes: Electrode Capacitance vs. Electrode Surface Area. Electrochim. Acta 2015, 176, 420–425. [Google Scholar] [CrossRef]
- Laxman, K.; Tay, M.; Myint, Z.; Bourdoucen, H.; Dutta, J. Enhancement in Ion Adsorption Rate and Desalination Efficiency in a Capacitive Deionization Cell through Improved Electric Field Distribution Using Electrodes Composed of Activated Carbon Cloth Coated with Zinc Oxide Nanorods. Appl. Mater. Interfaces 2014, 6, 10113–10120. [Google Scholar] [CrossRef]
- Wang, G.; Ling, Y.; Qian, F.; Yang, X.; Liu, X.X.; Li, Y. Enhanced Capacitance in Partially Exfoliated Multi-Walled Carbon Nanotubes. J. Power Sources 2011, 196, 5209–5214. [Google Scholar] [CrossRef]
- Keneko, K.; Ishii, C.; Ruike, M.; Kuwabara, H. Origin of Superhigh Surface Area and Microcrystalline Structures of Activated Carbons. Carbon 1992, 30, 1075–1088. [Google Scholar] [CrossRef]
- Zhao, R.; Biesheuvel, P.M.; Miedema, H.; Bruning, H.; Wal, A.V.D. Charge Efficiency: A Functional Tool to Probe the Double-Layer Structure Inside of Porous Electrodes and Application in the Modeling of Capacitive Deionization. J. Phys. Chem. Lett. 2010, 205–210. [Google Scholar] [CrossRef]
- Gallagher, K.G.; Fuller, T.F. Kinetic Model of the Electrochemical Oxidation of Graphitic Carbon in Acidic Environments. Phys. Chem. Chem. Phys. 2009, 11, 11557–11567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liang, P.; Bian, Y.; Sun, X.; Ma, J.; Jiang, Y.; Huang, X. Enhancement of Salt Removal in Capacitive Deionization Cell through PeriodicallySeparation and Purification Techn Alternated Oxidation of Electrodes. Sep. Purif. Technol. 2018, 194, 451–456. [Google Scholar] [CrossRef]
- Gao, X.; Omosebi, A.; Landon, J.; Liu, K. Enhancement of Charge Efficiency for a Capacitive Deionization Cell Using Carbon Xerogel with Modified Potential of Zero Charge. Electrochem. Commun. 2014, 39, 22–25. [Google Scholar] [CrossRef]
- Li, H.; Zaviska, F.; Liang, S.; Li, J.; He, L.; Yang, H.Y. A High Charge Efficiency Electrode by Self—Assembling Sulphonated Reduced Graphene Oxide onto Carbon Fibre: Towards Enhanced Capacitive Deionization. J. Mater. Chem. A 2014, 3484–3491. [Google Scholar] [CrossRef]
- Kim, T.; Dykstra, J.E.; Porada, S.; van der Wal, A.; Yoon, J.; Biesheuvel, P.M. Enhanced Charge Efficiency and Reduced Energy Use in Capacitive Deionization by Increasing the Discharge Voltage. J. Colloid Interface Sci. 2015, 446, 317–326. [Google Scholar] [CrossRef]
- Box, G.E.P. Robustness in the Strategy of Scientific Model Building. In Robustness in Statistics; Academic press Inc.: Waltham, MA, USA, 1979. [Google Scholar] [CrossRef]
- Fiorenti, S.; Guanetti, J.; Guezennec, Y.; Onori, S. Modeling and Experimental Validation of a Hybridized Energy Storage System for Automotive Applications. J. Power Sources 2013, 241, 112–120. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Hu, X.; Sun, F.; Dorrell, D.G. A Comparative Study of Equivalent Circuit Models of Ultracapacitors for Electric Vehicles. J. Power Sources 2015, 274, 899–906. [Google Scholar] [CrossRef]
- Masarapu, C.; Zeng, H.F.; Hung, K.H.; Wei, B. Effect of Temperature on the Capacitance of Carbon Nanotube Supercapacitors. ACS Nano 2009, 3, 2199–2206. [Google Scholar] [CrossRef]
- Dongowski, G.; Neubert, R.H.H.; Heinevetter, L. Permeation of Bile Acids across Artificial Lipid Membranes and Caco-2 Monolayers. Pharmazie 1999, 54, 517–520. [Google Scholar] [CrossRef]
- Brug, G.J.; van den Eeden, A.L.G.; Sluyters-Rehbach, M.; Sluyters, J.H. The Analysis of Electrode Impedances Complicated by the Presence of a Constant Phase Element. J. Electroanal. Chem. 1984, 176, 275–295. [Google Scholar] [CrossRef]
- Suss, M.E.; Baumann, T.F.; Worsley, M.A.; Rose, K.A.; Jaramillo, T.F.; Stadermann, M.; Santiago, J.G. Impedance-Based Study of Capacitive Porous Carbon Electrodes with Hierarchical and Bimodal Porosity. J. Power Sources 2013, 241, 266–273. [Google Scholar] [CrossRef]
- Brogioli, D.; La Mantia, F. Capacitive Energy Extraction from Double Layer Expansion (CDLE). In Fundamentals of the Method, 1st ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; Volume 24. [Google Scholar] [CrossRef]
- Gongadze, E.; Petersen, S.; Beck, U.; Van Rienen, U. Classical Models of the Interface between an Electrode and an Electrolyte. In Proceedings of the COMSOL Conference, Boston, MA, USA, 8–10 October 2009. [Google Scholar]
- Delgado, Á.V.; Ahualli, S.; Fernándex, M.M.; Iglesias, G.R.; Jiménez, M.L. Charge and Potential Distribution in the Electrical Double Layer of Porous Materials: Models. Interface Sci. Technol. 2018, 24, 3–18. [Google Scholar] [CrossRef]
- Biesheuvel, P.M.; Porada, S.; Levi, M.; Bazant, M.Z. Attractive Forces in Microporous Carbon Electrodes for Capacitive Deionization. J. Solid State Electrochem. 2014, 18, 1365–1376. [Google Scholar] [CrossRef]
- Porada, S.; Bryjak, M.; Van Der Wal, A.; Biesheuvel, P.M. Effect of Electrode Thickness Variation on Operation of Capacitive Deionization. Electrochim. Acta 2012, 75, 148–156. [Google Scholar] [CrossRef]
- Biesheuvel, P.M.; Fu, Y.; Bazant, M.Z. Electrochemistry and Capacitive Charging of Porous Electrodes in Asymmetric Multicomponent Electrolytes. Russ. J. Electrochem. 2012, 48, 580–592. [Google Scholar] [CrossRef]
- Biesheuvel, P.M. Activated Carbon Is an Electron-Conducting Amphoteric Ion Adsorbent. arXiv 2015, arXiv:1509.06354. [Google Scholar]
- Zhao, R.; Satpradit, O.; Rijnaarts, H.H.M.; Biesheuvel, P.M.; van der Wal, A. Optimization of Salt Adsorption Rate in Membrane Capacitive Deionization. Water Res. 2013, 47, 1941–1952. [Google Scholar] [CrossRef]
- Langmuir, I. The Adsorption of Gases on Plane Surfaces of Glass, Mica and Platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Azizian, S.; Eris, S.; Wilson, L.D. Re-Evaluation of the Century-Old Langmuir Isotherm for Modeling Adsorption Phenomena in Solution. Chem. Phys. 2018, 513, 99–104. [Google Scholar] [CrossRef]
- Li, H.; Pan, L.; Lu, T.; Zhan, Y.; Nie, C.; Sun, Z. A Comparative Study on Electrosorptive Behavior of Carbon Nanotubes and Graphene for Capacitive Deionization. J. Electroanal. Chem. 2011, 653, 40–44. [Google Scholar] [CrossRef]
- Ryoo, M.W.; Kim, J.H.; Seo, G. Role of Titania Incorporated on Activated Carbon Cloth for Capacitive Deionization of NaCl Solution. J. Colloid Interface Sci. 2003, 264, 414–419. [Google Scholar] [CrossRef]
- Gabelich, C.J.; Tran, T.D.; Suffet, I.H. “Mel.” Electrosorption of Inorganic Salts from Aqueous Solution Using Carbon Aerogels. Environ. Sci. Technol. 2002, 36, 3010–3019. [Google Scholar] [CrossRef]
- Wang, G.; Qian, B.; Dong, Q.; Yang, J.; Zhao, Z.; Qiu, J. Highly Mesoporous Activated Carbon Electrode for Capacitive Deionization. Sep. Purif. Technol. 2013, 103, 216–221. [Google Scholar] [CrossRef]
- Li, H.; Lu, T.; Pan, L.; Zhang, Y.; Sun, Z. Electrosorption Behavior of Graphene in NaCl Solutions. J. Mater. Chem. 2009, 19, 6773–6779. [Google Scholar] [CrossRef]
- Rudzinski, W.; Lee, S.L.; Panczyk, T.; Yan, C.C.S. A Fractal Approach to Adsorption on Heterogeneous Solids Surfaces. 2. Thermodynamic Analysis of Experimental Adsorption Data. J. Phys. Chem. B 2001, 105, 10857–10866. [Google Scholar] [CrossRef]
- Kowanga, K.D.; Gatebe, E.; Mauti, G.O.; Mauti, E.M. Kinetic, Sorption Isotherms, Pseudo-First-Order Model and Pseudo-Second-Order Model Studies of Cu (II) and Pb (II) Using Defatted Moringa Oleifera Seed Powder. J. Phytopharm. 2016, 5, 71–78. [Google Scholar]
- Wang, L.; Wang, M.; Huang, Z.-H.; Cui, T.; Gui, X.; Kang, F.; Wang, K.; Wu, D. Capacitive Deionization of NaCl Solutions Using Carbon Nanotube Sponge Electrodes. J. Mater. Chem. 2011, 21, 18295. [Google Scholar] [CrossRef]
- Li, H.; Pan, L.; Zhang, Y.; Zou, L.; Sun, C.; Zhan, Y.; Sun, Z. Kinetics and Thermodynamics Study for Electrosorption of NaCl onto Carbon Nanotubes and Carbon Nanofibers Electrodes. Chem. Phys. Lett. 2010, 485, 161–166. [Google Scholar] [CrossRef]
- Rudzinski, W.; Plazinski, W. Kinetics of Solute Adsorption at Solid/Solution Interfaces: A Theoretical Development of the Empirical Pseudo-First and Pseudo-Second Order Kinetic Rate Equations, Based on Applying the Statistical Rate Theory of Interfacial Transport. J. Phys. Chem. B 2006, 16514–16525. [Google Scholar] [CrossRef]
- Li, H.; Zou, L.; Pan, L.; Sun, Z. Using Graphene Nano-Flakes as Electrodes to Remove Ferric Ions by Capacitive Deionization. Sep. Purif. Technol. 2010, 75, 8–14. [Google Scholar] [CrossRef]
- Li, H.; Zou, L. Ion-Exchange Membrane Capacitive Deionization: A New Strategy for Brackish Water Desalination. Desalination 2011, 275, 62–66. [Google Scholar] [CrossRef]
- Ljung, L. ODE Parameter Estimation (Grey-Box Modeling). In System Identification Toolbox; TM User’s Guide; MathWorks: Natick, MA, USA, 2014. [Google Scholar]
- Rohatgi, A. WebPlotDigitizer. 2019. Available online: https://automeris.io/WebPlotDigitizer (accessed on 11 March 2019).
- Bouhadana, Y.; Ben-Tzion, M.; Soffer, A.; Aurbach, D. A Control System for Operating and Investigating Reactors: The Demonstration of Parasitic Reactions in the Water Desalination by Capacitive de-Ionization. Desalination 2011, 268, 253–261. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).